

Abstract
MicroRNAs (miRNAs) have great potential as biomarkers and therapeutic agents owing to their ability to control multiple genes and potential to influence cellular behavior. Here we identified that miR-23b is a methylation-silenced tumor suppressor in prostate cancer (PCa). We demonstrated that miR-23b expression is controlled by promoter methylation and has great promise as a diagnostic and prognostic biomarker in PCa. High levels of miR-23b expression are positively correlated with higher overall and recurrence-free survival in PCa patients. Further we elucidated the tumor suppressor role of miR-23b using in vitro and in vivo models. We demonstrated that proto-oncogene Src kinase and Akt are direct targets of miR-23b. Increased expression of miR-23b inhibited proliferation, colony formation, migration/invasion and triggered G0/G1 cell cycle arrest and apoptosis in PCa. Over-expression of miR-23b inhibited epithelial to mesenchymal transition (EMT) causing a decline in mesenchymal markers Vimentin and Snail and increasing the epithelial marker, E-cadherin. Depletion of Src by RNA interference conferred similar functional effects as that of miR-23b reconstitution. miR-23b expression caused a dramatic decrease in tumor growth in nude mice and attenuated Src expression in excised tumors compared to a control miR. These findings suggest that miR-23b is a methylation-silenced tumor suppressor that may be useful biomarker in PCa. Loss of miR-23b may confer proliferative advantage and promote PCa migration and invasion and re-expression of miR-23b may contribute to the epigenetic therapy for PCa.
Keywords: MicroRNA-23b, Src kinase, prostate cancer
Introduction
Prostate cancer (PCa) is the most frequently diagnosed malignant tumor and second-leading cause of cancer deaths in American men. It is estimated that 240, 890 newly diagnosed prostate cancer cases and 33, 720 attributed deaths will occur in 2011 (1). Current PCa treatments consisting of malignant prostate ablation by radical prostatectomy, radiotherapy, hormonal therapy and/or neo-adjuvant chemotherapy, are generally curative for the majority of patients diagnosed with localized and androgen-dependent PCa. However progression to androgen-independent and metastatic disease states is often accompanied by a recurrence of PCa and treatment remains a clinical challenge (2, 3).
MicroRNAs (miRNAs) are non-protein-coding sequences thought to regulate >90% of human genes (4). Growing evidence has strongly implicated the involvement of miRNAs in carcinogenesis (5, 6). From a clinical point of view, miRNAs have great potential as diagnostic and therapeutic agents. Microarray analysis has shown a general downregulation of miRNAs in tumors when compared with normal tissues (7). Owing to the tissue specificity of miRNAs, they have become a useful tool for defining the origin of tumors in poorly differentiated cancers (8). Prognosis and survival of patients depends on the cancer stage at diagnosis. For this reason, an important issue in clinical cancer research is to identify early biomarkers of the early tumorigenic process. miRNA signatures have been reported to be useful tools for early diagnosis of cancer (9, 10).
DNA hypermethylation of CpG sites within CpG islands is known to lead to the inactivation of many tumor-suppressive miRNAs (11). One of the most common causes of the loss for tumor-suppressor miRNAs is silencing of their primary transcripts by CpG-island hypermethylation (12–16). The DNA methylation profile of tumors is useful to define tumor type, clinical prognosis and treatment response (17, 18). Epigenetic silencing of miRNAs is also involved in the acquisition of an invasive phenotype and the development of metastasis (19). Inactivation of oncogenic miRNAs (20, 21) or restoration of tumor-suppressor miRNAs (12–14) has great potential for cancer treatment.
Here we report that (i) miR-23b is frequently silenced through tumor-specific DNA methylation in PCa, (ii) miR-23b acts as a tumor suppressor microRNA and has diagnostic/prognostic implication in PCa, (iii) miR-23b directly targets proto-oncogene Src kinase and Akt, (iv) miR-23b has anti-proliferative/-migratory/-invasive effects by downregulating molecules involved in these pathways, (v) miR-23b inhibits epithelial to mesenchymal transition (EMT) markers (vi) miR-23b decreased in-vivo tumor growth and Src kinase expression in nude mice xenografts.
Materials and Methods
Cell culture, plasmids and probes/primers
Human PCa cell lines PC3, Du145, LNCaP and a non-malignant prostate cell line RWPE1 were obtained from the American Type Culture Collection (ATCC) (Manassas, VA) and grown according to ATCC protocol. These human-derived cell lines were authenticated by DNA short-tandem repeat analysis by ATCC. The experiments with cell lines were performed within 6 months of their procurement/resuscitation. Plasmids pEZX-MT01 miRNA 3′UTR target expression clones for Src (HmiT017696-MT01), AKT (HmiT004995-MT01) and miRNA Target clone control vector for pEZX-MT01 (CmiT000001-MT01) (GeneCopoeia, Rockville, MD) were purchased. TaqMan probes for hsa-miR-23b (miR-23b) and negative control pre-miR were purchased from Applied Biosystems (Foster City, CA). siRNA duplexes ((Src (Human)-3 unique 27mer siRNA duplexes (SR304574)) were purchased from Origene (Origene Technologies, Inc., Rockville, MD).
