

Abstract
Scavenger receptor class B, type I (SR-BI) binds HDL and mediates the selective uptake of cholesteryl esters (CE). Although the extracellular domain of SR-BI is critical for function, the structural characteristics of this region remain elusive. Using sulfhydryl-labeling strategies, we report the novel finding that all six cysteine (Cys) residues in the extracellular domain of SR-BI are involved in disulfide bond formation that is intramolecular by nature. We hypothesize that an SR-BI conformation stabilized by extracellular disulfide bonds is a prerequisite of SR-BI-mediated cholesterol transport. Thus, single Cys mutant SR-BI receptors (C251S-, C280S-, C321S-, C323S-, C334S- and C384S-SR-BI), as well as Cys-less-SR-BI, a mutant SR-BI receptor void of all Cys residues, were created and plasma membrane localization was confirmed. Functional assays revealed that C280S-, C321S-, C323S-, C334S- and Cys-less SR-BI mutant receptors displayed reduced HDL binding and subsequent selective uptake of HDL-CE. However, only C323S-SR-BI and Cys-less-SR-BI were unable to mediate wild-type levels of efflux of free cholesterol (FC) to HDL. None of the Cys mutations disrupted SR-BI’s ability to redistribute plasma membrane FC. Taken together, the intramolecular disulfide bonds in the extracellular domain of SR-BI appear to maintain the receptor in a conformation integral to its cholesterol transport functions.
Keywords: disulfide bond, extracellular domain, selective uptake, efflux, conformation, cholesteryl ester
Cysteine (Cys)1 is unique among the twenty common amino acids due to its ability to form covalently-linked disulfide bonds. Intramolecular disulfide bonds, occurring between Cys residues on the same polypeptide chain, are the necessary reinforcements of protein architecture, aiding both in protein folding and conformational stability of secondary and tertiary protein structure [reviewed in (1–5)]. This is especially true of secreted and large transmembrane proteins such as receptors, transporters, and channels, including ATP-binding cassette transporter A1 (6), P2X1 receptors (7), rhodopsin (8, 9), β-adrenergic receptors (10, 11), rat serotonin transporters (12), cardiac Na+-Ca2+ exchangers (13), human transcobalamin II (14), and human vesicle monoamine transporters (15)—all of which require one or more intramolecular disulfide bonds for proper folding, conformation, and biological function. On the other hand, intermolecular disulfide bonds formed between Cys residues on separate peptide chains provide a link between two protein molecules and contribute to the formation of dimers or oligomers in proteins such as the human prostacyclin receptor (16), metabotropic glutamate receptors 1 and 5 (17–19), and CD36 (20, 21).
Scavenger receptor class B type I (SR-BI) is an 82-kDa glycosylated cell surface receptor that functions in the selective uptake of HDL-CE into cells (22), chiefly those of the liver and steroid-producing tissues (23, 24). The selective uptake process occurs in two steps: (i) binding of HDL to the extracellular domain of SR-BI and (ii) transfer of CE from HDL to the plasma membrane for hydrolysis (25–28), without endocytosis of the HDL particle (29, 30). In addition to mediating the selective uptake of HDL-CE, SR-BI also functions in promoting efflux of free cholesterol (FC) to HDL (31, 32), as well as enlarging the pool of plasma membrane FC sensitive to exogenous cholesterol oxidase (33, 34).
The predicted topology model of SR-BI consists of a large extracellular domain anchored by a transmembrane domain at both the N- and C-terminus [reviewed in (35)]. Although it is well-established that the extracellular domain is necessary for receptor function (29, 30, 36–38), little is known about the structural organization of this domain at the plasma membrane, including the role of the six Cys residues located in the C-terminal half of this domain. As with CD36 (39), a scavenger receptor with a similar predicted topology, it is assumed that these six Cys residues are involved in disulfide bond formation, although there is no direct evidence to support this notion.
We hypothesized that a pre-requisite of SR-BI-mediated cholesterol transport is an SR-BI conformation stabilized by the extracellular disulfide bonds. In this study, we provide the first evidence that all six extracellular Cys residues are indeed involved in the formation of intramolecular disulfide bonds. Our site-directed mutagenesis studies suggest that loss of at least one disulfide bond formation disrupts (i) binding of HDL, (ii) selective uptake of HDL-CE, and (iii) efflux of FC to HDL. However, disulfide bonds do not appear to be necessary for the ability of SR-BI to redistribute plasma membrane free cholesterol.
EXPERIMENTAL PROCEDURES
Materials
The following antibodies were used: polyclonal anti-SR-BI specific for the C-terminal or the extracellular domain (Novus Biologicals, Inc., Littleton, CO); anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore, Billerica, MA); peroxidase-conjugated goat anti-rabbit secondary IgG (Jackson ImmunoResearch Laboratories, West Grove, PA); peroxidase-conjugated sheep anti-mouse secondary IgG (Jackson ImmunoResearch Laboratories, West Grove, PA); FITC-conjugated goat anti-rabbit IgG (Fisher Scientific). Human HDL (1.063–1.21 g/mL) was purchased from Biomedical Technologies, Inc. [125I]Iodine was from Perkin-Elmer, while [3H]cholesterol and [3H]cholesteryl oleoyl ether (COE) were from GE Healthcare (Piscataway, NJ). Cholesterol oxidase (Streptomyces), acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor (Sandoz 58-035), cholesterol, 4-cholesten-3-one and cholesteryl oleate standards were purchased from Sigma. EZ-Link Sulfo-NHS-LC-biotin was purchased from Thermo Fisher Scientific (Rockford, IL). N-Biotinylaminoethyl methanethiosulfonate (MTSEA-biotin) was purchased from Toronto Research Chemicals, Inc. (North York, ON, Canada). N-(1-pyrene)maleimide was purchased from Invitrogen. All other reagents were of analytical grade.
Plasmids and sequencing
Site-directed mutations at C280, C321, and C334 were introduced into wild-type murine SR-BI [pSG5(SR-BI)] (29) using the QuikChange Site-Directed Mutagenesis kit (Stratagene) according manufacturer’s protocols. The following oligonucleotide primers (Integrated DNA Technologies) were used for mutagenesis: C280S, 5′ – GCC CGG AGG CAA GCA GGT CCA TGA AGC – 3′ and 5′ – GCT TCA TGG ACC TGC TTG CCT CCG GGC – 3′; C321S, 5′ – CCA CCC AAC GAA GGC TTC AGC CCA TGC CGA GAG TCT GGC – 3′ and 5′ – GCC AGA CTC TCG GCA TGG GCT GAA GCC TTC GTT GGG TGG – 3′; C334S, 5′ – GCA TTC AGA ATG TCA GCA CCA GCA GGT TTG GTG CGC C – 3′ and 5′ – GGC GCA CCA AAC CTG CTG GTG CTG ACA TTC TGA ATG C – 3′. All plasmids were purified using endotoxin-free Qiagen Maxi-Prep kits and sequenced through the coding region to verify the correct substitution and the absence of undesired mutations generated during the amplification steps. DNA sequencing was performed on an ABI 3100 at the Protein and Nucleic Acid Facility at the Medical College of Wisconsin. The C251S, C323S, C384S, and Cys-less mutant receptors were produced and sequenced by TOP Gene Technologies (Pointe-Claire, Quebec, Canada). Myc-SR-BI (36) and G420C-SR-BI (40) were previously described.
