

Abstract
Diazo compounds are in widespread use in synthetic organic chemistry, but have untapped potential in chemical biology. We report on the design and optimization of a phosphinoester that mediates the efficient conversion of azides into diazo compounds in phosphate buffer at neutral pH and room temperature. High yields are maintained in the presence of common nucleophilic or electrophilic functional groups, and reaction progress can be monitored by colorimetry. As azido groups are easy to install and maintain in biopolymers or their ligands, this new mode of azide reactivity could have substantial utility in chemical biology.
Diazo compounds are among the most versatile intermediates in organic synthesis. Due to their inherent dipolar nature, diazo compounds can readily participate in 1,3-dipolar cycloaddition reactions with a wide range of dipolarophiles.1 Moreover, C-protonation gives rise to diazonium ions, which are highly reactive alkylating agents.2 Thermal or photochemical generation of carbenes, along with transition metal-mediated carbenoid formation, facilitates addition to double bonds and insertion into C– H,O–H, and N–H bonds.3 This broad reactivity makes diazo compounds attractive for applications in chemical biology, having special promise in the labeling of proteins4,5 and as tunable reactants in 1,3-dipolar cycloaddition reactions with cycloalkynes.1,6
Although the first diazo compound was synthesized in the 19th century,7 there are still few methods for their construction: diazo transfer to an activated C–H acceptor, diazotization of an amine, decomposition or oxidation of a hydrazone, rearrangement of an N-acyl-N-nitrosoamine, and fragmentation of a triazene.5c,7,8 The harsh conditions required to access diazo compounds can provoke undesirable reactivity with common functional groups, and the need for organic solvents hampers the generation of diazo compounds in many molecules of biological interest.
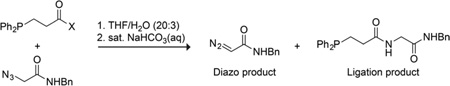
Recently, we reported that an azide can be converted into the corresponding diazo compound by the generation and subsequent decomposition of an acyl triazene.8f The phosphinoester reagent that mediates this deimidogenation is, however, nearly insoluble in water and unstable to hydrolysis, and the preferred solvent was THF/H2O (20:3). As azido groups have become a privileged functionality in chemical biology,9 a stable, water-soluble reagent that converts azides into their corresponding diazo compounds under physiological conditions and that tolerates the sensitive functionalities in biological molecules could have high utility.
According to our putative mechanism (Scheme 1),8f,10 azide deimidogenation begins with nucleophilic attack of the phosphine on the azide to form a phosphazide. Then, the reaction takes one of two possible pathways, depending on the ability of the pendant acyl group to trap the phosphazide intermediate prior to N2(g) extrusion. We hypothesized that this ability correlates with the pKa of the conjugate acid of the leaving group, and that more reactive acyl groups would favor diazo compound formation and less reactive groups would favor amides.
scheme 1.

To test our hypothesis, we surveyed the reactivity of a wide variety of potential phosphinoesters with α-azido-N-benzylacetamide. Reaction mixtures had equimolar reactants, which were incubated at room temperature for 4 h, quenched with saturated aqueous NaHCO3, and then stirred for another 8 h. We found that the diazo compound:amide product ratio does indeed correlate with the pKa of the conjugate acid of the leaving group (Table 1). When the pKa was ≥9.2, the intermediate phosphazide underwent rapid N2(g) extrusion rather than acyl transfer, yielding exclusively the Staudinger ligation product. As the pKa was lowered from 9.0 to 7.6, the product ratio began to increase. When the pKa was ≤7.1, the reaction produced the diazo compound, almost exclusively.
Table 1.
Phosphinoester Reactivity and Stability
 | |||||||
|---|---|---|---|---|---|---|---|
| Product Ratioa | % Decomposition at pH | ||||||
| XH | pKa | Diazo Compound |
Amide | 4.0 | 7.0 | 9.0 | 12.0 |
| methanol | 15.5 | 0 | 100 | — | — | — | — |
| ethanethiol | 10.6 | 0 | 100 | — | — | — | — |
| 4-fluorophenol | 10.0 | 0 | 100 | — | — | — | — |
| benzylmercaptan | 9.4 | 0 | 100 | — | — | — | — |
| 3-(dimethylamino)phenol | 9.2 | 0 | 100 | — | — | — | — |
| 3-chlorophenol | 9.0 | 33 | 67 | — | — | — | — |
| 3,5-difluorophenol | 8.7 | 63 | 27 | <5 | <5 | 5 | 21 |
| 3-hydroxypyridine | 8.5 | 67 | 33 | <5 | <5 | 12 | 62 |
| 2,2,2-trifluoroethanethiol | 7.6 | 83 | 17 | <5 | <5 | 13 | 35 |
| 4-nitrophenol | 7.1 | 97 | 3 | <5 | <5 | 10 | 46 |
| 2,4,6-trifluorophenol | 6.9 | 97 | 3 | <5 | <5 | <5 | 13 |
| N-hydroxysuccinimide | 6.0 | 98 | 2 | 12 | 40 | 54 | 98 |
| pentafluorophenol | 5.5 | 97 | 3 | <5 | <5 | <5 | 16 |
Determined by1H NMR spectroscopy: δ 4.73 (1H, diazo compound) and 3.95 (2H, amide).
