

Abstract
Wilson disease is a rare, inherited autosomal recessive disease of copper metabolism and may be more common where consanguinity is prevalent. Much has been known about the disease after it was first described by Kinnier Wilson as ‘progressive lenticular degeneration in 1912. Over 500 mutations of the ATP7B gene has been identified with no clear genotype to phenotype correlation. Loss of ATP7B function leads various grades of reduced biliary excretion of copper and reduced incorporation of copper into ceruloplasmin; accumulation and toxicity of copper in the liver, brain and other tissues results in liver toxicity and other myriad manifestations of the disease. The clinical features may vary from asymptomatic state to chronic liver disease, acute liver failure, neuropsychiatric manifestations and hemolytic anemia. Diagnosis is based on the combination of clinical sign's, biochemical features, histologic findings and mutation analysis of ATP7B gene. Subtle geographical differences exist with a disproportionate proportion of children presenting with acute liver failure. A high index of suspicion is needed for an early diagnosis. Ratios of biochemical indices for early diagnosis need validation across geographical regions and may not be particularly applicable in children. Better biomarkers or the need for tests for early detection of ALF persists. Drugs used in the treatment of Wilson disease include copper chelating agents such as d-Penicillamine, trientine and zinc salt. Untreated Wilson disease uniformly leads to death from liver disease or severe neurological disability. Early recognition and treatment has excellent prognosis. Liver transplantation is indicated in acute liver failure and end stage liver disease. Family screening in order to detect the disorder in the first-degree relatives is warranted. This review provides an overview of different aspects of Wilson disease including geographical differences in presentations and clinical management and the limitations of currently available tests.
Keywords: mutation, liver failure, ceruloplasmin, ATP7B, chelators
Abbreviations: CTR-1, copper transporter protein; CCS1, copper chaperone for superoxide dismutase 1; SOD1, superoxide dismutase; XIAP, X linked inhibitor of apoptosis; MRI, magnetic resonance imaging; CT, computerized tomography; ALF, acute liver failure; TM, tetrathiomolybdate; OLT, orthotropic liver transplantation; UNOS, United network for organ sharing
Wilson disease is an inherited autosomal recessive disease of copper metabolism resulting in copper toxicity. This was first described in 1912 by Kinnier Wilson as ‘progressive lenticular degeneration.1 Subsequently, in the early 1990's the role of copper in the pathogenesis of Wilson disease was established. The gene responsible for Wilson disease was identified in 1993.2–4 Approximately Wilson disease affects 1 in 30,000 to 40000 individuals.5–7 There is no community based incidence or prevalence studies in India, but this disease is more common where consanguinity is prevalent.8 At our center in a registry of Wilson disease maintained by us, approximately 15 de novo cases are diagnosed each year; more than two thirds are primarily liver related particularly in children and a considerable number present with acute liver failure.9 In a neighboring neurology referral institute with experience of nearly 400 patients approximately 15–20 new cases are registered annually primarily of neuropsychiatric presentations.10 Experience of centers with large number of cases highlight the ethnic, racial and geographical differences and similarities.
Copper metabolism
Copper is an essential trace element, and is an important cofactor for many enzymes required for cellular respiration, iron oxidation, pigment formation, neurotransmitter biosynthesis, antioxidant defense and connective formation.11,12 Although the recommended intake of copper is around 0.9 mg/day, the average diet supersedes this requirement with an intake of approximately 2–5 mg/day. Since the major route of copper elimination is by biliary excretion, the liver plays a critical role in copper metabolism by regulating biliary copper excretion. Pathways of copper homeostasis are summarized11,12 in Figures 1 and 2. Copper is absorbed in the proximal small intestine and is carried across the enterocyte by copper transporting enzyme, ATP7A into the portal circulation where copper is bound loosely into the albumin. Copper is transported into the hepatocytes via copper transporter protein (CTR-1), situated on the sinusoidal aspect of hepatocytes. Within the cell, as copper does not exist in the ionic form or free form it binds to metallochaperones, which are low molecular weight proteins that deliver copper into specific cellular targets. One such copper chaperone ATOX1 delivers the copper to Wilson disease protein, ATP7B by copper dependent protein–protein interaction. CCS1 (copper chaperone for superoxide dismutase 1) deliver the copper to superoxide dismutase (SOD1) a principally cytoplasmic defense against oxidant stress. ATP7B is present in the trans-Golgi network in case of normal and low copper state, and is important in the holoceruloplasmin synthesis. In case of copper excess, ATP7B moves toward the canalicular aspect, where it promotes biliary copper excretion. ATP7B dependent biliary copper excretion is the principal homeostatic mechanism for copper metabolism. Biliary excretion of copper also occurs by conjugation with glutathione; however it is a low affinity pathway in comparison to ATP7B dependent biliary copper excretion. Thus the liver utilizes some copper for its metabolic needs, including synthesis and secretion of ceruloplasmin (a copper containing protein) which is also involved in iron metabolism. The excess of copper is excreted in the bile in normal individuals and not in Wilson disease patients.
Figure 1.

Outline of copper homeostasis. Copper absorbed by the proximal small intestine is taken up by the liver that plays a central role in copper homeostasis by utilizing copper for metabolic needs and excreting excess copper into bile and thereby the gut, or exporting copper as copper containing ceruloplasmin used in iron metabolism and as non ceruloplasmin bound copper that may be used by or pathologically accumulated in other tissues or excreted into urine. Treatments for Wilson disease block copper absorption by the gut and increase fecal copper excretion (zinc) or increase urinary copper excretion (chelating agents D-Penicillamine and trientine). (Reproduced with permission from Thieme publication Semin Liver Dis 2011; 31(3):245–259).
Figure 2.
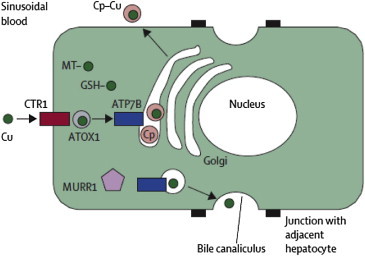
Pathways of copper metabolism in the hepatocyte. Cu = copper. CTR-1 = copper transporter 1. MT = metallothioneins. GSH = glutathione. Cp = ceruloplasmin (Reproduced with permission from Thieme publications Lancet 2007; 369:397–408).
Molecular pathogenesis
Wilson disease results from the mutation of ATP7B gene which is inherited as an autosomal recessive trait. ATP7B gene situated on the long arm of chromosome 13, encodes copper-transporting P-type ATPase which is situated intracellular. The ATP7B protein is involved in the incorporation of copper into the ceruloplasmin and biliary copper excretion. ATP7B mutation results in absent or non-functional ATPase with defective synthesis of ceruloplasmin and defective biliary copper excretion. The resulting copper accumulation in the hepatic and extrahepatic tissues leads to copper toxicity with myriad clinical features of Wilson disease. Being a prooxidant copper generates enough reactive oxygen species producing cellular damage. Copper also produces apoptosis due to the conformational change in the antiapoptotic protein, X linked inhibitor of apoptosis (XIAP). Figure 3 summarizes the pathogenesis of copper toxicity.11 More than 500 ATP7B mutations have been described in Wilson disease.13 Despite great efforts at detecting correlation a clear-cut genotype-phenotype pattern remains elusive although homozygote's with the H1069Q mutation are found frequently in patients of Eastern European origin and present more frequently at an older age with neurological symptoms.14 It is likely that epigenetic and other genetic factors including modifier genes may attenuate or amplify its effect on the mutated gene.11
Figure 3.
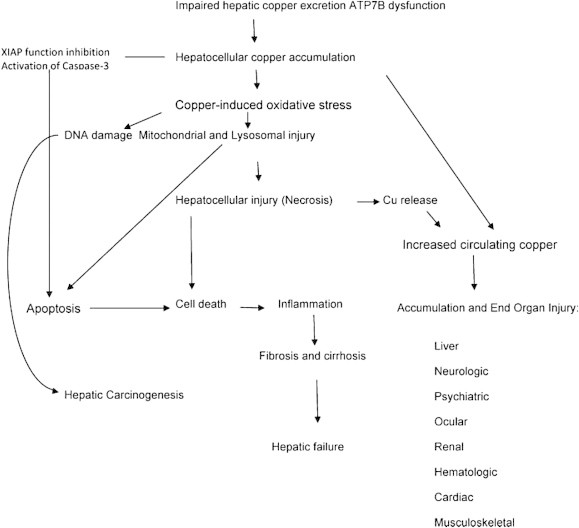
Pathogenesis of copper toxicity. The main pathways of copper overload/toxicity is through both a direct oxidative stress mechanism with lipid peroxidation of membranes, mitochondria, and DNA; and also from unregulated apoptosis resulting in cell death from copper-induced changes in the anti-apoptotic protein, X-linked inhibitor of apoptosis (XIAP), and its loss of inhibitory control of caspase-3 with copper accumulation (Reproduced with permission from Thieme Publications, Semin Liver Dis 2011; 31(3):245–259).