Cell culture and transfection
COS-7 cells were maintained in DMEM (Invitrogen), 10% calf serum (Invitrogen), 2 mM L-glutamine, 50 units/ml penicillin, 50 μg/ml streptomycin and 1 mM sodium pyruvate, and transfected as previously described (29). Cells were assayed 48 h post-transfection, with the exception of efflux assays in which cells were assayed 72 h post-transfection.
Cell lysis
COS-7 cells expressing either wild-type or mutant SR-BI were washed twice with PBS (pH 7.4) and lysed with 1% NP-40 cell lysis buffer (29) containing the following protease inhibitors: 1 μg/mL pepstatin, 0.2 mM phenylmethylsulfonyl fluoride, 1 μg/mL leupeptin, 10 μg/mL aprotinin. Protein concentrations were determined by the Lowry method as previously described (41).
PFO-PAGE
COS-7 cells expressing either wild-type or mutant SR-BI were washed twice with PBS (pH 7.4) and lysed with PBS containing the aforementioned protease inhibitors. Lysates were processed and PFO-PAGE was completed as previously described (42) using 4–15% polyacrylamide gradient gels.
Immunoblot analysis
Wild-type and mutant SR-BI, as well as GAPDH, were detected by immunoblot analysis as previously described (42).
Detection of free sulfhydryls by pyrene labeling of SR-BI
Myc-tagged SR-BI was immunoprecipitated from transiently transfected COS-7 cells (43) and labeled with a 2-fold molar excess of N-(1-pyrene) maleimide for 3 hr at 37°C in the dark in the presence or absence of 10 mM tris(2-carboxylethyl) phosphine (TCEP). DTT was labeled as above and served as a positive control. Pyrene fluorescence emission spectra (350–600 nm) were collected on a Photon Technology International (PTI) fluorimeter following excitation at 345 nm.
Immunofluorescence
Wild-type or mutant SR-BI cell surface expression in transiently-transfected COS7 cells was visualized by immunofluorescence as previously described (42, 44). Nuclei were stained with ToPro3 (Invitrogen) (45).
Cell surface biotinylation
COS-7 cells transiently expressing either wild-type or mutant SR-BI were washed twice with PBS (pH 7.4) and labeled with 1 mg/mL EZ-Link Sulfo-NHS-LC-biotin as previously described (42). To label free sulfhydryls at the cell surface, COS-7 cells expressing either wild-type or mutant SR-BI (in the presence and absence of 10 mM TCEP) were washed twice with PBS (pH 7.4) and labeled with 10 mM MTSEA-biotin for 15 min at room temperature in the dark and the reaction was stopped with 50 mM ammonium chloride. Cells were lysed as described above and biotinylated proteins were immunoprecipitated with streptavidin agarose. Proteins were separated by SDS-PAGE electrophoresis and visualized by immunoblot analysis.
Cell surface receptor expression levels by flow cytometry
Forty-eight hours post-transfection, COS-7 cells transiently expressing empty vector, wild-type SR-BI, or mutant SR-BI receptors were assayed for cell surface expression via flow cytometry. Cells were washed, pelleted, resuspended in PBS/0.5% BSA and incubated with an antibody directed against the extracellular domain of SR-BI (1:200 dilution) for 20 min on ice. Cells were then washed, resuspended with PBS/0.5% BSA and incubated with FITC-conjugated goat anti-rabbit IgG (1:1000 dilution) on ice for an additional 20 min. Cells were washed with PBS/0.5% BSA, resuspended in the same buffer, and fluorescence intensities were analyzed on a FACS® Calibur (Flow Cytometry Core, Medical College of Wisconsin).
HDL labeling
HDL was double-labeled with [125I]-dilactitol tyramine and non-hydrolyzable [3H]-COE as previously described (29). For the various preparations of radiolabeled HDL, average specific activities were 168.3 dpm/ng protein for [3H] and 204.1 dpm/ng protein for [125I].
Cell association of [125I]-HDL and selective uptake of [3H]-HDL-COE
COS-7 cells were transiently-transfected with empty pSG5 vector, wild-type SR-BI, or mutant SR-BI cDNA. Cell association of [125I]-HDL and selective uptake of non-hydrolyzable [3H]-COE were performed as previously described (29). Data presented are the average of two independent experiments and are representative of eight separate experiments, all performed in triplicate. Data were calculated as ng HDL/mg cell protein or ng HDL-COE/mg cell protein for binding and selective uptake experiments, respectively. The data were then normalized to cell surface expression as determined by flow cytometry described above. Vector (pSG5) values were subtracted from all wild-type and mutant values. The resulting values were expressed relative to wild-type SR-BI values, which were set at 100%. Statistical comparisons were determined using one-way ANOVA with Bonferroni post-tests for all groups.
Free cholesterol efflux and cholesterol oxidase sensitivity assays
Assays were performed as previously described (37), unless otherwise specified. Data represent the average of at least four separate experiments performed in quadruplicate. Statistical comparisons were determined using one-way ANOVA with Bonferroni post-tests for all groups.
RESULTS
All extracellular cysteine residues are involved in disulfide bond formation
The SR-BI receptor possesses a total of eight Cys residues in its amino acid sequence. Two of these residues, C462 and C470, are located at the junction of the C-terminal transmembrane domain and the C-terminal cytoplasmic domain (35). Both residues are postulated to be fatty acylated (46), yet they are dispensable for SR-BI function (36). The remaining six Cys residues (at positions 251, 280, 321, 323, 334 and 384) are located in the C-terminal half of the extracellular domain of SR-BI and are conserved among the human, mouse, rat, pig, bovine, dog, rabbit, hamster, horse and chicken species. While it is assumed that these six extracellular Cys residues in SR-BI are involved in disulfide bond formation, direct evidence to support this notion in the literature is not available. Therefore, we used two different strategies to ascertain whether the sulfhydryl groups of these extracellular Cys residues were free or involved in disulfide bond formation.
In the first strategy, myc-SR-BI was immunoprecipitated from transiently-transfected COS7 cell lysates and labeled with N-(1-pyrene)maleimide, a sulfhydryl specific reagent that fluoresces only when bound to a free sulfhydryl group (47). Pyrene labeling was performed in the presence (reducing conditions) or absence (oxidizing conditions) of TCEP (48). As shown in Figure 1, pyrene was unable to label SR-BI in the absence of TCEP, suggesting the lack of free sulfhydryl groups available for pyrene labeling under oxidizing conditions. However, the typical pyrene fluorescence spectrum, with emission peaks at 375 and 395 nm, was observed for SR-BI under reducing conditions in the presence of TCEP. DTT served as a positive control for pyrene labeling (Figure 1, inset).
Figure 1.

Pyrene labeling of free Cys residues. Myc-SR-BI was immunoprecipitated from COS-7 cell lysates and labeled with pyrene maleimide for 3 h at 37°C in the dark in the presence (solid line, reducing conditions) or absence (dotted line, oxidizing conditions) of 10 mM TCEP. DTT served as a positive control for pyrene labeling (inset). Fluorescence emission spectra were collected following excitation at 345 nm.
