

Abstract
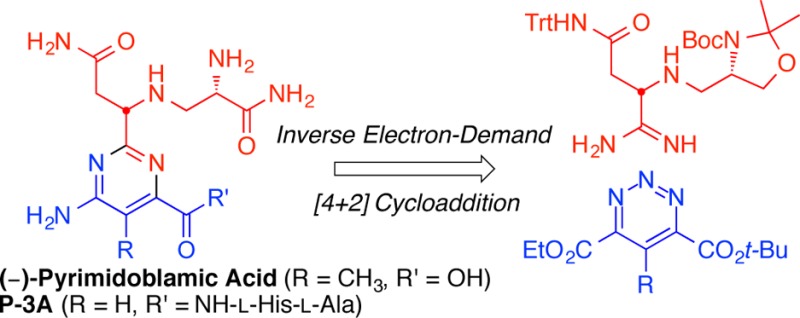
Total syntheses of (−)-pyrimidoblamic acid and P-3A are disclosed. Central to the convergent approach is a powerful inverse electron demand Diels–Alder reaction between substituted electron-deficient 1,2,3-triazines and a highly functionalized and chiral primary amidine, which forms the pyrimidine cores and introduces all necessary stereochemistry in a single step. Intrinsic in the convergent approach is the potential it provides for the late stage divergent synthesis of modified analogs bearing deep-seated changes in either the pyrimidine cores or the highly functionalized C2 side chain common to both natural products. The examination of the key cycloaddition reaction revealed that the inherent 1,2,3-triazine mode of cycloaddition (C4/N1 vs C5/N2) as well as the amidine regioselectivity were unaffected by introduction of two electron-withdrawing groups (−CO2R) at C4 and C6 of the 1,2,3-triazine even if C5 is unsubstituted (Me or H), highlighting the synthetic potential of the powerful pyrimidine synthesis.
Introduction
Bleomycin A2 (BLM, 1, Figure 1) is the major component (∼70%) of the clinical anticancer drug Blenoxane, which is currently used for the treatment of Hodgkin’s lymphoma, melanomas, head and neck carcinomas, and testicular cancers.1−4 BLM exerts its biological effects through a metal ion and oxygen dependent5−19 DNA cleavage that occurs selectively at 5′-GC or 5′-GT sites.4,7,12,15,20−25 Although capable of producing single-strand and double-strand breaks, the latter is believed to be the most biologically relevant event.26 Despite being an effective therapy for numerous malignancies, BLM treatment is often limited by dose-dependent pneumonitis, lung fibrosis, and skin toxicities, some of which have been attributed to the C-terminal bithiazole appendage.2,3,27,28 Thus, there remains an interest in identifying synthetic BLM-based analogs that lack off-target toxicity and maintain efficacy, which can be used as single component oncology drugs.
Figure 1.

Structure of bleomycin A2.
Each structural unit of BLM contributes an important role in the DNA binding and cleavage cascade. Systematic evaluation of its structure through single site deep-seated modifications prepared by total synthesis has delineated the function of each subunit and the role of their individual substituents.1,21,29−74 These studies, in conjunction with NMR-derived structural models of DNA bound BLM,75−83 continue to define the remarkable combination of functional, structural, and conformational properties integrated into the natural product. The pyrimidoblamic acid subunit not only participates in the metal chelation and O2 activation but also is responsible for the DNA cleavage selectivity via triplex-like hydrogen bonding to guanine at the 5′-GC and 5′-GT cleavage sites.76 In such studies, we have additionally shown that removal of the pyrimidine C5 methyl group, enlisting the functionalized pyrimidine found in P-3A, has no impact in the functional activity of the resulting BLM analog.56 From these studies, (−)-pyrimidoblamic acid (2, Figure 2) and the related peptide-derived natural product, P-3A (3, Figure 2), have been identified as key subunits for the continued preparation of improved or simplified bleomycin analogs. Total syntheses of (−)-pyrimidoblamic acid were originally described by Umezawa,32,42 Hecht,30,50 and later by us.60,64 Although elegant and pioneering in their own regards, these previous efforts have suffered from the inability to fully control the stereochemistry at the benzylic pyrimidine C2 tertiary center (Scheme 1). Similarly, our initial total synthesis of P-3A, whose pyrimidine core differs from that of pyrimidoblamic acid by only the lack of the C5 methyl group, relied on a late stage diastereoselective introduction of the benzylic pyrimidine C2 tertiary center that proceeded with modest diastereoselectivity (87:13).59,66
Figure 2.

Structures of (−)-pyrimidoblamic acid and P-3A.
Scheme 1. Prior Late Stage Installations of the Benzylic C2-Stereocenter of (−)-Pyrimidoblamic Acid.