Quantitative real-time PCR
Tissue samples from radical prostectomy were obtained from the Veterans Affairs Medical Center, San Francisco, CA, USA. Total RNA was extracted and assayed for mature miRNAs and mRNAs using the TaqMan MicroRNA Assays and Gene Expression Assays, respectively, in accordance with the manufacturer’s instructions (Applied Biosystems). All RT reactions were run in a 7500 Fast Real Time PCR System (Applied Biosystems). Relative expression was calculated using the comparative Ct.
Methylation analysis of miR-23b by quantitative methylation-specific PCR (qMSP)
To investigate the mechanism involved in reduced levels of miR-23b in PCa we performed methylation analysis in the 1.0 kb upstream sequence of miR-23b. Two CpG islands-CG-1 (−120 to −3bp) and CG-2 (−820 to −650bp) were located in 1.0 kb upstream sequence and further analyzed for methylation status in cell lines and tissue samples. DNA was available only for 38 (19 pairs) of laser captured microdisected tissue samples and these samples were from the same cohort of 118 samples for which RNA expression was available. DNA was bisulfite converted using EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. The converted DNA was amplified by PCR with 400 pM of either primer set F1/R1, or F2/R2, and HotStar Taq Plus DNA Polymerase (Qiagen, Valencia, CA, USA). PCR was performed by denaturation at 95°C for 5 minutes, followed by 15 cycles of 94°C for 30 seconds, 56°C for 30 seconds and 72°C for 30 seconds. 2μl of the PCR product was added to 40 μl solution containing 20 μl TaqMan Fast Universal PCR Master Mix (2X) (Applied Biosystems), 500 pM primers F1/R1 or F2/R2. The mixed solution was aliquoted evenly into two tubes, and was added 1 μl 5μM probe for methylation reaction (PM) for Methylation (M) reaction and 1μl 5μM probe for unmethylaiton reaction (PU) probe for Unmethylation (U) reaction, respectively. Methylation in CGI-1 and CGI-2 was measured by realtime quantitative PCR with an Applied Biosystems 7500 Fast Sequence Detection. For each sample, the percent of methylation was calculated by the difference of Ct in M reaction (Ct-M) and Ct in U reaction (Ct-U).
In Situ Hybridization
In situ hybridization was performed as described previously (22). Briefly cell lines and tissues were stained using DIG-labeled locked nucleic acid (LNA)-based probes specific for mir-23b and U6 following the manufacturer’s protocol (Exiqon, Inc Woburn, MA) and detected using anti-DIG-Fluorescein, Fab Fragments (Roche Applied Science, Indianapolis, IN) (for cell lines) and BM Purple AP Substrate (Roche Applied Science) (for tissues). In situ hybridization (ISH) results for tissue array were graded according to quick score (percent cells stained × intensity of stain) and normalized to U6 levels.
Flow cytometry, cell viability, migration, clonability and invasion assays
FACS analysis for cell cycle and apoptosis was done 72 hours post-transfection using nuclear stain DAPI for cell cycle analysis or ANNEXIN V-FITC /7-AAD KIT (Beckman Coulter, Inc. Fullerton, CA) for apoptosis analysis according to the manufacturer’s protocol. Cell viability was determined at 24, 48 and 72 h by using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI) according to the manufacturer’s protocol. For colony formation assay, cells were seeded at low density (1000 cells/plate or 200 cells/plate) and allowed to grow untill visible colonies appeared. Then, cells were stained with Giemsa and colonies were counted. Cytoselect 24-well cell migration and invasion assay kit (Cell Biolabs, Inc) was used for migration and invasion assays according to manufacturer’s protocol.
Immunoblotting and Immunofluorescence
Immunoblotting was performed as described previously (23). The antibodies used were specific for Src (2123; Cell Signaling), pSrc (2101; Cell Signaling), MEK1/2 (4694; Cell Signaling), pMEK1/2(Ser217/221) (9154; Cell Signaling), p44/42 MAPK (Erk1/2) (4695; Cell Signaling), p-p44/42 MAPK (Thr2021/Tyr204) (4370; Cell Signaling), STAT3 (9132; ), p-STAT3 (Tyr705) (9145; Cell Signaling), Akt (4685; Cell Signaling), p-Akt(Ser473) (4060; Cell Signaling), Bad (9292; Cell Signaling), p-Bad(Ser136) (4366; Cell Signaling), FAK (3285; Cell Signaling), c-Jun (9165; Cell Signaling) and GAPDH (sc-32233; Santa Cruz Biotechnology, Inc.). Blots were visualized using Western blotting luminal reagent (sc-2048; Santa Cruz Biotechnology, Inc.).
