

Abstract
Recent advances in the genetics of Autism Spectrum Disorders (ASD) are offering new valuable insights into molecular and cellular mechanisms of pathology. At the same time, the emerging data challenges long-standing diagnostic conventions and the notion of phenotypic specificity. This review addresses the particular issues that attend gene discovery in neuropsychiatric and neurodevelopmental disorders and ASD in particular; summarizes recent findings in human genetics broadly that are driving the reevaluation of the conventional wisdom regarding the allelic architecture of common psychiatric conditions; reviews selected discoveries in ASD and their relevance to models of pathology; highlights the conceptual and practical issues raised by the observation of a convergence of ASD genetic risks with distinct psychiatric disorders; and considers the important interplay of studies of neurobiology and genetics in clarifying and extending our understanding of social disability syndromes.
Introduction
Autism spectrum disorders (ASD) are defined by deficits in social communication, impaired language development, and the presence of highly restricted interests and/or stereotyped repetitive behaviors. As with all common neuropsychiatric conditions, the reliance on syndromic diagnoses comes as a consequence of lacking a better alternative, given a very limited understanding of underlying pathology. However, recent successes in both the genetics and genomics of ASD are promising to change this equation, and, along with the rapid pace of related neurobiological studies, are now allowing for a data-driven re-conceptualization of gene-brain-behavior relationships. This progress is already challenging long-standing dogma regarding the nature of the genetic variation thought to be contributing to ASD and is further calling into question the adequacy of the current psychiatric diagnostic nosology.
With the caveat that the field is just beginning to assimilate a flood of new data emerging from rapidly advancing genomic technologies, this review will highlight key issues that are arising as genetic investigations substantively inform the understanding of risks for social disability. We will not endeavor to provide a comprehensive recounting of the autism genetics literature here, but rather to highlight the particular challenges of gene discovery in human behavioral, cognitive and emotional phenotypes; to consider how recent empirical evidence is driving a reconsideration of the allelic architecture of common conditions including ASD, to highlight selected findings that are laying the foundation for the next steps in genetic and neurobiological studies; and to consider the ramifications of the apparent convergence of genetic risks among ASD and other quite distinct psychiatric conditions.
The Complexity of the Problem
Several decades of investigation have made clear that the difficulties attending gene discovery in ASD have arisen, in no small measure, from a combination of allelic (many variants at a single gene), locus (many genes) and phenotypic heterogeneity. In addition, the involvement of behavioral, social and cognitive domains of functioning presents it own challenges. Clinical diagnoses in the Diagnostic and Statistical Manual (DSM) typically rest on a series of binary descriptors: for example, with regard to ASD, the presence or absence of …”marked impairments in the use of multiple nonverbal behaviors such as eye-to-eye gaze, facial expression, body posture, and gestures to regulate social interaction1”. Yet these conditions involve domains that would more accurately be described in an ethologically relevant fashion using continuous measures, reflecting the underlying heterogeneity that exists in each of the relevant functional domains and their changing characteristics and trajectories over time. Nonetheless, despite considerable efforts to address this complexity through research diagnostic criteria and to identify relevant endophenotypes, there remains marked uncertainty regarding how to identify, a priori, useful phenotypic metrics, apart from categorical diagnoses, to support gene discovery in neuropsychiatric disorders.
Adding to the complexity, a half-century of basic research has revealed that the emergence of “higher order” functions disrupted in psychiatric disorders is influenced by neurodevelopmental processes that are guided by thousands of genes2. Complex functions are in turn mediated by hierarchically organized circuitries that include sensory and motor, autonomic regulatory, social-emotional and cognitive domains3. This layered complexity points to the considerable “distance” between distinct variations in the DNA code and the constellations of behaviors, emotions and experiences that psychiatrists confront in the clinic. Under the circumstances, it is not surprising that identifying the path from genotype to autism spectrum phenotype has not been an easy one.
Moreover, because of redundancy in the role of gene families in divergent biological processes, neuropsychiatric disorders may be more systemic in nature than generally appreciated. Phenomena that may be very frequently observed clinically, such as gastrointestinal complaints, seizure or sleep disorders in children with ASD4 do not presently contribute to categorical diagnosis. Whether these co-occurring conditions represent features that could distinguish subsets of affected individuals in genetically meaningful ways remains understudied, but may well be the case5, as discussed below.
Emerging Views of the Genetic Architecture of ASD
Challenges aside, the last several years of investigation have resulted in the identification of specific genetic variations contributing to these syndromes (see recent reviews in4,6,7). As these hard won successes begin to shed light on pathogenic mechanisms, they also have prompted a reappraisal of the conventional wisdom regarding the nature of the variation that is likely to be contributing to ASD and the predictability of the relationship between genetic variation and brain function and dysfunction.
For several decades, the predominant paradigm in psychiatric genetics generally has been the “common disease-common variant” (CDCV) hypothesis: namely that the majority of the risk for neuropsychiatric disorders will be found in a conspiracy of common alleles, each conferring modest risk – either for the overall phenotype or for subcomponents of a complex presentation8-10. However, despite both the intuitive attractiveness of the hypothesis and feasibility of undertaking case-control studies to search for associated common alleles in or near “candidate genes”, such approaches have struggled to provide replicable results for any common psychiatric condition.
Importantly, the fairly recent development of unbiased genome-wide association approaches, facilitated by the emergence of microarray technology, has changed this equation for much of medicine, and is now being applied with some success to neuropsychiatric disorders. Importantly, a host of reproducible results in other clinical areas, ranging from diabetes to inflammatory bowel disease to intracranial aneurysm, has provided important insights into the reasons for earlier difficulties11: Initial cohorts were markedly underpowered due to an overestimation of allelic effect sizes; the prior probability of choosing one or a small number of common alleles correctly from among the several million in the genome was fleetingly small, and genetic case-control studies were, and remain, highly sensitive to cryptic sources of mismatch, including for ancestry. Moreover, when viewed cumulatively, the common risk alleles reproducibly identified by GWAS studies have, in most instances, accounted for only a small fraction of the anticipated risk for common conditions. This has led to a preoccupation with “missing heritability” in complex disorders12,13,14, attributed variously to a combination of an initial over-estimation of the contribution of genetics, the involvement of hundreds to thousands of common risk variants of extremely small effect, and/or a significant role for alleles that are rare (minor allele frequency (MAF) <5%) or very rare (MAF<1%)15 in the population.