In the second strategy, we further confirmed the involvement of all extracellular Cys residues in disulfide bond formation by cell surface biotinylation experiments using sulfhydryl-specific, membrane-impermeable MTSEA-biotin (7, 49, 50). COS-7 cells transiently transfected with wild-type SR-BI were labeled with MTSEA-biotin in the presence and absence of TCEP. MTSEA-biotinylated SR-BI complexes were immunoprecipitated from cell lysates and analyzed by protein immunoblotting. Wild-type SR-BI was present in total lysates (Figure 2, Panel B), but was only biotinylated under reducing conditions (Figure 2, panel A, lane 1). The inability to detect SR-BI under oxidizing conditions suggests that there were no free sulfhydryl groups available for MTSEA-biotinylation. G420C-SR-BI harbors an extra Cys residue in the extracellular domain and therefore served as a positive control for MTSEA biotinylation. Together, these data confirm that all of the extracellular Cys residues are involved in disulfide bond formation. The inability of 10 mM iodoacetamide, a sulfhydryl alkylating agent (51), to react with SR-BI in cells prior to cell lysis confirmed that these bonds were pre-existing in live cells and not non-specific due to oxidation of -SH groups during the lysis procedure (data not shown).
Figure 2.
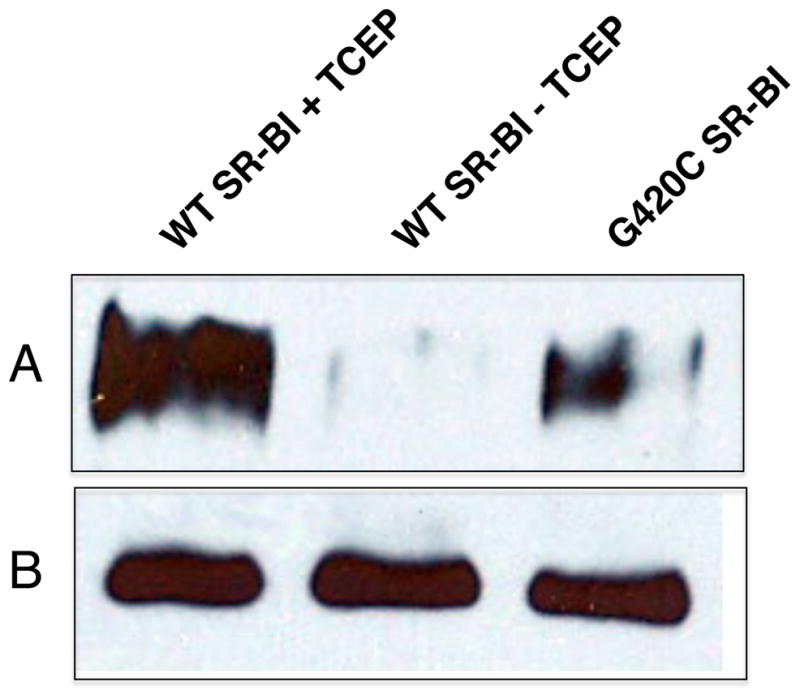
MTSEA-biotin labeling of free Cys residues. COS-7 cells expressing wild-type or Cys-less SR-BI were labeled with MTSEA-biotin for 15 min at room temperature under reducing (+ TCEP) and non-reducing (- TCEP) conditions. G420C-SR-BI served as a positive control for MTSEA labeling. Biotinylated complexes were immunoprecipitated from cell lysates using streptavidin agarose and samples were subjected to SDS-PAGE electrophoresis. Protein bands on the immunoblot were detected using an antibody directed against the C-terminal cytoplasmic tail of SR-BI. Panel A: biotinylated SR-BI (from ~200 μg of total lysate); panel B: total SR-BI expression in 20 μg of total cell lysates.
SR-BI possesses intramolecular disulfide bonds
Next, we sought to determine whether the extracellular disulfide bonds within SR-BI were intramolecular or intermolecular by nature. Since intramolecular disulfide bonds can affect protein conformation due to their role in protein folding and structural stability, polyacrylamide gel analysis often reveals a difference in migration between the oxidized and reduced forms of a protein (52–55). We compared the migration of SR-BI by 10% SDS-PAGE under oxidizing and reducing conditions. Our analysis revealed a slower electrophoretic mobility for SR-BI reduced with either 100 mM DTT or 10% β-mercaptoethanol (Figure 3, left panel). This observation (i) indicates the presence of intramolecular disulfide bonds and (ii) suggests that intramolecular disulfide bonding correlates with conformational changes within SR-BI’s secondary structure (18, 54–56).
Figure 3.
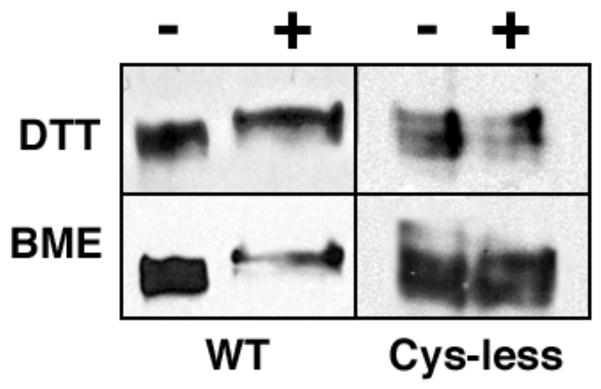
Reduction of intramolecular disulfide bonds by DTT and βME. COS-7 cells expressing wild-type (left panels) or Cys-less SR-BI (right panels) receptors were lysed and subjected to 10% SDS-PAGE electrophoresis in the presence and absence of either 10 mM DTT or 10% βME. Protein bands were detected using an antibody directed against the C-terminal cytoplasmic tail of SR-BI.
SR-BI is known to form dimers and higher order oligomers (43, 57–59). Since disulfide bonds can contribute to formation of multimeric complexes through the linkage of Cys residues on two separate peptide chains, we wished to determine whether any of the proposed disulfide bonds in the extracellular domain of SR-BI were intermolecular in nature. To this end, we created an SR-BI receptor void of Cys residues, herein referred to as Cys-less SR-BI. As expected, there were no differences in electrophoretic mobility of Cys-less SR-BI under reducing vs. oxidizing conditions (Figure 3, right panel). Protein complexes from COS-7 cell lysates transiently expressing either wild-type or Cys-less SR-BI were resolved by PFO-PAGE, an electrophoretic technique used to stabilize and separate existing oligomeric complexes (60). As shown in Figure 4, Cys-less SR-BI is still able to form dimers, despite the absence of Cys residues. These data support our initial findings that the disulfide bonds in the extracellular domain of SR-BI are most likely intramolecular by nature and that SR-BI oligomerization probably occurs via non-covalent interactions, as further evidenced by the presence of SR-BI dimers under reducing conditions (58). Further, it appears that the loss of disulfide bonds in Cys-less SR-BI has resulted in an extracellular domain with a conformation that can no longer support higher order oligomer formation as observed for wild-type SR-BI.
Figure 4.