For immunofluorescence, cells were transfected with precursors of miR-23b or cont-miR for 72 hours, washed and fixed with acetone-methanol (1:1) mixture and hybridized with the specific primary antibodies against EMT markers. Cells were washed and hybridized with fluorescein conjugated secondary antibody (1:1000) and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen-Life Technologies).
Luciferase Assays
The complimentary sites in 3′UTR of Src and Akt for miR-23b and mutated sequences are given in Supplemental Table 7. The Src, Akt and control vectors were purchased from GeneCopoeia and named Src-3′UTR and Akt 3′UTR. Mutated 3′UTR sequences of Src and Akt were cloned and named Mut Src3′UTR and Mut Akt3′UTR. For reporter assays, cells were transiently transfected with wild-type or mutated reporter plasmid and miR-23b or control-miR. Firefly luciferase activities were measured using the Dual Luciferase Assay (Promega, Madison, WI) 18 hr after transfection and the results were normalized with Renilla luciferase. Each reporter plasmid was transfected at least three times and each sample was assayed in triplicate.
In vivo intratumoral delivery of miR-23b and genistein
The antitumor effect of miR-23b was determined by local administration of miR-23b precursor in established tumors. Each mouse was injected sub-cutaneously with 5.0×106 PC3 prostate cancer cells. Once palpable tumors developed (average volume 55mm3), 6.25 μg of synthetic miRNA complexed with 1.6 μl siPORT Amine transfection reagent (Ambion, Austin, TX) in 50 μl PBS was delivered eight times intratumorally at 3-day intervals. Tumor growth was followed for 21 days from the first injection. All animal care was in accordance with the institutional guidelines.
Statistical analysis
Statistical analyses were performed with StatView for Windows (SAS Institute Inc. NC, USA), GraphPad-Pris-5 and MedCalc. All quantified data represents average of at least triplicate samples or as indicated. Error bars represent standard deviation of mean. All tests were performed two tailed and p-values <0.05 were considered statistically significant. Receiver operating curves (ROC) were calculated to determine potential of miR-23b or its methylation to discriminate between malignant and non-malignant samples. Chi-square tests were performed to determine correlation between miR-23b expression and clinicopathological characteristics. For disease progression analysis Kaplan-Meier approach (log-rank test) and Multiple Regression analysis were performed.
Results
microRNA-23b is significantly downregulated in PCa
To identify silenced microRNAs in PCa we performed microarray screening using cancer cell lines and a non-malignant cell line. Mir-23b was found to be a significantly downregulated microRNA in cancer compared to the non-malignant cell line. We validated the microarray data by miRNA-quantitative real time PCR (miR qRT-PCR) analysis. The results confirmed that miR-23b was downregulated in cancer cell lines (Figure 1A). In-situ hybridization also confirmed the presence of miR-23b expression (green signal) in RWPE1 cells compared to cancer cell lines (Figure 1B). Du145 cells express higher levels of miR-23b but the green signal is absent because we have to keep the exposure time same across the cell lines. To examine the biological significance of miR-23b, its expression was analyzed by quantitative real time PCR in 236 (118 pairs) laser-captured microdisected (LCM) matched tissue samples (Figure 1C, Supplemental Figure 1) and an unmatched group of 27 benign prostatic hyperplasia (BPH) and 20 tumor samples (Figure 1D) along with another cohort of 48 samples where expression was analyzed by in situ hybridization (Figure 1E). Among all samples miR-23b expression was significantly downregulated in cancer samples compared to normal or BPH (Figure 1C–E). These results indicate a putative tumor suppressor role of miR-23b in PCa.
Figure 1. miR-23b expression is downregulated in PCa.

A) Quantitative RT-PCR analysis of miR-23b. B) Fluorescence In-situ hybridization (FISH) in cell lines. C) Quantitative real time PCR analysis of mir-23b expression in 118 pairs of matched Laser-Captured Microdissected tissue samples. D) Box plot representation of mir-23b expression in a cohort of unmatched tissue samples. E) miR-23b in-situ hybridiztion (ISH) with prostate tissue microarrays. Box plot representation of ISH results of tissue samples. Representative pictures of miR-23b expression in normal, cancer and metastatic prostate tissues, U6 staining confirming the preservation of intact small RNAs in the same cases. (Ave-Average; T/N-Tumor/Normal).
