

Abstract
Purpose
Patients with 1p/19q codeleted anaplastic oligodendroglial tumors who participated in RTOG (Radiation Therapy Oncology Group) 9402 lived much longer after chemoradiotherapy (CRT) than radiation therapy (RT) alone. However, some patients with noncodeleted tumors also benefited from CRT; survival curves separated after the median had been reached, and significantly more patients lived ≥ 10 years after CRT than RT. Thus, 1p/19q status may not identify all responders to CRT.
Patients and Methods
Using trial data, we inquired whether an IDH mutation or germ-line polymorphism associated with IDH-mutant gliomas identified the patients in RTOG 9402 who benefited from CRT.
Results
IDH status was evaluable in 210 of 291 patients; 156 (74%) had mutations. rs55705857 was evaluable in 245 patients; 76 (31%) carried the G risk allele. Both were associated with longer progression-free survival after CRT, and mutant IDH was associated with longer overall survival (9.4 v 5.7 years; hazard ratio [HR], 0.59; 95% CI, 0.40 to 0.86; P = .006). For those with wild-type tumors, CRT did not prolong median survival (1.3 v 1.8 years; HR, 1.14; 95% CI, 0.63 to 2.04; P = .67) or 10-year survival rate (CRT, 6% v RT, 4%). Patients with codeleted mutated tumors (14.7 v 6.8 years; HR, 0.49; 95% CI, 0.28 to 0.85; P = .01) and noncodeleted mutated tumors (5.5 v 3.3 years; HR, 0.56; 95% CI, 0.32 to 0.99; P < .05) lived longer after CRT than RT.
Conclusion
IDH mutational status identified patients with oligodendroglial tumors who did (and did not) benefit from alkylating-agent chemotherapy with RT. Although patients with codeleted tumors lived longest, patients with noncodeleted IDH-mutated tumors also lived longer after CRT.
INTRODUCTION
Capitalizing on the many molecular alterations that cause cancer to develop more effective therapies and prescribe them individually is a formidable challenge. For gliomas, where initial treatment is often uniform, the first opportunity to consider a personalized approach to therapy, based on genotyping, arose in the context of chemoradiotherapy (CRT) for glioblastoma (GBM). In an analysis of the trial by Stupp et al,1 in which the addition of temozolomide (TMZ), an alkylating agent, to radiotherapy (RT) prolonged median and 2-year survival compared with RT alone, Hegi et al2 found that these benefits were associated with methylation of the O6-methylguanine-DNA methyltransferase (MGMT) promoter. Despite this finding, MGMT status is not used to guide initial treatment, because in the Stupp et al trial, some patients with unmethylated GBMs also lived longer after CRT. Contributing further to the status quo are tissue requirements for accurate MGMT testing, ease of use of TMZ, and absence of other effective chemotherapies for GBM. MGMT testing is important, although perhaps not precise enough to support a personalized treatment strategy for GBM.
A second opportunity to customize therapy has emerged for oligodendroglial cancers. Two randomized controlled trials (RCTs) found that CRT with procarbazine, lomustine, and vincristine (PCV)3 doubled the median survival of patients with 1p/19q codeleted tumors versus RT alone.4,5 CRT did not prolong the median survival of those with noncodeleted tumors in either trial, but in both, more patients with noncodeleted tumors had longer survival after CRT than RT. In the Radiation Therapy Oncology Group (RTOG) trial,4 25% of patients with noncodeleted tumors lived ≥ 10 years after PCV plus RT versus only 10% after RT alone, a significant difference. Thus, codeletion, although it predicts benefit from CRT, likely falls short of the precision expected of a test for personalizing treatment. Judging by experience with MGMT and GBM, oncologists will be reticent to rely solely on 1p/19q codeletion status to decide which patients with oligodendroglial tumors should be treated with CRT versus RT alone.