Clinical features
The clinical phenotype includes myriad presentations varying from asymptomatic state to chronic liver disease, neuropsychiatric manifestations or acute liver failure. Individuals usually becomes symptomatic between the ages of 5 and 35 years, which reflects the potential of the liver to store excess copper, but both younger individuals and older individuals have been well described.15 Liver disease manifestations are common in the younger age group usually the first and the second decade, while neurologic symptoms occur in the third decade of life.11 Wilson disease patients with neurological symptoms are assumed to have liver disease at the time of detection, which is usually asymptomatic.12 The natural history of the disease without treatment is almost always worsening of disease leading to death.16
Liver disease
The type of the liver disease can vary from biochemical abnormalities to any spectrum of liver disease including acute hepatitis, acute liver failure, and cirrhosis with portal hypertension. Presentations due to liver disease are common in children. Children may be asymptomatic and may present with hepatomegaly or abnormal serum aminotransferases.17 Some patients may have non-immune mediated (Coomb's negative hemolytic anemia) with transient episodes of jaundice or low-grade hemolysis, even when liver disease is asymptomatic. 17
Acute liver failure presentation is the most dramatic and may arise with catastrophic suddenness. It is considered rare, constituting only 3% of ALF cases (n = 9) in the pediatric ALF series in USA18 and associated with a high mortality reaching 95%.19 However, in some registries ALF presentation constitutes almost 30% of all cases of Wilson disease.20 Although females in general are overrepresented in patients with Wilson disease this is not universal. Literature from the Kings College Hospital series,21 our own series,20 and in an Egyptian series8 the disease was seen more often in boys than girls. Most patients in our series were prepubertal which may be an explanation for the higher incidence in boys. Both Coomb's negative hemolytic anemia and renal insufficiency may contribute to the clinical picture. This again has wide geographical differences. Coomb's negative hemolysis is often present and may be a distinctive diagnostic feature particularly in those presenting with jaundice or acute liver failure; in one large Japanese series hemolytic anemia was the sole presenting feature three of their 282 (1.0%) patients.22 However, hemolysis was present in 19 of their 77 patients (28%) who had jaundice at presentation.22 In patients with advancing acute liver failure, encephalopathy develops with cerebral edema.11 Previously treated patients who have stopped their medications, can also have acute presentation with rapid deterioration,19 which is a particular problem when patients enter the adolescence period or when it first occurs in teenagers.
Splenomegaly may be an important clinical clue to the diagnosis in patients who present with hypersplenism or those with signs of chronic liver disease.17,23 Hepatocellular carcinoma is less frequently associated with Wilson disease than with hemochromatosis but in a large series, it was seen in 1% of their Wilson's cohort during follow up.9 Table 1 summarizes the clinical features of Wilson disease patients with liver disease across 5 series.
Table 1.
Clinical symptoms in Wilson disease patients with liver disease.
| Author, Country [Ref] | Walshe, UK24 | Stremmel et al, Germany25 | Schilsky et al, USA26 | Scott et al, UK27 | Ferenci, Austria15 |
|---|---|---|---|---|---|
| N with liver disease (out of) | 87 (>250) | n.a (51) | 20a (320) | 17a (45) | 30 (64) |
| Presenting symptom | |||||
| Jaundice, anorexia, vomiting (%) | 44 | 14 | 15 | 41 | 37 |
| Ascites/edema (%) | 26 | 14 | 50 | 24 | 23 |
| Variceal hemorrhage (%) | 6 | 10 | 6 | 3 | |
| Hemorrhagic diathesis (%) | 8 | 3 | |||
| Hemolysis (%) | 20 | 10 | 5 | 10 | |
| Hepatomegaly/splenomegaly (%) | 16 | 49 | 15 | 29 | 17 |
| Acute liver failure (%) | n.a | n.a | n.a | n.a | 17 |
| Asymptomaticb (%) | 18 | 5 | 23 | ||
Only cases with chronic active hepatitis.
Elevated ALT at routine testing or accidental finding of cirrhosis or of Kayser-Fleischer rings (Reproduced with permission from Elsevier Publications J Hepatol. 2012; 56:671–85).
Neurological
Neurological assessment should be sought in all patients with Wilson disease. Neurological manifestations are the presenting features in 40–50% of patients with Wilson disease28 but are heavily biased by the referral system.29 The neurological manifestations are categorized as: (a) an akinetic-rigid syndrome similar to Parkinson's disease, (b) pseudosclerosis dominated by tremor, (c) ataxia, and (d) a dystonic syndrome.30 Behavioral changes, decrease in scholastic performance, or hand-eye discoordination can present before neurological signs.29 Micrographia, drooling, dysarthria, spasticity are other common neurological signs.31 The characteristic tremor in Wilson disease, similar to a wing beating appearance is described as coarse and irregular with proximal tremulousness. Dystonia are known to be focal, segmental or generalized, which often leads to severe contractures.32 In a large cohort of patients from Bangalore,29 the commonest neurological presentation includes parkinsonism 62.3%, dystonia 35.4%, cerebellar 28%, pyramidal signs 16%, chorea 9%, athetosis 2.2%, myoclonus 3.4% and behavioral abnormalities 16%. In patients with advanced liver disease, neurological presentations can be misdiagnosed as hepatic encephalopathy.33
Psychiatry
Psychiatric abnormalities can present before neurological or hepatic signs in one-third of patients.33 Decreased academic performance or personality changes like impulsiveness, labile mood, sexual exhibitionism, and inappropriate behavior can be seen in children, while psychotic features resembling paranoia, schizophrenia or depression can be seen in older patients.32 Affective disorder, major depression, and dysthymia were commoner psychiatric diagnosis in Wilson disease patients in a series from Bangalore.34
Infrequent presentations include gigantism, lunule cerulea, renal abnormalities such as aminoaciduria nephrolithiasis, hypercalciuria and nephrocalcinosis, cardiomyopathy, myopathy, chondrocalcinosis and osteoarthritis, hypoparathyroidism, and pancreatitis.33 Infertility or repeated miscarriages as presentations are rare although diagnosed patients have these as associations.33
Diagnosis
Early diagnosis and prompt treatment of Wilson disease is critical for complete recovery which is otherwise quite fatal. Hence, it is important to consider the disease in any unexplained hepatic and neurological presentation.33 The working party at the 8th International Meeting on Wilson disease, Leipzig 2001 (Table 2)15,33 proposed a diagnostic score, which provides a good accuracy. Table 3 shows the routine tests done for diagnosis of Wilson disease. With a score of >4 the diagnosis of WD can be made with reasonable accuracy.15
Table 2.
Scoring system developed at the 8th International Meeting on Wilson's disease, Leipzig 2001.33
| Symptoms | |
| Kayser–Fleischer rings | |
| Present | 2 |
| Absent | 0 |
| Neurologic symptoms or typical imaging at brain magnetic resonance imaging | |
| Severe | 2 |
| Mild | 1 |
| Absent | 0 |
| Serum ceruloplasmin | |
| Normal (>0.2 g/L) | 0 |
| 0.1–0.2 g/L | 1 |
| <0.1 g/L | 2 |
| Coombs negative hemolytic anemia | |
| Present | 1 |
| Absent | 0 |
| Other tests | |
| Liver copper (in the absence of cholestasis) | |
| >5x ULN (>4 μmol/g) | 2 |
| 0.8–4 μmol/g | 1 |
| Normal (<0.8 μmol/g) | −1 |
| Rhodanine-positive granulesa | 1 |
| Urinary copper (in the absence of acute hepatitis) | |
| Normal | 0 |
| 1-2x ULN | 1 |
| >2 ULN | 2 |
| Normal, but >5x ULN after D-Penicillamine | 2 |
| Mutation analysis | |
| On both chromosome detected | 4 |
| On 1 chromosome detected | 1 |
| No mutations detected | 0 |
| Total score | Evaluation: |
| 4 or more | Diagnosis established |
| 3 | Diagnosis possible, more tests needed |
| 2 or less | Diagnosis very unlikely |
ULN: upper limit of normal.
If no quantitative liver copper available.
Reproduced with permission from Elsevier Publications J Hepatol. 2012; 56:671–85.
Table 3.
Routine tests for diagnosis of Wilson disease.
| Test | Typical finding | False negative | False positive |
|---|---|---|---|
| Serum ceruloplasmin | Decreased by 50% of lower normal value | Normal levels in patients with marked hepatic inflammation. Overestimation by immunologic assay Pregnancy, estrogen therapy |
Low levels in:
|
| 24 h urinary copper | >100 mcg/24 h >40 mcg/24 h in children |
Normal
|
Increased
|
| Serum free copper | >200 mcg/L | Normal if ceruloplasmin overestimated by immunologic assay | |
| Hepatic copper | >250 mcg/g dry weight | Due to regional variation
|
Cholestatic syndromes |
| Kayser–Fleischer rings by slit-lamp examination | Present | Absent
|
Primary biliary cirrhosis |
Reproduced with permission from Elsevier Publications J Hepatol. 2012; 56:671–85.
Kayser–Fleischer ring
Kayser–Fleischer (K–F) ring (Figure 4), are considered to be the hallmark of WD, which are corneal copper deposits within descement membrane, appearing as granular golden-greenish layer near the limbus. Often they are quite obvious to the trained eye, but are best seen by slit-lamp examination. The frequency of K–F rings in children presenting with liver disease and young asymptomatic patients is variable and reported to be low,35 but may be high in certain populations and may be seen in up to 80–90% of patients.8,9 However, they are almost universal in neurological presentations and may be seen in up to 98% of patients with neurological and psychiatric presentation.36 It may rarely be found in other chronic cholestatic syndromes including primary biliary cirrhosis, cryptogenic cirrhosis, chronic active hepatitis, and neonatal hepatitis.37 but these disorders can easily be differentiated clinically from Wilson disease.
Figure 4.
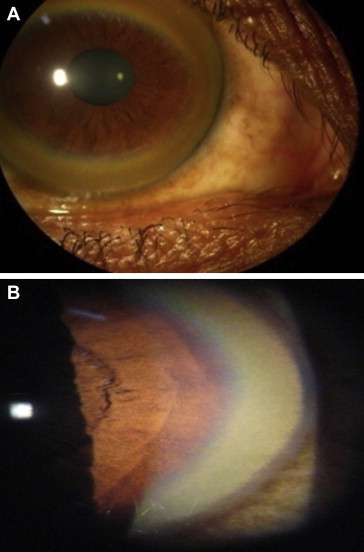
(A): Kayser–Fleischer ring on naked eye examination, (B): Kayser–Fleischer ring on slit-lamp examination.