Indeed the potential contribution of rare genetic variation has garnered tremendous interest of late, driven both by the experience with GWAS as well as by new genomic technologies, including next generation sequencing, that make increasingly feasible the investigation of low frequency alleles on a genome-wide scale. In practice, the search for rare variants in common disease typically focuses on one or both of two strategies: identifying extremely rare, Mendelian, examples of common disorders and/or investigating the cumulative contribution of rare mutation to common phenotypes. The driving rationale for the former is that gene discovery, even in the most rare examples, has the potential to illuminate key molecular and cellular mechanisms leading, in turn, to novel opportunities for intervention. The latter seeks to account for a significant proportion of population risk, based on the notion that individually rare alleles, given a sufficiently high degree of genetic heterogeneity, may account for most or all of the risk for commonly occurring illnesses.
The notion that rare mutations may underlie a common syndrome such as ASD may at first blush seem counterintuitive, but in fact a number of considerations would argue strongly otherwise. First, purifying selection would indeed be expected to drive down the population frequency of highly deleterious transmitted alleles, particularly for early onset conditions that impact reproductive fitness (Figure 1). In this context, one might also expect a significant number of sporadic cases due to de novo mutation, something that is well described in the ASD literature 16-22. Clearly, if rare variants were to account for a substantial portion of the risk for ASD, the number of potential gene targets across the genome would be expected to be large and yet to converge on a coherent set of biological processes. In support of this, the structural variation and single gene mutations so far identified have pointed to convergent neurodevelopment and molecular pathways (discussed below) and no single recurrent variation has so far been found in more than about 1% of the affected population.
Figure 1. Allele frequency and effect size in ASD.
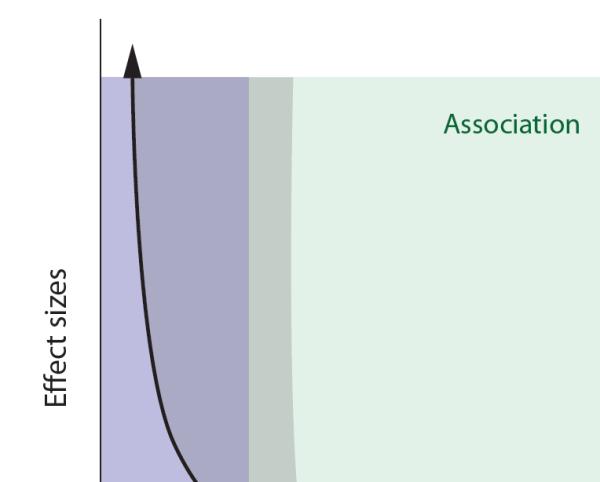
The graph illustrates the relationship of allele frequency to effect size for ASD-related variants. Rare mutations, as expected, have been found to carry larger effects than common variations. Consistent with the broad medical literature, the scale of effects for common alleles implicated in ASD has been quite modest. The linkage (blue) and association (green) sections illustrate the historically divergent approaches to demonstrating the role of rare versus common variation in disease; rare alleles have been expected to show an approximately 1:1 relationship with a given phenotype, often demonstrated via linkage, while common polymorphisms have been expected to show more probabilistic outcomes, typically demonstrated by some type of case-control association design. The shaded area illustrates that the range of effect sizes for rare alleles identified in ASD to date have been broader than those expected for strictly Mendelian disorders, and that association approaches have been necessary to confirm the relationship of genotype to phenotype.
Rare and Common Alleles in ASD
In fact, while the common and rare variant perspectives have tended to be offered as stark counterpoints, the evidence suggests that both are likely to contribute to risk, and, given the rudimentary understanding of pathophysiology of ASD, both have clear potential to offer novel and important insights.
Common Variation
As noted above, until early in the current century, most studies of common variation selected candidate gene(s) based on their biological plausibility and evaluated the contribution of one or several common alleles within or near these loci. As was the case for almost every common condition studied, the majority of the resulting findings did not replicate convincingly (ref). In ASD, only a handful of common variants identified in this pre-GWAS-era yielded solid evidence for association through studies in large samples, independent replications, and the demonstration of alleles influencing gene expression and/or protein function. Two findings in particular, involving regulatory SNPs in the receptor tyrosine kinase MET23-25 and SNPs mapping to the interval corresponding to the gene CNTNAP226,27 (discussed in a subsequent section) continue to generate considerable interest.
Family cohort and case-control studies23-25,28 have reported ASD-associated variants in MET, a gene encoding a tyrosine kinase receptor that promotes neuronal growth and synaptogenesis29-31. Three different 5’ regulatory alleles, and deletion copy number variants (CNVs) in 3 individuals have been identified that are not seen in controls (for review 32). Moreover, MET signals through the same intracellular pathway containing mutations in other genes implicated in idiopathic and syndromic ASD28,33,34. The initially characterized MET promoter allele is functional, causing a 50% reduction in gene transcription24. Met expression at synapses peaks during their formation, and is limited to forebrain limbic and neocortical structures involved in emotional and social behavior regulation35,36. Consistent with these findings, recent studies of functional deletion of Met in the mouse reveal altered cortical dendrite and spines29 and a functional phenotype implicated in ASD 37- electrophysiologically defined local hyperconnectivity38.