With the knowledge that molecular alterations that initiate cancers can sometimes be targets for highly effective drug therapies,6 we inquired whether the ability to predict survival benefit from neoadjuvant PCV using codeletion status could be enhanced by incorporating or substituting markers of earlier events in the genesis of oligodendrogliomas. We focused on two biomarkers of potential utility: somatic mutations of isocitrate dehydrogenase (IDH), the earliest known molecular alteration in oligodendroglial tumors,7 and the G (v A) allele of rs55705857, a germ-line polymorphism associated with a six-fold risk of developing IDH-mutated glioma.8,9 We reanalyzed the RTOG 9402 trial, paying particular attention to the proportion of long-term survivors after CRT in the noncodeleted anaplastic oligodendroglioma (AO)/oligoastrocytoma (AOA) subset.4
PATIENTS AND METHODS
Data Source, Study Eligibility, Protocol Therapy, and Codeletion Status
Data were obtained from an RCT of PCV plus RT versus RT for AO and AOA.4,10 Eligible patients were age ≥ 18 years with AO/AOA and Karnofsky performance score (KPS) ≥ 60. Diagnoses were confirmed by central review.10 Patients consented to random assignment and blood and tumor collection, and each center had institutional review board approval. Patients were stratified by age at diagnosis, KPS, and degree of anaplasia and were randomly assigned within 8 weeks to an intensive PCV regimen followed by RT (ie, CRT) or to RT alone.10 Baseline testing, surveillance assessments, and toxicity criteria have been reported.10 Chromosomes 1p and 19q were assessed by fluorescence in situ hybridization.11
IDH Mutation and G Allele Analyses
IDH1 and IDH2 were evaluated by immunohistochemistry (antihuman IDH1R132H H09) and sequencing, respectively.9 Presence of the G (v A) allele of rs55705857 at 8q24.21 was assessed by custom genotyping of DNA from study-patient blood or Epstein-Barr virus–transformed leukocytes. GoldenGate assays were performed at the Mayo Clinic Genotyping Core Facility (Rochester, MN) using the VeraCode platform designed by Illumina (San Diego, CA).9 Samples were analyzed in 96-well plates with replicates, as described previously.8,9 In patients with noncodeleted tumors harboring an IDH mutation, ATRX expression was assessed by immunohistochemistry (No. HPA001906; Sigma-Aldrich, St Louis, MO), as described elsewhere.12
Statistical Methods
The interactions of an IDH mutation or a germ-line risk allele (ie, homozygous or heterozygous for high-risk G allele v homozygous for standard-risk A allele) with survival after PCV plus RT or RT were examined independently and in association with 1p/19q codeletion status. The strength of association was assessed by odds ratios. Survival was analyzed using the Kaplan-Meier method with two-sided log-rank statistics. Univariable and multivariable Cox hazards models were fitted to assess the independent effects of treatment, demography, and clinical and genetic variables on survival. Those items with a P value less than .1 in the univariable analysis were included in the multivariable analysis with step-wise selection. A global test for the interaction of treatment with covariates was computed using a χ2 statistic.13 The c-index was also used to compare the predictive value of the model with and without IDH status.14 χ2 tests were used to compare clinical and molecular features by treatment. All P values were two tailed and unadjusted for multiple comparisons.