Diagnostic and prognostic significance of miR-23b in PCa
Clinical demographics of the study cohort are summarized in Supplemental Table 1. Receiver operating curve (ROC) analyses were performed to evaluate the ability of miR-23b expression to discriminate between normal and tumor cases using 151 pairs of tissue samples. An area under the ROC curve (AUC) of 0.975 (P<0.0001; 95% CI=0.950 to 0.999) (Figure 2A) was obtained suggesting that miR-23b expression can discriminate between malignant and non-malignant samples and hence can be used as a diagnostic marker for PCa. To determine whether miR-23b has any prognostic significance, we divided 151 cases into low miR-23b (expression T/N<0.8 fold) and high miR-23b (expression T/N>0.8 fold) groups and performed Kaplan-Meier survival analysis and Multiple regression analysis. In Kaplan-Meier analysis, the miR-23b high group displayed significantly higher overall survival probability compared to the miR-23b low group (Logrank Test p<0.0001, HR=3.3, 95% CI=4–19) (Figure 2B). Kaplan-Meier survival analysis for recurrence free survival was performed using 105 cases. Cases with high miR-23b expression had better recurrence free survival than with low miR-23b expression cases (Logrank Test p<0.002, HR=6, 95% CI=3–13) (Figure 2C). We performed multiple regression analysis for the same set of patients using Entry, Forward, Backward and Stepwise methods one by one (Supplemental Tables 2–5). Multiple regression analysis revealed that miR-23b expression is an independent predictor of biochemical recurrence (p<0.02) as determined in entry, forward, backward and stepwise methods (Supplemental Table 2). We also determined the correlation of miR-23b expression with clinicopathological variables such as Gleason grade, pathological stage (pT) and biochemical recurrence (Figure 2D) and details are described in detail in Supplemental Results. Correlation tests revealed that cases with low miR-23b expression increased from low grade, low pathological stage to high grade and high pathological stage (Figure 2D). Patients that had PSA recurrence also had significantly low miR-23b expression. These findings suggest that miR-23b has a potential to be a diagnostic and prognostic marker for predicting the biochemical recurrence of PCa patients though addition of more samples may strengthen these results.
Figure 2. Diagnostic and prognostic significance of miR-23b in PCa.
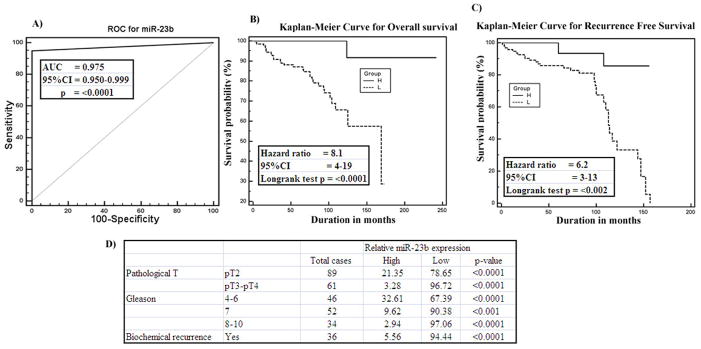
A) ROC curve analysis showing performance of miR-23b expression to discriminate between malignant and non-malignant tissue samples. B–C) Kaplan-Meier analysis for overall survival and recurrence free survival based on miR-23b expression. D) Chi-square test showing correlation of clinicopathological characteristics with miR-23b expression. Group H L-miR-23b High or Low.
Mechanism of silencing of miR-23b in PCa is through CpG hypermethylation. Correlation between miR-23b methylation status with expression and use as diagnostic marker
Two CpG islands-CG-1 and CG-2 were located in 1.0 kb upstream sequence (Figure 3A). CG-1 was the methylation ‘hot spot’ that was methylated in cancer cell lines and in tumors. Our results showed a hypermethylated sequence in matched tumor tissue samples compared to their normal counterparts (Figure 3B). Similarly PCa cell lines were also hypermethylated compared to the non-malignant RWPE-1 cell line (Figure 3C). These results indicate that miR-23b is silenced by hypermethylation in PCa. To investigate whether miR-23b methylation status affects its expression in PCa cell lines, we treated cells with the demethylating agent 5-Aza-deoxycitidine (5Aza, 1μM) for 1 week and then determined methylation status and miR-23b expression by methylation specific quantitative real time PCR and quantitative real-time PCR respectively. The methylation percentage in 5Aza treated cells was significantly decreased compared to untreated controls along with a concomitant increase in miR-23b expression (Figure 3C–D). To determine the correlation between percent methylation of miR-23b and expression levels of its target Src kinase, we analyzed the expression of miR-23b, Src kinase and the methylation of miR-23b in a different set of matched patient samples (12 pairs). A negative correlation was observed between miR-23b and Src expression (p<0.01) where as a positive correlation was found between percent methylation of miR-23b and Src expression (p<0.03) (Figure 3H) in the same patient samples.
Figure 3. Methylation status of miR-23b in PCa.

A) miR-23b gene and CpG islands within the 1.0 kb region upstream of miR-23b gene. CG1-2 CpG islands 1 and 2, F1-2-Froward primers, R1-2-Reverse primers, M1-2/U1-2 Methylation and unmethylation specific probes. Sequences of primers and probes are given in Supplemental Table 7. B) Mir-23b methylation percentage in matched tissue samples. C) Mir-23b methylation percentage and demethylation by 5Aza treatment in cell lines. D) Induction of miR-23b expression by 5Aza treatment in prostate cell lines. E) Expression of miR-23b in the same samples in which methylation was analyzed to show that miR-23b expression is inversely correlated to percent methylation. F–G) ROC for percent methylation in tissues and cell lines.