Yet even with this convergent data, there remain interpretive challenges. For example, so far a significant association of MET has not been identified in any of the three published GWAS studies of ASD. However, as discussed below, there have so far been no common risk variants significantly associated with ASD in more than a single genome-wide study. It is possible that the MET results reflect type 1 error, but this seems unlikely given independent replication of the functional allele in other (but not all) cohorts (reviewed in 32), altered transcript and protein expression in independent ASD samples of postmortem tissue39,40, and neurobiological evidence for relevance in forebrain circuit development 29,38. Another likely alternative is that the lack of agreement across studies reflects patient cohorts that remain markedly underpowered to detect common variants with now-plausible effect sizes in ASD41. Moreover, in the case of MET it is noteworthy that once the first functional risk variant was found to be associated with ASD, further analyses demonstrated the allele was enriched in multiplex compared to simplex families and in affected individuals who also exhibit co-occurring gastro-intestinal conditions5, a common clinical problem in individuals with ASD42. In this regard, it is important to recall that common functional variants are likely to be modulators of ASD relevant phenotypes, not disorder-causing per se. Simply stated, some of these alleles may be disorder-associated only in certain subpopulations and there is likely to be a diversity of common alleles influencing the trajectories of disorder-relevant phenotypes apart from categorical diagnoses.
These observations point to the likelihood that genetic mechanisms through which associations occur may help redefine and stratify disorders by exhibiting enrichment in subgroups with a common diagnosis but with distinguishing phenotypic features. However, this possibility must be viewed simultaneously with the strong evidence that specific genetic risks for ASD also lead to highly divergent, non-overlapping clinical phenomena 43,44. This renders the task of identifying, a priori, component phenotypes that will enhance genetic homogeneity sufficiently to drive gene discovery efforts extremely challenging. It suggests the more likely and productive scenario will involve hypothesis-driven investigations undertaken subsequent to a definitively established relationship between a particular gene, variation, or molecular pathway and ASD.
As noted, the shift from candidate gene to genome wide studies of association has been accompanied by an increasingly sophisticated appreciation of the plausible effect sizes of common alleles, an attendant focus on large cohorts, and careful attention to and correction for confounds including occult differences in ancestry among cases and controls, leading to a host of reproducible findings in other areas of medicine. In ASD, the application of rigorous GWAS methodology has so far led to the identification of three disorder-associated alleles that meet accepted discovery criteria: one mapping to an intragenic region on chromosome 5p between the neuronal adhesion molecules Cadherin 9 (CDH9) and CDH1045 a second mapping within 80kb of Semaphorin 5A46 and a third mapping within the locus encoding the gene MACROD247. However, despite justifiable excitement over the emergence of alleles demonstrating genome-wide significance and surviving internal replication prior to publication, there remain important uncertainties: each study has so far failed to replicate the significant findings from either of the others, and a joint evaluation of all three investigations has suggested that the combined data decreases evidence for association for all of the identified risk alleles41. As noted above, it seems likely that sample sizes in ASD are still considerably underpowered. And while it is certainly possible that common alleles will not be found to contribute to ASD risk, given recent findings, particularly with regard to schizophrenia41, it seems far more likely that there are indeed multiple common variants of very small effect remaining to be identified.
When such alleles are definitively replicated, translating the initial mapping into potentially targetable neurobiological mechanism will constitute an additional challenge. Through ‘guilt-by-proximity’, those nearest to the current GWAS alleles have tended to be characterized as new risk genes. However, this short hand may obscure a much more complex situation. For instance, the 5p common variant45 noted above mapped between two molecules that are implicated in histogenic neural events, but resides approximately 1MB from either gene and is not in strong linkage disequilibrium with these Cadherin loci. This highlights several pressing questions that arise subsequent to an initial GWAS finding, regardless of whether an allele is found in coding, intra- or inter-genic regions, that must be answered experimentally: 1) whether nearby transcript(s) exhibit disorder-related alterations in expression or function; 2) whether there may be other cryptic protein-coding or other transcripts within association intervals that may prove relevant biologically; and 3) for non-coding changes, how alterations in gene regulation relate to neural processes underlying ASD. In this context, the small overall increment in genetic risk associated with the vast majority of alleles identified so far by GWAS, including with regard to ASD, suggests that observable effects in experimental assays also could be quite modest.
Nonetheless, in our view it would be a mistake to equate the importance of a common variant finding with its effect size just as it would be short sighted to judge the impact of a rare variant finding on the overall frequency of the mutation in the general population. Both approaches, when successful, provide important avenues to illuminate the etiology, molecular and cellular biology and genetic architecture of ASD.
Rare variation
While the emphasis on the contribution of rare variation has grown of late, in fact there is a comparatively long history is autism and related conditions. For example, studies of Mendelian single gene disorders over the past decade have offered key insights into the molecular mechanisms of cognitive and behavioral syndromes and have already extended into the realm of social disability. Several such disorders, best exemplified at present with regard to Fragile X and tuberous sclerosis (TSC)48, demonstrate a clear increase in risk for ASD. Moreover, syndrome-defining mutations have not infrequently been found in probands previously diagnosed with idiopathic ASD. These observations suggest that the illumination of neurobiological mechanisms will have implications far beyond the confines of the syndromes themselves.
This is not to suggest that there is unanimity with regard to the value of studying the coincidence of ASD and known genetic disorders. The findings of subtle differences in the social phenotypes relative to idiopathic autism and the observation of a correlation between lower IQ and the diagnosis of autistic features 48,49 has led to some skepticism that studying single gene disorders will translate into an improved understanding of “pure” social disability49.