Other rare ophthalmological changes such as sunflower cataracts, found by slit-lamp examination, represent deposits of copper in the lens and do not affect vision.17 The effect of medical treatment and liver transplantation can lead to disappearance of these ophthalmological findings, but it does not correspond with subsidence of clinical symptoms.17 The non-compliance of medical therapy can lead to reoccurrence of either of these ophthalmologic findings.38 Night blindness, exotropic strabismus, optic neuritis, and optic disc pallor are rarer ophthalmological manifestations.12
Serum ceruloplasmin
Ceruloplasmin, a 132 kDa protein, is produced mainly in the liver.17 It is a major carrier of copper in the blood and it contains six copper atoms per molecule (holoceruloplasmin) but may be present just as the protein without the copper (apoceruloplasmin). Ceruloplasmin is an acute phase reactant possessing a nitric oxide oxidase property, levels of which may be measured enzymatically by its activity toward specific substrates.17 It can also be measured by antibody dependent assays such as radioimmunoassay, radial immunodiffusion, or nephelometry. Enzymatic assays are better than immunologic assays as the latter may overestimate ceruloplasmin concentrations.39 In normal individuals, levels are very low from early infancy to the age of 6 months, followed by higher value in early childhood (at approximately 0.3–0.5 g/L), before dropping down to the adult range.17 By the enzymatic assay, normal values varies among laboratories with a lower limit of detection ranging between 0.15 and 0.2 g/L and lower value may be seen in approximately 20% of heterozygotes.33 In patients with neurological presentation it may be typically decreased, whereas in 50% of active liver disease patients it may be found in the low normal range.33 It is low in other conditions like marked renal or enteric protein loss, malabsorption syndromes or with severe end stage liver disease of any etiology,40 while it is increased by acute inflammation, hyperestrogenemic states such as pregnancy and estrogen supplementation.17 Patients with aceruloplasminemia who lack the protein entirely due to mutations may exhibit hemosiderosis but do not have copper accumulation.17 In a prospective cohort of liver disease patients, subnormal ceruloplasmin showed positive predictive value of only 6% as a screening test.41 Korman et al reported a poor predictive value of ceruloplasmin for diagnosis of Wilson disease in acute liver failure.42 Low serum ceruloplasmin heightens suspicion of Wilson disease but is rarely used in isolation and is often used in conjunction with other test such as 24 h urinary copper and K–F ring. Values below 10 mg/dl strongly favor a diagnosis of Wilson disease; such values are unlikely in carriers with Wilson disease.7
Serum copper (non ceruloplasmin bound copper)
Inspite of being a disease of copper overload, the total serum copper is usually reduced in proportion to the decreased ceruloplasmin in the circulation, whereas in acute liver failure, it may even be higher due to the sudden release of copper from liver tissue damage. Increased or normal serum copper levels inspite of decreased ceruloplasmin levels, suggests increased concentration of copper not bound to ceruloplasmin (non ceruloplasmin bound copper or free copper). Free copper or non ceruloplasmin serum copper can be calculated by subtracting ceruloplasmin bound copper from the total serum copper concentration (i.e. total serum copper in mcg/L—3.15 × ceruloplasmin levels in mg/L).43,44 Free copper is suggested as a useful diagnostic test for Wilson disease but is limited by its dependency on the adequacy of the methods for measuring both serum copper and ceruloplasmin on the same sample and also by the gross variability in the values.7 In most untreated patients, free copper is elevated above 200 mcg/L (normal reference < 150 mcg/L),17 but may also be elevated in acute liver failure of any etiology, chronic cholestasis, or copper intoxication.45 It is of more importance in monitoring of pharmacotherapy,33 where levels less than 50 mcg/L may indicate systemic copper depletion that can occur with prolonged treatment.17 Walshe demonstrated lack of correlation between the serum and urinary copper and found the term free copper misleading and suggesting it be replaced by the accurate term “non ceruloplasmin bound copper”.46 More recently, exchangeable copper, which indicates labile copper, was found to be analytically reliable with good sensitivity and specificity in diagnosing Wilson disease. Relative Exchangeable Copper calculated by the equation (REC = ratio of exchangeable copper/total copper) was suggested as a new biomarker for diagnosing Wilson disease.47
Urinary copper excretion
The 24 h urinary copper excretion may be useful for the diagnosis of Wilson disease and for monitoring treatment. In untreated patients, it reflects the amount of free copper in the circulation and depends on urine volume and the total creatinine excretion per 24 h for accurate determination. The test is inaccurate for determination in case of renal failure. The 24 h urinary copper excretion greater than 100 mcg is considered diagnostic in symptomatic Wilson disease patients.48–50 In a study comparing 40 mcg/24 h vs. 100 mcg/24 h found the lower value best for diagnostic accuracy.36 It is insignificant in healthy individuals, but excretion above 40 mcg/24 h can be suggestive of Wilson disease in asymptomatic children49 and therefore may be used in asymptomatic siblings of affected patients.33 Urinary copper excretion is higher in heterozygote's than in controls but it rarely exceeds the normal values. Interpretation of excretion values can be arduous due to the overlap with other liver disease like acute liver failure of any etiology, autoimmune hepatitis, chronic active liver disease or cholestasis.33 In pediatric population, urinary copper excretion with d-Penicillamine administration is a useful diagnostic test but was inaccurate in the diagnosis of asymptomatic siblings and is not recommended in adults. In children a 500 mg of d-Penicillamine is given orally at the beginning and again 12 h later during 24 h urinary excretion, irrespective of body weight. When the excretion is more than 1600 mcg/24 h, it clearly discriminates Wilson disease from other liver disease such as autoimmune hepatitis, primary sclerosing cholangitis, and acute liver failure of other etiology.51
Liver biopsy
Increased hepatic copper accumulation is the characteristic finding of Wilson disease and may be the single test that readily differentiates Wilson disease from other non-cholestatic chronic liver disease. Histochemical evaluation with special stains like rhodamine or orcein detects only the lysosomal copper deposition, revealing focal copper stores in less than 10% of patients. Hence, quantification of copper concentration in hepatic parenchyma is diagnostic method of choice. The accuracy of measurement is improved with adequate specimen size of at least 1 cm core length and placed dry in copper free container. A copper content > 250 mcg/g dry weight is considered diagnostic for Wilson disease, but lowering the threshold to 70 mcg/g dry weight improved sensitivity from 83.3% to 96.5% but decreased specificity (98.6% vs. 95.4%).52 A copper content < 50 μg/g dry weight is helpful in eliminating diagnosis of Wilson disease in untreated patients.53 Inhomogeneous distribution of copper within the liver in later stages of disease may lead to underestimation.33 The higher levels of hepatic copper content can also be seen in idiopathic copper toxicosis syndromes (Indian childhood cirrhosis).33 Long standing cholestatic disorders such as primary biliary cirrhosis, biliary atresia, prolonged extrahepatic biliary obstruction and sclerosing cholangitis may have increased copper accumulation but may be easily distinguished clinically and rarely lead to diagnostic confusion.36
Liver histology may be a useful adjunct for diagnosing indeterminate cases and also in staging the presence and degree of fibrosis, since evidence of fibrosis is seen in 90% of asymptomatic patients.54 The histological abnormalities in early stages include mild steatosis (Figure 5), glycogenated nuclei in hepatocytes, and focal hepatocellular necrosis, whereas, progressive parenchyma damage leads to fibrosis (Figure 6) and consequently develops into cirrhosis. Even in acute liver failure, marked hepatocellular degeneration via apoptosis and parenchyma collapse is noticed on the background of cirrhosis making Wilson disease a prime example of acute on chronic liver failure. Ultrastructural analysis reveals mitochondrial abnormalities (Figure 7) such as increased intracristal space with dilatation of the tips of the cristae, resulting in a cystic appearance which is often are considered pathognomic.55 These changes may regress with prolonged therapy.56
Figure 5.
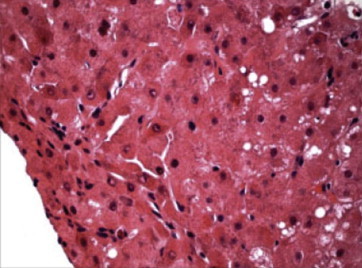
Liver biopsy showing mild steatosis and hepatocellular swelling. (Reproduced with permission from Thieme Medical Publishers. Semin Liver Dis. 2011;31:239–244).
Figure 6.
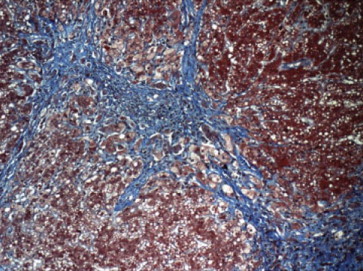
Liver biopsy showing pericellular fibrosis in evolving cirrhosis. (Reproduced with permission from Thieme Medical Publishers. Semin Liver Dis. 2011;31:239–244).
Figure 7.
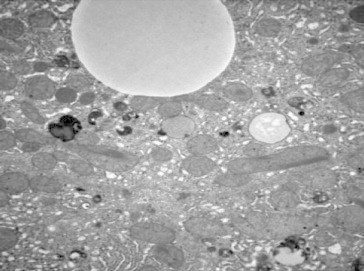
Liver biopsy showing mitochondria abnormalities in Wilson disease. (Reproduced with permission from Thieme Medical Publishers. Semin Liver Dis. 2011;31:239–244).
