

Summary
Transparency in the eye lens is maintained via specific, functional interactions among the structural βγ- and chaperone α-crystallins. Here we report the structure and α-crystallin binding interface of the G18V variant of human γS-crystallin (γS-G18V), which is linked to hereditary childhood-onset cortical cataract. Comparison of the solution NMR structures of wild-type and G18V γS-crystallin, both presented here, reveal that the increased aggregation propensity of γS-G18V results from neither global misfolding nor the solvent exposure of a hydrophobic residue, but instead involves backbone rearrangement within the N-terminal domain. αB-crystallin binds more strongly to the variant, via a well-defined interaction surface that represents the first such interface directly observed between a variant structural crystallin and α-crystallin. In the context of the αB-crystallin structure and the finding that it forms heterogeneous multimers, our structural studies suggest a potential mechanism for cataract formation via the depletion of the finite αB-crystallin population of the lens.
Introduction
The crystallins are the primary protein components of the eye lens, reaching concentrations higher than 400 mg/mL in humans (Tardieu et al., 1992). Short-range ordered interactions between crystallins at high concentrations are thought to maintain transparency while providing the refractive index gradient required to focus light on the retina (Delaye and Tardieu, 1983; Ponce et al., 2006). Perturbations to the inter-crystallin interactions concomitant with the formation of high molecular weight aggregates can lead to lens opacification during aging and cataractogenesis. Characterizing the interactions between members of the two crystallin superfamilies, α-, and βγ-, is critical to understanding cataract formation, because insoluble aggregates of crystallins from both have been found in cataractous lenses (Takemoto and Sorensen, 2008). The α-crystallins (αA and αB) act as holdase chaperones, binding to but not refolding structural βγ-crystallins for which solubility is compromised due to damage or mutation. Because of the extremely low protein turnover in lens fiber cells, the inter-crystallin interactions are thus the first line of defense against aggregation. Mutations in either the α-, or βγ crystallin genes can alter these critical attractive interactions (Fu and Liang, 2003). In the case of γC-crystallin, α-crystallins do not recognize all known disease-related variants (Moreau and King, 2012). Likewise, in γD-crystallin, both the E107A and R76S variants are implicated in early-onset cataract, but the former exhibits increased α-crystallin attraction (Banerjee et al., 2011), while α-crystallin interactions with the latter remain unchanged (Ji et al., 2012). Here we focus on interactions between αB- and γS-crystallins; αB is the more versatile chaperone, abundantly expressed in tissues outside the eye lens (Iwaki et al., 1990), up-regulated by various stressors (Klemenz et al., 1991), and implicated in several neuropathological diseases (Iwaki et al., 1989; van Noort et al., 1995; Ousman et al., 2007), whereas γS is the most abundant of the structural βγ-crystallins in the human lens cortex and is highly conserved across several species (Chang and Chang, 1987; Quax-Jeuken et al., 1985; van Rens et al., 1991; van Rens et al., 1989).
At present, there are four known cataractogenic mutations in human γS-crystallin: the γS-V42M variant, which distorts the compact β-sheet packing in the core of the N-terminal domain and causes severe congenital cataract in children (Vendra et al., 2013); the Coppock cataract-associated γS-D26G variant, which leads to decreased protein stability but apparently has little effect on the overall molecular architecture (Karri et al., 2013); the γS-S39C variant, which is linked to microcornea and cataract (Devi et al., 2008) and which is hypothesized to have an exposed cysteine available for disulfide crosslinkage and aggregation; and the γS-G18V variant, which is implicated in childhood-onset cortical cataract (Sun et al., 2005). Although its decreased thermodynamic stability relative to wild-type (γS-WT) (Ma et al., 2009) has been established, additional experiments indicated that γS-G18V is aggregation-prone well below its unfolding temperature, suggesting an aggregation mechanism more complex than simple denaturation (Brubaker et al., 2011). In order to better understand how structural changes in the cataract-related G18V variant of γS-crystallin lead to altered intermolecular interactions, we have solved the solution NMR structures of human wild-type and γS-G18V and elucidated the binding interface between αB-crystallin and γS-G18V.