However, as the molecular underpinnings of these syndromes have been elaborated, they have tended to converge on alterations in the assembly and functioning of synapses33,50 and these discoveries have been strikingly consistent with the earliest molecular genetic findings in studies of idiopathic ASD. For instance, in 2004 Neuroligin 4 (NLGN4), encoding a neuronal adhesion molecule present in the post synaptic density of excitatory synapses, was the first transcript for which rare coding ASD-related mutations were identified in non-syndromic individuals 16,51. Subsequent studies have reproducibly identified rare functional mutations (both sequence and structural) in the genes Neurexin 118,52-54, a presynaptic binding partner for NLGNs, as well as SHANK322,55 and more recently SHANK256,57, postsynaptic scaffolding molecules that interact with PSD95.
The convergence of data from studies of rare variation in syndromic and idiopathic ASD is similarly reflected in recent findings with regard to Contactin and associated molecules. Contactin 4 (CNTN4) was first identified as having a role in social and intellectual disability in the context of studies of a recurrent deletion syndrome 58,59 and heterozygous mutations have subsequently been identified in well-characterized patients with idiopathic ASD60,61. Similarly, very rare homozygous protein-truncating mutations in Contactin Associated Protein 2 (CNTNAP2) have been described in consanguineous pedigrees demonstrating intractable epilepsy and ASD 62 , and heterozygous rare mutations have been found in patients with idiopathic social disability17,26,63 and schizophrenia 64. Common variants in CNTNAP2 have been associated with ASD and language delay26,65 as well as altered functional connectivity 66, selective mutism and anxiety67.
These data point to a promising areas of investigation, though also underscore key challenges in elaborating mechanism in the absence of definitive human genetic findings. For example, there are several lines of evidence for the involvement of CNTNAP2 in idiopathic ASD, but neither a strict replication of a common associated allele nor consistent statistical evidence for an excess burden of rare variants has so far materialized. In addition, hypotheses regarding the underlying histogenic disruptions relevant to ASD are just beginning to emerge. In the case of FMRP and NLGN4 and related molecules, the issues of specificity, location and timing of dysfunction and their relationship to phenotype remain something of a puzzle. Even less is known about the normal functions of CNTNAP2 in the brain. However, interestingly, the rare opportunity to examine pathological specimens from human temporal lobe resections62 in consanguineous families with epilepsy, ASD and loss-of-function mutations in CNTNAP2 points to abnormalities in cortical neuronal morphology and migration. Whether these reflect an alternative pathway to ASD or these mutations simultaneously lead to disruptions in the formation and/or functioning of excitatory/glutamatergic synapses remains to be elucidated.
Arguably the most important recent milestone in the study of rare variation in ASD has been the emergence of copy number variation (CNV) analyses, affording the first opportunity to conduct unbiased surveys for rare variation genome-wide at sub-microscopic resolution. The first such analyses demonstrated that rare de novo structural variations were overrepresented in simplex families (those with only one affected offspring) as compared both to controls as well as to families in which there were multiple affected individuals17. This excess of de novo CNVs in cases versus controls has now been repeatedly replicated18,61,68, but it is not as clear yet whether there is a significant difference in the contribution of these events in simplex versus multiplex ASD57.
Studies of increasingly large samples have delved more deeply into the relationship of de novo CNVs and ASD risk: For example, two recent independent analyses of a comprehensively assessed simplex cohort, the Simons Simplex Collection, involving more than 1000 families and including unaffected siblings, demonstrated that the risk attributable to individually rare de novo CNVs encompassing multiple genes is many times greater than the effect sizes suggested for any common ASD variant, with odds ratios of ~5-6 20,21.
It is noteworthy that though the resolution of detection of array platforms has increased markedly over the last several years, the overall burden of these de novo variants in ASD populations has remained fairly constant, with between 5-10% of affected individuals in simplex families carrying at least one de novo CNV. Based on recent data, it would seem that ASD risk is most pronounced (or detectable) for large (>100kb) multi-genic de novo CNVs. Whether this is a consequence of such events covering more genomic territory and thus being more likely to disrupt a single gene of particular relevance, or whether, as we suspect, it suggests that the simultaneous disruption of multiple genes and regulatory elements in genomic intervals carries particular risk, remains to be clarified.
Several studies also have identified and replicated specific recurrent structural variations strongly associated with ASD. A cumulative analysis of confirmed de novo variants reported across four large genome-wide case-control studies demonstrates that deletions and duplications at 16p11.2, when considered either independently or combined; duplications at 15q11-13; deletions and duplications combined at 22q11.2, deletions at the Neurexin 1 locus , and duplications at 7q11.23, all reach genome wide significance21 . Multiple additional intervals, involving both recurrent rare de novo and transmitted CNVs, including at 17q 69 and 1q2170, have been observed to be over-represented in cases compared to controls and, with the additional power afforded by larger samples, appear poised to cross this threshold as well. Along these lines another recent large-scale study looking both at transmitted and de novo CNVs highlighted the important point that the former also contribute to ASD risk, but with an overall effect that is somewhat more attenuated than for de novo CNVs alone 68.
Several recent specific findings are particularly notable: 16p11.2 17-19,71 and 15q11-13 CNVs are so far the most frequently seen in idiopathic ASD, with deletions and duplications of the former identified cumulatively in ~1% of cases. Moreover, the finding that both deletions and duplications at 16p11.2 are independently associated with ASD is striking. It was not at all clear initially that both increases and decreases in copy number (and presumably in gene dosage) at a single locus should account for similar phenotypes. In the case of 16p11.2, the additional observation that duplications carry somewhat smaller risk for ASD compared to deletions, but then also increase the liability for schizophrenia is similarly quite intriguing. Finally, the diversity of possible outcomes of reciprocal changes in copy number is underscored by the recent association of duplications at 7q11.23. Deletions of this interval result in Williams-Beuren Syndrome, characterized, in part, by a social, highly affiliative and empathic personality72 This contrast with ASD suggests that a dosage sensitive gene or genes, most likely mapping within this region, plays a critical role in the modulation of human social behavior.