Neuroimaging
Neurologic evaluation should be performed in patients with neuropsychiatric manifestations and may also be carried out in presymptomatic and hepatic Wilson disease. It is particularly useful when distinction between causes for early onset extrapyramidal disorders is required57 where magnetic resonance imaging (MRI) or computerized tomography (CT) of the brain may detect structural abnormalities which include increased density on CT or hyperintensity on T2 magnetic resonance imaging in the region of the basal ganglia, tectal-plate and central pons.58 The face of the giant panda sign, characteristic finding in Wilson disease is seen in minority of patients.59 Other neuroimaging techniques as magnetic resonance spectroscopy, single-photon emission computed tomography, transcranial brain parenchyma sonography, auditory-evoked brainstem potentials might be helpful in diagnosing early brain damage in Wilson disease.33
Diagnostic dilemma in Acute liver failure (ALF)
The clinical features of ALF are essentially the same across many aetiologies. Some distinct features that suggest a diagnosis of ALF due to Wilson disease include Coomb’s-negative hemolytic anemia, coagulopathy unresponsive to vitamin K administration, renal failure, relative modest rises in serum aminotransferase, elevated serum copper, elevated 24 h urine copper and presence of K–F rings.17 Presence of ascites and splenomegaly are subtle clues about the diagnosis of Wilson disease.20,23 In ALF, the serum ceruloplasmin may be falsely elevated, and together with the 24 h urine copper studies may have a turnaround time of several days thus delaying the diagnosis. Moreover K–F rings may be absent in more than 50% of patients.12 Hence, several investigators have tried to investigate alternative diagnostic clues for early diagnosis of Wilson disease and ALF. Berman and colleagues described a ratio of alkaline phosphatase to total bilirubin < 2 and AST/ALT > 4 in their series of 6 patients with Wilson disease and compared it to 43 patients with non-Wilsonian FHF.60 The ratio of ALP/TB < 2 provided a sensitivity and specificity 100% in identifying fulminant Wilson disease.60 However, Sallie and coworkers61 could not replicate this finding and the distinction between fulminant WD and other causes of FHF was not possible. Furthermore, this ratio is not helpful in children given the high alkaline phosphatase contributed by growing bone.61 More recently Korman42 et al used a ratio of ALP/Total bilirubin < 4 and an AST/ALT ratio > 2.2 in early identification of Wilson disease producing ALF. They reported an excellent sensitivity of 94%, 94% and 100%for the ratio of AP/TB, AST/ALT, and AP/TB plus AST/ALT respectively.42 A recent study investigating the same indices demonstrated a sensitivity of 21.7%, 36.7% and 38.3% for patients with WD pediatric ALF.20 This discrepancy could be attributed to the fact that Korman's series consisted of patients with advanced liver failure with a MELD score of 40 and may hence not be considered “early”. This view was supported by O'Brien and Williams who cautioned about the generalizability of this result including in the pediatric population where ALF is more common.43 All this underscores the need for better biomarkers or tests for the early detection of WD in the setting of ALF. The diagnosis of WD and ALF should be whenever possible, since it may be helpful to screen asymptomatic siblings.33
Genetic testing
Genetic tests are useful to ascertain the diagnosis in suspected patients and to screen asymptomatic siblings. Detection of ATP7B mutations is assigned the highest weight in the scoring system for Wilson disease.3 However, with >500 ATP7B mutations reported and with no common set of mutation across regions and clinical manifestations, its role in the diagnosis of index cases is limited. In studies of Indian patients, common mutations include C813A in 19%,62 G3182A in 16%,63 C813A in 12%,63 T33053 in 6%,64 C2975A in 6%,64 2977insA in 6%,64 whereas in the Caucasians population, the most common mutation is H1069Q which accounts for 37–63%12,15 of cases and is the basis for screening. H1069 mutation was not identified in an Indian series.63
Family screening
In Wilson disease, all first-degree relatives must be screened with biochemical markers including liver function tests, serum copper and ceruloplasmin concentration, urinary copper analysis and slit-lamp examination for identification of K–F ring. There is 25% probability of finding a homozygote amongst the siblings.11,33 In indeterminate cases, liver biopsy for hepatic copper quantification can be useful. In families, when both the mutations have been found in index patient molecular genetic analysis can be useful, whereas when mutations are not found haplotype analysis may be helpful.12
Differential diagnosis
Wilson disease should be considered in the differential diagnosis of any young patient with unexplained liver disease, or concomitant liver and neuropsychiatric illness, or presentation with ALF. Non-alcoholic fatty liver disease, steatohepatitis, and autoimmune hepatitis may have common findings in the initial stage of the disease, but when this stage goes unnoticed and cirrhosis is the presentation, all other aetiologies of liver disease should be excluded.11 Other disorders like fatty acid oxidation metabolism, mitochondrial disease, or non-Wilsonian copper overload syndromes (such as Indian childhood cirrhosis) must also be considered particularly in children. Indian childhood cirrhosis has waned in incidence and afflicts particularly young children <3–5 years.65
Prognosis
Patients with Wilson disease have an excellent prognosis provided they are compliant with therapy. With chelation or liver transplantation, prolonged survival can be achieved.25,49,66 Untreated Wilson's disease is uniformly fatal, with most patients dying from liver disease or from complications of progressive neurologic disease. Patients who discontinue treatment are at high risk of fulminant hepatic failure as reported in one study where 8 of the 11 patients died with an average survival of only 2.6 years.67 Hence all guidelines recommend treatment for life which should not be discontinued, unless liver transplantation has been performed17,33 (AASLD Class I, Level B, EASL-GRADE II-1, B, 1).
Various prognostic indices have been developed for patients with acute liver failure to identify patients who require liver transplantation. Nazer et al68 developed a score of the following: serum bilirubin, serum aspartate aminotransferase and prolongation of prothrombin time above normal. A score of greater than 7 connotes a high risk of mortality. Later Dhawan et al21 from the same group modified the scoring system. A score greater than 11 was nearly always fatal. Petrasek69 and coworkers from Czeck republic found the revised Kings College criteria useful whereas Fischer et al70 sounded caution in the application of the score.
Treatment
Treatment for Wilson disease focuses on achieving a negative copper balance either with chelators (drugs that promote cupriuresis) or zinc which reduces absorption or both. Liver transplantation is indicated in patients with acute liver failure, unresponsive to medical treatment or those with end stage liver disease. Drugs available for the treatment of Wilson disease include d-Penicillamine, Trientine, Ammonium tetrathiomolybdate and Zinc (Table 4). It should be remembered that these drugs have not been subjected to randomized controlled trials,71 and trials are underway to clarify these issues.
Table 4.
Drugs used in treatment of Wilson disease.3,7,17
| Drug | Mode of action | Adverse effects | Dose | Monitoring adequacy |
|---|---|---|---|---|
| D-Penicillamine | Chelates copper and induces copper excretion | Early: Fever, rash, lymphadenopathy, cytopenias, proteinuria Late: Nephrotoxicity, lupus like syndrome, good Pasteur syndrome, bone marrow toxicity, dermatological toxicity (elastosis perforans serpiginosa) Very late: Nephrotoxicity, myasthenia gravis, polymyositis |
Initial: Adult 750 mg–1500 mg BID—QID Children—20 mg/kg/d BID—QID Maintenance 15 mg/kg/day |
24 h urinary copper Nonceruloplasmin bound copper |
| Trientine | Chelator and induces copper excretion | Gastritis, aplastic anemia, sideroblastic anemia | Initial: Adult 750 mg-1500 mg BID—QID Children—20 mg/kg/d BID—QID Maintenance 15 mg/kg/day |
24 h urinary copper Nonceruloplasmin bound copper |
| Zinc | Stimulates metallothionein in intestine and liver and, inhibits intestinal absorption of copper | Gastritis, biochemical pancreatitis, doubtful immunosuppressant effects | Adults (>50 kg): 150 mg/day of elemental iron in three divided doses Children (<50 kg): 75 mg/day of elemental iron in three divided doses | 24 h urinary copper Nonceruloplasmin bound copper 24 h urine zinc level |
| Tetrathiomolybdate | Tripartite complexes with protein and copper and renders it unabsorbable | Anemia, neutropenia, rarely hepatotoxicity | Still an experimental therapy and not commercially available. 20 mg 3× daily with meals, 20 mg 3× daily between meals | |
d-Penicillamine
Dimercaprol was used as chelating agent until 1956 when d-Penicillamine was introduced.72 Since then, this has been widely evaluated and preferred drug in the treatment of Wilson disease. Penicillamine is the breakdown product of Penicillin with free sulphydryl group which acts as the copper chelator. d-Penicillamine may also act by inducing metallothionein thereby enhancing cupriuresis and sequestering free copper.17
d-Penicillamine has a double-peaked curve73–75 for intestinal absorption with oral bioavailability of 40–70%.74,76 Major route of excretion is by kidney. d-Penicillamine is started at dose of 250–500 mg/day, increased by 250 mg every 4–7 days to a maximum of 1000–1500 mg/day in 2–4 divided dosages. Maintenance dose is usually 750–1000 mg/day administered in two divided doses.17 Food inhibits the absorption of the drug and hence it is best recommended 1 h before or 2 h after the meal.17
According to a systematic review analyzing one randomized and twelve observational studies, d-Penicillamine is probably the most effective drug in hepatic presentation of Wilson disease.71 Numerous studies have shown the efficacy of d-Penicillamine in liver disease.77–82 Improvement in clinical symptoms (jaundice and ascites) and synthetic functions of the liver (albumin and INR) usually occurs after 2–6 months of treatment. Failure of compliance can lead to progression of liver failure in next 1–12 months.67
Worsening of neurological symptoms may be noticed in 10–50% of the patients during the initial phase of treatment.83,84 The propensity for the neurological deterioration is highest for d-Penicillamine among the chelator. Other side effect profiles are depicted in Table 4. Since cytopenias and renal involvement are the most common adverse events, these need to be monitored at regular intervals. d-Penicillamine tends to interfere with pyridoxine action and hence supplemental pyridoxine is still advocated.85 Penicillamine also interferes with collagen cross-linking86 because of which the dose is reduced in the latter months of pregnancy.
Trientine
Trientine was introduced in 1982 as an alternative to d-Penicillamine in view of its side effect profile.87 However, Trientine is still not available in various parts of the world. It is a chelator with polyamine like structure and lacks sulphydryl group. It chelates by forming a stable complex with the four constituent nitrogens in a planar ring. Trientine also chelates iron, and co-administration of trientine and iron should be avoided because the complex with iron is toxic.17
Data are scarce regarding the pharmacokinetics of Trientine. Trientine is particularly preferred in patients who are intolerant to d-Penicillamine and in patients with neurological symptoms as the worsening of the neurological symptoms are less common compared to d-Penicillamine.67,88
Trientine is a better tolerated drug with fewer side effect profiles as mentioned in Table 4. There are encouraging data regarding the use of Trientine in patients with severe liver disease.89,90 Typical initial dose of trientine is 750–1500 mg/day in two or three divided doses, with 750–1000 mg/day used for maintenance therapy.17
Zinc
Zinc was first introduced in 1961 and subsequently developed for the treatment of Wilson disease.91,92 Zinc acetate received approval by U.S. FDA in 1997.7 In other parts of the world zinc sulfate is used and is equally effective.7,92 Zinc acts by inhibiting the copper uptake from the gastrointestinal mucosa. Zinc induces enterocyte metallothionein, a cysteine-rich protein that has greater affinity for copper than for zinc thereby inhibiting copper entry into the portal circulation. The bound copper is lost when the enterocyte is shed during normal cell turnover.93 Zinc also induces copper binding metallothionein synthesis in the hepatocytes, thereby reducing the damaging effects of free copper.94–96
Zinc has very few side effects as indicated in Table 4. Neurological deterioration is uncommon with Zinc.93 Although zinc is currently reserved for maintenance treatment, it has been used as first-line therapy, most commonly for asymptomatic or presymptomatic patients. It appears to be equally effective as penicillamine but much better tolerated.71 Combination treatment with chelators (trientine or penicillamine) in which the chelator and the zinc are given at widely spaced intervals during the day has been advocated but not yet reported in rigorously designed series.