To confirm that promoter methylation is an important mechanism of miR-23b suppression in PCa patients, we investigated the correlation between methylation status of miR-23b and its expression in the same samples. There was a significant inverse correlation between percentage of methylation and miR-23b expression such that cases with higher miR-23b expression had a lower percentage of methylation (p<0.01) (Figure 3B, E, Supplemental Table 6). Sequences of primers and probes are given in Supplemental Table 7. We also performed ROC analysis to evaluate the percentage of miR-23b methylation to distinguish malignant from normal cases in matched tissue samples and cell lines. For tissues the AUC was 0.742 (p<0.001, 95% CI=0.575 to 0.870) (Figure 3G), whereas for cell lines (PC3 and DU145 vs RWPE1), the AUC was a perfect 1.00 (p<0.000; 95% CI= 0.664 to 1.000) (Figure 3H) indicating that miR-23b methylation can distinguish between disease and normal cases.
MicroRNA-23b overexpression suppresses PCa cell proliferation, migration/invasion and colony formation and induces G0/G1 cell cycle arrest and apoptosis
Next we determined the functional significance of miR-23b overexpression PCa. A significant decrease in cell proliferation was observed over time in miR-23b transfected PC3 and Du145 cells (Figure 4A) as compared to cells expressing cont-miR. miR-23b transfected cells also had low colony formation ability as the number of foci in miR-23b expressing cells were decreased when compared with cont-miR transfected cells (Figure 4B, Supplemental Figure 2). Less absorbance was observed at 560 nm with miR-23b transfected cells compared to cont-miR in the migration assay (Figure 4C) and miR-23b overexpression also significantly reduced the invasiveness of cancer cells (Figure 4D). FACS (fluorescence activated cell sorting) analysis revealed that re-expression of miR-23b leads to a significant increase (22–27%) in the number of cells in the G0-G1 phase of the cell cycle while the S-phase population decreased from 19–20% to 4% suggesting that miR-23b causes a G0-G1 arrest in miR-23b transfected cells compared to controls (Cont-miR) (Figure 4E). FACS analysis for apoptosis was performed using Annexin-V-FITC-7-AAD dye. The percentage of total apoptotic cells (early apoptotic + apoptotic) was significantly increased (13–22%) in response to miR-23b overexpression compared to cont-miR (3–4%) with a corresponding 13–20% decrease in the viable cell population (Figure 4F). Functional assays were also performed in androgen dependent LNCaP cells and the results were consistent (Supplemental Figure 3). These results indicate a tumor suppressor role for miR-23b in PCa.
Figure 4. Transient transfection of miR-23b inhibits PCa cell proliferation, colony formation, migration, invasion, and induces apoptosis and cell cycle arrest.

A) Proliferation of PC3 and Du145 cells after miR-23b transfection was significantly reduced compared to cont-miR. B) miR-23b overexpression significantly inhibits colony forming ability of PCa cells. C) Migration assays of PC3 and Du145 cells transfected with miR-23b. D) Invasion assay shows a significant decrease in the number of invading PC3 and Du145 cells transfected with miR-23b. E) Cell cycle analysis showing an increase in the G0/G1 phase of PC3 and Du145 cells overexpressing miR-23b. F) Apoptosis assay showing induction of apoptosis by miR-23b. *p<0.05, ± SD.
miR-23b directly represses proto-oncogene Src kinase and its down-stream genes involved in proliferation/survival/invasion and migration pathways in prostate carcinoma
We investigated whether Src kinase is a direct functional target of miR-23b in PCa. Complimentary sites in 3′UTR of Src kinase for miR-23b are given in Supplemental Table 7. We chose Src kinase because it has been reported to be a central stage molecule in cancer with pleiotropic effects due to the multiple signalling pathways engaged by Src kinase (Figure 5A). In PCa Src kinase has been reported to be involved in proliferation, migration and invasion. Our results showed that Src kinase is overexpressed in PCa cell lines compared to RWPE-1 cells (Figure 5B). Transient transfection of human PCa cells with Src 3′UTR plasmids along with miR-23b led to a significant decrease in relative luciferase units when compared with Src Mut3′UTR vector and cont-miR or Src Mut3′UTR vector and miR-23b (Figure 5C). These results indicate that Src kinase is a direct target of miR-23b in PCa.
Figure 5. miR-23b directly targets Src kinase and regulates downstream pathway genes and EMT.

A) Src is a center stage molecule involved in various pathways. B) Src kinase protein expression is high in PCa cell lines compared to non-malignant RWPE1 cells. C) Luciferase assays showing decreased reporter activity after co-transfection of either wild type Src-3′UTR or its mutated 3′UTR with miR-23b in PC3 and Du145 cells. Mut Src3′UTR-Mutated 3′UTR sequence. D) Western blot analysis showing miR-23b represses Src kinase translationally. E) Western blot analysis showing decreased expression of Src kinase downstream genes. F–G) Western blot and immunefluorescence showing miR-23b downregulates EMT markers.