Results
The G18V Mutation Causes Structural Perturbation
Like the highly homologous murine protein (Wu et al., 2005) and other mammalian structural crystallins, γS-WT has a double Greek key fold. A comparison of the γS-WT and γS-G18V structures reveals local shifts in the backbone but little change in the overall fold. The average heavy-atom RMSD between the two structures is 1.62 Å for the N-terminal domain and 1.13 Å for the C-terminal domain. (Figure 1, Table 1, Supplementary Figures S1, S2). Because G18 is located on a surface-exposed loop, solvent exposure of the valine sidechain might be expected to provide a plausible mechanism for the solubility impact of this mutation; however the structural data indicate that it is buried, with the backbone occupying an unusual conformation at this position. Many examples of residues stabilized by hydrogen bonds in unfavorable Ramachandran angles (Jia et al., 1993; Gunasekaran et al., 1996) have been found in the context of enzyme active sites in which the conformation is required for activity (Jia et al., 1993; Pal and Chakrabarti, 2002). Here, the configuration of V18 is stabilized in part by the pi-stacking interactions between R20 and F16 (Figure 1c). The dihedral angles shift from φ = 79.3°, ψ = −146.4° for G18 to ϕ = 103.0°, ψ = −134.0° for V18. Presumably because of steric clashes with sidechains on the opposing side of the affected loop, the methyls of V18 are angled toward the C-terminal end of the polypeptide chain, locking the R19 amide proton into place centered between the V18 methyls (Figure 1b). Additional structural calculations indicate that the V18 side chain remains buried even upon exclusion of all distance restraints to V18, and the adoption of a favorable backbone dihedral angle configuration would require the elimination of restraints from several surrounding residues, producing an extensive structural disruption to the surrounding loop region that is not supported by the NMR data (Supplementary Figure S3). The effect propagates down the polypeptide chain through more than half of the N-terminal domain of γS-crystallin. Despite these local differences, the overall folds of both structures are very similar (Figure 1a), consistent with previous CD and UV-fluorescence data. Both γS-WT and γS-G18V were monomeric under the conditions used for structural NMR (Supplementary Figure S4).
Figure 1.

Structural detail of wild-type and variant γ-S crystallin. (a) A licorice depiction of the average solution NMR structures of γS-WT (green) and γS-G18V (blue). Both proteins are highly structured, with the double Greek key fold typical of structural crystallins, although the variant protein displays structural changes in the N-terminal domain (left) relative to wild-type. The red circle indicates the region that is shown in greater detail in Panel (b). Here the affected loop remains essentially intact; in γS-WT (left) the α proton from G18 is angled slightly askew from the R19 amide proton. In γS-G18V the orientation of the V18 methyls forces the amide proton into alignment with the valine sidechain, altering the V18 ψ angle. Panel (c) shows the overlaid structures of γS-WT (green) and γS-G18V (blue) with the sidechains from F16, V18, and R20 shown, indicating the details of selected structural changes, particularly the dramatic shift in the position of R20. (d) The addition of V18 and its effect on the backbone angles the former β-strand outward and twists it, moving R20 inward, where it displaces Y11 and forces the tyrosine away from F16, placing it flat against the surface of the first Greek key motif. Although each of these structural changes is minor and has primarily local impact, taken in aggregate they result in significant perturbations to the N-terminal domain, potentially providing sites for altered intermolecular interactions and recognition by αB-crystallin. See also Figures S1–S4 and Supplementary Tables 1 and 2.
Table 1.
A tabular summary of the NMR structures for γS-WT and γS-G18V structures, calculated using full restraint sets as described in the Methods section. See also Figures S1–S4 and Supplementary Tables 1 and 2.
| Restraint Summary | γS-WT | γS-G18V |
|---|---|---|
| Total NOE restraints: | 7444 | 4682 |
| intraresidue: | 1547 | 1329 |
| interresidue: | ||
| sequential (|i − j| = 1): | 1559 | 972 |
| medium-range (|i − j| ≤ 5): | 1286 | 709 |
| long-range (|i − j| ≥ 6): | 3052 | 1672 |
| Total angular restraints: | 408 | 450 |
| RDCs: | 156 | 147 |
| dihedral (3J coupling): | -- | 57 |
| dihedral (TALOS+): | 252 | 246 |
| H-bond restraints: | 46 | 90 |
| Structure statistics (20 lowest-energy structures) | ||
| Restraint violations | ||
| NOE > 0.3 Å | 0.6 ± 0.8 | 0.7 ± 1.1 |
| dihedral > 5° | 1.1 ± 1.3 | 1.6± 0.9 |
| RMSD from ideal covalent geometry | ||
| bonds (Å) | 0.005 ± 0.000 | 0.004 ± 0.000 |
| angles (°) | 0.582 ± 0.011 | 0.537 ± 0.016 |
| impropers (°) | 0.503 ± 0.013 | 0.481 ± 0.016 |
| Restraint RMSD | ||
| NOE (Å) | 0.034 ± 0.025 | 0.024 ± 0.002 |
| dihedral (°) | 0.914 ± 0.137 | 0.893 ± 0.114 |
| RDC (Hz) | 0.238 ± 0.021 | 0.188 ± 0.018 |
| Average pairwise RMSD (residues 5–178) | ||
| backbone RMSD (Å) | 0.4 | 0.5 |
| heavy atom RMSD (Å) | 0.8 | 0.9 |
| RDC Statistics | ||
| R-factor (%) | 0.642 ± 0.055 | 0.459 ± 0.046 |
| R-factor (free) (%) | 0.817 ± 0.073 | 0.556 ± 0.052 |
αB-crystallin and γS-G18V Interact to Form Large Complexes
Because γS-G18V is implicated in early-onset cataract formation and has an altered structure in solution, we hypothesized that the molecular chaperone αB would interact more strongly with the disease-related variant than with wild-type γS. In order to assess the extent of binding, dynamic light scattering (DLS) measurements were performed at 25 °C on samples of αB, γS-WT and γS-G18V individually and as mixtures at pH 6.9. The DLS data are shown in Supplementary Figure S5. These results are consistent with previous findings; human αB-crystallin spontaneously forms spherical multimers 80 to 180 Å in diameter with a variable number (~24–32) of subunits (Haley et al., 1998; Jehle et al., 2010). γS-WT has an average hydrodynamic diameter of 50.40 ± 0.28 Å, which is in agreement with reported values for other monomeric γ-crystallins (Liu et al., 1998). γS-G18V forms large multimers with diameters up to 289.2 ± 8.8 Å at pH 6.9. Mixtures of αB and γS-WT or γS-G18V have apparent hydrodynamic diameters up to 155.88 ± 0.46 and 478.2 ± 2.3 Å, respectively.