Finally, two recent CNV analyses have used the observations of the frequency and distribution of de novo events to estimate the likely number of such loci contributing to ASD20,21. Both arrived, via independent methods, at ~300 ASD-related rare de novo risk CNV regions in the human genome, providing additional strong evidence of locus heterogeneity in ASD and pointing to the opportunities for discovery that still remain in the study of rare structural variation.
As with every other area of inquiry related to ASD, however, the next steps are likely to confront considerable obstacles. The replication of risk regions is a cause for celebration in the psychiatric genetics community, but the requirement to move from the identification of an associated region to a risk gene or genes is pressing. The tendency for the identified de novo CNVs to be large and encompass many genes, coupled with recent evidence supporting a multiple-rare-hit mechanism70, suggests that there is considerable work to be done to further clarify the neurobiological substrates of these reproducible genomic findings.
Further Challenges in Demonstrating and Clarifying Risk
It is axiomatic that the stronger the genetic evidence relating a variation to a clinical outcome, the more robust will be the conclusions that emerge from subsequent neurobiological studies. In turn, these discoveries will offer the possibility of providing the necessary traction to undertake translation to the clinic. Consequently, it is worth considering the challenges that remain, in light of recent findings, in establishing a clear relationship between genotype and phenotype and to anticipate how these obstacles are likely to play out with the advent of next generation sequencing.
Risk versus Causal Alleles
Recent success in rare variant discovery has, along with recent GWAS results, forced a re-examination of long-held views regarding the manner in which specific classes of variation contribute to pathology (Figure 1) and consequently how such a relationship may be established. For instance, the expectation that rare, functionally deleterious disease alleles of large effect will show a 1:1 correspondence (or nearly so) with a given phenotype has been repeatedly challenged. For example with regard to 16p11.2, risk CNVs have often not been found among affected first degree relatives within nuclear families and conversely, have been present in unaffected family members18,19,73. A cursory look at these pedigrees would tend to argue against the relevance of these variants based on Mendelian expectations. In contrast, the strength of the replicated population association underscores the importance of rethinking notions of causality and risk for rare variants in ASD.
Along similar lines, as common alleles have been found to carry much smaller risks than anticipated, the range of plausible effect sizes attributable to rare and very rare alleles has necessarily expanded. As a consequence, investigators are increasingly required to demonstrate risk probabilities for rare alleles that are neither necessary nor sufficient to lead to the phenotype (figure 1). In this regard, for both common and rare variant studies, the importance of stringent statistical thresholds, control for known confounds including population stratification, technical artifact and multiple comparisons, and independent replication cannot be overstated. Moreover, despite recent successes with regard to CNVs, it is important to recall that though effect sizes are considerably larger than those thought plausible for common variant, the low frequency of the events in question demand that discovery efforts rely on very large samples, rivaling those required for association of common alleles. Finally, the substantial number of disease-neutral rare variations in each individual’s genome, and difficulties in distinguishing these from the functional variations of interest, further complicates discovery efforts. In fact, studies in other areas of medicine have suggested that it may require specific knowledge of the protein in question to identify true disease associations74.
These considerations cumulatively point to important challenges for the analysis of whole-exome and whole genome sequencing data. Indeed even in the case of variations carrying relatively large effects and using so-called collapsing methods that tally rare variations within a gene unit, genome wide case-control cohorts are not likely to yet be sufficiently well-powered to drive initial gene discovery efforts genome wide.
Alternative strategies are already being explored. For example, in contrast to rare transmitted alleles that are quite numerous, rare coding de novo missense and nonsense single nucleotide changes are seen infrequently, occurring at less than once per exome per parent-child trio. Consequently, the observation of multiple recurrences among independent cases at a single gene is very unlikely to be a chance event. We exploited similar properties with regard to recurrent de novo structural variation in ASD to generate sufficient power from a sample of approximately 1000 cases and 1000 controls to exceed a rigorous genome-wide significance threshold for CNVs21 carrying moderate to large effects. Similar approaches may prove extremely useful with regard to exome-wide and genome-wide sequence analyses. Moreover, once a potential sentinel event is identified, the ability to investigate rare variation in specific transcript in many thousands of individuals is now plausible due to the rapidly declining costs of next generation sequencing.
Phenotypic Specificity
Another source of complexity in establishing the relationship between genetic variation and psychiatric disorder derives from the recent observations that identical mutations may be associated with highly divergent clinical phenomena. Again, 16p11.2 is a striking case in point. Deletions at this locus have been strongly associated with ASD, intellectual disability, and obesity75while duplications at this region have been convincingly shown to be associated with a wide range of behavioral phenotypes, including ASD and schizophrenia 44 and perhaps attention deficit disorder and bipolar disorder as well19.
It is worth considering the range of plausible explanations for these and similar findings: first, they could simply reflect differences in labeling. For example, social impairment could be defined in one study as ASD, while in another, as schizophrenia prodrome or negative symptoms; second, they may reflect the contribution of age-dependent penetrance. If children with ASD are also at higher risk for later developing psychotic symptoms, their ascertainment at an early age would yield evidence for association with the former, while, in adulthood, association with the latter; third, these could reflect epiphenomenon, resulting from individuals with intellectual impairment having impaired ability to compensate for limitations in, for instance, social functioning. Based on this view, gene discovery in cohorts that have both ID and ASD would be more likely to identify highly penetrant alleles and molecular mechanisms relevant to the former, rather than clarifying the latter49; fourth, these could be mediated through apparently identical, but functionally distinct alleles, for example, having gain of function versus loss of function point mutations at a single locus or, alternatively, reflecting epigenetic phenomenon, as is the case with regard to 15q11-q13 deletions leading alternative to Angelman or Prader Willi syndromes; fifth, these could reflect the involvement of shared behavioral endophenotypes among distinct disorders, for example language functioning or attention; and, finally, type 1 error combined with publication bias must be considered.