In addition to promoting diazo-compound formation, a useful reagent must exhibit high chemical stability. We were concerned about this attribute because esters formed with alcohols having pKa ≤7.1 can be unstable to hydrolysis. Accordingly, we assessed stability by stirring a reagent in phosphate buffer containing 40% v/v THF (to enhance solubility) for 19 h, and evaluating decomposition by1H NMR spectroscopy. Hydrolysis was not observed at pH 7.0 (Table 1), except for the N-hydroxysuccinimide (NHS) ester, which was the object of our previous study.8f Most of the phosphinoesters were likewise stable at pH 9.0. Based on these reactivity/stability screens, ease-of-synthesis, and chromogenicity, the 4-nitrophenyl ester was chosen as ideal.
Next, we sought a reagent that was soluble in water. We reported previously that N,N-dimethylamino groups imparted water solubility to phosphinothioesters and enabled the traceless Staudinger ligation in water.11 N,N-Dimethylamino groups, however, led to rapid decomposition of the phosphinoesters used here-in. After screening other functional groups that confer water solubility on similar phosphines,11 we settled on methoxyethoxymethyl (MEM) as a preferred group.12
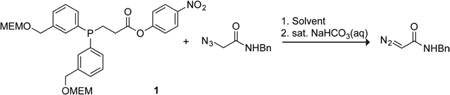
The synthesis of an optimized reagent for the conversion of azides into diazo compounds in water began from 3-bromobenzyl alcohol that was MEM-protected, converted into the corresponding Grignard compound, and added to diethyl phosphite to give the bis-aryl phosphine oxide (Scheme 2). Reduction of the phosphine oxide and subsequent protection with borane dimethylsulfide gave the borane-protected phosphine.13 Direct deprotonation of the protected phosphine and subsequent conjugate addition with methyl acrylate followed by hydrolysis afforded the phosphine-carboxylic acid.8f,14 Removing the borane protecting group with a mild methanol reflux,15 and installing the 4-nitrophenylester using standard coupling conditions provided phosphinoester 1 in 6 steps and 45% overall yield.
scheme 2.

Synthetic route to phosphinoester 1
The ability of phosphinoester 1 to convert azides into their corresponding diazo compounds was assessed first in phosphate buffer (Table 2). α-Azido-N-benzylacetamide was treated with phosphinoester 1 in buffers of various pH for 24 h, and the ensuing diazo compound was isolated by chromatography on silica gel. The reaction was found to be efficient under the conditions screened, with the optimal yield being achieved at neutral pH without the addition of an organic co-solvent (entry 3). This result bodes well for biological applications, as the optimal conversion was attained near physiological conditions. Under those conditions, reaction progress can be monitored by quantifying the yellow 4-nitrophenolate anion.16
Table 2.
Effect of pH and Organic Co-solvent on Diazo Compound Formation by Phosphinoester 1
 | |||||
|---|---|---|---|---|---|
| Entry | Solventa | Yield (%)b | Entry | Solventc | Yield (%)b |
| 1 pH | 5.0 | 69 | 5 | 50% v/v MeOH | 80 |
| 2 pH | 6.0 | 88 | 6 | 50% v/v CH3CN | 74 |
| 3 pH | 7.0 | 91 | 7 | 50% v/v DMF | 75 |
| 4 pH | 8.0 | 71 | 8 | 1% v/v DMSO | 74 |
| 9 | 20% v/v E.G. | 78 | |||
10 mM sodium phosphate buffer.
Isolated yield.
In 10 mM sodum phosphate buffer, pH 7.0. E.G. = ethylene glycol.