The size of the particles in the αB + γS-WT mixture is similar to that of αB alone, likely because there are only weak interactions between the two proteins, and the DLS size of the mixture thus reflects the much larger αB complexes. Conversely, the particle size of the αB + γS-G18V mixture is much larger than either that of γS-G18V or αB alone. This result indicates that αB is interacting more strongly with γS-G18V than with γS-WT and is consistent with the conclusions drawn by Abgar et al. (Abgar et al., 2001) that α-crystallin binds destabilized proteins to prevent non-specific aggregation and that the resulting complex reorganizes into large particles in order to remain soluble.
αB-crystallin Interacts More Strongly With γS-G18V
To localize the regions of γS-crystallin involved in interactions with αB, heteronuclear single quantum coherence (HSQC) NMR spectroscopy was performed on mixtures of 15N-labeled γS-WT and γS-G18V with αB at pH 6.9. Solution NMR is sensitive to small changes in the electronic environment of the detected nuclei, therefore binding interactions between αB and γS should result in shifts or disappearances of relevant cross peaks. The addition of αB to 15N-labeled γS-WT leads only to minor chemical shift changes in the 15N-1H HSQC spectrum recorded at 25 °C (Figure 2a). Resi dues corresponding to cross peaks that undergo minor chemical shift changes include S35, W47, E66, G92, F122, and H123; all surface residues. Furthermore, the lack of cross peak disappearance in the presence of the molecular chaperone supports the conclusion that αB only weakly interacts with native γS in dilute solution, consistent with the results of a past 1H NMR spectroscopic study of bovine α- and γS-crystallin (Cooper et al., 1994). In contrast, upon addition of αB to 15N-labeled γS-G18V, nearly 50% of the cross peaks broadened below the noise threshold of the 15N-1H HSQC, presumably due to the formation of large αB/γS-G18V complexes in solution (expected to be > 300 kDa). Using 15N-1H TROSY-HSQC spectroscopy, we acquired a 2D spectrum of the αB/γS-G18V mixture. Analysis of the resulting TROSY-HSQC of γS-G18V (Figure 2b) reveals the disappearance of several N-H cross peaks; T9, Y11, D13, N15, F16, R19, Y21, C23, C25, C27, Y33*, L34, S35*, R36, C37, N38, I40, W47ε, G65, Y67, S82, S85, and G91. These residues are located in the N-terminal domain except for G91, which is situated in the linker between the two domains. Interestingly, the majority of cross peaks that lose signal intensity correspond to γ-G18V residues that occupy different positions (and whose cross peaks therefore shift) with respect to γS-WT. Y33* and S35* correspond to cross peaks from a minor alternate conformation of γS-G18V, also previously assigned (Brubaker and Martin, 2011). There are several cross peaks that slightly shift in the γS-G18V/αB mixture, including Y33, A56, G57, W73ε, S105, E110, I118, Q121, M124, G147, I161, W163ε, A165, and V170. These peaks are distributed throughout the protein and may shift due to altered surface interactions with αB and γS or due to changes in the local chemical environment resulting from the binding of αB and the associated formation of larger complexes.
Figure 2.
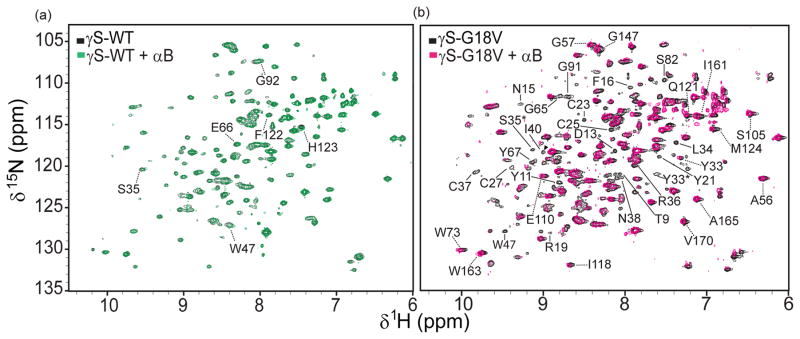
15N-1H HSQCs of (a) 15N -labeled γS-WT with (green) and without (black) αB and (b) 15N -labeled γS-G18V with (pink) and without (black) αB. Dashed lines indicate cross peaks that shift while solid lines indicate cross peaks that disappear with respect to the samples without αB. * Indicate cross peaks that belong to the alternate structure of γS-G18V. (Brubaker and Martin, 2011) See also Table S1.