Dosing is based in milligrams of elemental zinc. Larger children and adults require 150 mg/day is administered in three divided doses and smaller children (<50 kg) require 75 mg/day.17
Ammonium tetrathiomolybdate (TM)
TM acts by forming a tripartite complex with copper and protein both in the gut and blood rendering the complex non absorbable and non toxic7 Administered with meals TM acts by inhibiting copper uptake and administered between the meals it acts by binding copper from the plasma.7,97–99 Clinical experience with this drug is limited as it is not commercially available. Adverse events are indicated Table 4.
In a randomized double blind study comparing tetrathiomolybdate and trientine in patients with the neurologic presentation, 6 of 23 patients in the trientine arm and 1 of 25 patients in the tetrathiomolybdate arm developed neurologic deterioration. Neurologic and speech recovery during a 3-year follow-up period were good.100 The greatest utility of TM will be in the treatment of neuro-psychiatrically disorders especially when d-Penicillamine therapy is not tolerated and results in worsening of the condition.7 TM has only been evaluated in initial therapy and its utility in maintenance treatment awaits further trials.
Diet
Foods with very high concentrations of copper (shellfish, nuts, chocolate, mushrooms, and organ meats) should be avoided, more so in the first year of treatment. (EASL: GRADE II-3, B, 2, AASLD Class I, Level C) Well water or water delivery systems to households commonly made from copper should be checked for copper levels and a water purifying system may be advisable. Copper containers should be avoided while cooking.17,33 However the role of diet is probably overstated.7 The earlier methods that measured copper in important foods overestimated the copper contents due to methodological issues and also because food processing has changes.7 The copper content in the livers of domestic animals and shellfish are high in copper and need to be avoided.7 However, the copper content in older tables show a higher copper content than is actually present and may need to be modified in the light of more recent data.101 One group does not restrict chocolates, mushrooms or nuts as they do not find them particularly high in copper.7
Other treatments
As oxidative stress plays a key role in the pathogenesis of Wilsons disease, antioxidants and Vitamin E102–106 have been investigated for its role in Wilson disease. No well controlled studies are available to advocate its routine use.
Specific situations
Asymptomatic/Presymptomatic Cohorts
According to a systematic review, d-Penicillamine and Zinc both were both effective in 100% of presymptomatic cohorts. However, d-Penicillamine had a higher incidence of side effects.73 In a randomized study comparing d-Penicillamine and Zinc with 12 years follow up, the long term effectiveness was similar with more side effects reported in the d-Penicillamine arm.82 Hence both the AASLD and EASL recommend usage of either chelator or Zinc with the latter preferred in children under the age of 3 years. (AASLD Class I Level B, EASL Grade II-1, B,1).17,33
Symptomatic Liver Disease Patients
Randomized clinical trials comparing the various decopper agents are lacking.71 A recent review found d-Penicillamine to be effective in 42 ⁄ 57 (73.7%) patients, whereas zinc therapy had a positive effect in 5 ⁄ 9 (55.6%) of patients treated.71 Another study of 17 patients with severe hepatic insuffiency without encephalopathy, early administration of d-Penicillamine was associated with improved survival without transplantation.107 The AASLD and the EASL recommend initial treatment for symptomatic patients with Wilsons disease should include a chelating agent (d-Penicillamine or trientine). Trientine may be better tolerated (AASLD Class I, Level B, EASL GRADE II-1, B, 1).17,33
Once the adequate treatment is achieved with the initiated chelator as assessed by clinical parameters or synthetic function, maintenance therapy can be achieved by reduced doses of either chelator or with by Zinc (AASLD Class I, Level B, EASL GRADE II-1, B, 1).17,33
Merle and coworkers retrospectively studied 163 patients with Wilson disease, of whom 138 patients were treated initially with d-Penicillamine, 9 with trientine, and 13 with zinc salts. Three underwent liver transplantation for acute liver failure. Among patients on d-Penicillamine, 39 were changed to Trientine and 34 to Zinc due to side effects. More than three fourth patients had stable or improved course of the disease. The mean follow up of the study was 16.7years.49
In another study from Czech republic 117 patients with WD were followed up for an average period of 15 ± 10 years.108 The patients were treated with d-Penicillamine (81%) or zinc salts (17%). The majority of patients improved with during follow up. The long term survival of these patients was similar to that of the general population without liver disease underlining the efficacy of drugs in the long term management of patients.108
Decompensated Cirrhosis
Studies demonstrating the efficacy of chelators with or without Zinc in patients with decompensated liver disease have been published.21,90,109 In such a scenario, a chelator and Zinc should be spaced 5–6 h apart in order to avoid negation of the effect of respective drugs. Those who respond may then be maintained on chelator or Zinc monotherapy. However non-responders to chelation should be promptly evaluated for liver transplantation. (AASLD Class I Level B, EASL Grade II-2, B, 1).17,33
Acute Liver Failure
Patients with acute liver failure and particularly those with encephalopathy often require liver transplantation. Spontaneous recovery is very rare with mortality approaching 100%. Chelation is rarely effective due to time lag needed for the onset of action and hence the need to identify such patients and transfer them to a facility with advanced care or liver transplantation becomes critical.
Liver transplantation
Orthotropic liver transplantation (OLT) is indicated in patients with acute liver failure and decompensated liver disease unresponsive to medical management.110 Liver transplantation corrects the underlying hepatic metabolic defects of Wilson disease.111 Patients undergoing liver transplantation have a slightly overall less favorable outcome when compared to transplantations performed for other etiologies.26,112–114
In a study by Schilsky and coworkers,26 55 patients underwent liver transplantation; 33 for decompensated cirrhosis, 21 for acute liver failure and one for intractable neurological Wilson disease. The median survival after OLT was 2.5 years, the longest survival time after OLT was 20 years. Survival at 1 year was 79%. In another Chinese retrospective study consisting of 36 patients, the 1, 3, and 5 years post-transplant survival was 91.7%, 83.3% and75% respectively.112 Although there are anecdotal reports of improvement in neurological.
Wilson after OLT, neurological deficits are rarely eliminated and liver transplantation as a cure for neurological disease is not advocated.115–118 Arnon and coworkers reviewed the outcome in 400 adults and 170 children who underwent liver transplantation over a 21 year period from the UNOS (United network for organ sharing) database. This included transplantation for Wilson disease with ALF and chronic disease. They found the outcome much better in adults than children and also for transplantation for chronic disease than ALF.119
Pregnancy
Patients contemplating pregnancy should have their copper status optimized before pregnancy. Patients should be frequently monitored during pregnancy. Although there is some concern of teratogenicity of d-Penicillamine, benefits of continuing treatment outweigh risks of withdrawal of treatment.120 Interrupting treatment during pregnancy can result in acute liver failure.121 While the dosage of Zinc can be maintained without any change the dose of chelators such as d-Penicillamine and Trientine may be reduced by 25–50%. (AASLD Class I, Level C, EASL Grade II-2, B, 1).17,33 Wound healing may be impaired after Caesarean section or episiotomy due to chelators.122 Therefore the dose of d-Penicillamine may be reduced toward the latter weeks of pregnancy in order to facilitate better wound healing after pregnancy. Regarding contraception, only spermicidal, barrier methods and progesterone only preparations are advisable as estrogens may interfere with biliary copper excretion and some intrauterine devices contain copper.33,123
Monitoring of treatment
Monitoring of treatment is required to confirm clinical and biochemical improvement, ensure compliance with therapy, and identify adverse events.17,33 This is particularly so in during maintenance therapy rather than the initial period and more so during adolescence. Appearance or reappearance of KF ring in whom it was absent should trigger suspicion of non-compliance or non-response .17,33
Patients taking d-Penicillamine or trientine may have to monitor their 24 h urinary copper more frequently in the first year and yearly thereafter. The 24 h urinary copper excretion should be more than 200–500 μg/day (3–8 μmol/day). Urine copper excretion below 200 μg/day (3.2 μmol/day) may indicate either nonadherence to therapy or overtreatment and excess copper removal.17,33 This can be differentiated by non ceruloplasmin bound coppers which is elevated in nonadherence (>15 μg/dL or >150 μg/L) or low (<5 g/dl or <50 g/L) when overtreated.17,33 Development of anemia or leukopenia after a period of stabilization may indicate copper deficiency and treatment should be temporarily discontinued.7 24 h urinary copper excretion levels for patients on zinc and penicillamine should be no more than 75 μg/day (1.2 μmol/day). For patients on zinc alone, and suspected to be non-compliant the 24 h urinary excretion of zinc should be more than below 2 mg/day and the urinary copper will begin to increase.17,33
24-h urinary copper excretion values for patients on zinc should be no more than 75 μg/day (1.2 μmol/day). 24 h urinary excretion of zinc may be measured to check the compliance (should be on the order of 2 mg/day).17,33
For routine monitoring, serum copper and ceruloplasmin, liver enzymes and international normalized ratio, complete blood count and urine analysis as well as physical and neurological examinations should be performed regularly, at least twice annually (AASLD Class I, Level C, EASL GRADE II-2, B, 1).17,33
The 24 h urinary copper excretion on medication and after 2 days of cessation of therapy should be measured at least yearly The estimated serum non ceruloplasmin bound copper may be useful parameter to control therapy (AASLD Class I, Level C EASL GRADE II-3, B, 1).17,33
Conclusions
Wilson disease is an inherited metabolic disease which results in copper accumulation affecting mainly the liver and neurological systems. It should be suspected especially in children and young adults presenting with jaundice, acute liver failure or chronic liver disease. Diagnosis of Wilson disease can be made using the scoring system incorporating routinely available laboratory tests. ATP 7 B gene mutation when identified in a proband is helpful in identifying pre-symptomatic siblings and is the highest weighted variable in the Leipzig score. Haplotype analysis or direct sequencing is useful for screening of siblings. However, if one can establish a diagnosis on clinical grounds alone, there may not be a need for genetic testing.124 Wilson disease carries an excellent prognosis if diagnosed and treated early and if compliance with treatment is maintained. The outcome may be less desirable in those with neurologic disabilities. Drugs have excellent efficacy but has to be monitored at regular intervals. Liver transplantation may be indicated in those who present with acute liver failure or those unresponsive to medical management. Siblings need to be screened and started on treatment preferably with Zinc to prevent disease manifestation.