We next determined whether overexpression of miR-23b regulates Src kinase at translational level and alters downstream signaling events. Transient transfection of PCa cells with miR-23b significantly downregulated total Src and active Src (pSrcY416) protein expression (Figure 5D). Western blot analysis showed reduced levels of downstream molecules that are involved in proliferation, migration and invasion (Figure 5E) in cells with suppressed Src expression following miR-23b overexpression. Akt is an important downstream gene of Src. We found complimentary miR-23b binding sequence in its 3′UTR (Supplemental Table 7). Our results showed that miR-23b can also directly target Akt (Supplemental Figure 4). This further compliments our results that miR-23b exerts its effects in prostate cancer at-least partly through Src-Akt pathway axis. We do not rule out the involvement of other target genes of miR-23b action in prostate cancer, though our results do indicate that Src-Akt axis pathway is partly involved.
miR-23b downregulates EMT markers and suppresses cell migration and invasion independent of proliferation
To further demonstrate that the effect of miR-23b on cell migration and invasion is an event independent of cell proliferation, we examined the effect of overexpression of miR-23b on EMT markers. A decrease in Vimentin and Slug (mesenchymal markers) and an increase in E-cadherin (epithelial marker) was observed in miR-23b transfected cells compared to the cont-miR (Figure 5F–G). The process of EMT is involved in epithelial-derived tumors causing them to become invasive and metastatic. These results show that miR-23b suppressed EMT markers along with the other migratory/invasive genes such as Src and FAK and thus confirm that effect of miR-23b on invasion/migration is independent of proliferation.
Depletion of Src by RNA interference mimics miR-23b reconstitution in PCa
Phenocopy experiments were also performed by siRNA inhibition of Src (Figure 6, Supplemental Figure). We used a validated siRNA that resulted in 80–90% Src gene knockdown at the mRNA and protein levels (Figure 6A) for further experiments. Our results showed that siRNA inhibition of Src increased G0/G1 cell cycle population (16–21%), whereas there was a decrease of 11–12% in S-phase cell population (Figure 6B, C). Approximately 15–18% of the cells were in the apoptotic fraction in Src siRNA transfected cells compared to 4% in non-specific control (Figure 6D, E). These results suggest that inhibition of Src (that in turn depletes the downstream Akt axis) by miR-23b over-expression is partly responsible for the observed phenotype in PCa cells and siRNA depletion of Src mimics the effect of miR-23b over-expression. We further performed experiments with antimiR-23b and antimiR-Neg-Control to determine if the protein expressions of the all the downregulated genes (by miR-23b) are rescued. For this experiment we chose Du145 cell line since it expresses higher levels of miR-23b than PC3. Indeed the protein expression of all genes was rescued by antimiR-23b (Figure 6F).
Figure 6. Depletion of Src kinase by siRNA mimics miR-23b over-expression.

A) Src protein levels were significantly attenuated with 50nM Src-siRNA duplex (Si) compared to a nonsilencing siRNA duplex (Con), (P-PC3, D-Du145). B–C) Cell cycle analysis showing an increase in the G0/G1 phase of PC3 and DU145 cells transfected with siRNA. D–E) Apoptosis assay showing induction of apoptosis after Src knockdown by siRNA in PC3 and Du145 cells. *p<0.05. F) Western blot analysis showing the protein expressions of Src and its downstream genes are rescued by antimiR-23b transfection.
Intra-tumoral delivery of miR-23b suppresses tumorigenecity in vivo
We also performed in-vivo growth suppression experiments to determine the tumor suppressive effect of miR-23b after local administration in established tumors. Tumor growth was significantly suppressed by miR-23b over the course of experiment compared to the cont-miR. Average tumor volume in Cont-miR was 850mm3 compared to the average tumor volume of 38mm3 in mice that received miR-23b (Figure 7A) at the termination of the experiment. We also checked the expression of miR-23b or Src kinase in harvested tumors. Our results showed that miR-23b expression was significantly high with a corresponding significant decrease in the target Src kinase gene expression in miR-23b treated tumors compared to controls (Figure 7B–C). These results confirm the tumor suppressor effect of miR-23b in a prostate xenograft model.
Figure 7. miR-23b inhibit tumor growth in vivo.