Recently, we defined the cycloaddition scope and productive reactivity of substituted electron-deficient 1,2,3-triazines with various dienophiles, including the first report of their ability to participate in previously unexplored [4 + 2] cycloaddition reactions with amidine or imidate heterodienophiles to provide highly substituted pyrimidines.84,85 These latter studies provided the basis for the potential development of second generation asymmetric total syntheses of (−)-pyrimidoblamic acid and P-3A, in which all necessary stereochemistry is installed prior to a late stage [4 + 2] cycloaddition introduction of the pyrimidine, addressing the common limitation of previously reported routes, permitting the late stage divergent modification of their structures, and further expanding the scope of the 1,2,3-triazine inverse electron demand Diels–Alder reaction to encompass the use of chiral amidines.
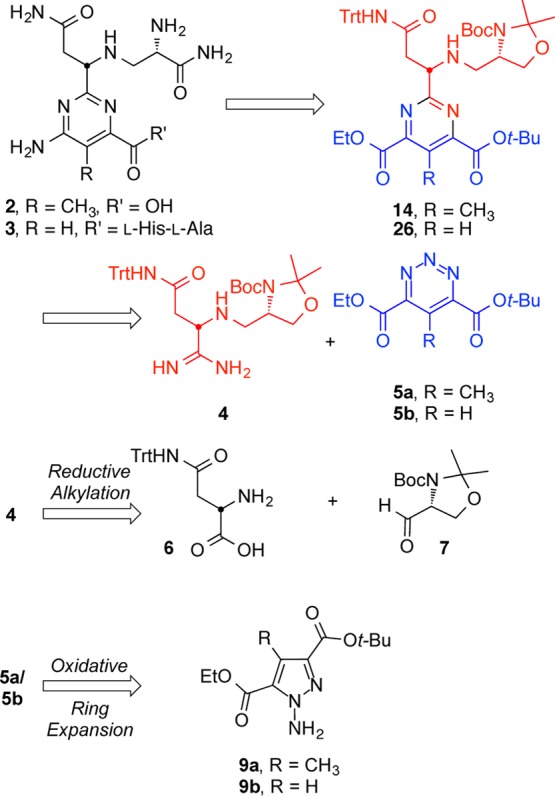
We envisioned that the pyrimidine core of (−)-pyrimidoblamic acid would arise from an inverse electron demand Diels–Alder reaction between the highly functionalized amidine 4 and 1,2,3-triazine 5a (Scheme 2). Additionally, the same amidine 4 and its cycloaddition with 1,2,3-triazine 5b would produce the pyrimidine core found in P-3A.This late stage convergent assemblage of the pyrimidine cores not only allows the early stage synthesis of the stereochemically rich C2 side chain common to both pyrimidoblamic acid and P-3A but also permits the late stage divergent synthesis of the natural products utilizing two different 1,2,3-triazines. Amidine 4 was expected to arise from the reductive alkylation product of N-(triphenylmethyl)-l-asparagine (6) with aldehyde 7. Key to the construction of 4 is the manipulation of intermediates, especially the desired free-based amidine, without sacrificing the stereochemical integrity of the α-stereocenter, as similar amidines have demonstrated a propensity to epimerize.86,87 This concern dictated the late stage amidine introduction in order to minimize the potential impact of amidine epimerization. 1,2,3-Triazines 5a and 5b, the electron-deficient heterocyclic azadienes for use in the key cycloaddition reaction, were anticipated to arise from an oxidative ring expansion of N-aminopyrazoles 9a and 9b.88−91 A key caveat to the approach is that although the two electron-withdrawing substitutents on the 1,2,3-triazines would be expected to enhance their cycloaddition reactivity, it was unknown whether their placement at the C4 and C6 positions would sterically slow the reaction and electronically redirect the mode of cycloaddition (C4/N1 vs C5/N2) with amidines.
Scheme 2. Retrosynthetic Analysis.
Results and Discussion
Synthesis of 1,2,3-Triazines 5a and 5b
The requisite N-aminopyrazole 9a for oxidative ring expansion to the 1,2,3-triazine 5a was prepared from 8a that was accessed by a palladium-mediated tandem cross-coupling/electrocyclization reaction of commercially available ethyl diazoacetate and the vinyl triflate derived from t-butyl acetoacetate (Scheme 3).92N-Amination of 8a was accomplished upon treatment with O-4-nitrobenzoylhydroxylamine in the presence of potassium t-butoxide (t-BuOK) in N-methylpyrrolidone (NMP), which yielded N-aminopyrazole 9a in 73% yield in a near 1:1 mixture of inconsequential regioisomers.93 In an analogous fashion and with similar yield (72%), the requisite N-aminopyrazole 9b used to access 1,2,3-triazine 5b was prepared from pyrazole 8b, which was generated from a [3 + 2] dipolar cycloaddition between ethyl diazoacetate and t-butyl propiolate.94
Scheme 3. Synthesis of Requisite N-Aminopyrazoles.