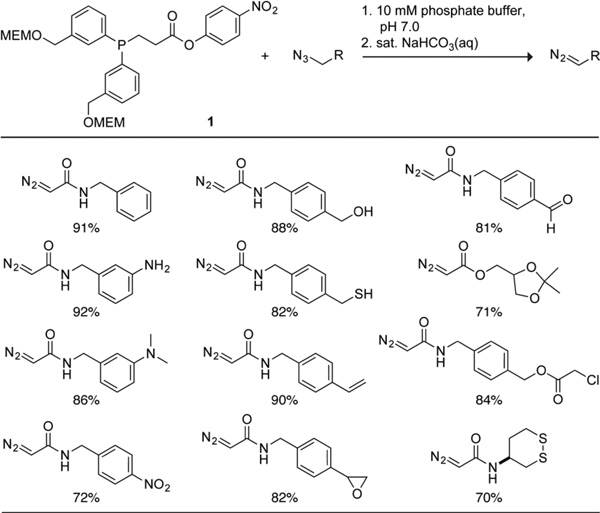
Then, we probed functional group compatibility using derivatives of azido-glycine. The initial test was to probe the tolerance of azide deimidogenation by phosphinoester 1 to strong nucleophiles, such as alcohols, amines, and thiols, all of which are capable of undergoing acyl transfer reactions with 4-nitrophenyl esters.17 We found that acyl transfer was not competitive with deimidogenation in phosphate buffer at pH 7.0 (Table 3).
Table 3.
Chemoselectivity of Diazo-Compound Formation in Water by Phosphinoester 1a
 |
Isolated yields.
The next test was to probe the tolerance of electrophiles such as aldehydes,18 α-chloroesters,19 and disulfide bonds,20 and epoxides,21 in the presence of the nucleophilic phosphorus of phosphinoester 1. Again, we found phosphinoester 1 to be highly chemoselective, converting azido-glycine derivatives into their corresponding diazo compounds without notable side reactivity (Table 3). Moreover, both acetals (which are hydrolyzed readily) and styrenes (which are prone to polymerization) were quite tolerant of the reaction conditions.
The final but critical test was to assess the compatibility of the deimidogenation reaction with actual biomolecules. We found that α-azido-N-benzylacetamide was converted to a diazo compound with a high isolated yield (79%) in the presence of 20 equiv of oxidized l-glutathione, which is an abundant cellular component that contains both nucleophilic (amino and carboxyl) and electrophilic (disulfide) functional groups. The yield was likewise high (87% by1H NMR spectroscopy) in the presence of bovine pancreatic (RNase A)—a well-known model protein22—at 14 mg/mL. Moreover, RNase A was not modified covalently by the procedure according to mass spectrometry and retained full enzymatic activity, indicative of its maintaining a proper three-dimensional conformation.
In conclusion, we have developed a phosphinoester that mediates the efficient conversion of azides into diazo compounds in phosphate buffer at neutral pH. This conversion is tolerant to the functional groups relevant to chemical biology. No other method for generating diazo compounds has these attributes. Azido groups, which can be introduced with a simple SN2 reaction, have found widespread use in chemical biology.9 Accordingly, a reagent capable of converting azides into their smaller and even more versatile diazo congeners in water could have substantial utility.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dr. N. A. McGrath, Dr. E. Myers, and C. H. Eller for contributive discussions and assistance. This work was supported by grant R01 GM044783 (NIH). This study made use of the National Magnetic Resonance Facility at Madison, which is supported by grants P41 RR002301 and P41 GM066326 (NIH).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures and spectral data for novel compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
REFERENCES
- 1.(a) Padwa A, editor. Hoboken, NJ: John Wiley & Sons; 1984. 1,3-Dipolar Cycloaddition Chemistry. [Google Scholar]; (b) Padwa A, Pearson WH, editors. Hoboken, NJ: John Wiley & Sons; 2002. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products. [Google Scholar]
- 2.(a) Dahn H, Diderich G. Helv. Chim. Acta. 1971;54:1950–1960. [Google Scholar]; (b) Regitz M, Maas G. Diazo Compounds: Properties and Synthesis. New York: Academic Press; 1986. [Google Scholar]; (c) Johnston JN, Muchalski H, Troyer TL. Angew. Chem. Int. Ed. 2010;49:2290–2298. doi: 10.1002/anie.200904828. [DOI] [PubMed] [Google Scholar]
- 3.(a) Padwa A, Krumpe KE. Tetrahedron. 1992;48:5385–5453. [Google Scholar]; (b) Padwa A, Weingarten MD. Chem. Rev. 1996;96:223–269. doi: 10.1021/cr950022h. [DOI] [PubMed] [Google Scholar]; (c) Doyle MP, Forbes DC. Chem. Rev. 1998;98:911–935. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; (d) Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861–2903. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (e) Wee AGH. Curr. Org. Synth. 2006;3:499–555. [Google Scholar]; (f) Ferreira VF. Curr. Org. Synth. 2007;11:177–193. [Google Scholar]