All of these explanations are reasonable, but none convincingly account for the entirety of recent observations. For example, while distinguishing ASD and schizophrenia may be difficult in some cases, their natural histories are strikingly different in most, making it unlikely that diagnostic substitution would be sufficiently common to explain the observed overlap. Similarly, while there are certainly cases in which children with ASD go on to develop psychosis in adulthood, review of the longitudinal data, though limited, does not suggested a significant increase in risk for psychotic illness in adulthood among children presenting initially with ASD76. Importantly, given the number of regions that have been found to carry risks for both, these questions may now be evaluated directly via prospective longitudinal studies of genetically homogenous subjects.
Recent data also should temper arguments that studies of rare mutations are likely to find ID genes, with ASD appearing as a secondary phenomenon. For instance, while large de novo CNVs carry marked risks for an ASD diagnoses, we found that within the Simons Simplex Collection, they correlate very modestly with lower IQ in males and show no such effect in females21. The converse was also true, lower IQ served as a poor predictor for an individual carrying an ASD de novo risk CNV. These findings, which are consistent with a recent population based twin study, showing only a modest correlation of autistic traits and lower IQ77, suggests that the co-occurrence of ID and ASD does not necessarily imply a predominant biological role for the former.
A final and we think important possibility is that the divergent phenotypes emerging from identical genetic variation(s) are a consequence of the combination of pleiotropy and locus heterogeneity (Figure 2). As considered in more detail below, this model would suggest that mutation(s) at hundreds of targets could converge on a much smaller number of molecular, cellular and anatomical pathways critical to the development and functioning of the CNS. Subtle alterations in these basic processes would then set the stage for developmental trajectories that could encompass a wide array of behavioral, emotional and social phenomena. The emergence of a complex behavioral phenotype would consequently be influenced by multiple inputs (not addressed in detail here) beyond the initial genetic “insult” including environmental factors, epigenetic mechanisms, stochastic events, and additional genetic variations, either in the form of multiple rare alleles or additional modulatory common variants.
Figure 2. Convergent rare risks and common modulators leading to divergent neuropsychiatric disorders.

The figure illustrates one explanatory model for the combination of specific rare variations, carrying large effects for ASD, with common functional variants modulating the risk for clinically distinct phenomenon. The ideogram on the left represents the human genome and the black bars, the high degree of locus heterogeneity underlying ASD; the dotted lines link divergent loci to rare mutations that disrupt protein function and/or structure and sub-serve coherent molecular pathways. Here this is illustrated as multiple mutations in synaptic molecules, though as noted in the text, it is likely that a variety of distinct neural-developmental processes are altered by ASD-related mutations. In this model, alterations to basic cellular and molecular processes are expressed through increasingly complex layers of organization, represented in the figure by the diffusion tensor imaging data showing long-range neural pathways. Moreover, the manifestations of these altered molecular and cellular processes play out across development and are influenced by a variety of factors, including, importantly, common functional genetic variation (most likely regulatory in nature), as well as stochastic effects, environmental inputs and epigenetic phenomenon (shown in the text box at the bottom of the figure). The graph on the far right further illustrates that particular genetic variations may launch and simultaneously influence diverse developmental trajectories (lines of similar colors reflect rare mutations in the same CNV or gene) that may ultimately correspond to a range of distinct clinical phenomenon (in this example ASD and schizophrenia (Scz)) or to typical development.
What is clear at present is that the field has not yet arrived at a new coherent understanding of the relationship of genotype and phenotype in ASD. The conflicting observations of: profound genetic heterogeneity; relative coherence at the phenomenological level (as exemplified in replicable neuroimaging, eye-tracking and other neuropsychological findings across patient cohorts); and, highly divergent outcomes from apparently identical rare risks, presents a serious conceptual challenge. The next big hurdle, in the wake of the current exciting era of gene discovery, will be to not only clarify how common risks emerge as distinct phenomenon but to integrate this understanding into the development of treatment targets and the reconceptualization of approaches to psychiatric diagnosis.
Top Down Versus Bottom Up – The hard work of ‘next steps’
Given the sheer amount of genomic, transcriptomic and proteomic data already available, combined with an ever-increasing, detailed understanding of the organization of local and long distance circuits involved in higher social-emotional and cognitive processes, the task of extending beyond correlational relationships in childhood onset neurodevelopmental disorders is daunting. Studies that delve deeply into single risk factors currently remain the most tractable for investigators. However, these require more than the observation of a genetic variation accompanied by a descriptive analysis of behavioral outcomes. Risk must be connected to specific neurodevelopmental phenomena to make findings relevant and “translatable.”
Yet, given that there are literally thousands of genes involved in the histogenic processes of neuronal and glial specialization, migration, axon targeting, synaptogenesis and activity-dependent stabilization and pruning, the classic bottom-up approach to decipher the involvement of disease-relevant genes in clinical populations is not without its difficulties. Confidently assigning a phenotypic dimension to a single gene, which may code for multiple protein isoforms and underpin multiple highly complex functions at different times in development, may in some cases prove an intractable problem. Even in syndromic disorders, there have proven to be substantial challenges to defining genotype-phenotype relationships. For example, in the case of Rett syndrome, mosaicism of X-inactivation, multiple, distinct mutations 78, and likely other genetic and epigenetic modifiers, appear to contribute to a variable relationship between mutation and clinical presentation.
It is consequently reassuring then to note that progress is being made in syndromic disorders that have well-delineated genetic causes, such as Fragile X, TSC or Rett syndromes 79,80 with findings leading to novel therapeutic strategies that are being translating into clinical trials81. These suggest that a somewhat reductionist approach, in the face of all the aforementioned complexity, may still be productive.