Conflicts of interest
All authors have none to declare.
References
- 1.Wilson S.A.K. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34:295–507. doi: 10.1093/brain/awp193. [DOI] [PubMed] [Google Scholar]
- 2.Bull P.C., Thomas G.R., Rommens J.M. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 3.Tanzi R.E., Petrukhin K., Chernov I. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 4.Yamaguchi Y., Heiny M.E., Gitlin J.D. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem Biophys Res Commun. 1993;197:271–277. doi: 10.1006/bbrc.1993.2471. [DOI] [PubMed] [Google Scholar]
- 5.Thomas G.R., Roberts E.A., Walshe J.M. Haplotypes and mutations in Wilson disease. Am J Hum Genet. 1995;56:1315–1319. [PMC free article] [PubMed] [Google Scholar]
- 6.Kim G.H., Yang J.Y., Park J.Y. Estimation of Wilson’s disease incidence and carrier frequency in the Korean population by screening ATP7B major mutations in newborn filter papers using the SYBR green intercalator method based on the amplification refractory mutation system. Genet Test. 2008;12:395–399. doi: 10.1089/gte.2008.0016. [DOI] [PubMed] [Google Scholar]
- 7.Brewer G.J. Kluwer Academic Publishers; Boston, MA: 2001. Wilson’s Disease: A Clinician’s Guide to Recognition, Diagnosis, and Management. [Google Scholar]
- 8.Abdel Ghaffar T.Y., Elsayed S.M., Elnaghy S. Phenotypic and genetic characterization of a cohort of pediatric Wilson disease patients. BMC Pediatr. 2011;11:56. doi: 10.1186/1471-2431-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devarbhavi H., Singh R., Adarsh C.K. The clinical, laboratory characteristics, natural history and outcome in 201 patients with Wilson disease. Hepatology. 2012;56(suppl S1):826A. [Abstract] [Google Scholar]
- 10.Taly A.B., Prashanth L.K., Sinha S. Wilson’s disease: an Indian perspective. Neurol India. 2009;57:528–540. doi: 10.4103/0028-3886.57789. [DOI] [PubMed] [Google Scholar]
- 11.Rosencrantz R., Schilsky M. Wilson disease: pathogenesis and clinical considerations in diagnosis and treatment. Semin Liver Dis. 2011;31:245–259. doi: 10.1055/s-0031-1286056. [DOI] [PubMed] [Google Scholar]
- 12.Ala A., Walker A.P., Ashkan K. Wilson's disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 13.http://www.wilsondisease.med.ualberta.ca/database.asp
- 14.Stapelbroek J.M., Bollen C.W., van Amstel J.K. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta-analysis. J Hepatol. 2004;41:758–763. doi: 10.1016/j.jhep.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 15.Ferenci P., Caca K., Loudianos G. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 16.Brewer G.J. Zinc and tetrathiomolybdate for the treatment of Wilson's disease and the potential efficacy of anticopper therapy in a wide variety of diseases. Metallomics. 2009;1:199–206. doi: 10.1039/b901614g. [DOI] [PubMed] [Google Scholar]
- 17.Roberts E.A., Schilsky M.L. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47:2089–2111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- 18.Squires R.H., Jr., Shneider B.L., Bucuvalas J. Acute liver failure in children: the first 348 patients in the pediatric acute liver failure study group. J Pediatr. 2006;148:652–658. doi: 10.1016/j.jpeds.2005.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walshe J.M., Dixon A.K. Dangers of non-compliance in Wilson’s disease. Lancet. 1986;12:845–847. doi: 10.1016/s0140-6736(86)90949-9. [DOI] [PubMed] [Google Scholar]
- 20.Devarbhavi H., Singh R., Adarsh C.K. A simple prognostic model predicting outcome in children and adolescents with Wilson disease and acute liver failure and comparison with Wilson disease specific models. Hepatology. 2012;56(suppl S1):962A. [Abstract] [Google Scholar]
- 21.Dhawan A., Taylor R.M., Cheeseman P. Wilson's disease in children: 37-year experience and revised King's score for liver transplantation. Liver Transpl. 2005;11:441–448. doi: 10.1002/lt.20352. [DOI] [PubMed] [Google Scholar]
- 22.Saito T. Presenting symptoms and natural history of Wilson disease. Eur J Pediatr. 1987;146:261–265. doi: 10.1007/BF00716470. [DOI] [PubMed] [Google Scholar]
- 23.Riordan S.M., Williams R. The Wilson's disease gene and phenotypic diversity. J Hepatol. 2001;34:165–171. doi: 10.1016/s0168-8278(00)00028-3. [DOI] [PubMed] [Google Scholar]
- 24.Walshe J.M. Wilson’s disease presenting with features of hepatic dysfunction: a clinical analysis of eighty-seven patients. Q J Med. 1989;70:253–263. [PubMed] [Google Scholar]
- 25.Stremmel W., Meyerrose K.W., Niederau C. Wilson’s disease: clinical presentation, treatment, and survival. Ann Intern Med. 1991;115:720–726. doi: 10.7326/0003-4819-115-9-720. [DOI] [PubMed] [Google Scholar]
- 26.Schilsky M.L., Scheinberg I.H., Sternlieb I. Liver transplantation for Wilson’s disease: indications and outcome. Hepatology. 1994;19:583–587. doi: 10.1002/hep.1840190307. [DOI] [PubMed] [Google Scholar]
- 27.Scott J., Gollan J.L., Samourian S. Wilson's disease, presenting as chronic active hepatitis. Gastroenterology. 1978;74:645–651. [PubMed] [Google Scholar]
- 28.Walshe J.M. Wilson’s disease. The presenting symptoms. Arch Dis Child. 1962;37:253–256. doi: 10.1136/adc.37.193.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taly A.B., Meenakshi- S.S., Sinha S. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine. 2007;82:112–121. doi: 10.1097/MD.0b013e318045a00e. [DOI] [PubMed] [Google Scholar]
- 30.Svetel M., Kozic D., Stefanova E., Semnic R., Dragasevic N., Kostic V.S. Dystonia in Wilson‘s disease. Mov Disord. 2001;16:719–723. doi: 10.1002/mds.1118. [DOI] [PubMed] [Google Scholar]
- 31.Del Rosario M.A., Davis M.M., Chong S.K. Wobbly handwriting. Lancet. 1998;351:336. doi: 10.1016/S0140-6736(97)10254-9. [DOI] [PubMed] [Google Scholar]
- 32.Svetel M., Potrebic A., Pekmezovic T. Neuropsychiatric aspects of treated Wilson’s disease. Parkinsonism Relat Disord. 2009;15:772–775. doi: 10.1016/j.parkreldis.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 33.European Association for Study of Liver EASL clinical practice guidelines: Wilson's disease. J Hepatol. 2012;56:671–685. doi: 10.1016/j.jhep.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Shanmugiah A., Sinha S., Taly A.B. Psychiatric manifestations in Wilson’s disease: a cross-sectional analysis. J Neuropsychiatry Clin Neurosci. 2008;20:81–85. doi: 10.1176/jnp.2008.20.1.81. [DOI] [PubMed] [Google Scholar]
- 35.Giacchino R., Marazzi M.G., Barabino A. Syndromic variability of Wilson’s disease in children. Clinical study of 44 cases. Ital J Gastroenterol Hepatol. 1997;29:155–161. [PubMed] [Google Scholar]
- 36.Nicastro E., Ranucci G., Vajro P. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010 Dec;52:1948–1956. doi: 10.1002/hep.23910. [DOI] [PubMed] [Google Scholar]
- 37.Frommer D., Morris J., Sherlock S. Kayser–Fleischer-like rings in patients without Wilson’s disease. Gastroenterology. 1977;72:1331–1335. [PubMed] [Google Scholar]
- 38.Esmaeli B., Burnstine M.A., Martonyi C.L. Regression of Kayser–Fleischer rings during oral zinc therapy: correlation with systemic manifestations of Wilson’s disease. Cornea. 1996;15:582–588. [PubMed] [Google Scholar]
- 39.Merle U., Eisenbach C., Weiss K.H. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson’s disease. J Hepatol. 2009;51:925–930. doi: 10.1016/j.jhep.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 40.Walshe J.M. Diagnostic significance of reduced serum caeruloplasmin concentration in neurological disease. Mov Disord. 2005;20:1658–1661. doi: 10.1002/mds.20628. [DOI] [PubMed] [Google Scholar]
- 41.Perman J.A., Werlin S.L., Grand R.J. Laboratory measures of copper metabolism in the differentiation of chronic active hepatitis and Wilson disease in children. J Pediatr. 1979;94:564–568. doi: 10.1016/s0022-3476(79)80011-6. [DOI] [PubMed] [Google Scholar]
- 42.Korman J.D., Volenberg I., Balko J. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology. 2008;48:1167–1174. doi: 10.1002/hep.22446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Brien A., Williams R. Rapid diagnosis of Wilson disease in acute liver failure: no more waiting for the ceruloplasmin level? Hepatology. 2008;48:1030–1032. doi: 10.1002/hep.22587. [DOI] [PubMed] [Google Scholar]