- 4.(a) Chibnall AC, Mangan JL, Rees MW. Biochem. J. 1958;68:114–118. doi: 10.1042/bj0680114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Doscher MS, Wilcox PE. J. Biol. Chem. 1961;236:1328–1337. [PubMed] [Google Scholar]; (c) Riehm JP, Scheraga HA. Biochemistry. 1965;4:772–782. doi: 10.1021/bi00880a023. [DOI] [PubMed] [Google Scholar]
- 5.(a) Antos JM, Francis MB. J. Am. Chem. Soc. 2004;126:10256–10257. doi: 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]; (b) Antos JM, McFarland JM, Iavarone AT, Francis MB. J. Am. Chem. Soc. 2009;131:6301–6308. doi: 10.1021/ja900094h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xiao Q, Zhang Y, Wang J. Acc. Chem. Res. 2012;46:236–247. doi: 10.1021/ar300101k. [DOI] [PubMed] [Google Scholar]; (d) Ball ZT. Acc. Chem. Res. 2012;46:560–570. doi: 10.1021/ar300261h. [DOI] [PubMed] [Google Scholar]; (e) Chen Z, Vohidov F, Coughlin JM, Stagg LJ, Arold ST, Ladbury JE, Ball ZT. J. Am. Chem. Soc. 2012;134:10138–10145. doi: 10.1021/ja302284p. [DOI] [PubMed] [Google Scholar]
- 6.McGrath NA, Raines RT. Chem. Sci. 2012;3:3237–3240. doi: 10.1039/C2SC20806G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Curtius T. Ber. Dtsch. Chem. Ges. 1890;23:3023–3033. [Google Scholar]; (b) Curtius T. J. Prakt. Chem. 1894;50:275–294. [Google Scholar]
- 8.(a) Curtius T. Ber. Dtsch. Chem. Ges. 1883;16:2230–2231. [Google Scholar]; (b) Baum JS, Shook DA, Davies HML, Smith HD. Synth. Commun. 1987;17:1709–1716. [Google Scholar]; (c) Holton TL, Shechter H. J. Org. Chem. 1995;60:4725–4729. [Google Scholar]; (d) Furrow ME, Myers AG. J. Am. Chem. Soc. 2004;126:12222–12223. doi: 10.1021/ja0459779. [DOI] [PubMed] [Google Scholar]; (e) Fulton JR, Aggarwal VK, de Vicente J. Eur. J. Org. Chem. 2005:1479–1492. [Google Scholar]; (f) Myers EL, Raines RT. Angew. Chem. Int. Ed. 2009;48:2359–2363. doi: 10.1002/anie.200804689. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Maas G. Angew. Chem. Int. Ed. 2009;48:8186–8195. doi: 10.1002/anie.200902785. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kolb HC, Sharpless KB. Drug Discov. Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; (b) Debets MF, van der Doelen CW, Rutjes FP, van Delft FL. ChemBioChem. 2010;11:1168–1184. doi: 10.1002/cbic.201000064. [DOI] [PubMed] [Google Scholar]; (c) Jewett JC, Bertozzi CR. Chem. Soc. Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schilling CI, Jung N, Biskup M, Schepers U, Bräse S. Chem. Soc. Rev. 2011;40:4840–4871. doi: 10.1039/c0cs00123f. [DOI] [PubMed] [Google Scholar]; (e) El-Sagheer AH, Brown T. Acc. Chem. Res. 2012;45:1258–1267. doi: 10.1021/ar200321n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Staudinger H, Meyer J. Helv. Chim. Acta. 1919;2:635–646. [Google Scholar]; (b) Nilsson BL, Kiessling LL, Raines RT. Org. Lett. 2000;3:9–12. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]; (c) Nilsson BL, Kiessling LL, Raines RT. Org. Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 11.(a) Tam A, Soellner MB, Raines RT. J. Am. Chem. Soc. 2007;129:11421–11430. doi: 10.1021/ja073204p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tam A, Raines RT. Bioorg. Med. Chem. 2009;17:1055–1063. doi: 10.1016/j.bmc.2008.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Kremers JA, Meijer EW. J. Org. Chem. 1994;59:4262–4266. [Google Scholar]; (b) Wuts PGM, Greene TW. Greene’s Protective Groups in Organic Synthesis. 4th ed. Hoboken, NJ: John Wiley & Sons; 2006. [Google Scholar]
- 13.Stankevič M, Pietrusiewicz KM. Synlett. 2003:1012–1016. [Google Scholar]
- 14.(a) Imamoto T, Oshiki T, Onozawa T, Kusumoto T, Sato K. J. Am. Chem. Soc. 1990;112:5244–5252. [Google Scholar]; (b) Enders D, Saint-Dizier A, Lannou M-I, Lenzen A. Eur. J. Org. Chem. 2006;2006:29–49. [Google Scholar]
- 15.Van Overschelde M, Vervecken E, Modha SG, Cogen S, Van der Eycken E, Van der Eycken J. Tetrahedron. 2009;65:6410–6415. [Google Scholar]
- 16.4-Nitrophenol has a pH-dependent extinction coefficient. At pH 7 0, ε ≈ 1 × 104 M−1cm−1 at 410 nm. Biggs AI. Trans. Faraday Soc. 1954;50:800–802. Levine MN, Lavis LD, Raines RT. Molecules. 2008;13:204–211. doi: 10.3390/molecules13020204.