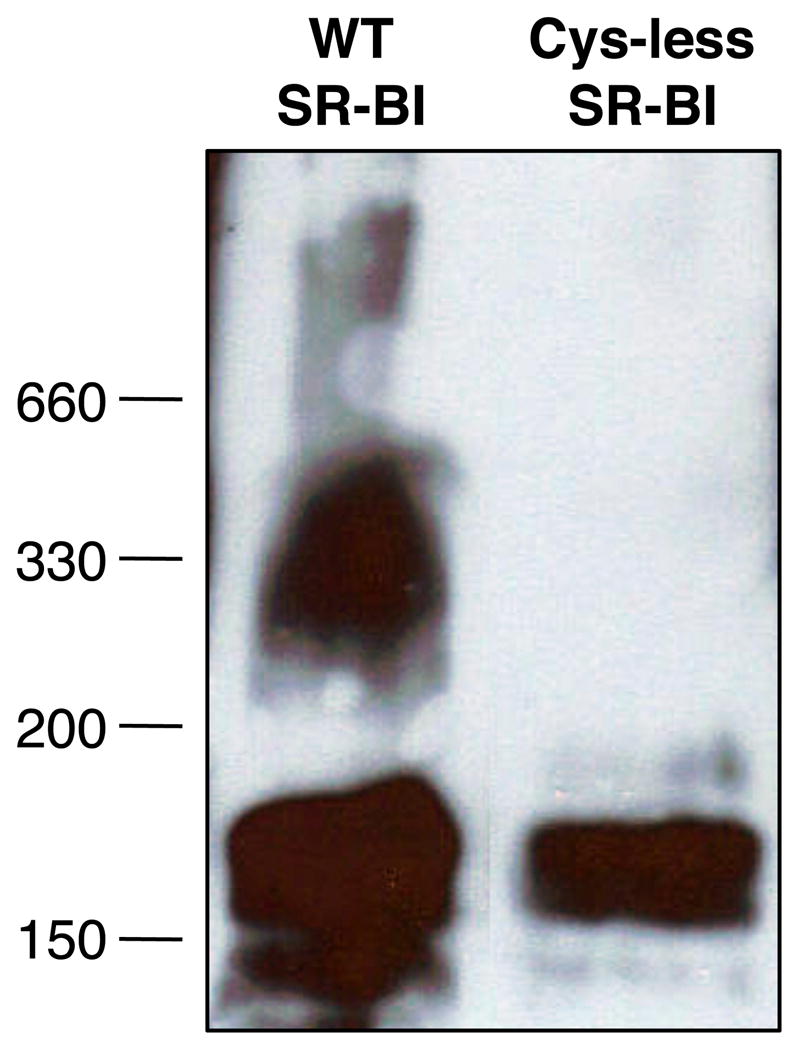
Oligomerization patterns of wild-type and Cys-less SR-BI. COS-7 cell lysates expressing either wild-type or Cys-less SR-BI were examined by PFO-PAGE as previously described (42) using 4–15% polyacrylamide gradient gels. SR-BI was detected using an antibody directed against the C-terminal cytoplasmic tail of SR-BI.
Wild-type and mutant SR-BI receptors are expressed at the cell surface
In order to determine the functional role of the Cys residues in the extracellular domain of SR-BI, in addition to Cys-less SR-BI, we generated a panel of single point mutations in which each Cys residue was mutated to serine (Ser, S) to generate the following receptors: C251S-, C280S-, C321S-, C323S-, C334S-, and C384S-SR-BI. Since loss of Cys residues can lead to impaired receptor trafficking to the plasma membrane (61, 62), COS-7 cells transiently expressing wild-type or mutant SR-BI receptors were analyzed for plasma membrane localization by fluorescent microscopy. All mutant receptors displayed similar staining patterns at the cell surface and cell extensions as wild-type SR-BI (44) (Figure 5). No staining was observed in cells transfected with empty vector or in non-transfected cells. In conjunction with these studies, immunoblot analysis from MTSEA-biotinylation of single-Cys SR-BI mutant receptors supported the cell surface localization of these receptors and further confirmed the presence of a free sulfhydryl group (Figure 6). Densitometry analyses did not reveal statistically significant differences in cell surface expression for all receptors tested (ANOVA, p>0.05; data not shown). Sulfo-NHS-LC-biotinylation also verified the cell surface localization of Cys-less SR-BI (Figure 6, right panels), although it tended to express less than wild-type SR-BI. Expression of all mutant SR-BI receptors in total cell lysates is also shown (Figure 6B). Therefore, loss of a single extracellular Cys residue, and the corresponding loss of a single disulfide bond, did not affect trafficking of SR-BI to the cell surface.
Figure 5.

Cell surface staining patterns of wild-type and mutant SR-BI receptors. Twenty-four h post-transfection, COS-7 cells expressing wild-type or mutant SR-BI receptors were plated onto glass coverslips. Cells were fixed and stained with an antibody directed against the extracellular domain of SR-BI followed by an Alexa 568-conjugated secondary antibody. Cells expressing empty vector stained with primary antibody (pSG5) or SR-BI without primary antibody staining (SR-BI, no prim) are also shown. Nuclei were stained with ToPro.
Figure 6.
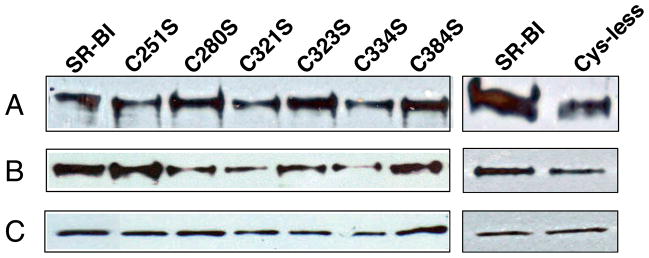
Cell surface expression of mutant SR-BI receptors. COS-7 cells expressing wild-type (in the presence of reducing TCEP) or single mutant SR-BI receptors were incubated with MTSEA-biotin. In a separate experiment, cells expressing wild-type and Cys-less SR-BI receptors were incubated with Sulfo-NHS-LC-Biotin. All biotinylated proteins were immunoprecipitated as previously described (42). Immunoblot analysis of SR-BI expression at (A) the cell surface by biotinylation (from ~200 μg of total lysate) and (B) in 20 μg of total cell lysate is shown. Protein bands were detected at ~82 kDa using an antibody directed against the C-terminal cytoplasmic tail of SR-BI. (C) GAPDH was detected at ~37 kDa as a loading control. Data are representative of three independent transfections.
Several mutant receptors display a reduced ability to bind HDL and mediate the selective uptake of HDL-COE
In order to determine whether the loss of Cys residues affected SR-BI function, wild-type and mutant SR-BI receptors were tested for their ability to bind HDL and mediate selective uptake of HDL-COE following transient transfection in COS-7 cells. Flow cytometry using antibodies directed against the extracellular domain of SR-BI revealed that all receptors expressed at similar levels at the cell surface (p > 0.05; data not shown). Upon normalization to cell surface expression, our analysis revealed that C251S- and C384S-SR-BI mediated wild-type levels of HDL binding and selective uptake of HDL-COE. However, for C280S-, C321S-, C323S-, and C334S-SR-BI mutant receptors, their reduced ability to bind HDL (28%, 48%, 41%, and 43% of wild-type, respectively, Figure 7A) was accompanied by an impaired ability to mediate the selective uptake of HDL-COE (35%, 45%, 40%, and 46% of wild-type, respectively, Figure 7B). Cys-less SR-BI displayed an even greater reduction in binding of HDL and selective uptake of HDL-COE (11% and 4% of wild-type, respectively). These data demonstrate the requirement of disulfide bonds associated with residues C280, C321, C323 ad C334 for efficient CE uptake.
Figure 7.