- 44.Gaffney D., Fell G.S., O’Reilly D.S. ACP Best Practice No 163. Wilson’s disease: acute and presymptomatic laboratory diagnosis and monitoring. J Clin Pathol. 2000;53:807–812. doi: 10.1136/jcp.53.11.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gross J.B., Jr., Ludwig J., Wiesner R.H. Abnormalities in tests of copper metabolism in primary sclerosing cholangitis. Gastroenterology. 1985;89:272–278. doi: 10.1016/0016-5085(85)90326-9. [DOI] [PubMed] [Google Scholar]
- 46.Walshe J.M. Serum 'free' copper in Wilson disease. QJM. 2012;105:419–423. doi: 10.1093/qjmed/hcr229. [DOI] [PubMed] [Google Scholar]
- 47.El Balkhi S., Trocello J.M., Poupon J. Relative exchangeable copper: a new highly sensitive and highly specific biomarker for Wilson's disease diagnosis. Clin Chim Acta. 2011;412:23–24. doi: 10.1016/j.cca.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 48.Ferenci P., Członkowska A., Merle U. Late onset Wilson disease. Gastroenterology. 2007;132:1294–1298. doi: 10.1053/j.gastro.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 49.Merle U., Schaefer M., Ferenci P. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut. 2007;56:115–120. doi: 10.1136/gut.2005.087262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tu J.B., Blackwell R.Q. Studies on levels of penicillamine-induced cupriuresis in heterozygotes of Wilson’s disease. Metabolism. 1967;16:507–513. doi: 10.1016/0026-0495(67)90079-0. [DOI] [PubMed] [Google Scholar]
- 51.Martins da Costa C., Baldwin D., Portmann B. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson’s disease. Hepatology. 1992;15:609–615. doi: 10.1002/hep.1840150410. [DOI] [PubMed] [Google Scholar]
- 52.Ferenci P., Steindl-Munda P., Vogel W. Diagnostic value of quantitative hepatic copper determination in patients with Wilson's Disease. Clin Gastroenterol Hepatol. 2005;3:811–818. doi: 10.1016/s1542-3565(05)00181-3. [DOI] [PubMed] [Google Scholar]
- 53.Weiss K.H., Stremmel W. Evolving perspectives in Wilson disease: diagnosis, treatment and monitoring. Curr Gastroenterol Rep. 2012;14:1–7. doi: 10.1007/s11894-011-0227-3. [DOI] [PubMed] [Google Scholar]
- 54.Rosencrantz R.A., Schilsky M.L. Mining for a diagnosis of wilson’s disease in children: genetics, score and ore. Hepatology. 2010;52:1872–1874. doi: 10.1002/hep.24054. [DOI] [PubMed] [Google Scholar]
- 55.Sternlieb I. Mitochondrial and fatty changes in hepatocytes of patients with Wilson’s disease. Gastroenterology. 1968;55:354–367. [PubMed] [Google Scholar]
- 56.Johncilla M., Mitchell K.A. Pathology of the liver in copper overload. Semin Liver Dis. 2011;31:239–244. doi: 10.1055/s-0031-1286055. [DOI] [PubMed] [Google Scholar]
- 57.Prashanth L.K., Sinha S., Taly A.B. Do MRI features distinguish Wilson’s disease from other early onset extrapyramidal disorders? An analysis of 100 cases. Mov Disord. 2010;25:672–678. doi: 10.1002/mds.22689. [DOI] [PubMed] [Google Scholar]
- 58.Van Wassenaer-van Hall H.N., Van den Heuvel A.G., Algra A. Wilson disease: findings at MR imaging and CT of the brain with clinical correlation. Radiology. 1996;198:531–536. doi: 10.1148/radiology.198.2.8596862. [DOI] [PubMed] [Google Scholar]
- 59.Jacobs D.A., Markowitz C.E., Liebeskind D.S. The ‘‘double panda sign’’ in Wilson’s disease. Neurology. 2003;61:969. doi: 10.1212/01.wnl.0000085871.98174.4e. [DOI] [PubMed] [Google Scholar]
- 60.Berman D.H., Leventhal R.I., Gavaler J.S. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology. 1991;100:1129–1134. doi: 10.1016/0016-5085(91)90294-u. [DOI] [PubMed] [Google Scholar]
- 61.Sallie R., Katsiyiannakis L., Baldwin D. Failure of simple biochemical indexes to reliably differentiate fulminant Wilson's disease from other causes of fulminant liver failure. Hepatology. 1992;16:1206–1211. [PubMed] [Google Scholar]
- 62.Rajkumar R., Kashyap V.K. Genetic structure of four socioculturally diversified caste populations of southwest India and their affinity with related Indian and global groups. BMC Genet. 2004;5:23. doi: 10.1186/1471-2156-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santhosh S., Shaji R.V., Eapen C.E. ATP7B mutations in families in a predominantly Southern Indian cohort of Wilson’s disease patients. Indian J Gastroenterol. 2006;25:277–282. [PubMed] [Google Scholar]
- 64.Gupta A., Aikath D., Neogi R. Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Hum Genet. 2005;118:49–57. doi: 10.1007/s00439-005-0007-y. [DOI] [PubMed] [Google Scholar]
- 65.Pandit A., Bavdekar A.R., Bhave S.A. Wilson’s disease. Indian J Pediatr. 2002;69:785–791. doi: 10.1007/BF02723693. [DOI] [PubMed] [Google Scholar]
- 66.Czlonkowska A., Tarnacka B., Litwin T. Wilson’s disease—cause of mortality in 164 patients during 1992–2003 observation period. J Neurol. 2005;252:698–703. doi: 10.1007/s00415-005-0720-4. [DOI] [PubMed] [Google Scholar]
- 67.Scheinberg I.H., Jaffe M.E., Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson’s disease. N Engl J Med. 1987;317:209–213. doi: 10.1056/NEJM198707233170405. [DOI] [PubMed] [Google Scholar]
- 68.Nazer H., Ede R.J., Mowat A.P. Wilson’s disease: clinical presentation and use of prognostic index. Gut. 1986;27:1377–1381. doi: 10.1136/gut.27.11.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petrasek J., Jirsa M., Sperl J. Revised King's College score for liver transplantation in adult patients with Wilson's disease. Liver Transpl. 2007;13:55–61. doi: 10.1002/lt.20920. [DOI] [PubMed] [Google Scholar]
- 70.Fischer R.T., Soltys K.A., Squires R.H.J. Prognostic scoring indices in Wilson disease: a case series and cautionary tale. J Pediatr Gastroenterol Nutr. 2011;52:466–469. doi: 10.1097/MPG.0b013e31820b0211. [DOI] [PubMed] [Google Scholar]
- 71.Wiggenlinkhuizen M., Tilanus M.E.C., Bollen C.W. Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharmacol Ther. 2009;29:947–958. doi: 10.1111/j.1365-2036.2009.03959.x. [DOI] [PubMed] [Google Scholar]
- 72.Walshe J.M. Wilson’s disease. New oral therapy. Lancet. 1956;267:25–26. doi: 10.1016/s0140-6736(56)91859-1. [DOI] [PubMed] [Google Scholar]
- 73.Perrett D. The metabolism and pharmacology of d-penicillamine in man. J Rheumatol. 1981;7:41–50. [PubMed] [Google Scholar]
- 74.Wiesner R.H., Dickson E.R., Carlson G.L. The pharmacokinetics of d-penicillamine in man. J Rheumatol. 1981;8:51–55. [PubMed] [Google Scholar]
- 75.Bergstrom R.F., Kay D.R., Harkcom T.M. Penicillamine kinetics in normal subjects. Clin Pharmacol Ther. 1981;30:404–413. doi: 10.1038/clpt.1981.180. [DOI] [PubMed] [Google Scholar]
- 76.Kukovetz W.R., Beubler E., Kreuzig F. Bioavailability and pharmacokinetics of d-penicillamine. J Rheumatol. 1983;10:90–94. [PubMed] [Google Scholar]
- 77.Falkmer S., Samuelson G., Sjolin S. Penicillamine-induced normalization of clinical signs, and liver morphology and histochemistry in a case of Wilson’s disease. Pediatrics. 1970;45:260–268. [PubMed] [Google Scholar]
- 78.Walshe J.M. Copper chelation in patients with Wilson’s disease. A comparison penicillamine triethylene tetramine dihydrochloride. Q J Med. 1973;42:441–452. [PubMed] [Google Scholar]
- 79.Sass-Kortsak A. Wilson’s disease. A treatable liver disease in children. Pediatr Clin North Am. 1975;22:963–984. [PubMed] [Google Scholar]
- 80.Grand R.J., Vawter G.F. Juvenile Wilson disease: histologic and functional studies during penicillamine therapy. J Pediatr. 1975;87:1161–1170. doi: 10.1016/s0022-3476(75)80131-4. [DOI] [PubMed] [Google Scholar]
- 81.Sternlieb I. Copper and the liver. Gastroenterology. 1980;78:1615–1628. [PubMed] [Google Scholar]
- 82.Czlonkowska A., Gajda J., Rodo M. Effects of long-term treatment in Wilson’s disease with D-penicillamine and zinc sulphate. J Neurol. 1996;243:269–273. doi: 10.1007/BF00868525. [DOI] [PubMed] [Google Scholar]
- 83.Brewer G.J., Terry C.A., Aisen A.M. Worsening of neurologic syndrome in patients with Wilson’s disease with initial penicillamine therapy. Arch Neurol. 1987;44:490–493. doi: 10.1001/archneur.1987.00520170020016. [DOI] [PubMed] [Google Scholar]
- 84.Walshe J.M., Yealland M. Chelation treatment of neurological Wilson’s disease. Q J Med. 1993;86:197–204. [PubMed] [Google Scholar]
- 85.Gibbs K.R., Walshe J.M. Penicillamine and pyridoxine requirements in man. Lancet. 1966;309:175–179. doi: 10.1016/s0140-6736(66)90700-8. [DOI] [PubMed] [Google Scholar]
- 86.Siegel R.C. Collagen cross-linking effect of d-penicillamine on crosslinking in vitro. J Biol Chem. 1977;252:254–259. [PubMed] [Google Scholar]
