

Abstract
Inducing an intramolecular reaction is a powerful means of accelerating reactions. Though this mechanism of catalysis is common in enzymes, it is underutilized in synthetic catalysts. This article outlines our group’s recent efforts to use reversible covalent bonding to induce an intramolecular reaction, allowing for rate acceleration as well as control of the selectivity in the desymmetrization of 1,2-diols.
Keywords: asymmetric catalysis, desymmetrization, organocatalysis, diols, enantioselectivity
The development of new catalysts is a constantly evolving process. To design a new catalytic system requires an understanding of how reactions are accelerated. In general terms, there are two fundamental means for catalysts to accelerate reactions: 1) activation of the substrate(s) 2) preorganization of the substrate(s). For synthetic catalysts a significant amount of attention has been placed on the activation of substrates through the generation of reactive intermediates (e.g., Lewis acid or base activation, enamine formation, metal complexation).1 In contrast, nature has focused on accelerating reactions in large part through preorganization of substrates.2 Using substrate-binding forces, enzymes can position substrates near catalytic residues effectively turning multimolecular steps into pseudo-intramolecular transformations. Through substrate-binding forces, enzymes make a down payment in entropy that can be used to accelerate a subsequent step. Generically, bimolecular elementary steps require 15–35 eu in activation entropy; therefore, turning a biomolecular step into a unimolecular step has the potential to provide a rate enhancement of 104–108 for 1 M reactants at room temperature.3
This SynPact article will outline our inspirations and recent efforts to develop catalysts that tap into this mode of rate acceleration.
The design and application of bifunctional catalysts is one approach that chemists have employed to capture the rate acceleration provided by induced intramolecularity. In many cases the catalysts are designed to have a Lewis acidic and basic site allowing for dual activation of an electrophile and nucleophile.4 Some early examples of bifunctional catalysis in the asymmetric additions to carbonyls are shown in Figure 1 from the Corey,5 Noyori,6 and Shibasaki laboratories.4f Subsequent to this work bifunctional catalysts have been designed for both metal andnonmetal catalysts.7 The majority of the examples focus on the activation of two reactants in close proximity to one another; however, in terms of induced intramolecularity, activation of the substrate is not required to gain rate acceleration, only proximity. A paragon of this mode of catalysis was reported by the Kelly group, in which they demonstrated that an SN2 reaction can be accelerated through a templating catalyst (Equation 1).8
 |
Equation 1 |
Figure 1.

Proposed intermediates with bifunctional catalysts
Recently our group has capitalized on using proximity as a means of accelerating reactions by developing organic scaffolds that colocate a catalyst and starting material. The generic structure of a scaffolding catalyst has a catalyst-binding site (or bound catalytic residue) and a substrate-binding site (Scheme 1). The key insight for this concept is that substrate binding does not require simultaneous activation of a functional group; therefore, the scaffolds can be applied potentially to a wide range of transformations. In order for these scaffolds to be used catalytically, a reversible interaction is needed between substrate and scaffold (Scheme 1); we chose to use reversible covalent bonding in order to make a rigid interaction with the aim of improving selectivity in the desired reactions.9
Scheme 1.

Catalytic cycle for a scaffolding catalyst
Inspired by the works of catalytic directing groups in transition-metal catalysis,10 we initially applied this idea to regio-,11 diastereo-,12 and enantioselective hydroformylation reactions.13 In these cases a phosphorous-based ligand (2, Scheme 2) was used as the scaffold, where the phosphorous atom serves to coordinate to the metal catalyst, and the carbon in the orthoformate oxidation state binds to an array of organic functional groups. Having the substrate bound to the catalyst generates a chelating ligand for the metal allowing for both rate enhancement as well as control of the selectivity. For example, we employed 2 in the regioselective hydroformylation of 1,1-disubstituted olefins towards the formation of quaternary carbon centers.14
Scheme 2.