Although a range of oxidants have been reported for transforming N-aminopyrazoles into 1,2,3-triazines, we have found that the optimal reagent for this oxidative ring expansion reaction is substrate dependent. The ring expansion is often suggested to occur through a nitrene intermediate, which initiates N–N bond migration and inserts the central nitrogen of the 1,2,3-triazine.90 Adaptation of conditions developed by Ohsawa and colleagues, in which an N-aminopyrazole is treated with iodine (I2) in the presence of aqueous potassium bicarbonate (KHCO3), provided the 1,2,3-triazine 5a in excellent yield (75%).91 These conditions also proved effective for the production of 5b, yielding the 1,2,3-triazine in 68% yield. Notably, 1,2,3-triazines 5a and 5b proved stable to extended storage at room temperature and purification by flash chromatography using silica gel. As summarized in Figure 3, the most commonly used oxidants for this transformation (i.e., Pb(OAc)4, Ni2O3, and MnO2)88−91 proved unsuccessful in converting 9a to the desired 1,2,3-triazine 5a, instead resulting in N–N bond cleavage and reversion to the starting pyrazole 8a.
Figure 3.

Oxidative ring expansion survey.
Silver(I) nitrate, a commonly utilized nitrene transfer reagent in aziridination,95,96 alone failed to provide 5a but produced 5a in modest yield (34%) when used in the presence of phenyliodine diacetate (PIDA) as a co-oxidant. Similarly, sodium periodate (NaIO4),84 which promotes the oxidative ring expansion efficiently for 4-substituted N-aminopyrazoles, afforded the 1,2,3-triazine 5a in moderate yield (28%) with a majority (70%) of the material isolated being a mixture of unreacted starting material (9a) and free pyrazole (8a). Addition of a phase transfer catalyst improved the conversions to as high as 70%, but was less consistent, and the use of tetrabutylammonium periodate failed to generate any desired compound.
Synthesis of Amidine 4
Amidine 4 was prepared from commercially available N-(triphenylmethyl)-l-asparagine (6) and aldehyde 7 (Scheme 4). Reductive alkylation of 6 with 7 in the presence of excess NaBH(OAc)3 provided 10 in 84% yield. Treatment of 10 with 1-hydroxybenzotriazole (HOBt) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) followed by NH3/THF, provided the primary amide 11. Dehydration of 11 to provide the nitrile 12 was accomplished by treatment with N-propanephosphonic acid anhydride (T3P) in the presence of Hunig’s base (i-Pr2NEt). Treatment of 12 with 50% aqueous NH2OH cleanly afforded amidoxime 13. All intermediates up to this stage, including 13, exhibited a robust stability and no epimerization was observed throughout the reaction sequence. Treatment of 13 with Raney Nickel (Ra–Ni) in the presence of acetic acid (AcOH) in MeOH generated the acetate salt of the desired amidine, which could be filtered through Celite, redissolved in CH2Cl2, and treated with 20% aqueous NaOH to yield the free-based amidine 4. Chromatographic purification of the amidine 4, as either the acetate salt or the free base, resulted in partial epimerization. As a result, the crude amidine was used without purification and produced immediately prior to its use. Notably, the free-based amidine epimerized much more readily than the protonated amidine in solution, and if the adjacent secondary amine was protected as the t-butylcarbamate (Boc), even more significant slow epimerization of the α-stereocenter occurred upon amidine formation. Therefore, the free secondary amine is essential to the observed stereochemical stability of the adjacent amidine chiral center.
Scheme 4. Synthesis of Chiral Amidine 4.

[4 + 2] Cycloaddition Reaction of 1,2,3-Triazine 5a with Amidine 4
Effective access to the requisite intermediates 4 and 5a set the stage for examination of the key inverse electron demand Diels–Alder reaction. Consistent with the electron-deficient character of the 1,2,3-triazine, the cycloaddition reaction was found to proceed at room temperature or lower. Significantly, the intrinsic regioselectivity of the amidine cycloaddition and 1,2,3-triazine cycloaddition mode were unaltered by the C4/C6 substitution with electron-withdrawing groups, and the amidine was found to add exclusively across C4/N1 (C6/N3) with no evidence of a redirected C5/N2 mode of cycloaddition. By design and because of the pseudo-C2-symmetric nature of the product pyrimidine, the cycloaddition across C4/N1 versus C6/N3 are indistinguishable, permitting the differential ester protection. Although not all conditions surveyed are included, Figure 4 summarizes representative examples in the optimization of the reaction between 4 and 5a to provide the desired pyrimidine 14. In accordance with our previous studies,84,85 which utilized 2.0 equiv of amidine, the investigation began with this reaction stoichiometry. As summarized in Figure 4, an increase in reaction time and temperature significantly increases the overall yield (entries 1–4). However, increasing the reaction temperature diminished the diastereomeric purity of the product, resulting from competitive amidine epimerization. Notably, we found that the reaction produces a single diastereomer of 14 when conducted at 5 °C in acetonitrile (entry 5). Because the 1,2,3-triazine 5a is the simpler of the two reaction components, we focused on identifying conditions that employed amidine 4 as the limiting reagent (entries 8 and 9 vs 6 and 7). When the reaction was conducted with 1.0 equiv of amidine 4 and 2.0 equiv of the 1,2,3-triazine 5a, the yield increased to a respectable 46%. Finally, if the reaction was allowed to stir at 5 °C for 14 h and then warmed to 25 °C for 6 h, 14 was obtained as a single diastereomer in 54% yield.