However, with regard to complex multi-genic disorders, these types of investigation will undoubtedly need to be complemented by top-down approaches that begin to elaborate the combinatorial nature of risk. For instance, recent studies of FOXP2 have identified this protein as a clear transcriptional regulatory hub influencing three implicated genes – CNTNAP282, MET83 and PLAUR84 providing the potential for synthetic investigations of ASD risk that otherwise might have been missed. Systems biological approaches are already being applied to genes within de novo CNVs85 and differential gene expression in post mortem brain40, in an effort to identify relevant molecular and neural networks and the nature of associated pathological changes. Clearly, it will be the integration of foundational neurobiological knowledge, combined with replicable genetic and genomic findings, and multi-disciplinary and computational approaches86-88, that will ultimately illuminate the pathophysiology of ASD.
Acknowledgments
This work has been supported in part by grants from the Simons Foundation (to MWS and PL) and the National Institute of Mental Health (MH067842 and MH080759 (PL) and MH089956 and MH081754 (MWS))
Footnotes
Conflicts of Interest: Dr. State co-holds a patent pertaining to rare variation in the gene Contactin Associated Protein 2 and the risk for autism spectrum disorders.
Literature Cited
- 1.American Psychiatric Association. & American Psychiatric Association . Diagnostic and statistical manual of mental disorders : DSM-IV-TR. American Psychiatric Association; Washington, DC: 2000. Task Force on DSM-IV. [Google Scholar]
- 2.Rakic P. Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci. 2009;10:724–735. doi: 10.1038/nrn2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammock EAD, Levitt P. The discipline of neurobehavioral development: the emerging interface that builds processes and skills. Human Development. 2006;49:294–309. [Google Scholar]
- 4.Geschwind DH. Advances in autism. Annu Rev Med. 2009;60:367–380. doi: 10.1146/annurev.med.60.053107.121225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell DB, et al. Distinct Genetic Risk Based on Association of MET in Families With Co-occurring Autism and Gastrointestinal Conditions. in Pediatrics. 2009;123:1018–1024. doi: 10.1542/peds.2008-0819. [DOI] [PubMed] [Google Scholar]
- 6.Ivleva EI, et al. Genetics and intermediate phenotypes of the schizophrenia-bipolar disorder boundary. in Neuroscience and Biobehavioral Reviews. 2010;34:897–921. doi: 10.1016/j.neubiorev.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 7.State MW. The genetics of child psychiatric disorders: focus on autism and Tourette syndrome. Neuron. 2010;68:254–269. doi: 10.1016/j.neuron.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakravarti A. Population genetics--making sense out of sequence. Nature genetics. 1999;21:56–60. doi: 10.1038/4482. [DOI] [PubMed] [Google Scholar]
- 9.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 10.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends in genetics : TIG. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 11.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Institute, N.H.G.R. A catalogue of published genome-wide association studies. http://www.genome.gov/gwastudies(2010)
- 13.Goldstein DB. Common genetic variation and human traits. The New England journal of medicine. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 14.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 16.Jamain S, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 20.Levy D, et al. Rare De Novo and Transmitted Copy-Number Variation in Autistic Spectrum Disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 21.Sanders SJ, et al. Multiple recurrent de novo copy number variations (CNVs), including duplications of the 7q11.23 Williams-Beuren syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durand CM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sousa I, et al. MET and autism susceptibility: family and case-control studies. in Eur J Hum Genet. 2009;17:749–758. doi: 10.1038/ejhg.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell DB, et al. A genetic variant that disrupts MET transcription is associated with autism. in Proc Natl Acad Sci USA. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson PB, et al. Further evidence that the rs1858830 C variant in the promoter region of the MET gene is associated with autistic disorder. in Autism research : official journal of the International Society for Autism Research. 2009;2:232–236. doi: 10.1002/aur.87. [DOI] [PubMed] [Google Scholar]
- 26.Alarcon M, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arking DE, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell DB, Li C, Sutcliffe JS, Persico AM, Levitt P. Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. in Autism Res. 2008;1:159–168. doi: 10.1002/aur.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Judson MC, Eagleson KL, Wang L, Levitt P. Evidence of cell-nonautonomous changes in dendrite and dendritic spine morphology in the Met signaling-deficient mouse forebrain. J Comp Neurol in press. 2010 doi: 10.1002/cne.22467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyndall SJ, Patel SJ, Walikonis RS. Hepatocyte growth factor-induced enhancement of dendritic branching is blocked by inhibitors of N-methyl-D-aspartate receptors and calcium/calmodulin-dependent kinases. J Neurosci Res. 2007;85:2343–2351. doi: 10.1002/jnr.21390. [DOI] [PubMed] [Google Scholar]
- 31.Gutierrez H, Dolcet X, Tolcos M, Davies A. HGF regulates the development of cortical pyramidal dendrites. Development. 2004;131:3717–3726. doi: 10.1242/dev.01209. [DOI] [PubMed] [Google Scholar]
- 32.Judson MC, Eagleson KL, Levitt P. A new synaptic player leading to autism risk: Met receptor tyrosine kinase. J Neurodev Disord. 2011 doi: 10.1007/s11689-011-9081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levitt P, Campbell DB. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. in J. Clin. Invest. 2009;119:747–754. doi: 10.1172/JCI37934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bill BR, Geschwind DH. Genetic advances in autism: heterogeneity and convergence on shared pathways. Curr Opin Genet Dev. 2009;19:271–278. doi: 10.1016/j.gde.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Judson MC, Bergman MY, Campbell DB, Eagleson KL, Levitt P. Dynamic gene and protein expression patterns of the autism-associated met receptor tyrosine kinase in the developing mouse forebrain. J Comp Neurol. 2009;513:511–531. doi: 10.1002/cne.21969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Judson MC, Amaral DG, Levitt P. Conserved Subcortical and Divergent Cortical Expression of Proteins Encoded by Orthologs of the Autism Risk Gene MET. Cerebral cortex. 2010 doi: 10.1093/cercor/bhq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Qiu S, Anderson CT, Levitt P, Shepherd GM. Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated met receptor tyrosine kinase. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:5855–5864. doi: 10.1523/JNEUROSCI.6569-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campbell DB, et al. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol. 2007;62:243–250. doi: 10.1002/ana.21180. [DOI] [PubMed] [Google Scholar]