Thermally Stressed γS-WT Does Not Recruit αB-crystallin
Previous studies investigating the solubilizing capabilities of α-crystallin using γ-crystallin mixtures purified from bovine lenses have shown that α-crystallin can prevent the thermal aggregation of γ-crystallins (Horwitz, 1992). A similar study noted that the rate of protein aggregation was dependent on α-crystallin concentration and that with a 3:2 α-:γ-crystallin ratio, using combinations of bovine α-crystallin and bovine γA-D, aggregation of the mixture occurred after heating at 72 °C (Wang and Spector, 1995).
In light of these results and the finding that only weak binding was observed between human αB and γS-WT at 22 °C, we set out to characterize the inter actions between αB and γ-WT upon heating. An 15N-1H HSQC spectrum was taken every 5 °C as γS-WT was heated between 22 – 47 °C, with and without αB. Sample precipitation and loss of signal occurred at temperatures above 47 °C, consistent with our previous observation that wild-type γS-crystallin forms aggregates well below its unfolding temperature (Brubaker et al., 2011). In the spectra obtained for both γS-WT and the mixture of γS-WT + αB, (shown overlaid in Figure 3) several cross-peaks of γS-WT shift due to temperature change. A direct comparison between the spectra of γS-WT in the presence and absence of αB (Figure 3a) reveals slight shifts and significant line broadening of the cross peaks, which decrease with increasing temperature. However, no disappearance of cross peaks is observed. The line broadening is due to weak transient interactions between αB and γS-WT, and its decrease with increasing temperature is attributed to accelerated molecular tumbling. Our observations of protein precipitation in the γS-WT/αB mixture at higher temperatures and the lack of disappearing cross peaks in the HSQCs indicates that αB-crystallin does not recognize thermally unfolded γS-crystallin. Human αB crystallin retains its native secondary structure up to 70 °C, although its chaperone activity is greatest near physiological temperatures (van Boekel et al., 1999; Reddy et al., 2000; Datta and Rao, 1999). This finding also suggests that thermal denaturation of native γS-crystallin is not a realistic model for cataract formation in this system.
Figure 3.

Solution NMR data indicating the minor conformational changes in γS-WT upon heating, and its weak, non-specific interaction with αB-crystallin. (a) 15N-1H HSQC temperature series from 22–47 °C of γS-WT (top) and γS-WT + αB (bottom). (b) Overlay of the γS-WT (black) and γS-WT + αB (blue) temperature series. (c) Change in 15N and 1H resonances by residue of γS-WT between 22 and 47 °C.
Discussion
Several three-dimensional structures of crystallins have been reported in the last decade, revealing the double Greek key fold as a common feature of βγ-crystallins (Jaenicke and Slingsby, 2001; Mills et al., 2007). The high stability and solubility of the crystallins is critical to their function because protein degradation and synthesis do not occur in mature differentiated lens fiber cells. Our results indicate that the relatively minor structural changes in γS-G18V result in a perturbation to the delicately balanced set of weak interactions between crystallins. The large body of structural investigations and interaction studies on β- and γ-crystallins suggests that specific interactions between them are functionally important (Slingsby et al., 1991). Weak interactions of individual βγ-clusters with α-crystallins may be relevant at the high protein concentrations in the eye lens, coupling these clusters to the structural dynamics of the polydisperse α-crystallin oligomers and preventing the formation of insoluble aggregates. Our observation of weak interactions between wild-type γS-crystallin and αB supports this hypothesis, because interactions should be weak even at millimolar concentrations to ensure an optimal level of exchange dynamics in the eye lens.
The spectra of the αB/γS-WT sample exhibited only minor chemical shift changes, and the affected residues are widely distributed throughout the structure (Figure 4a and b), confirming the presence of non-specific interactions. In this context, we expect the protein interfaces to exhibit very weak binding at the relatively low concentrations investigated here. Conversely, αB interacts more strongly with the disease-related variant γS-G18V as evidenced by loss of cross peak intensities and shifts due to the interactions with the chaperone. More quantitative comparison of the binding affinities presents experimental challenges, because binding to wild-type γS-crystallin is mainly characterized by line broadening rather than chemical shift differences. Furthermore, because αB-crystallin forms polydisperse oligomers alone and upon binding to aggregation-prone substrates, its binding to wild-type and G18V γS-crystallin cannot be correctly described by a single dissociation constant. However, based on the NMR data the interaction surface of γS-G18V that is recognized by αB-crystallin can be determined and is shown in Figure 4c and d. Residues of γS-G18V whose cross peaks lose intensity are localized to the N-terminal domain, coincident with the greatest structural changes due to the V18 substitution. The additional weak interactions may play an important role in maintaining the solubility of the larger αB/γS-G18V complex.