Branch-selective hydroformylation to form quaternary carbon centers
Formation of quaternary carbon centers has traditionally been challenging in hydroformylation due to the strong preference to place the aldehyde on the least hindered carbon.15 Using the scaffolding catalyst, we form the desired product in high regioselectivity and good yield (Scheme 2). Notably, a control reaction with Ph3P shows no conversion to hydroformylation products; furthermore, employing the methyl ether substrate (which can not bind to 2) also shows no conversion to product (Equation 2). These results demonstrate that substrate binding to the scaffold both accelerates the reaction and controls the selectivity.
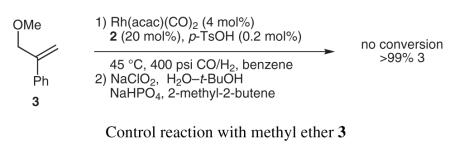 |
Equation 2 |
Having successfully applied the scaffolding strategy to hydroformylation, we looked to expand the concept to other transformations. In order to demonstrate the broad utility of the idea, we applied our strategy to the area of organocatalysis. Previous work by Hoveyda and Snapper had shown that 1,2- and 1,3-diols can be efficiently desymmetrized using an amino acid based catalyst (Equation 3).16–18 Using the Hoveyda/Snapper catalyst (4) as a starting point, we synthesized catalyst 5 guided by our basic design principle of having a catalytic site adjacent to a covalent substrate-binding site (Scheme 3).
 |
Equation 3 |
Scheme 3.
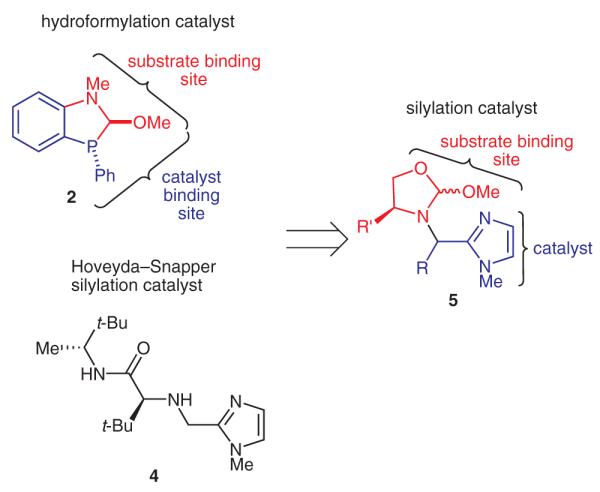
Design of organic catalyst 5
Optimization of core structure 5 led to the identification of 5a as an efficient catalyst for the desymmetrization of ciscyclopentane-1,2-diol (94% ee and 92% yield, Equation 4).19 An interesting feature of 5a is that the stereocenter at the exchange site likely epimerizes under the reaction conditions, yet only a single diastereomer is observed by 1H NMR. An X-ray crystal structure of 5c, formed by exchange of 5a with 4-bromobenzyl alcohol, shows that the alcohol binds syn to the i-Pr group on the oxazolidine ring (Figure 2). These data suggest that the external stereocenter is effectively gearing the stereo-center at the exchange site. As evidence that the reaction occurs through a reversible covalent bond, we tested catalyst 5b, which does not contain a substrate-binding site. Catalyst 5b provides both poor yield (5%) and enantioselectivity (4% ee), consistent with the idea that covalent bonding of the substrate to the catalyst is necessary for both selectivity and activity (Equation 4).
 |
Equation 4 |
Figure 2.

X-ray crystal structure of 5c
With the optimal catalyst in hand, we extended the substrate scope to include both cyclic and acyclic 1,2-diols. These substrates generally afford practical levels of yield (79–93%) and enantioselectivity (86–95% ee, Table 1). Though a range of substrates work in the desymmetrization reaction, the rate of reaction is highly dependent on the nature of the substrate. In general, five- and six-membered rings form product the fastest, with larger rings and acyclic substrates reacting at significantly slower rates. To improve on the practicality of the reaction, we found that use of TESCl in place of TBSCl shortened the reaction times and also allowed for the lowering of the equivalence of silyl chloride relative to substrate. In the case of 2,3-butane diol, the optimal conditions with TBSCl (4 equiv) require a reaction time of 36 hours at 0 °C, whereas TESCl (1.2 equiv) can be performed at room temperature in less than four hours with comparable yield (84%) and ee (92%, Equation 5).
 |
Equation 5 |
Table 1.
Desymmetrization of 1,2-Diols
| Entry | Product | Yield (%) | ee (%) |
|---|---|---|---|
| 1 |