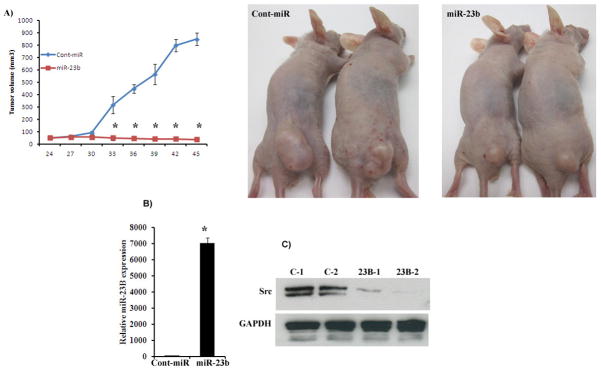
*p<0.05 A) Tumor volume following intratumoral injection of Cont-miR or miR-23b precursor into established tumors. *p<0.05. B) Average expression of miR-23b in excised tumors. D) Depletion of Src kinase by miR-23b in excised tumors. Protein was extracted from two tumors that received Cont-miR (C-1-2) or miR-23b precursor (23B-1-2).
Discussion
In this study we found miR-23b to be significantly silenced/downregulated in human prostate tumor samples when compared to adjacent normal samples. The downregulation of miR-23b expression was also observed in PCa cell lines when compared to a non-malignant cell line. This is consistent with a previous microRNA profiling analysis of 9 prostate carcinoma samples and various cell lines that showed downregulation of miR-23b (24). The suppression of miR-23b expression in tumors and cancer cell lines suggests a tumor suppressor role in PCa. However, neither the functional role nor the diagnostic or prognostic implications of miR-23b in PCa have been previously defined.
DNA methylation–mediated downregulation of miRNAs by proximal CpG islands has been described by a number of groups (15, 25), and identification of other targets for methylation may clarify the specific molecular events involved in PCa progression, enabling the prevention, diagnosis, and treatment of PCa to be approached at a molecular level. Here, we show that miR-23b is frequently silenced through tumor-specific DNA methylation in PCa tissues and cell lines and the methylation can be used as a diagnostic marker to distinguish malignant from normal cases. MicroRNAs possess several features that make them attractive candidates as new prognostic biomarkers and powerful tools for the early diagnosis of cancer (26). In this study, we found that miR-23b was predictive of overall survival and recurrence free survival. Multiple regression analysis also showed that miR-23b is an independent predictor of biochemical recurrence. MicroRNA-23b expression also distinguished malignant from normal tissues indicating the diagnostic significance of miR-23b in PCa.
An obstacle to understanding miRNA function has been the relative lack of experimentally validated targets. We validated Src kinase to be a direct target of miR-23b in PCa. Src kinase requires phophorylation within a segment of the kinase domain termed the activation loop for full catalytic activity and this auto phosphorylation site is Tyrosine 416 (27). Src kinase has been reported to be activated in prostate cancer (28, 29, 30). We demonstrated that miR-23b inhibits Src kinase at protein levels by directly binding to its 3′UTR complimentary sequences. Src is involved in multiple signaling pathways including Ras/Raf/ERK1/2, PI3K/AKT, β-catenin/c-Myc/cyclinD1 and FAK/p130CAS/MMP-9 and increased Src activity correlates with more aggressive phenotypes (28, 29, 31, 32). We observed that inhibition of Src by miR-23b overexpression reduced signaling via the MEK/ERK pathway. A previous study by Chang et al. (28) have shown that Src inhibition by small molecule inhibitors induced apoptosis and cell cycle arrest at the G0/G1 phase of the cell cycle in PCa cell lines. Our results revealed that inhibition of Src by miR-23b overexpression induced apoptosis and G0/G1 arrest in PCa cells. This effect on the cell cycle and apoptosis prompted us to study effect on Src downstream target genes AKT, BAD and STAT3, a Src target and key transcriptional factor for c-Myc and cyclin D1 (33) that are involved in the G0/G1 phase of cell cycle. We found all these genes were downregulated at the protein level. Inhibition of Src has been found to decrease the invasion and migration of PCa cells (29) through selective inhibition of Src substrates, such as focal adhesion kinase (FAK). Our results indicate that miR-23b inhibited prostate cell migration and invasion and also downregulated cJun and FAK. Overexpression of miR-23b caused a decline in the mesenchymal markers Vimentin and Snail whereas there was an increase in the expression of E-cadherin, an epithelial marker. These results suggest that miR-23b suppresses migration and invasion independent of cell proliferation and that it has an anti-metastatic effect on PCa cells. To determine whether Src inhibition is atleast partly responsible for the changes observed after miR-23b over-expression, we performed phenocopy experiments after inhibiting Src by siRNA. Our results showed that inhibition of Src was responsible for G0/G1 cell cycle arrest whereas there was a decrease in S-phase cell population. Induction of apoptosis was also observed in Src siRNA transfected cells compared to non-specific control. These results were similar to that of miR-23b overexpression assays. We further determined that the protein expressions of the genes that were downregulated by miR-23b were rescued by antimiR-23b. These results strongly indicate that the tumor suppressive effect of miR-23b is atleast partly mediated by Src-Akt axis inhibition in PCa.
The anti-proliferative effects of miR-23b observed in this study were confirmed in prostate tumor xenograft models. In conclusion, our study demonstrates that miR-23b has an important tumor suppressor role both in vitro and in vivo.