Figure 4.

Optimization of the [4 + 2] cycloaddition reaction.
Total Synthesis of (−)-Pyrimidoblamic Acid
With an effective route to 14 in hand, the synthesis of (−)-pyrimidoblamic acid was completed (Scheme 5). The secondary amine of 14 was protected as the t-butylcarbamate to provide 15 to eliminate oxidation risks later in the synthesis. Chemoselective hydrolysis of 15 with 1 M aqueous NaOH in THF:MeOH (3:1) was followed with a Curtius rearrangement to provide 16 (78% yield over two steps). Removal of the acetonide protecting group with p-toluenesulfonic acid monohydrate (p-TsOH·H2O) in MeOH yielded the Boc-protected amino alcohol 17, which was subsequently treated with Jones’ reagent to give the carboxylic acid 18. Conversion of 18 to amide 19 was accomplished upon treatment with HOBt and EDCI followed by NH3/THF, which provided the fully functionalized and protected (−)-pyrimidoblamic acid 19 in 96% yield. Although attempts to promote a global deprotection with 4 M HCl in EtOAc proved unsuccessful, as the tritylated carboxamide exhibited an unusual stability to these traditional reaction conditions, treatment of 19 with a 3:1 mixture of trifluoroacetic acid (TFA) and CH2Cl2 followed by deliberate counterion exchange with a 1 M aqueous HCl workup provided (−)-pyrimidoblamic acid (2) in quantitative yield and identical in all respects with authentic material.29,64
Scheme 5. Completion of the Total Synthesis of (−)-Pyrimidoblamic Acid.

Generality of the 1,2,3-Triazine/Amidine [4 + 2] Cycloaddition
Although the similarity in the pyrimidine cores of (−)-pyrimidoblamic acid and P-3A is striking and may suggest that extrapolation of the approach to P-3A is straightforward, it was not clear what the impact of the C5 substituent might be. With electron-withdrawing groups at the C4- and C6-positions of 5a and 5b, it was still unknown whether the C5 methyl group affects the efficiency of the cycloaddition reaction of 5a and if its absence in 5b would alter the mode of cycloaddition. Thus, before embarking on the synthesis of P-3A, we first examined the reactions of the two 1,2,3-triazines 5a and 5b in parallel. Remarkably, the less substituted 1,2,3-triazine 5b was even more reactive than 5a, providing the product pyrimidine at a faster rate and in a higher yield in its reaction at room temperature with the aliphatic amidine substrate, where good conversion to the product was observed even within 5 min (Figure 5). Slower and a more comparable reactivity between 5a and 5b were observed with the aryl amidines. Most significant in these studies is the fact that 5b showed no evidence of a potentially competitive cycloaddition across C5/N2 (vs C4/N1) despite the lack of a C5 substituent. With this knowledge in hand, the total synthesis of P-3A was pursued.
Figure 5.

Comparison of the cycloaddition reactions between 5a or 5b and amidines.
Total Synthesis of P-3A
In accordance with the model substrates, the cycloaddition reaction between 4 and 5b (25 °C, CH3CN, 12 h) yielded the desired pyrimidine 26 in 76% yield as a single diastereomer (Scheme 6). In contrast to the reaction between 4 and 5a, which requires 5 °C for 12 h prior to warming to 25 °C to prevent competitive epimerization and preserve the amidine stereochemical integrity, the faster reaction between 4 and 5b can be conducted at 25 °C without compromising the diastereomeric purity of the product and provided the stereochemically pure pyrimidine 26 in even higher yield (76%).
Scheme 6. Total Synthesis of P-3A.