Figure 4.

Residues of γS-WT involved in weak transient interactions with αB are shown in blue on the surface of γS in two views: (a) from the front and (b) with the N-terminal domain rotated forward. Residues of γS-G18V involved in binding interactions (orange) and transient interactions (blue) with αB shown on the surface of γS in two views: (c) from the front and (d) with the N-terminal domain rotated forward.
In recognition of the results obtained with heat-denatured γS as well as recent mass spectrometry analyses (Lampi et al., 2012), it appears that during the lifetime of the organism, the chaperone activity of α-crystallins is required to account for accumulated post-translational modifications, such as deamidation or oxidation, rather than protein unfolding per se. Naturally occurring disease-related mutations represent a good model for this type of aggregation. Here, αB-crystallin interacts only weakly with γS-WT but more strongly and specifically with γS-G18V as indicated by the differences in the NMR spectra between the two mixtures.
The stronger binding of αB to the variant is consistent with the hypothesis that the mechanism of cataractogenesis from the G18V mutation in γS-crystallin may be related to the depletion of the finite amount of α-crystallin in the eye lens. Given the deoptimization of binding strength in the γS-WT interactions, we speculate that many disease-related modifications or mutations cause the formation of tighter complexes with α-crystallins. In this way, α-crystallins perform a holdase chaperone function, preventing unfavorable interactions among β- and γ-crystallins. Additionally, disease onset may be accelerated due to both larger γS-G18V and αB–γS-G18V particles which may be more prone to aggregation and precipitation. Similar effects were observed by others, reporting an increase in particle size after mixtures of α-crystallin and γ-crystallins from D. mawsoni, T. obesus, and B. taurus were heated extensively (Kiss et al., 2004). The exact mechanism that leads to this increase is yet unknown, but recruiting larger amounts of αB to the ‘sick’ protein is likely, since an aggregation-prone or stability-relevant area requires additional protection by α-crystallin on top of any normal interactions. In this model, one role α-crystallins may play in the eye lens is contributing a polydispersity principle, preventing formation of larger, more regular, and ultimately insoluble aggregates.
Experimental Procedures
Sample preparations
γS-WT, γS-G18V (Brubaker et al., 2011; Brubaker and Martin, 2011), and αB-crystallin (Jehle et al., 2010) were produced in E. coli and purified as described previously. NMR samples for structural work were at protein concentrations of 2.11 mM and 1.50 mM, respectively, in 10 mM acetate buffer pH 4.5, 10% D2O, 0.05% sodium azide, and 2 mM TMSP. Samples of γS-WT and γS-G18V under these conditions have remained stable and monomeric for over a year, showing no change in NMR spectra, when stored at 4 °C. Samples for NMR studies with αB were at protein concentrations of 1.5 mM for both γS-WT and γS-G18V in 10 mM phosphate buffer pH 6.9, 10% D2O, 0.05% sodium azide. Mixed samples with αB-crystallin consisted of an αB:γS molar ratio of 2:1. All experiments except for 15N IPAP spectra and the spectra observing the αB/γS thermal interactions were collected at 22 °C.
For RDC measurements the DIOTPC/DIOHPC bicelle system (Ottiger and Bax, 1999) at 10% (w/v) lipid concentration was used to align the protein samples. DIOTPC and DIOHPC disolved in chloroform (Avanti Polar Lipids) were mixed at a molar ratio of 3:1 DIOTPC:DIOHPC and dried under a stream of nitrogen gas. The residual chloroform was removed by lyophilization. A 270 μL protein sample in 10 mM acetate buffer pH 4.5, 10% D2O, 0.05% sodium azide, and 2 mM TMSP was added to the lyophilized lipids. The sample was cycled several times between an ice bath and room temperature over the course of a few hours with gentle mixing between each incubation in order to fully rehydrate the lipids and mix the bicelle sample.
H-D exchange samples were prepared by concentrating the γS samples to saturation in centrifugal concentrator columns (with γS at a concentration of approximately 270 mg/mL at saturation) in a volume of approximately 60 μL and adding 99.9% D2O to a total volume of 300 μL immediately before starting the data collection.
NMR experiments
NMR experiments were performed on a Varian UnityINOVA system operating at 800 MHz equipped with a 1H/13C/15N 5 mm tri-axis PFG triple-resonance probe. Decoupling of 15N nuclei was performed by GARP sequences. 1H shifts were referenced to TMSP, 13C and 15N shifts were referenced indirectly to TMSP. Heated samples of αB/γS mixtures were equilibrated for several minutes before data acquisition. NMR data were processed using NMRPipe and analyzed using Sparky. H-D exchange spectra were taken at intervals of approximately 30 minutes for the first 4 hours following the addition of D2O, and then at intervals of 1 hour for the next 20 hours. Additional spectra were collected at intervals of several days. For each collection period a 1-dimensional proton spectrum was collected and non-exchanging methyls in the protein were used to adjust for differences in shimming between data collections.