Accumulating evidence indicates that modulation of miRNA also represents an attractive strategy for therapeutic gene silencing. miRNAs potentially influence cellular behavior through the regulation of extensive gene expression networks. Therapeutic modulation of a single miRNA may therefore affect many pathways simultaneously to achieve clinical benefit. Our study is the first report demonstrating that miR-23b has diagnostic/prognostic significance and inhibits the Src-Akt axis genes, a center stage pathway that regulates proliferation, survival, migration/invasion in cancer. It also highlights the therapeutic potential of miR-23b in the treatment of PCa.
Supplementary Material
Acknowledgments
We thank Dr. Roger Erickson for his support and assistance with the preparation of the manuscript. This research was supported by the National Center for Research Resources of the National Institutes of Health through Grant Number RO1CA 138642, RO1CA130860, RO1CA160079, T32DK007790 and VA Merit Review and VA Program Project.
Footnotes
Conflict of Interest: The authors disclose no potential conflicts of interest.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 61:212–36. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Albertsen PC, Hanley JA, Fine J. 20-year outcomes following conservative management of clinically localized prostate cancer. JAMA. 2005;293:2095–101. doi: 10.1001/jama.293.17.2095. [DOI] [PubMed] [Google Scholar]
- 3.Mimeault M, Batra SK. Recent advances on multiple tumorigenic cascades involved in prostatic cancer progression and targeting therapies. Carcinogenesis. 2006;27:1–22. doi: 10.1093/carcin/bgi229. [DOI] [PubMed] [Google Scholar]
- 4.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–17. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 5.Shi XB, Tepper CG, White RW. MicroRNAs and prostate cancer. J Cell Mol Med. 2008;12:1456–65. doi: 10.1111/j.1582-4934.2008.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 8.Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, Zepeniuk M, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462–9. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 9.Heinzelmann J, Henning B, Sanjmyatav J, Posorski N, Steiner T, Wunderlich H, et al. Specific miRNA signatures are associated with metastasis and poor prognosis in clear cell renal cell carcinoma. World J Urol. 29:367–73. doi: 10.1007/s00345-010-0633-4. [DOI] [PubMed] [Google Scholar]
- 10.Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 101:2087–92. doi: 10.1111/j.1349-7006.2010.01650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–9R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 12.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 13.Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–9. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 14.Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008;105:13556–61. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, et al. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008;68:4123–32. doi: 10.1158/0008-5472.CAN-08-0325. [DOI] [PubMed] [Google Scholar]
- 16.Huang YW, Liu JC, Deatherage DE, Luo J, Mutch DG, Goodfellow PJ, et al. Epigenetic repression of microRNA-129-2 leads to overexpression of SOX4 oncogene in endometrial cancer. Cancer Res. 2009;69:9038–46. doi: 10.1158/0008-5472.CAN-09-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 17:330–9. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 19.Lujambio A, Esteller M. How epigenetics can explain human metastasis: a new role for microRNAs. Cell Cycle. 2009;8:377–82. doi: 10.4161/cc.8.3.7526. [DOI] [PubMed] [Google Scholar]
- 20.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 467:86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 21.Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 43:371–8. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majid S, Saini S, Dar AA, Hirata H, Shahryari V, Tanaka Y, et al. MicroRNA-205 inhibits Src-mediated oncogenic pathways in renal cancer. Cancer Res. 71:2611–21. doi: 10.1158/0008-5472.CAN-10-3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majid S, Dar AA, Saini S, Yamamura S, Hirata H, Tanaka Y, et al. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer. doi: 10.1002/cncr.25488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67:6130–5. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 25.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 26.Schaefer A, Jung M, Mollenkopf HJ, Wagner I, Stephan C, Jentzmik F, et al. Diagnostic and prognostic implications of microRNA profiling in prostate carcinoma. Int J Cancer. 126:1166–76. doi: 10.1002/ijc.24827. [DOI] [PubMed] [Google Scholar]
- 27.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 28.Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27:6365–75. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185–9. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 30.Fizazi K. The role of Src in prostate cancer. Ann Oncol. 2007;18:1765–73. doi: 10.1093/annonc/mdm086. [DOI] [PubMed] [Google Scholar]
- 31.Lee LF, Louie MC, Desai SJ, Yang J, Chen HW, Evans CP, et al. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene. 2004;23:2197–205. doi: 10.1038/sj.onc.1207344. [DOI] [PubMed] [Google Scholar]
- 32.Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, et al. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5:621–9. doi: 10.1158/1535-7163.MCT-05-0268. [DOI] [PubMed] [Google Scholar]
- 33.Prathapam T, Tegen S, Oskarsson T, Trumpp A, Martin GS. Activated Src abrogates the Myc requirement for the G0/G1 transition but not for the G1/S transition. Proc Natl Acad Sci U S A. 2006;103:2695–700. doi: 10.1073/pnas.0511186103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.