Following a synthetic route modeled on that used to complete the synthesis of (−)-pyrimidoblamic acid, compound 26 was advanced (Scheme 6). Boc-protection of the secondary amine provided compound 27, which was subjected to saponification of the ethyl ester. However, attempts to selectively hydrolyze the ethyl ester by treatment with 1 M aqueous NaOH in THF/MeOH provided a mixture of compounds, which included those derived from transesterification or hydrolysis of the t-butyl ester. By substituting t-butanol (t-BuOH) for MeOH as the reaction cosolvent the competitive reactions were avoided and the reaction cleanly provided the requisite carboxylic acid, which was subjected to Curtius rearrangement conditions to provide 28 in good yield (82% over two steps). Removal of the acetonide protecting group with p-TsOH·H2O in MeOH afforded the amino alcohol 29, which could be transformed to the primary carboxamide 31 in two steps. Global deprotection of 31 was effected by treatment with TFA:CH2Cl2 (3:1) to provide 32, a key analog of (−)-pyrimidoblamic acid lacking only the C5 methyl group, in quantitative yield. Additionally, treatment of 31 with 1 M aqueous NaOH in THF:MeOH (3:1) results in t-butyl ester hydrolysis and provided carboxylic acid (33), which was coupled with N(Im)-Boc-l-His-l-Ala-O(t-Bu) to provide the fully assembled but protected P-3A (34). Notably, this coupling reaction enlisted a more highly protected derivative of 32 than our prior efforts60,64 and proceeded more smoothly, providing higher yields of the product 34. Global deprotection of 34 with TFA:CH2Cl2 (3:1) followed by counterion exchange with a 1 M aqueous HCl workup provided P-3A (3), in quantitative yield and spectroscopically identical in all comparable respects with authentic material.66,97
Conclusions
Convergent total syntheses of (−)-pyrimidoblamic acid and P-3A were detailed based on the early stage preparation of the chiral highly functionalized C2 side chain and subsequent late stage, divergent construction of the pyrimidine cores through use of a powerful inverse electron demand amidine/1,2,3-triazine [4 + 2] cycloaddition reaction conducted at ≤25 °C. In addition to permitting full control of the natural product stereochemistry, the approach provides the opportunity for the late stage divergent synthesis of modified analogs bearing deep-seated changes in either the pyrimidine core (C4 and C5) or the highly functionalized C2 side chain. Such investigations are in progress and will be reported in due course. The examination of the key 1,2,3-triazine cycloaddition reaction with amidines defined nonobvious 1,2,3-triazine substituent effects that maintain or enhance the heterocyclic azadiene reactivity without altering the intrinsic regioselectivity or mode of cycloaddition. These observations with 1,2,3-triazines extend the utility of the inverse electron demand cycloaddition reactions of electron-deficient heterocyclic azadienes that includes the complementary 1,2,4- and 1,3,5-triazines,52,98−105 1,2,4,5-tetrazines,106−113 and 1,2-diazines114,115 in the synthesis of highly substituted and functionalized heterocycles found in complex natural products.116,117
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056). A.S.D. is an American Cancer Society postdoctoral fellow (PF-12-122-01-CDD).
Supporting Information Available
Full experimental details including NMR results and graphs. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Umezawa H. Pure Appl. Chem. 1971, 28, 665. [DOI] [PubMed] [Google Scholar]
- Umezawa H.; Takita T.; Saito S.; Muraoka Y.; Takahashi K.; Ekimoto H.; Minamide S.; Nishikawa K.; Fukuoka T.; Nakatani T.; Fujii A.; Matsuda A.. Bleomycin Chemotherapy; Academic Press: Orlando, FL, 1985. [Google Scholar]
- Mir L. M.; Tounekti O.; Orlowski S. Gen. Pharmacol. 1996, 27, 745. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Cai H. Angew. Chem., Int. Ed. 1999, 38, 448. [DOI] [PubMed] [Google Scholar]