Restraints
NOE restraints were assembled by manually picking slices from 13C-filtered and 15N -filtered NOESY experiments corresponding to the 13C and 15N HSQC crosspeaks. The manually picked peaks were assigned in a “binned” fashion using three sets of chemical shift tolerances of decreasing stringency to minimize NOE crosspeak assignment ambiguity, and restraints were generated in CCPNMR Analysis. Duplicate and redundant restraints were eliminated from the exported restraint lists, and only unambiguous NOE restraints were used in the final structure calculations.
In-phase, anti-phase (IPAP) spectra of isotropic γS-WT and aligned γS-G18V were acquired at 32 °C and 30 °C. Each IPAP dataset was processed into two spectra each containing only one of the two doublet peaks so that all peaks could be easily resolved. For the RDCs, peaks were manually picked for all crosspeaks in the spectra and the the difference between the J-splittings and the J+D splittings were computed for each resonance using a spreadsheet program. The starting error for all of the RDCs in the angular restraint table was set as the standard deviation of the measured J-splittings, 2.47 Hz and 3.01 Hz for γS-WT and γS-G18V, respectively. RDC experiments yielded 156 and 147 couplings for γS-WT and -G18V, respectively, including the sidechain tryptophan Nε protons, corresponding to nearly every visible peak in the 1H-15N HSQC, which are included in the restraints deposited in the PDB. See Supplementary Tables 1 and 2.
H-D exhange experiments yielded a total of 46 and 90 hydrogen bonding restraints used to refine the final structures for γS-WT and -G18V, respectively.
Dihedral angle restraints were calculated using the TALOS+ program (Shen et al., 2009) for γS-WT and for both the major and minor chemical shift sets of γS-G18V.
3J HN-HA couplings were calculated from peaks in a 3D H-N-HA experiment and were used as restraints in structure calculations for the γS-WT and γS-G18V structures (Table 1).
Supplementary Material
Acknowledgments
We thank Wytze van der Veer for assistance with optical data collection and Doug Tobias and Melanie Cocco for helpful discussions. This work was supported by NIH grant 1R01EY021514 (to R.W.M) and by a DFG grant within the frame of the SFB 740 (to H.O).
Abbreviations
- αB
human αB-crystallin
- γS
human γS-crystallin
- DLS
dynamic light scattering
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum coherence spectroscopy
- TROSY
transverse relaxation-optimized spectroscopy
Footnotes
C. N. K. and W. D. B. prepared protein samples, performed NMR and DLS experiments, analyzed NMR and DLS data, and wrote the manuscript. These authors contributed equally. S. M., A.D., and A. J. B. prepared protein samples. H. O. and R. W. M. designed the experiments, analyzed data, and wrote the manuscript.
Accession Numbers
The solution NMR structures of γS-WT and γS-G18V have been deposited in the Protein Data Bank, with PDBIDs 2M3T and 2M3U, respectively.
In this appendix we describe the calculations used to determine the NMR structures of γS-WT and γS-G18V and report the 20 lowest-energy structures (Supplementary Figures S1 and S2). Additional structure calculations to confirm the position of valine-18 in the γS-G18V structure are also reported here (Supplementary Figure S3). DLS data showing the particles sizes of the NMR samples are also presented here to show that both γS-WT and γS-G18V are monomeric under the NMR sample conditions (Supplementary Figure S4). Supplementary Figure S5 contains the full DLS data of αB, γS-WT, γS-G18V, and mixtures thereof as determined by dynamic light scattering.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abgar S, Vanhoudt J, Aerts T, Clauwaert J. Study of the chaperoning mechanism of bovine lens α-crystallin, a member of the alpha-small heat shock superfamily. Biophys J. 2001;80:1986–1995. doi: 10.1016/S0006-3495(01)76168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee PR, Pande A, Patrosz J, Thurston GM, Pande J. Cataract-associated mutant E107A of human γD-crystallin shows increased attraction to α-crystallin and enhanced light scattering. Proc Natl Acad Sci U S A. 2011;108:574–579. doi: 10.1073/pnas.1014653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker WD, Freites JA, Golchert KJ, Shapiro RA, Morikis V, Tobias DJ, Martin RW. Separating instability from aggregation propensity in γS-crystallin variants. Biophys J. 2011;100:498–506. doi: 10.1016/j.bpj.2010.12.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker WD, Martin RW. 1H, 13C, and 15N assignments of wild-type human γS-crystallin and its cataract-related variant γS-G18V. Biomolec. NMR Assign. 2011;6:63–67. doi: 10.1007/s12104-011-9326-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T, Chang WC. Cloning and sequencing of a capr beta s-crystallin cDNA. Biochem Biophys Acta. 1987;910:89–92. doi: 10.1016/0167-4781(87)90098-4. [DOI] [PubMed] [Google Scholar]