HDL binding and selective uptake of HDL-COE in cells expressing wild-type and mutant SR-BI receptors. COS-7 cells transiently expressing wild-type or mutant SR-BI receptors were incubated at 37°C for 1.5 h with double-labeled [125I]DLT/[3H]COE-labeled HDL (10 μg/mL). Cells were then processed and analyzed as previously described (29). Cell associated HDL (A) and selective uptake of HDL-COE (B) are shown. For both panels, data are normalized to cell surface receptor expression (as determined by flow cytometry of cells from parallel wells) and expressed relative to wild-type SR-BI values, which were set at 100%, following subtraction of empty vector values. Flow cytometry did not reveal any significant differences in cell surface expression between all receptors tested (ANOVA, p>0.05; data not shown). Values represent means ± SD of six replicates from two independent experiments and are representative of eight independent experiments overall. ***p < 0.001 as determine by one-way ANOVA.
Mutant receptors do not display major changes in cholesterol efflux and oxidase-sensitivity of membrane free cholesterol
SR-BI not only mediates flux of CE from HDL into cells, but it can also stimulate the flux of FC out of cells to HDL and other acceptor particles (31–33). In order to establish whether this cholesterol transport function was affected by our SR-BI mutations, COS-7 cells expressing either wild-type or mutant SR-BI receptors were assayed for their ability to stimulate efflux of FC to HDL. Interestingly, with the exception of C323S-SR-BI, all single Cys mutant receptors displayed wild-type levels of cholesterol efflux (Figure 8). Cys-less-SR-BI exhibited a similar muted ability to efflux FC to HDL as C323S-SR-BI. Control FC efflux to 0.5% BSA was minimal (1.0–1.8%) and did not differ between wild-type SR-BI and mutant receptors (data not shown).
Figure 8.

Efflux of [3H]-cholesterol to HDL by wild-type and mutant SR-BI receptors. COS-7 cells transiently expressing wild-type or mutant SR-BI receptors were pre-labeled with [3H]cholesterol and incubated with 50 μg/mL HDL for 4 h to measure the efflux of [3H]cholesterol to the HDL acceptor. Values represent means ± SD of six independent experiments, each performed in quadruplicate. ***p < 0.001 as determined by one-way ANOVA.
SR-BI also increases the pool of plasma membrane FC available for oxidation by exogenous cholesterol oxidase, as judged by a higher membrane content of cholestenone (34). In order to determine whether our mutations affected the distribution of FC in the plasma membrane, COS-7 cells expressing either wild-type or mutant SR-BI receptors were treated with exogenous cholesterol oxidase 48 h post-transfection. Lipids were extracted, separated by thin layer chromatography, and assessed for cholestenone production. Surprisingly, none of the mutant receptors, including Cys-less SR-BI, showed a decrease in FC accessibility, as indicated by wild-type levels of cholestenone production (Figure 9). This observation suggests that the sensitivity of plasma membrane FC pools to exogenous oxidase is not dependent on Cys residues, nor perhaps a specific conformation of SR-BI.
Figure 9.

Sensitivity of cells expressing wild-type and mutant SR-BI receptors to cholesterol oxidase. COS-7 cells transiently expressing wild-type or mutant SR-BI receptors were pre-labeled with [3H]cholesterol and incubated with exogenous cholesterol oxidase (0.5 U/mL) for 4 hr to measure the production of cholestenone from free cholesterol. Values represent means ± SD of five independent experiments, each performed in quadruplicate.
DISCUSSION
The absence of a high-resolution structure of SR-BI remains an obstacle in understanding the molecular mechanisms of SR-BI-mediated cholesterol transport and how the molecular architecture of the extracellular domain contributes to this process (29, 30, 36–38). In this study, we sought to understand the role of the six evolutionarily-conserved Cys residues that are located in the extracellular domain of SR-BI. We provide the first known confirmation of the involvement of these Cys residues in disulfide bond formation and show that these bonds are most likely intramolecular. We also investigated the impact of these disulfide bonds on SR-BI function using in vitro assays. Our data revealed that Cys-less SR-BI, as well as four single Cys mutant receptors—C280S-, C321S-, C323S-, and C334S-SR-BI—displayed a reduced ability to bind HDL and mediate the selective uptake of HDL-CE. Further, C323S-SR-BI and Cys-less SR-BI displayed a defect in efflux of FC to HDL. Taken together, we conclude that a specific disulfide bonding pattern is required to maintain SR-BI in a conformation that supports these cholesterol transport functions.
An interesting finding from our experiments is that Cys 323 is critical for SR-BI function, as it not only disrupted HDL-CE selective uptake, but it was also the only single-Cys mutant receptor that revealed a deficiency in FC efflux to HDL. Cys-less SR-BI displayed a similarly blunted level of efflux capacity. It is likely that the lack of the C323 disulfide bond negatively affects the conformation of the extracellular domain, thus supporting the notion that SR-BI function is compromised if proper protein folding is not maintained. Alternatively, it is also possible that mutation of C323 may have caused disulfide shuffling or reorganization to form new “promiscuous” disulfide bonds, as reported for mutants of the P2X1 receptor (7). The resulting “alternate” protein conformation may be unfavorable for efflux of FC to HDL.
An unexpected finding from our studies was that sensitivity of plasma membrane FC to exogenous cholesterol oxidase was not affected by any of the Cys mutations. The various functions of SR-BI (e.g. FC efflux vs. oxidase sensitivity) are considered to be separable, and dependent on individual subdomains within the extracellular domain (38, 63, 64). Therefore, it is possible that the region of the extracellular domain that spans Cys 251 to Cys 384 is located within a sub-domain that is not responsible for mediating the redistribution of plasma membrane FC. Indeed, we recently published data that demonstrates the importance of several hydrophobic regions within the N-terminal half of the extracellular domain of SR-BI that influence the cholesterol-oxidase sensitivity of plasma membrane FC (42).
We took advantage of the known changes in electrophoretic migration of proteins based on oxidation/reduction states of the sulfhydryls (52–55) to demonstrate that the extracellular Cys residues, all of which are located in the C-terminal half of the receptor, are most likely involved in intramolecular disulfide bonding. This result was not surprising since the presence of a monomeric SR-BI population in cell lysates is indicative of intramolecular bonding (43). This bonding pattern has considerable impact on the structural organization of the region spanning residues 240–400 of the extracellular domain of SR-BI and most likely maintains this region in a conformation that supports productive complex formation (65), where both the HDL ligand and the receptor are precisely aligned and/or have the capacity to undergo appropriate conformational changes in order for efficient lipid transport to occur. Based on the data obtained from the single Cys mutant SR-BI receptors, mutation of C280, C321 or C334 is still able to mediate between 35–46% of HDL-CE selective uptake, suggesting that loss of a single disulfide bond may not have major effects on disruption of a functional conformation. However, mutation of C323 only, or all the Cys residues in Cys-less SR-BI, results in much greater decreases in the ability of SR-BI to mediate selective uptake of HDL-CE and free cholesterol efflux to HDL, thus highlighting the important contribution of all the disulfide bonds, in particular the bond linked to C323, to maintaining SR-BI’s extracellular domain in a conformation that supports its functions.
As our previous studies demonstrated that SR-BI dimerizes via regions within the C-terminal half of its extracellular domain (59), it is possible that the extracellular Cys residues could contribute to SR-BI oligomerization by mediating receptor-receptor interactions via intermolecular disulfide bonds. However, as shown in Figure 4, PFO-PAGE analysis of Cys-less SR-BI eliminated this possibility, as this mutant receptor was still able to form dimers, similar to wild-type SR-BI. Our data suggest that self-association between C-terminal regions of SR-BI is most likely mediated via non-covalent interactions between SR-BI monomers (Hanson and Sahoo, unpublished data). More recently, a glycine dimerization motif was also identified in the N-terminal transmembrane domain of SR-BI (66).