- Ishida R.; Takahashi T. Biochem. Biophys. Res. Commun. 1975, 66, 1432. [DOI] [PubMed] [Google Scholar]
- Sausville E. A.; Peisach J.; Horwitz S. B. Biochem. Biophys. Res. Commun. 1976, 73, 814. [DOI] [PubMed] [Google Scholar]
- D’Andrea A. D.; Haseltine W. A. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausville E. A.; Peisach J.; Horwitz S. B. Biochemistry 1978, 17, 2740. [DOI] [PubMed] [Google Scholar]
- Takeshita M.; Grollman A. P.; Ohtsubo E.; Ohtsubo H. Proc. Natl. Acad. Sci. U. S. A. 1978, 1978, 5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povirk L. C. Biochemistry 1979, 18, 3989. [DOI] [PubMed] [Google Scholar]
- Burger R. M.; Peisach J.; Horwitz S. B. J. Biol. Chem. 1981, 256, 11636. [PubMed] [Google Scholar]
- Sugiura Y.; Suzuki T. J. Biol. Chem. 1982, 257, 10544. [PubMed] [Google Scholar]
- Burger R. M.; Kent T. A.; Horwitz S. B.; Munck E.; Peisach J. J. Biol. Chem. 1983, 258, 1559. [PubMed] [Google Scholar]
- Ehrenfeld G. M.; Shipley J. B.; Heimbrook D. C.; Sugiyama T.; Long E. C.; van Boom J. H.; van der Marel G. A.; Oppenheimer N. J.; Hecht S. M. Biochemistry 1987, 26, 931. [DOI] [PubMed] [Google Scholar]
- Kuwahara J.; Sugiura Y. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long E. C.; Hecht S. M.; van der Marel G. A.; van Boom J. H. J. Am. Chem. Soc. 1990, 112, 5272. [Google Scholar]
- Natrajan A.; Hecht S. M.; van der Marel G. A.; van Boom J. H. J. Am. Chem. Soc. 1990, 112, 3997. [Google Scholar]
- Carter B. J.; Reddy K. S.; Hecht S. M. Tetrahedron 1991, 47, 2463. [Google Scholar]
- Burger R. M.; Tian G.; Drlica K. J. Am. Chem. Soc. 1995, 117, 1167. [Google Scholar]
- Murray V.; Martin R. F. J. Biol. Chem. 1985, 260, 10389. [PubMed] [Google Scholar]
- Carter B. J.; Murty V. S.; Reddy K. S.; Wang S.; Hecht S. M. J. Biol. Chem. 1990, 265, 4193. [PubMed] [Google Scholar]
- Dedon P. C.; Goldberg I. H. Chem. Res. Toxicol. 1992, 5, 311. [DOI] [PubMed] [Google Scholar]
- Kane S. A.; Natrajan A.; Hecht S. M. J. Biol. Chem. 1994, 269, 10899. [PubMed] [Google Scholar]
- Zuber G.; Quada J. C. Jr.; Hecht S. M. J. Am. Chem. Soc. 1998, 120, 9365. [Google Scholar]
- Tse W. C.; Boger D. L. Chem. Biol. 2004, 11, 1607. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Kozarich J. W.; Wu W.; Vanderwall D. E. Acc. Chem. Res. 1996, 73, 133. [Google Scholar]
- Hecht S. M. J. Nat. Prod. 2000, 63, 158. [DOI] [PubMed] [Google Scholar]
- Fyfe A. J.; McKay P. Publ. – R. Coll. Physicians Edinburgh 2010, 40, 213. [DOI] [PubMed] [Google Scholar]
- Yoshioka T.; Muraoka Y.; Takita T.; Maeda K.; Umezawa H. J. Antibiot. 1972, 25, 625. [PubMed] [Google Scholar]
- Arai H.; Hagmann W. K.; Suguna H.; Hecht S. M. J. Am. Chem. Soc. 1980, 102, 6631. [Google Scholar]
- Fukuoka T.; Muraoka Y.; Fujii A.; Naganawa H.; Takita T.; Umezawa H. J. Antibiot. 1980, 33, 114. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Otsuka M.; Narita M.; Ohno M. J. Am. Chem. Soc. 1980, 102, 6630. [Google Scholar]
- Umezawa Y.; Morishima H.; Saito S.; Takita T.; Umezawa H. J. Am. Chem. Soc. 1980, 102, 6630. [Google Scholar]
- Takita T.; Umezawa Y.; Saito S.; Morishima H.; Umezawa H. Tetrahedron Lett. 1981, 22, 671. [Google Scholar]
- Aoyagi Y.; Katano K.; Suguna H.; Primeau J.; Chang L.; Hecht S. M. J. Am. Chem. Soc. 1982, 104, 5537. [Google Scholar]
- Aoyagi Y.; Suguna H.; Murugesan N.; Ehrenfeld G. M.; Chang L.; Ohgi T.; Shekhani M. S.; Kirkup M. P.; Hecht S. M. J. Am. Chem. Soc. 1982, 104, 5237. [Google Scholar]
- Henichart J.; Houssin R.; Bernier J.; Catteau J. J. Chem. Soc. Chem. Commun. 1982, 1295. [Google Scholar]
- Saito S.; Umezawa Y.; Morishima H.; Takita T.; Umezawa H. Tetrahedron Lett. 1982, 23, 529. [Google Scholar]
- Takita T.; Umezawa Y.; Saito S.; Morishima H.; Naganawa H.; Umezawa H. Tetrahedron Lett. 1982, 23, 521. [Google Scholar]
- Otsuka M.; Kittaka A.; Takamasa I.; Yakashita H.; Kobayashi S.; Ohno M. Chem. Pharm. Bull. 1985, 33, 509. [DOI] [PubMed] [Google Scholar]
- Otsuka M.; Kobayashi S.; Ohno M.; Umezawa Y.; Morishima H.; Umezawa H. Chem. Pharm. Bull. 1985, 33, 515. [DOI] [PubMed] [Google Scholar]