- Cooper PG, Carver JA, Aquilina JA, Ralston GB, Truscott RJ. A 1H NMR spectroscopic comparison of γS- and γB-crystallins. Exp Eye Res. 1994;59:211–220. doi: 10.1006/exer.1994.1099. [DOI] [PubMed] [Google Scholar]
- Datta SA, Rao CM. Differential temperature-dependent chaperone-like activity of αA- and αB-crystallin homoaggregates. J Biol Chem. 1999;274:34773–34778. doi: 10.1074/jbc.274.49.34773. [DOI] [PubMed] [Google Scholar]
- Delaye M, Tardieu A. Short-range order of crystallin proteins accounts for eye lens transparency. Nature. 1983;302:425–417. doi: 10.1038/302415a0. [DOI] [PubMed] [Google Scholar]
- Devi RR, Yao W, Vijayalakshmi P, Sergeev YV, Sundaresan P, Hejtmancik JF. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis. 2008;14:1157–1170. [PMC free article] [PubMed] [Google Scholar]
- Fu L, Liang JJN. Alteration of protein-protein interactions of congenital cataract crystallin mutants. Invest Ophthalmol Vis Sci. 2003;44:1155–1159. doi: 10.1167/iovs.02-0950. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K, Ramakrishnan C, Balaram P. Disallowed Ramachandran Conformations of Amino Acid Residues in Protein Structures. J Mol Biol. 1996;264:191–198. doi: 10.1006/jmbi.1996.0633. [DOI] [PubMed] [Google Scholar]
- Haley DA, Horowitz J, Sterwart PL. The small heat-shock protein, αB-crystallin, has variable quaternary structure. J Mol Biol. 1998;20:27–35. doi: 10.1006/jmbi.1997.1611. [DOI] [PubMed] [Google Scholar]
- Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci U S A. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki T, Kume-Iwaki A, Goldman JE. Cellular distribution of alpha B-crystallin in non-lenticular tissues. J Histochem Cytochem. 1990;38:31–39. doi: 10.1177/38.1.2294148. [DOI] [PubMed] [Google Scholar]
- Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE. Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell. 1989;57:71–78. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- Jaenicke R, Slingsby C. Lens crystallins and their microbial homologs: structure, stability, and function. Crit Rev Biochem Molec Biol. 2001;36:435–499. doi: 10.1080/20014091074237. [DOI] [PubMed] [Google Scholar]
- Jehle S, Rajagopal P, Bardiaux B, Markovic S, Ku hne R, Stout JR, Higman VA, Klevit RE, van Rossum BJ, Oschkinat H. Solid-state NMR and SAXS studies provide a structural basis for the activation of alpha B-crystallin oligomers. Nat Struct Mol Biol. 2010;17:1037–1043. doi: 10.1038/nsmb.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji F, Jung Y, Gronenborn AM. Structural and biochemical characterization of the childhood cataract-associated R76S mutant of human γD-crystallin. Biochemistry. 2012;51:2588–2596. doi: 10.1021/bi300199d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Vandonselaar M, Quail JW, Delbaere LT. Active-centre torsion-angle strain revealed in 1.6 A-resolution structure of histidine-containing phosphocarrier protein. Nature. 1993;361:94–97. doi: 10.1038/361094a0. [DOI] [PubMed] [Google Scholar]
- Karri S, Kasetti RB, Vendra VPR, Chandani S, Balasubramanian D. Structural analysis of the mutatant protein D26G of human γS-crystallin, associated with Coppock cataract. Mol Vis. 2013;19:1231–1237. [PMC free article] [PubMed] [Google Scholar]
- Kiss AJ, Mirarefi AY, Ramakrishnan S, Zukoski CF, DeVries AL, Cheng CHC. Cold-stable eye lens crystallins of the Antarctic nototheniid toothfish Dissoctichus mawsoni Norman. J Exp Biol. 2004;207:4633–4649. doi: 10.1242/jeb.01312. [DOI] [PubMed] [Google Scholar]
- Klemenz R, Fröhli E, Steiger RH, Schäfer R, Aoyama A. Alpha B-crystallin is a small heat shock protein. Proc Natl Acad Sci U S A. 1991;88:3652–3656. doi: 10.1073/pnas.88.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampi KJ, Fox CB, David LL. Changes in accessibility of wild-type and deamidated βB2-crystallin follwing complex formation with αA-crystallin. Exp Eye Res. 2012;104:48–58. doi: 10.1016/j.exer.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Pande J, Lomakin A, Ogun O, Benedek GB. Aggregation in aqueous solutions of lens γ-crystallins: special role of γS-crystallin. Invest Ophthalmol Vis Sci. 1998;39:1609–1619. [PubMed] [Google Scholar]