- 40.Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011:>1–7. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devlin B, Melhem N, Roeder K. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain research. 2011;1380:78–84. doi: 10.1016/j.brainres.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buie T, et al. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: a consensus report. in Pediatrics. 2010;125(Suppl 1):S1–18. doi: 10.1542/peds.2009-1878C. [DOI] [PubMed] [Google Scholar]
- 43.Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 2009;25:528–535. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCarthy SE, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anney R, et al. A genome-wide scan for common alleles affecting risk for autism. Human molecular genetics. 2010;19:4072–4082. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res. 2009;53:852–873. doi: 10.1111/j.1365-2788.2009.01197.x. [DOI] [PubMed] [Google Scholar]
- 49.Skuse DH. Rethinking the nature of genetic vulnerability to autistic spectrum disorders. Trends Genet. 2007;23:387–395. doi: 10.1016/j.tig.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Bourgeron T. A synaptic trek to autism. in Curr Opin Neurobiol. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 51.Laumonnier F, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szatmari P, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bachman P, et al. Abnormally high EEG alpha synchrony during working memory maintenance in twins discordant for schizophrenia. Schizophr Res. 2008;103:293–297. doi: 10.1016/j.schres.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ching MS, et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:937–947. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gauthier J, et al. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
- 56.Berkel S, et al. Mutations detected in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010 doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- 57.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez T, et al. Disruption of Contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. 2008;82:1385. doi: 10.1016/j.ajhg.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandez T, et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. 2004;74:1286–1293. doi: 10.1086/421474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roohi J, et al. Disruption of contactin 4 in three subjects with autism spectrum disorder. J Med Genet. 2009;46:176–182. doi: 10.1136/jmg.2008.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glessner JT, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strauss KA, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 63.Bakkaloglu B, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friedman JI, et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Molecular psychiatry. 2008;13:261–266. doi: 10.1038/sj.mp.4002049. [DOI] [PubMed] [Google Scholar]
- 65.Vernes SC, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359:2337–2345. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scott-Van Zeeland AA, et al. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci Transl Med. 2010;2:56ra80. doi: 10.1126/scitranslmed.3001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stein MB, et al. A Common Genetic Variant in the Neurexin Superfamily Member CNTNAP2 Is Associated with Increased Risk for Selective Mutism and Social Anxiety-Related Traits. Biological psychiatry. 2011;69:825–831. doi: 10.1016/j.biopsych.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010 doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moreno-De-Luca D, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. American journal of human genetics. 2010;87:618–630. doi: 10.1016/j.ajhg.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mefford HC, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. The New England journal of medicine. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar RA, et al. Association and mutation analyses of 16p11.2 autism candidate genes. PLoS One. 2009;4:e4582. doi: 10.1371/journal.pone.0004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pober BR. Williams-Beuren syndrome. The New England journal of medicine. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 73.Kumar RA, et al. Recurrent 16p11.2 microdeletions in autism. Human Molecular Genetics. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 74.Ji W, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nature genetics. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bochukova EG, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–670. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.King BH, Lord C. Is schizophrenia on the autism spectrum? Brain research. 2011;1380:34–41. doi: 10.1016/j.brainres.2010.11.031. [DOI] [PubMed] [Google Scholar]
- 77.Hoekstra RA, Happe F, Baron-Cohen S, Ronald A. Association between extreme autistic traits and intellectual disability: insights from a general population twin study. The British journal of psychiatry : the journal of mental science. 2009;195:531–536. doi: 10.1192/bjp.bp.108.060889. [DOI] [PubMed] [Google Scholar]
- 78.Scala E, et al. MECP2 deletions and genotype-phenotype correlation in Rett syndrome. in Am J Med Genet A. 2007;143A:2775–2784. doi: 10.1002/ajmg.a.32002. [DOI] [PubMed] [Google Scholar]
- 79.Ehninger D, Li W, Fox K, Stryker MP, Silva AJ. Reversing Neurodevelopmental Disorders in Adults. in Neuron. 2008;60:950–960. doi: 10.1016/j.neuron.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. in Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krab LC, et al. Effect of Simvastatin on Cognitive Functioning in Children With Neurofibromatosis Type 1: A Randomized Controlled Trial. in JAMA: The Journal of the American Medical Association. 2008;300:287–294. doi: 10.1001/jama.300.3.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vernes SC, et al. A functional genetic link between distinct developmental language disorders. The New England journal of medicine. 2008;359:2337–2345. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mukamel Z, et al. Regulation of MET by FOXP2, genes implicated in higher cognitive dysfunction and autism risk. J. Neurosci. 2011 doi: 10.1523/JNEUROSCI.0181-11.2011. IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roll P, et al. Molecular networks implicated in speech-related disorders: FOXP2 regulates the SRPX2/uPAR complex. Human Molecular Genetics. 2010;19:4848–4860. doi: 10.1093/hmg/ddq415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gilman SR, et al. Rare De Novo Variants Associated with Autism Implicate a Large Functional Network of Genes Involved in Formation and Function of Synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Akil H, et al. Medicine. The future of psychiatric research: genomes and neural circuits. in Science. 2010;327:1580–1581. doi: 10.1126/science.1188654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ayhan Y, Sawa A, Ross CA, Pletnikov MV. Animal models of gene-environment interactions in schizophrenia. Behav Brain Res. 2009;204:274–281. doi: 10.1016/j.bbr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Geschwind DH, Konopka G. Neuroscience in the era of functional genomics and systems biology. in Nature. 2009;461:908–915. doi: 10.1038/nature08537. [DOI] [PMC free article] [PubMed] [Google Scholar]