- 87.Walshe J.M. Treatment of Wilson's disease with trientine (triethylene tetramine) dihydrochloride. Lancet. 1982;1:643–647. doi: 10.1016/s0140-6736(82)92201-2. [DOI] [PubMed] [Google Scholar]
- 88.Walshe J.M. The management of Wilson’s disease with trienthylene tetramine 2HC1 (Trien 2HC1) Prog Clin Biol Res. 1979;34:271–280. [PubMed] [Google Scholar]
- 89.Saito H., Watanabe K., Sahara M. Triethylene-tetramine (trien) therapy for Wilson’s disease. Tohoku J Exp Med. 1991;164:29–35. doi: 10.1620/tjem.164.29. [DOI] [PubMed] [Google Scholar]
- 90.Santos Silva E.E., Sarles J., Buts J.P. Successful medical treatment of severely decompensated Wilson disease. J Pediatr. 1996;128:285–287. doi: 10.1016/s0022-3476(96)70412-2. [DOI] [PubMed] [Google Scholar]
- 91.Hoogenraad T.U., Koevoet R., de Ruyter Korver E.G. Oral zinc sulphate as long-term treatment in Wilson’s disease (hepatolenticular degeneration) Eur Neurol. 1979;18:205–211. doi: 10.1159/000115077. [DOI] [PubMed] [Google Scholar]
- 92.Hoogenraad T.U., Van Hattum J., Van den Hamer C.J. Management of Wilson’s disease with zinc sulphate. Experience in a series of 27 patients. J Neurol Sci. 1987;77:137–146. doi: 10.1016/0022-510x(87)90116-x. [DOI] [PubMed] [Google Scholar]
- 93.Brewer G.J., Yuzbasiyan-Gurkan V., Young A.B. Treatment of Wilson’s disease. Semin Neurol. 1987;7:209–220. doi: 10.1055/s-2008-1041420. [DOI] [PubMed] [Google Scholar]
- 94.Cousins R.J. Absorption, transport and hepatic metabolism of copper and zinc: special reference to metallothionein and ceruloplasmin. Physiol Rev. 1985;65:238–309. doi: 10.1152/physrev.1985.65.2.238. [DOI] [PubMed] [Google Scholar]
- 95.Hill G.M., Brewer G.J., Prasad A.S. Treatment of Wilson’s disease with zinc. I. Oral zinc therapy regimens. Hepatology. 1987;7:522–528. doi: 10.1002/hep.1840070318. [DOI] [PubMed] [Google Scholar]
- 96.Schilsky M., Blank R.R., Czaja M.J. Hepatocellular copper toxicity and its attenuation by zinc. J Clin Invest. 1989;84:1562–1568. doi: 10.1172/JCI114333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McQuaid A., Mason J. A comparison of the effects of penicillamine, trientine, and trithiomolybdate on [35S]-labeled metallothionein in vitro; implications for Wilson's disease therapy. J Inorg Biochem. 1991;41:87–92. doi: 10.1016/0162-0134(91)80002-y. [DOI] [PubMed] [Google Scholar]
- 98.Brewer G.J., Dick R.D., Yuzbasiyan-Gurkin V. Initial therapy of patients with Wilson's disease with tetrathiomolybdate. Arch Neurol. 1991;48:42–47. doi: 10.1001/archneur.1991.00530130050019. [DOI] [PubMed] [Google Scholar]
- 99.Ogra Y., Suzuki K.T. Targeting of tetrathiomolybdate on the copper accumulating in the liver of LEC rats. J Inorg Biochem. 1998;70:49–55. doi: 10.1016/s0162-0134(98)00012-9. [DOI] [PubMed] [Google Scholar]
- 100.Brewer G.J., Askari F., Lorincz M.T. Treatment of Wilson disease with ammonium tetrathiomolybdate. IV: comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63:521–527. doi: 10.1001/archneur.63.4.521. [DOI] [PubMed] [Google Scholar]
- 101.Brewer G.J., Yubasiyan-Gurkan V. Wilson disease. Medicine. 1992;71:139–164. doi: 10.1097/00005792-199205000-00004. [DOI] [PubMed] [Google Scholar]
- 102.Von Herbay A., de Groot H., Hegi U. Low vitamin E content in plasma of patients with alcoholic liver disease, hemochromatosis and Wilson’s disease. J Hepatol. 1994;20:41–46. [PubMed] [Google Scholar]
- 103.Sokol R.J., Twedt D., McKim J.M., Jr. Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. Gastroenterology. 1994;107:1788–1798. doi: 10.1016/0016-5085(94)90822-2. [DOI] [PubMed] [Google Scholar]
- 104.Ogihara H., Ogihara T., Miki M. Plasma copper and antioxidant status in Wilson’s disease. Pediatr Res. 1995;37:219–226. doi: 10.1203/00006450-199502000-00016. [DOI] [PubMed] [Google Scholar]
- 105.Sinha S., Christopher R., Arunodaya G.R. Is low serum tocopherol in Wilson’s disease a significant symptom? J Neurol Sci. 2005;228:121–123. doi: 10.1016/j.jns.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 106.Nagasaka H., Inoue I., Inui A. Relationship between oxidative stress and antioxidant systems in the liver of patients with Wilson disease: hepatic manifestation in Wilson disease as a consequence of augmented oxidative stress. Pediatr Res. 2006;60:472–477. doi: 10.1203/01.pdr.0000238341.12229.d3. [DOI] [PubMed] [Google Scholar]
- 107.Durand F., Bernuau J., Giostra E. Wilson’s disease with severe hepatic insufficiency: beneficial effects of early administration of d-penicillamine. Gut. 2001;48:849–852. doi: 10.1136/gut.48.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bruha R., Marecek Z., Pospisilova L. Long-term follow-up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int. 2011;31:83–91. doi: 10.1111/j.1478-3231.2010.02354.x. [DOI] [PubMed] [Google Scholar]
- 109.Askari F.K., Greenson J., Dick R.D. Treatment of Wilson’s disease with zinc. XVIII. Initial treatment of the hepatic decompensation presentation with trientine and zinc. J Lab Clin Med. 2003;142:385–390. doi: 10.1016/S0022-2143(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 110.Khanna A., Jain A., Eghtesad B., Rakela J. Liver transplantation for metabolic liver diseases. Surg Clin North Am. 1999;79:153–162. doi: 10.1016/s0039-6109(05)70012-8. [DOI] [PubMed] [Google Scholar]
- 111.Groth C.G., Dubois R.S., Corman J. Metabolic effects of hepatic replacement in Wilson’s disease. Transplant Proc. 1973;5:829–833. [PMC free article] [PubMed] [Google Scholar]
- 112.Cheng F., Li G.Q., Zhang F. Outcomes of living related liver transplantation for Wilson’s disease: a single-center experience in China. Transplantation. 2009;87:751–757. doi: 10.1097/TP.0b013e318198a46e. [DOI] [PubMed] [Google Scholar]
- 113.Yoshitoshi E.Y., Takada Y., Oike F. Long-term outcomes for 32 cases of Wilsons disease after living-donor liver transplantation. Transplantation. 2009;87:261–267. doi: 10.1097/TP.0b013e3181919984. [DOI] [PubMed] [Google Scholar]
- 114.Schumacher G., Platz K.P., Mueller A.R. Liver transplantation in neurologic Wilsons disease. Transplant Proc. 2001;33:1518–1519. doi: 10.1016/s0041-1345(00)02578-1. [DOI] [PubMed] [Google Scholar]
- 115.Bax R.T., Hassler A., Luck W. Cerebral manifestation of Wilsons disease successfully treated with liver transplantation. Neurology. 1998;51:863–865. doi: 10.1212/wnl.51.3.863. [DOI] [PubMed] [Google Scholar]
- 116.Lui C.C., Chen C.L., Cheng Y.F. Recovery of neurological deficits in a case of Wilson’s disease after liver transplantation. Transplant Proc. 1998;30:3324–3325. doi: 10.1016/s0041-1345(98)01048-3. [DOI] [PubMed] [Google Scholar]
- 117.Stracciari A., Tempestini A., Borghi A. Effect of liver transplantation on neurological manifestations in Wilson disease. Arch Neurol. 2000;57:384–386. doi: 10.1001/archneur.57.3.384. [DOI] [PubMed] [Google Scholar]
- 118.Wu J.C., Huang C.C., Jeng L.B. Correlation of neurological manifestations and MR images in a patient with Wilson’s disease after liver transplantation. Acta Neurol Scand. 2000;102:135–139. doi: 10.1034/j.1600-0404.2000.102002135.x. [DOI] [PubMed] [Google Scholar]
- 119.Arnon R., Annunziato R., Schilsky M. Liver transplantation for children with Wilson disease: comparison of outcomes between children and adults. Clin Transplant. 2011;25(1):E52–E56. doi: 10.1111/j.1399-0012.2010.01327.x. [DOI] [PubMed] [Google Scholar]
- 120.Ferenci P. Wilson’ s disease. In: Bacon B., editor. Comprehensive Clinical Hepatology. Elsevier Mosby; Maryland Heights, Miss. USA: 2005. pp. 351–367. [Google Scholar]
- 121.Shimono N., Ishibashi H., Ikematsu H. Fulminant hepatic failure during perinatal period in a pregnant woman with Wilson’s disease. Gastroenterol Jpn. 1991;26:69–73. doi: 10.1007/BF02779512. [DOI] [PubMed] [Google Scholar]
- 122.Scheinberg I.H., Sternlieb I. Pregnancy in penicillamine-treated patients with Wilson’s disease. N Engl J Med. 1975;293:1300–1302. doi: 10.1056/NEJM197512182932507. [DOI] [PubMed] [Google Scholar]
- 123.Haimov-Kochman R., Ackerman Z., Anteby E.Y. The contraceptive choice for a Wilson’s disease patient with chronic liver disease. Contraception. 1997;56:241–244. doi: 10.1016/s0010-7824(97)00141-8. [DOI] [PubMed] [Google Scholar]
- 124.Pagon presentation. www.ncbi.nlm.nih.gov/projects/genetests/Roberta; Accessed 18.03.13.