- Otsuka M.; Narita M.; Yoshida M.; Kobayashi S.; Ohno M.; Umezawa Y.; Morishima H.; Saito S.; Takita T.; Umezawa H. Chem. Pharm. Bull. 1985, 33, 520. [DOI] [PubMed] [Google Scholar]
- Otsuka M.; Kittaka A.; Ohno M. Tetrahedron Lett. 1986, 27, 3639. [Google Scholar]
- Kittaka A.; Sugano Y.; Otsuka M.; Ohno M. Tetrahedron 1988, 44, 2811. [Google Scholar]
- Kittaka A.; Sugano Y.; Otsuka M.; Ohno M. Tetrahedron 1988, 44, 2821. [Google Scholar]
- Lomis T. J.; Siuda J. F.; Shepherd R. E. J. Chem. Soc. Chem. Commun. 1988, 290. [Google Scholar]
- Shipley J. B.; Hecht S. M. Chem. Res. Toxicol. 1988, 1, 25. [DOI] [PubMed] [Google Scholar]
- Brown S. J.; Hudson S. E.; Stephan D. W.; Mascharak P. K. Inorg. Chem. 1989, 28, 468. [Google Scholar]
- Suga A.; Sugiyama T.; Sugano Y.; Kittaka A.; Otsuka M.; Ohno M.; Sugiura Y.; Maeda K. Synlett 1989, 70. [Google Scholar]
- Aoyagi Y.; Chorghade M. S.; Padmapriya A. A.; Suguna H.; Hecht S. M. J. Org. Chem. 1990, 55, 6291. [Google Scholar]
- Otsuka M.; Masuda T.; Haupt A.; Ohno M.; Shiraki T.; Sugiura Y.; Maeda K. J. Am. Chem. Soc. 1990, 112, 838. [Google Scholar]
- Boger D. L.; Dang Q. J. Org. Chem. 1992, 57, 1631. [Google Scholar]
- Boger D. L.; Menezes R. F. J. Org. Chem. 1992, 57, 4331. [Google Scholar]
- Boger D. L.; Menezes R. F.; Dang Q. J. Org. Chem. 1992, 57, 4333. [Google Scholar]
- Boger D. L.; Menezes R. F.; Dang Q.; Yang W. Bioorg. Med. Chem. Lett. 1992, 2, 261. [Google Scholar]
- Boger D. L.; Yang W. Bioorg. Med. Chem. Lett. 1992, 2, 1649. [Google Scholar]
- Owa T.; Haupt A.; Otsuka M.; Kobayashi S.; Tomioka N.; Itai A.; Ohno M. Tetrahedron 1992, 48, 1193. [Google Scholar]
- Tan J. D.; Hudson S. E.; Brown S. J.; Olmstead M. M.; Mascharak P. K. J. Am. Chem. Soc. 1992, 114, 3841. [Google Scholar]
- Boger D. L.; Honda T. Tetrahedron Lett. 1993, 34, 1567. [Google Scholar]
- Boger D. L.; Menezes R. F.; Honda T. Angew. Chem., Int. Ed. 1993, 32, 273. [Google Scholar]
- Guajardo R. J.; Hudson S. E.; Brown S. J.; Mascharak P. K. J. Am. Chem. Soc. 1993, 115, 7971. [Google Scholar]
- Boger D. L.; Colletti S. L.; Honda T.; Menezes R. F. J. Am. Chem. Soc. 1994, 116, 5607. [Google Scholar]
- Boger D. L.; Honda T. J. Am. Chem. Soc. 1994, 116, 5647. [Google Scholar]
- Boger D. L.; Honda T.; Dang Q. J. Am. Chem. Soc. 1994, 116, 5619. [Google Scholar]
- Boger D. L.; Honda T.; Menezes R. F.; Colletti S. L. J. Am. Chem. Soc. 1994, 116, 5631. [Google Scholar]
- Boger D. L.; Honda T.; Menezes R. F.; Colletti S. L.; Dang Q.; Yang W. J. Am. Chem. Soc. 1994, 116, 82. [Google Scholar]
- Boger D. L.; Colletti S. L.; Teramoto S.; Ramsey T. M.; Zhou J. Bioorg. Med. Chem. 1995, 3, 1281. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Teramoto S.; Honda T.; Zhou J. J. Am. Chem. Soc. 1995, 117, 7338. [Google Scholar]
- Boger D. L.; Ramsey T. M.; Cai H. Bioorg. Med. Chem. 1996, 4, 195. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Teramoto S.; Cai H. Bioorg. Med. Chem. 1996, 4, 179. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Ramsey T. M.; Cai H.; Hoehn S. T.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1998, 120, 53. [Google Scholar]
- Boger D. L.; Ramsey T. M.; Cai H.; Hoehn S. T.; Stubbe J. J. Am. Chem. Soc. 1998, 120, 9149. [Google Scholar]
- Boger D. L.; Ramsey T. M.; Cai H.; Hoehn S. T.; Stubbe J. J. Am. Chem. Soc. 1998, 120, 9139. [Google Scholar]
- Boger D. L.; Aquila B. M.; Tse W. C.; Searcey M. Tetrahedron Lett. 2000, 41, 9493. [Google Scholar]
- Lehmann T. E.; Ming L.; Rosen M. E.; Que L. Jr. Biochemistry 1997, 36, 2807. [DOI] [PubMed] [Google Scholar]
- Wu W.; Vanderwall D. E.; Teramoto S.; Lui S. M.; Hoehn S. T.; Tang X.; Turner C. J.; Boger D. L.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1998, 120, 2239. [Google Scholar]
- Manderville R. A.; Ellena J. F.; Hecht S. M. J. Am. Chem. Soc. 1994, 116, 10851. [Google Scholar]
- Wu W.; Vanderwall D. E.; Stubbe J.; Kozarich J. W.; Turner C. J. J. Am. Chem. Soc. 1994, 116, 10843. [Google Scholar]