|
79 | 89 |
| 2 |

|
87 | 90 |
| 3 |

|
88 | 95 |
| 4 |

|
86 | 92 |
| 5 |

|
82 | 90 |
| 6 |

|
93 | 86 |
| 7 |

|
78 | 90 |
As is often the case some of the more interesting results are the substrates that provide poor yields or selectivities. For example, (R,S)-pentane-2,4-diol and trans-cyclohexane-1,2-diol affords less than 5% yield of product. Furthermore, cis-4-cyclopentane-1,3-diol yields 26% of the product in 15% ee. Notably, cyclic cis-1,3-diols are effectively desymmetrized by the Hoveyda–Snapper catalyst 4 (Equation 3). We believe the drastic change in selectivity is a function of the mechanism of stereoselection for 5a. The covalent nature of the interaction between the scaffold and substrate leads to significant restriction of the spatial location of the substrate relative to the imidazole group. The net effect is that proximity plays a significant role in whether the silyl group can be intramolecularly transferred. Therefore, the location of the free hydroxyl affects both the selectivity and reactivity of a substrate. We are currently looking to exploit this effect to develop next-generation catalysts that will recognize specific functional-group displays, which can be applied to reactions requiring site selectivity.
The application of induced intramolecularity is a powerful means of accelerating reactions. Intriguingly, this mode of acceleration is orthogonal to traditional modes of catalysis that rely on the generation of reactive intermediates. Consequently, the two forms of catalysis can be used in conjunction, as demonstrated by most bifunctional catalysts. Generation of a reactive intermediate is not required to exploit intramolecularity in catalysis; therefore, incorporating binding sites into catalysts can be a means of accelerating reactions that use functional groups that are difficult to activate. Using covalent bonding between substrate and catalyst provides a confined structure that can be used to control a variety of selectivities, including site-, regio-, and stereoselectivity. In the case of silylation catalyst 5a, we believe a significant portion of the enantio-selectivity arises from proximity of the substrate and catalytic residue, which is enforced through the covalent bonding between catalyst and scaffold. We envision that through proper design of a molecular scaffold that binding selectivity and proximity can be exploited to achieve reactions that would be challenging using noncovalent interactions.
Acknowledgment
We thank the ACS-PRF (DNI-5001400) and NIGMS (RO1GM087581) for funding this research. A.D.W. was supported by a LaMattina graduate fellowship. Mass spectrometry instrumentation at Boston College is supported by funding from the NSF (DBI-0619576). The Boston College X-ray facility is supported by funding from the NSF (CHE-0923264).
Biography
 Kian Tan (left) was born in Virginia Beach, VA, USA. In 1999 he received his BSc degree in chemistry from the University of Virginia, and in 2004, he earned his PhD in chemistry from the University of California, Berkeley. After a two-year post-doctoral fellowship in the laboratories of Eric Jacobsen at Harvard University, he began his independent career as an assistant professor at Boston College.
Kian Tan (left) was born in Virginia Beach, VA, USA. In 1999 he received his BSc degree in chemistry from the University of Virginia, and in 2004, he earned his PhD in chemistry from the University of California, Berkeley. After a two-year post-doctoral fellowship in the laboratories of Eric Jacobsen at Harvard University, he began his independent career as an assistant professor at Boston College.
Amanda Worthy (center) was born in Petersburg, WV, USA. She received her BA at Goucher College, and is currently a 5th year PhD student in the chemistry department at Boston College.
Xixi Sun (right) was born in Chongqing, China. He received his BSc at the University of Science and Technology of China, and is currently a 4th year PhD student in the chemistry department at Boston College.
References
- (1) (a).Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito: 2010. [Google Scholar]; (b) Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito: 2009. [Google Scholar]; (c) Yamamoto H, Ishihara K. Acid Catalysis in Modern Organic Synthesis. Wiley-VCH; Weinheim: 2008. [Google Scholar]; (d) Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:1560. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- (2) (a).Blow D. Structure Fold Des. 2000;8:R77. doi: 10.1016/s0969-2126(00)00125-8. [DOI] [PubMed] [Google Scholar]; (b) Jencks WP. Annu. Rev. Biochem. 1997;66:1. doi: 10.1146/annurev.biochem.66.1.1. [DOI] [PubMed] [Google Scholar]; (c) Jencks WP. Catalysis in Chemistry and Enzymology. 2nd ed Dover; New York: 1975. [Google Scholar]