In addition to the maintenance of protein structure, disulfide bond formation has been reported to be important for proper protein targeting to the plasma membrane (61, 62). Since immunofluorescence and biotinylation studies confirmed the cell surface localization of all single Cys mutant SR-BI receptors, as well as Cys-less SR-BI, we are confident that Cys residues in SR-BI are not required for protein trafficking to the plasma membrane. It should be noted, however, that as Cys-less SR-BI expression in total cell lysates (by immunoblotting) was generally slightly lower than wild-type SR-BI expression, loss of all disulfide bonds may have minor effects on protein stability or rates of receptor trafficking to the cell surface. Therefore, unlike other transporters such as sucrose permease (67), ACAT1 (68), mouse organic anion transporter 1(61) and sulfate transporter SHST1 (69), that display normal membrane trafficking and function despite loss of all Cys residues, we believe the loss of function in our mutant receptors correlates to the requirement of Cys residues for the structural integrity of the extracellular domain of SR-BI.
As the disruption of selective uptake of HDL-CE correlates with the loss of disulfide bonds in SR-BI’s extracellular domain, assigning a bonding pattern to each pair of Cys residues will be critical to gaining insight as to how the structural organization of the C-terminal half of the extracellular domain influences SR-BI function. Our data suggest that single substitution of Cys residues at position 251 and 384 had no effect on SR-BI function. These results implicate that a disulfide bond most likely exists between C251 and C384, and this bond is not necessary to support a receptor conformation that supports the cholesterol transport-related functions of SR-BI. It is also possible that these residues may be in separate disulfide bonds, but are able to substitute for one another if one of the residues is absent, as reported for other proteins (6, 14). However, this is an unlikely scenario as Ser substitution of Cys residues at positions 280, 321, 323, or 334 causes a significant reduction in SR-BI function, suggesting that these four Cys residues are combined in such a way to form two disulfide bonds. Experiments are currently underway to map each extracellular disulfide bond in SR-BI. These data will be valuable in understanding how the conformational restraints afforded by these covalent bonds impact the molecular architecture of this portion of the extracellular domain that is critical for SR-BI function.
Acknowledgments
The authors thank Kay Nicholson for excellent technical assistance and Jeff Woodliff for performing the flow cytometry experiments. We are grateful to Drs. Victor Drover and Krishna Rajarathnam for a critical review of the manuscript.
Footnotes
This work was supported by a National Institute of Health grant (HLR0158012) to D.S. and an American Heart Association Predoctoral Fellowship (10PRE2790023) to G.A.P.
ABBREVIATIONS: ACAT, acyl-CoA: cholesterol acyltransferase; BME, β-mercaptoethanol; CE, cholesteryl ester; COE, cholesteryl oleyl ether; Cys, C, cysteine; FC, free cholesterol; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HDL, high density lipoprotein; MTSEA, N-biotinylaminoethyl methanethiosulfonate; PFO, perfluorooctanoic acid; Ser, S, serine; SR-BI, scavenger receptor class B type I; TCEP, tris(2-carboxyethyl)phosphine
References
- 1.Brosnan JT, Brosnan ME. The sulfur-containing amino acids: an overview. J Nutr. 2006;136:1636S–1640S. doi: 10.1093/jn/136.6.1636S. [DOI] [PubMed] [Google Scholar]
- 2.Raina S, Missiakas D. Making and breaking disulfide bonds. Annu Rev Microbiol. 1997;51:179–202. doi: 10.1146/annurev.micro.51.1.179. [DOI] [PubMed] [Google Scholar]
- 3.Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- 4.Swaisgood HE. The importance of disulfide bridging. Biotechnol Adv. 2005;23:71–73. doi: 10.1016/j.biotechadv.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. Disulfide bonds and protein folding. Biochemistry. 2000;39:4207–4216. doi: 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 6.Hozoji M, Kimura Y, Kioka N, Ueda K. Formation of two intramolecular disulfide bonds is necessary for ApoA-I-dependent cholesterol efflux mediated by ABCA1. J Biol Chem. 2009;284:11293–11300. doi: 10.1074/jbc.M900580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ennion SJ, Evans RJ. Conserved cysteine residues in the extracellular loop of the human P2X(1) receptor form disulfide bonds and are involved in receptor trafficking to the cell surface. Mol Pharmacol. 2002;61:303–311. doi: 10.1124/mol.61.2.303. [DOI] [PubMed] [Google Scholar]
- 8.Davidson FF, Loewen PC, Khorana HG. Structure and function in rhodopsin: replacement by alanine of cysteine residues 110 and 187, components of a conserved disulfide bond in rhodopsin, affects the light-activated metarhodopsin II state. Proc Natl Acad Sci U S A. 1994;91:4029–4033. doi: 10.1073/pnas.91.9.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwa J, Reeves PJ, Klein-Seetharaman J, Davidson F, Khorana HG. Structure and function in rhodopsin: further elucidation of the role of the intradiscal cysteines, Cys-110, -185, and -187, in rhodopsin folding and function. Proc Natl Acad Sci U S A. 1999;96:1932–1935. doi: 10.1073/pnas.96.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moxham CP, Ross EM, George ST, Malbon CC. Beta-adrenergic receptors display intramolecular disulfide bridges in situ: analysis by immunoblotting and functional reconstitution. Mol Pharmacol. 1988;33:486–492. [PubMed] [Google Scholar]
- 11.Noda K, Saad Y, Graham RM, Karnik SS. The high affinity state of the beta 2-adrenergic receptor requires unique interaction between conserved and nonconserved extracellular loop cysteines. J Biol Chem. 1994;269:6743–6752. [PubMed] [Google Scholar]
- 12.Chen JG, Liu-Chen S, Rudnick G. External cysteine residues in the serotonin transporter. Biochemistry. 1997;36:1479–1486. doi: 10.1021/bi962256g. [DOI] [PubMed] [Google Scholar]
- 13.Santacruz-Toloza L, Ottolia M, Nicoll DA, Philipson KD. Functional analysis of a disulfide bond in the cardiac Na(+)-Ca(2+) exchanger. J Biol Chem. 2000;275:182–188. doi: 10.1074/jbc.275.1.182. [DOI] [PubMed] [Google Scholar]
- 14.Kalra S, Li N, Seetharam S, Alpers DH, Seetharam B. Function and stability of human transcobalamin II: role of intramolecular disulfide bonds C98-C291 and C147–C187. Am J Physiol Cell Physiol. 2003;285:C150–160. doi: 10.1152/ajpcell.00496.2002. [DOI] [PubMed] [Google Scholar]
- 15.Thiriot DS, Sievert MK, Ruoho AE. Identification of human vesicle monoamine transporter (VMAT2) lumenal cysteines that form an intramolecular disulfide bond. Biochemistry. 2002;41:6346–6353. doi: 10.1021/bi015779j. [DOI] [PubMed] [Google Scholar]