- Manderville R. A.; Ellena J. F.; Hecht S. M. J. Am. Chem. Soc. 1995, 117, 7891. [Google Scholar]
- Wu W.; Vanderwall D. E.; Lui S. M.; Tang X.; Turner C. J.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1996, 118, 1268. [Google Scholar]
- Wu W.; Vanderwall D. E.; Turner C. J.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1996, 118, 1281. [Google Scholar]
- Lui S. M.; Vanderwall D. E.; Wu W.; Tang X.; Turner C. J.; Kozarich J. W.; Stubbe J. J. Am. Chem. Soc. 1997, 119, 9603. [DOI] [PubMed] [Google Scholar]
- Vanderwall D. E.; Lui S. M.; Wu W.; Turner C. J.; Kozarich J. W.; Stubbe J. Chem. Biol. 1997, 4, 373. [DOI] [PubMed] [Google Scholar]
- Anderson E. D.; Boger D. L. J. Am. Chem. Soc. 2011, 133, 12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. D.; Boger D. L. Org. Lett. 2011, 13, 2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield M. K.; Woolley D. W. J. Org. Chem. 1964, 30, 1548. [DOI] [PubMed] [Google Scholar]
- Kenda B. M.; Matagne A. C.; Talaga P. E.; Pasau P. M.; Differding E.; Lallemand B. I.; Frycia A. M.; Moureau F. G.; Klitgaard H. V.; Gillard M. R.; Fuks B.; Michel P. J. Med. Chem. 2004, 47, 530. [DOI] [PubMed] [Google Scholar]
- Ohsawa A.; Arai H.; Ohnishi H.; Igeta H. J. Chem. Soc. Chem. Commun. 1980, 1182. [Google Scholar]
- Ohsawa A.; Arai H.; Ohnishi H.; Itoh T.; Kaihoh T.; Okada M.; Igeta H. J. Org. Chem. 1985, 26, 5520. [Google Scholar]
- Boulton A. J.; Fruttero R.; Kalenga Saka J. D. J. Chem. Soc. Perkin Trans. I 1986, 1249. [Google Scholar]
- Ohsawa A.; Kaihoh T.; Itoh T.; Okada M.; Kawabata C.; Yamaguchi K.; Igeta H. Chem. Pharm. Bull. 1988, 36, 3838. [Google Scholar]
- Babinski D. J.; Aguilar H. R.; Still R.; Frantz D. E. J. Org. Chem. 2011, 76, 5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlanti L.; Discordia R. P.; Hynes J. Jr.; Miller M. M.; O’Grady H.; Shi Z. Org. Lett. 2007, 9, 3821. [DOI] [PubMed] [Google Scholar]
- Cheung K. M. J.; Reynisson J.; McDonald E. Tetrahedron Lett. 2010, 51, 5915. [Google Scholar]
- Li Z.; Capretto D. A.; He C. Prog. Inorg. Chem. 2009, 56, 1. [Google Scholar]
- Wentrup C. Acc. Chem. Res. 2011, 44, 393. [DOI] [PubMed] [Google Scholar]
- Iitaka Y.; Nakatani T.; Muraoka Y.; Fujii A.; Takita T.; Umezawa H. J. Antibiot. 1978, 31, 1070. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Panek J. S. J. Org. Chem. 1981, 46, 2179. [Google Scholar]
- Boger D. L.; Panek J. S. J. Org. Chem. 1982, 47, 3763. [Google Scholar]
- Boger D. L.; Panek J. S.; Meier M. M. J. Org. Chem. 1982, 47, 895. [Google Scholar]
- Boger D. L.; Schumacher J.; Mullican M. D.; Patel M.; Panek J. S. J. Org. Chem. 1982, 47, 2673. [Google Scholar]
- Boger D. L.; Panek J. S. Tetrahedron Lett. 1984, 25, 3175. [Google Scholar]
- Boger D. L.; Panek J. S.; Yasuda M. Org. Synth. 1987, 66, 142. [Google Scholar]
- Boger D. L.; Dang Q. Tetrahedron 1988, 44, 3379. [Google Scholar]
- Boger D. L.; Kochanny M. J. J. Org. Chem. 1994, 59, 4950. [Google Scholar]
- Boger D. L.; Panek J. S. Tetrahedron Lett. 1983, 24, 4511. [Google Scholar]
- Boger D. L.; Coleman R. S.; Panek J. S. J. Org. Chem. 1985, 50, 5377. [Google Scholar]
- Boger D. L.; Sakya S. M. J. Org. Chem. 1988, 53, 5377. [Google Scholar]
- Boger D. L.; Panek J. S.; Patel M. Org. Synth. 1992, 70, 79. [Google Scholar]
- Sakya S. M.; Groskopf K. K. Tetrahedron Lett. 1997, 38, 3805. [Google Scholar]
- Boger D. L.; Schaum R. P.; Garbaccio R. M. J. Org. Chem. 1998, 63, 6329. [DOI] [PubMed] [Google Scholar]
- Soenen D. R.; Zimpleman J. M.; Boger D. L. J. Org. Chem. 2003, 68, 3593. [DOI] [PubMed] [Google Scholar]
- Hamasaki A.; Ducray R.; Boger D. L. J. Org. Chem. 2006, 71, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Coleman R. S. J. Org. Chem. 1984, 49, 2240. [Google Scholar]
- Kessler S. N.; Wegner H. A. Org. Lett. 2010, 12, 4062. [DOI] [PubMed] [Google Scholar]
- Boger D. L. Tetrahedron 1983, 39, 2869. [Google Scholar]
- Boger D. L. Chem. Rev. 1986, 86, 781. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.