- (3) (a).Pascal R. Eur. J. Org. Chem. 2003:1813. [Google Scholar]; (b) Page MI, Jencks WP. Proc. Natl. Acad. Sci. U.S.A. 1971;68:1678. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4) (a).For reviews on bifunctional catalysis, see: Rowlands GJ. Tetrahedron. 2001;57:1865.. Ma JA, Cahard D. Angew. Chem. Int. Ed. 2004;43:4566. doi: 10.1002/anie.200300635.. Kanai M, Kato N, Ichikawa E, Shibasaki M. Synlett. 2005:1491.. Ikariya T, Murata K, Noyori R. Org. Biomol. Chem. 2006;4:393. doi: 10.1039/b513564h.. Paull DH, Abraham CJ, Scerba MT, Alden-Danforth E, Lectka T. Acc. Chem. Res. 2008;41:655. doi: 10.1021/ar700261a.. Shibasaki M, Kanai M, Matsunaga S, Kumagai N. Acc. Chem. Res. 2009;42:1117. doi: 10.1021/ar9000108.
- (5).Corey EJ, Helal CJ. Angew. Chem. Int. Ed. 1998;37:1987. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- (6).Noyori R, Kitamura M. Angew. Chem. Int. Ed. 1991;30:49. [Google Scholar]
- (7) (a).For reviews on bifunctional organic catalysts, see Bhadury PS, Song BA, Yang S, Hu DY, Xue W. Curr. Org. Synth. 2009;6:380. Connon SJ. Chem. Commun. 2008:2499. doi: 10.1039/b719249e. Wei Y, Shi M. Acc. Chem. Res. 2010;43:1005. doi: 10.1021/ar900271g. Georgiou I, Ilyashenko G, Whiting A. Acc. Chem. Res. 2009;42:756. doi: 10.1021/ar800262v.
- (8).Kelly TR, Bridger GJ, Zhao C. J. Am. Chem. Soc. 1990;112:8024. [Google Scholar]
- (9).Tan KL. ACS Catal. 2011;1:877. [Google Scholar]
- (10) (a).For reviews of catalytic directing groups in transition-metal catalysis, see Park YJ, Park JW, Jun CH. Acc. Chem. Res. 2008;41:222. doi: 10.1021/ar700133y. Rousseau G, Breit B. Angew. Chem. Int. Ed. 2011;50:2450. doi: 10.1002/anie.201006139.
- (11) (a).Worthy AW, Gagnon MM, Dombrowski MT, Tan KL. Org. Lett. 2009;11:2764. doi: 10.1021/ol900921e. [DOI] [PubMed] [Google Scholar]; (b) Lightburn TE, De Paolis OA, Cheng KH, Tan KL. Org. Lett. 2011;13:2686. doi: 10.1021/ol200782d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lightburn TE, Dombrowski MT, Tan KL. J. Am. Chem. Soc. 2008;130:9210. doi: 10.1021/ja803011d. [DOI] [PubMed] [Google Scholar]
- (13) (a).Worthy AD, Joe CL, Lightburn TE, Tan KL. J. Am. Chem. Soc. 2010;132:14757. doi: 10.1021/ja107433h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joe CL, Tan KL. J. Org. Chem. 2011;76:7590. doi: 10.1021/jo201328d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sun X, Frimpong K, Tan KL. J. Am. Chem. Soc. 2010;132:11841. doi: 10.1021/ja1036226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15) (a).Keulemans AIM, Kwantes A, van Bavel T. Recl. Trav. Chim. Pays-Bas. 1948;67:298. [Google Scholar]; (b) Breit B, Seiche W. Synthesis. 2001:1. [Google Scholar]
- (16).For an early example of a desymmetrization reaction with an amino acid based catalyst, see Oriyama T, Imai K, Sano T, Hosoya T. Tetrahedron Lett. 1998;39:3529.
- (17) (a).For reviews of asymmetric Si–O bond formation, see Weickgenannt A, Mewald M, Oestreich M. Org. Biomol. Chem. 2010;8:1497. doi: 10.1039/b925722e. Rendler S, Oestreich M. Angew. Chem. Int. Ed. 2008;47:248. doi: 10.1002/anie.200704210.
- (18) (a).Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature (London) 2006;443:67. doi: 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]; (b) Zhao Y, Mitra AW, Hoveyda AH, Snapper ML. Angew. Chem. Int. Ed. 2007;46:8471. doi: 10.1002/anie.200703650. [DOI] [PubMed] [Google Scholar]; (c) You Z, Hoveyda AH, Snapper ML. Angew. Chem. Int. Ed. 2009;48:547. doi: 10.1002/anie.200805338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rodrigo JM, Zhao Y, Hoveyda AH, Snapper ML. Org. Lett. 2011;13:3778. doi: 10.1021/ol2010819. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Isobe T, Fukuda K, Araki Y, Ishikawa T. Chem. Commun. 2001:243. [Google Scholar]; (f) Rendler S, Auer G, Oestreich M. Angew. Chem. Int. Ed. 2005;44:7620. doi: 10.1002/anie.200502631. [DOI] [PubMed] [Google Scholar]; (g) Weickgenannt A, Mewald M, Muesmann TWT, Oestreich M. Angew. Chem. Int. Ed. 2010;49:2223. doi: 10.1002/anie.200905561. [DOI] [PubMed] [Google Scholar]; (h) Sheppard CI, Taylor JL, Wiskur SL. Org. Lett. 2011;13:3794. doi: 10.1021/ol2012617. [DOI] [PubMed] [Google Scholar]
- (19).Sun X, Worthy AD, Tan KL. Angew. Chem. Int. Ed. 2011;50:8167. doi: 10.1002/anie.201103470. [DOI] [PMC free article] [PubMed] [Google Scholar]