- 16.Giguere V, Gallant MA, de Brum-Fernandes AJ, Parent JL. Role of extracellular cysteine residues in dimerization/oligomerization of the human prostacyclin receptor. Eur J Pharmacol. 2004;494:11–22. doi: 10.1016/j.ejphar.2004.04.041. [DOI] [PubMed] [Google Scholar]
- 17.Ray K, Hauschild BC. Cys-140 is critical for metabotropic glutamate receptor-1 dimerization. J Biol Chem. 2000;275:34245–34251. doi: 10.1074/jbc.M005581200. [DOI] [PubMed] [Google Scholar]
- 18.Romano C, Miller JK, Hyrc K, Dikranian S, Mennerick S, Takeuchi Y, Goldberg MP, O’Malley KL. Covalent and noncovalent interactions mediate metabotropic glutamate receptor mGlu5 dimerization. Mol Pharmacol. 2001;59:46–53. [PubMed] [Google Scholar]
- 19.Romano C, Yang WL, O’Malley KL. Metabotropic glutamate receptor 5 is a disulfide-linked dimer. J Biol Chem. 1996;271:28612–28616. doi: 10.1074/jbc.271.45.28612. [DOI] [PubMed] [Google Scholar]
- 20.Daviet L, Malvoisin E, Wild TF, McGregor JL. Thrombospondin induces dimerization of membrane-bound, but not soluble CD36. Thromb Haemost. 1997;78:897–901. [PubMed] [Google Scholar]
- 21.Thorne RF, Meldrum CJ, Harris SJ, Dorahy DJ, Shafren DR, Berndt MC, Burns GF, Gibson PG. CD36 forms covalently associated dimers and multimers in platelets and transfected COS-7 cells. Biochem Biophys Res Commun. 1997;240:812–818. doi: 10.1006/bbrc.1997.7755. [DOI] [PubMed] [Google Scholar]
- 22.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 23.Glass C, Pittman RC, Civen M, Steinberg D. Uptake of high-density lipoprotein-associated apoprotein A-I and cholesterol esters by 16 tissues of the rat in vivo and by adrenal cells and hepatocytes in vitro. J Biol Chem. 1985;260:744–750. [PubMed] [Google Scholar]
- 24.Pittman RC, Steinberg D. Sites and mechanisms of uptake and degradation of high density and low density lipoproteins. J Lipid Res. 1984;25:1577–1585. [PubMed] [Google Scholar]
- 25.Connelly MA, Kellner-Weibel G, Rothblat GH, Williams DL. SR-BIdirected HDL-cholesteryl ester hydrolysis. J Lipid Res. 2003;44:331–341. doi: 10.1194/jlr.M200186-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Delamatre JG, Carter RM, Hornick CA. Evidence that a neutral cholesteryl ester hydrolase is responsible for the extralysosomal hydrolysis of high-density lipoprotein cholesteryl ester in rat hepatoma cells (Fu5AH) J Cell Physiol. 1993;157:164–168. doi: 10.1002/jcp.1041570121. [DOI] [PubMed] [Google Scholar]
- 27.Shimada A, Tamai T, Oida K, Takahashi S, Suzuki J, Nakai T, Miyabo S. Increase in neutral cholesteryl ester hydrolase activity produced by extralysosomal hydrolysis of high-density lipoprotein cholesteryl esters in rat hepatoma cells (H-35) Biochim Biophys Acta. 1994;1215:126–132. doi: 10.1016/0005-2760(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 28.Sparrow CP, Pittman RC. Cholesterol esters selectively taken up from high-density lipoproteins are hydrolyzed extralysosomally. Biochim Biophys Acta. 1990;1043:203–210. doi: 10.1016/0005-2760(90)90297-b. [DOI] [PubMed] [Google Scholar]
- 29.Connelly MA, Klein SM, Azhar S, Abumrad NA, Williams DL. Comparison of class B scavenger receptors, CD36 and scavenger receptor BI (SR-BI), shows that both receptors mediate high density lipoprotein-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J Biol Chem. 1999;274:41–47. doi: 10.1074/jbc.274.1.41. [DOI] [PubMed] [Google Scholar]
- 30.Gu X, Trigatti B, Xu S, Acton S, Babitt J, Krieger M. The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J Biol Chem. 1998;273:26338–26348. doi: 10.1074/jbc.273.41.26338. [DOI] [PubMed] [Google Scholar]
- 31.Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, Tall AR. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 32.Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599–5606. doi: 10.1074/jbc.273.10.5599. [DOI] [PubMed] [Google Scholar]
- 33.de la Llera-Moya M, Rothblat GH, Connelly MA, Kellner-Weibel G, Sakr SW, Phillips MC, Williams DL. Scavenger receptor BI (SR-BI) mediates free cholesterol flux independently of HDL tethering to the cell surface. J Lipid Res. 1999;40:575–580. [PubMed] [Google Scholar]
- 34.Kellner-Weibel G, de La Llera-Moya M, Connelly MA, Stoudt G, Christian AE, Haynes MP, Williams DL, Rothblat GH. Expression of scavenger receptor BI in COS-7 cells alters cholesterol content and distribution. Biochemistry. 2000;39:221–229. doi: 10.1021/bi991666c. [DOI] [PubMed] [Google Scholar]
- 35.Krieger M. Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem. 1999;68:523–558. doi: 10.1146/annurev.biochem.68.1.523. [DOI] [PubMed] [Google Scholar]
- 36.Connelly MA, de la Llera-Moya M, Monzo P, Yancey PG, Drazul D, Stoudt G, Fournier N, Klein SM, Rothblat GH, Williams DL. Analysis of chimeric receptors shows that multiple distinct functional activities of scavenger receptor, class B, type I (SR-BI), are localized to the extracellular receptor domain. Biochemistry. 2001;40:5249–5259. doi: 10.1021/bi002825r. [DOI] [PubMed] [Google Scholar]
- 37.Connelly MA, De La Llera-Moya M, Peng Y, Drazul-Schrader D, Rothblat GH, Williams DL. Separation of lipid transport functions by mutations in the extracellular domain of scavenger receptor class B, type I. J Biol Chem. 2003;278:25773–25782. doi: 10.1074/jbc.M302820200. [DOI] [PubMed] [Google Scholar]
- 38.Temel RE, Trigatti B, DeMattos RB, Azhar S, Krieger M, Williams DL. Scavenger receptor class B, type I (SR-BI) is the major route for the delivery of high density lipoprotein cholesterol to the steroidogenic pathway in cultured mouse adrenocortical cells. Proc Natl Acad Sci U S A. 1997;94:13600–13605. doi: 10.1073/pnas.94.25.13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasmussen JT, Berglund L, Rasmussen MS, Petersen TE. Assignment of disulfide bridges in bovine CD36. Eur J Biochem. 1998;257:488–494. doi: 10.1046/j.1432-1327.1998.2570488.x. [DOI] [PubMed] [Google Scholar]
- 40.Parathath S, Darlington YF, de la Llera Moya M, Drazul-Schrader D, Williams DL, Phillips MC, Rothblat GH, Connelly MA. Effects of amino acid substitutions at glycine 420 on SR-BI cholesterol transport function. J Lipid Res. 2007;48:1386–1395. doi: 10.1194/jlr.M700086-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 42.Papale GA, Nicholson K, Hanson PJ, Pavlovic M, Drover VA, Sahoo D. Extracellular hydrophobic regions in scavenger receptor BI play a key role in mediating HDL-cholesterol transport. Arch Biochem Biophys. 2010;496:132–139. doi: 10.1016/j.abb.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahoo D, Darlington YF, Pop D, Williams DL, Connelly MA. Scavenger receptor class B Type I (SR-BI) assembles into detergent-sensitive dimers and tetramers. Biochim Biophys Acta. 2007;1771:807–817. doi: 10.1016/j.bbalip.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Peng Y, Akmentin W, Connelly MA, Lund-Katz S, Phillips MC, Williams DL. Scavenger receptor BI (SR-BI) clustered on microvillar extensions suggests that this plasma membrane domain is a way station for cholesterol trafficking between cells and high-density lipoprotein. Mol Biol Cell. 2004;15:384–396. doi: 10.1091/mbc.E03-06-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Hooijdonk CA, Glade CP, Van Erp PE. TO-PRO-3 iodide: a novel HeNe laser-excitable DNA stain as an alternative for propidium iodide in multiparameter flow cytometry. Cytometry. 1994;17:185–189. doi: 10.1002/cyto.990170212. [DOI] [PubMed] [Google Scholar]