- Ma Z, Piszczek G, Wingfield P, Sergeev Y, Hejtmancik J. The G18V CRYGS mutation associated with human cataracts increases γS-crystallin sensitivity to thermal and chemical stress. Biochemistry. 2009;48:7334–7341. doi: 10.1021/bi900467a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills IA, Flaugh SL, Kosinski-Collins MS, King JA. Folding and stability of the isolated Greek key domains of the long-lived human lens proteins gamma D-crystallin and gamma S-crystallin. Protein Sci. 2007;16:2427–2444. doi: 10.1110/ps.072970207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau KL, King JA. Cataract-causing defect of a mutant γ-crystallin proceeds through an aggregation pathway which bypasses recognition by the α-crystallin chaperone. PloS ONE. 2012;7:e37256. doi: 10.1371/journal.pone.0037256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottiger M, Bax A. Bicelle-based liquid crystals for NMR-measurement of dipolar couplings at acidic and basic pH values. J Biomol NMR. 1999;13:187–191. doi: 10.1023/a:1008395916985. [DOI] [PubMed] [Google Scholar]
- Ousman SS, Tomooka BH, Noort JMV, Wawrousek EF, O’Connor KC, Halfler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for αB-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- Pal D, Chakrabarti P. On residues in the disallowed region of the Ramachandran map. Biopolymers. 2002;63:195–206. doi: 10.1002/bip.10051. [DOI] [PubMed] [Google Scholar]
- Ponce A, Sorensen C, Takemoto L. Role of short-range protein interactions in lens opacifications. Mol Vis. 2006;12:879–884. [PubMed] [Google Scholar]
- Quax-Jeuken Y, Driessen H, Leunissen J, Quax W, de Jong W, Bloemendal H. Beta s-crystallin: structure and evolution of a distinct member of the beta gamma-superfamily. EMBO J. 1985;4:2597–2602. doi: 10.1002/j.1460-2075.1985.tb03976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GB, Das KP, Petrash JM, Surewicz WK. Temperature-dependent chaperone activity and structural properties of human αA- and αB-crystallins. J Biol Chem. 2000;275:4565–4570. doi: 10.1074/jbc.275.7.4565. [DOI] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slingsby C, Simpson A, Ferszt A, Bateman OA, Nalini B. Molecular interactions in the eye lens. Biochem of the Eye. 1991;19:853–858. doi: 10.1042/bst0190853. [DOI] [PubMed] [Google Scholar]
- Sun H, Ma Z, Li Y, Liu B, Li Z, Ding X, Gao Y, Ma W, Tang X, Li X, Shen Y. Gamma-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans. J Med Genet. 2005;42:706–710. doi: 10.1136/jmg.2004.028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto L, Sorensen CM. Protein-protein interactions and lens transparency. Exp Eye Res. 2008;87:496–501. doi: 10.1016/j.exer.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieu A, Vérétout F, Krop B, Slingsby C. Protein interactions in the calf eye lens: interactions between beta-crystallins are repulsive whereas in gamma-crystallins they are attractive. Eur Biophys J. 1992;21:1–12. doi: 10.1007/BF00195438. [DOI] [PubMed] [Google Scholar]
- van Boekel MA, de Lange F, de Grip WJ, de Jong WW. Eye lens αA- and αB-crystallin: complex stability versus chaperone-like activity. Biochimica Et Biophysica Acta. 1999;1434:114–123. doi: 10.1016/s0167-4838(99)00178-8. [DOI] [PubMed] [Google Scholar]
- van Noort JM, Sechel ACV, Bajramovic JJ, Ouagmiri ME, Polman CH, Lassmann H, Ravid R. The small heat-shock protein αB-crystallin as candidate autoantigen in multiple sclerosis. Nature. 1995;375:798–801. doi: 10.1038/375798a0. [DOI] [PubMed] [Google Scholar]
- van Rens GL, de Jong WW, Bloemendal H. One member of the gamma-crystallin gene family, gamma s, is expressed in birds. Exp Eye Res. 1991;53:135–138. doi: 10.1016/0014-4835(91)90156-9. [DOI] [PubMed] [Google Scholar]
- van Rens GL, Raats JM, Driessen HP, Oldenburg M, Wijnen JT, Khan PM, de Jong WW, Bloemendal H. Structure of the bovine eye lens gamma s-crystallin gene (formerly beta s) Gene. 1989;78:225–233. doi: 10.1016/0378-1119(89)90225-4. [DOI] [PubMed] [Google Scholar]
- Vendra VP, Chandani S, Balasubramanian D. The mutation V42M distorts the compact packing of the human gamma-S-crystallin molecule, resulting in congenital cataract. PLoS One. 2013;7:e51401. doi: 10.1371/journal.pone.0051401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Spector A. α-Crystallin can act as a chaperone under conditions of oxidative stress. Investigative Ophthalmology & Visual Science. 1995;36:311–321. [PubMed] [Google Scholar]
- Wu Z, Delaglio F, Wyatt K, Wistow G, Bax A. Solution structure of (gamma)S crystallin by molecular fragment replacement NMR. Protein Sci. 2005;14:2142–2143. doi: 10.1110/ps.051635205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.