- 17.(a) Fourteau L, Benoist E, Dartiguenave M. Synlett. 2001;1:126–128. [Google Scholar]; (b) Ishikawa F, Tsumuraya T, Fujii I. J. Am. Chem. Soc. 2008;131:456–457. doi: 10.1021/ja808792x. [DOI] [PubMed] [Google Scholar]
- 18.(a) Yam M, Chong JH, Tsang C-W, Patrick BO, Lam AE, Gates DP. Inorg. Chem. 2006;45:5225–5234. doi: 10.1021/ic060236p. [DOI] [PubMed] [Google Scholar]; (b) Bates JI, Patrick BO, Gates DP. New J. Chem. 2010;34:1660–1666. [Google Scholar]
- 19.(a) Yavari I, Alizadeh A, Anary-Abbasinejad M. Tetrahedron Lett. 2003;44:2877–2879. [Google Scholar]; (b) Castañeda F, Aliaga C, Acuña C, Silva P, Bunton CA. Phosphorus Sulfur Silicon Relat. Elem. 2008;183:1188–1208. [Google Scholar]; (c) Wube AA, Hüfner A, Thomaschitz C, Blunder M, Kollroser M, Bauer R, Bucar F. Bioorg. Med. Chem. Lett. 2011;19:567–579. doi: 10.1016/j.bmc.2010.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pettersson B, Hasimbegovic V, Bergman J. J. Org. Chem. 2011;76:1554–1561. doi: 10.1021/jo101864n. [DOI] [PubMed] [Google Scholar]
- 20.(a) Rüegg UT, Rudinger J. Methods Enzymol. 1977;47:111–116. doi: 10.1016/0076-6879(77)47012-5. [DOI] [PubMed] [Google Scholar]; (b) Cline DJ, Redding SE, Brohawn SG, Psathas JN, Schneider JP, Thorpe C. Biochemistry. 2004;43:15195–15203. doi: 10.1021/bi048329a. [DOI] [PubMed] [Google Scholar]; (c) Scales CW, Convertine AJ, McCormick CL. Biomacromolecules. 2006;7:1389–1392. doi: 10.1021/bm060192b. [DOI] [PubMed] [Google Scholar]; (d) Hanusek J, Russell MA, Laws AP, Jansa P, Atherton JH, Fettes K, Page MI. Org. Biomol. Chem. 2007;5:478–484. doi: 10.1039/b616298c. [DOI] [PubMed] [Google Scholar]; (e) Jones MW, Strickland RA, Schumacher FF, Caddick S, Baker JR, Gibson MI, Haddleton DM. J. Am. Chem. Soc. 2011;134:1847–1852. doi: 10.1021/ja210335f. [DOI] [PubMed] [Google Scholar]
- 21.(a) Fox DL, Robinson AA, Frank JB, Salvatore RN. Tetrahedron Lett. 2003;44:7579–7582. [Google Scholar]; (b) Azizi N, Saidi MR. Tetrahedron Lett. 2003;44:7933–7935. [Google Scholar]; (c) El-Sawi EA, Mostafa TB, Radwan HA. Chem. Heterocycl. Compd. 2009;45:981–989. [Google Scholar]; (d) Fernández-Pérez H, Donald SMA, Munslow IJ, Benet-Buchholz J, Maseras F, Vidal-Ferran A. Chem. Eur. J. 2010;16:6495–6508. doi: 10.1002/chem.200902915. [DOI] [PubMed] [Google Scholar]
- 22.(a) Raines RT. Chem. Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]; (b) Marshall GR, Feng JA, Kuster DJ. Biopolymers. 2008;90:259–277. doi: 10.1002/bip.20845. [DOI] [PubMed] [Google Scholar]; (c) Cuchillo CM, Nogués MV, Raines RT. Biochemistry. 2011;50:7835–7841. doi: 10.1021/bi201075b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.