- 46.Babitt J, Trigatti B, Rigotti A, Smart EJ, Anderson RG, Xu S, Krieger M. Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J Biol Chem. 1997;272:13242–13249. doi: 10.1074/jbc.272.20.13242. [DOI] [PubMed] [Google Scholar]
- 47.Wu CW, Yarbrough LR. N-(1-pyrene)maleimide: a fluorescent cross-linking reagent. Biochemistry. 1976;15:2863–2868. doi: 10.1021/bi00658a025. [DOI] [PubMed] [Google Scholar]
- 48.Burns JA, Butler JC, Moran J, Whitesides GM. Selective reduction of disulfides by tris(2-carboxyethyl)phosphine. J Org Chem. 1991;56:2648–2650. [Google Scholar]
- 49.Chen JG, Liu-Chen S, Rudnick G. Determination of external loop topology in the serotonin transporter by site-directed chemical labeling. J Biol Chem. 1998;273:12675–12681. doi: 10.1074/jbc.273.20.12675. [DOI] [PubMed] [Google Scholar]
- 50.Hu YK, Kaplan JH. Site-directed chemical labeling of extracellular loops in a membrane protein. The topology of the Na, K-ATPase alpha-subunit. J Biol Chem. 2000;275:19185–19191. doi: 10.1074/jbc.M000641200. [DOI] [PubMed] [Google Scholar]
- 51.Smythe CV. The reaction of iodoacetate and iodoacetamide with various sulfydryl groups, with urease, and with yeast preparations. J Biol Chem. 1936;114:601–612. [Google Scholar]
- 52.Betakova T, Moss B. Disulfide bonds and membrane topology of the vaccinia virus A17L envelope protein. J Virol. 2000;74:2438–2442. doi: 10.1128/jvi.74.5.2438-2442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y, Yang Y, Qi J, Peng H, Zhang JT. Effect of cysteine mutagenesis on the function and disulfide bond formation of human ABCG2. J Pharmacol Exp Ther. 2008;326:33–40. doi: 10.1124/jpet.108.138115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moxham CP, Malbon CC. Fat cell beta 1-adrenergic receptor: structural evidence for existence of disulfide bridges essential for ligand binding. Biochemistry. 1985;24:6072–6077. doi: 10.1021/bi00343a007. [DOI] [PubMed] [Google Scholar]
- 55.Silva CM, Cidlowski JA. Direct evidence for intra- and intermolecular disulfide bond formation in the human glucocorticoid receptor. Inhibition of DNA binding and identification of a new receptor-associated protein. J Biol Chem. 1989;264:6638–6647. [PubMed] [Google Scholar]
- 56.Raghu P, Ghosh S, Soundarya K, Haseeb A, Aruna B, Ehtesham NZ. Dimerization of human recombinant resistin involves covalent and noncovalent interactions. Biochem Biophys Res Commun. 2004;313:642–646. doi: 10.1016/j.bbrc.2003.11.156. [DOI] [PubMed] [Google Scholar]
- 57.Landschulz KT, Pathak RK, Rigotti A, Krieger M, Hobbs HH. Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J Clin Invest. 1996;98:984–995. doi: 10.1172/JCI118883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reaven E, Cortez Y, Leers-Sucheta S, Nomoto A, Azhar S. Dimerization of the scavenger receptor class B type I: formation, function, and localization in diverse cells and tissues. J Lipid Res. 2004;45:513–528. doi: 10.1194/jlr.M300370-JLR200. [DOI] [PubMed] [Google Scholar]
- 59.Sahoo D, Peng Y, Smith JR, Darlington YF, Connelly MA. Scavenger receptor class B, type I (SR-BI) homo-dimerizes via its C-terminal region: fluorescence resonance energy transfer analysis. Biochim Biophys Acta. 2007;1771:818–829. doi: 10.1016/j.bbalip.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramjeesingh M, Huan LJ, Garami E, Bear CE. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem J. 1999;342(Pt 1):119–123. [PMC free article] [PubMed] [Google Scholar]
- 61.Tanaka K, Zhou F, Kuze K, You G. Cysteine residues in the organic anion transporter mOAT1. Biochem J. 2004;380:283–287. doi: 10.1042/BJ20031724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pajor AM, Krajewski SJ, Sun N, Gangula R. Cysteine residues in the Na+/dicarboxylate co-transporter, NaDC-1. Biochem J. 1999;344(Pt 1):205–209. [PMC free article] [PubMed] [Google Scholar]
- 63.Rodrigueza WV, Thuahnai ST, Temel RE, Lund-Katz S, Phillips MC, Williams DL. Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J Biol Chem. 1999;274:20344–20350. doi: 10.1074/jbc.274.29.20344. [DOI] [PubMed] [Google Scholar]
- 64.Williams DL, de La Llera-Moya M, Thuahnai ST, Lund-Katz S, Connelly MA, Azhar S, Anantharamaiah GM, Phillips MC. Binding and cross-linking studies show that scavenger receptor BI interacts with multiple sites in apolipoprotein A-I and identify the class A amphipathic alpha-helix as a recognition motif. J Biol Chem. 2000;275:18897–18904. doi: 10.1074/jbc.M002411200. [DOI] [PubMed] [Google Scholar]
- 65.Liu T, Krieger M, Kan HY, Zannis VI. The effects of mutations in helices 4 and 6 of ApoA-I on scavenger receptor class B type I (SR-BI)-mediated cholesterol efflux suggest that formation of a productive complex between reconstituted high density lipoprotein and SR-BI is required for efficient lipid transport. J Biol Chem. 2002;277:21576–21584. doi: 10.1074/jbc.M112103200. [DOI] [PubMed] [Google Scholar]
- 66.Gaidukov L, Nager AR, Xu S, Penman M, Krieger M. Glycine dimerization motif in the N-terminal transmembrane domain of the HDL receptor SR-BI required for normal receptor oligomerization and lipid transport. J Biol Chem. 2011 doi: 10.1074/jbc.M111.229872. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sahin-Toth M, Frillingos S, Lawrence MC, Kaback HR. The sucrose permease of Escherichia coli: functional significance of cysteine residues and properties of a cysteine-less transporter. Biochemistry. 2000;39:6164–6169. doi: 10.1021/bi000124o. [DOI] [PubMed] [Google Scholar]
- 68.Lu X, Lin S, Chang CC, Chang TY. Mutant acyl-coenzyme A:cholesterol acyltransferase 1 devoid of cysteine residues remains catalytically active. J Biol Chem. 2002;277:711–718. doi: 10.1074/jbc.M109427200. [DOI] [PubMed] [Google Scholar]
- 69.Howitt SM. The role of cysteine residues in the sulphate transporter, SHST1: construction of a functional cysteine-less transporter. Biochim Biophys Acta. 2005;1669:95–100. doi: 10.1016/j.bbamem.2005.01.002. [DOI] [PubMed] [Google Scholar]