RESULTS
Survival Analysis and Results of RTOG 9402
Overall survival (OS) was the primary end point. Analyses were performed on case-eligible (n = 291) and all-case (n = 299) bases with similar results.4 Arms were balanced for clinical features, AO versus AOA, and steroid use.10 The status of 1p or 19q was known in 91% of patients.4 Median follow-up was 11.3 years. One hundred forty-eight patients were randomly assigned to PCV plus RT, and 143 to RT. Median survivals were similar: 4.6 years for PCV plus RT versus 4.7 years for RT (HR, 0.79; 95% CI, 0.60 to 1.04; P = .1); however, the adjusted results favored PCV plus RT (HR, 0.67; 95% CI, 0.50 to 0.91; P = .01).4 Those with codeleted tumors had long survival regardless of therapy, which was much longer after PCV plus RT than RT (14.7 v 7.3 years; HR, 0.59; 95% CI, 0.37 to 0.95; P = .03).4 Median survivals were similar in those with noncodeleted tumors (2.6 v 2.7 years; HR, 0.85; 95% CI, 0.58 to 1.23; P = .39), but there were significantly more 10-year survivors after PCV plus RT than RT alone (25% v 10%; P < .05).4
IDH Status, Survival, and Treatment Effects
IDH status was assessable in 210 of 291 patients (72%; Table 1). In 81 similar patient cases, either testing failed or there was no tissue (Appendix Table A1, online only). Mutations were seen in 156 (74%; IDH1, n = 154; IDH2, n = 2) and associated with younger age, good KPS, accessibility to resection, AO pathology, single focus, and moderate anaplasia. Patients with mutated tumors lived longer than those with tumors that did not have a detectable mutation (PCV plus RT: 9.4 v 1.3 years; HR, 0.28; 95% CI, 0.17 to 0.46; P < .001; RT: 5.7 v 1.8 years; HR, 0.38; 95% CI, 0.23 to 0.64; P < .001; survival curves not shown); moreover, their survival differed by treatment.
Table 1.
Pretreatment Characteristics by IDH Mutation Status
| Characteristic | Not Mutated (n = 54) |
Mutated (n = 156) |
P | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Age, years | .003 | ||||
| < 50 | 27 | 50.0 | 113 | 72.4 | |
| ≥ 50 | 27 | 50.0 | 43 | 27.6 | |
| Sex | .07 | ||||
| Male | 38 | 70.4 | 88 | 56.4 | |
| Female | 16 | 29.6 | 68 | 43.6 | |
| KPS | .03 | ||||
| 60-70 | 10 | 18.5 | 12 | 7.7 | |
| 80-100 | 44 | 81.5 | 144 | 92.3 | |
| Surgery | .02* | ||||
| Biopsy | 11 | 20.4 | 13 | 8.3 | |
| Partial resection | 31 | 57.4 | 89 | 57.1 | |
| Total resection | 12 | 22.2 | 52 | 33.3 | |
| Unknown | 0 | 0.0 | 2 | 1.3 | |
| Neurologic function | .92† | ||||
| No symptoms | 17 | 31.5 | 48 | 30.8 | |
| Minor symptoms | 26 | 48.1 | 82 | 52.6 | |
| Moderate symptoms | 11 | 20.4 | 26 | 16.7 | |
| Histology | < .001‡ | ||||
| AO | 16 | 29.6 | 95 | 60.9 | |
| AOA | 38 | 70.4 | 61 | 39.1 | |
| Grade | .01 | ||||
| Moderately anaplastic | 21 | 38.9 | 92 | 59.0 | |
| Highly anaplastic | 33 | 61.1 | 64 | 41.0 | |
| Multifocal disease | < .001§ | ||||
| No (or unknown) | 44 | 81.5 | 150 | 96.2 | |
| Yes | 10 | 18.5 | 6 | 3.8 | |
| Steroid use at baseline | .40 | ||||
| No | 19 | 35.2 | 65 | 41.7 | |
| Yes | 35 | 64.8 | 91 | 58.3 | |
| Assigned treatment | .44 | ||||
| Chemotherapy and RT | 31 | 57.4 | 80 | 51.3 | |
| RT alone | 23 | 42.6 | 76 | 48.7 | |
Abbreviations: AO, anaplastic oligodendroglioma; AOA, anaplastic oligoastrocytoma; KPS, Karnofsky performance score; RT, radiotherapy.
Biopsy versus partial plus total.
No symptoms versus other.
AO versus AOA.
No versus yes.
An IDH mutation was identified in 80 (72%) of 111 patients randomly assigned to PCV plus RT and 76 (77%) of 99 randomly assigned to RT alone. For those with IDH-mutated tumors, survival was significantly longer after PCV plus RT than RT (9.4 v 5.7 years; HR, 0.59; 95% CI, 0.40 to 0.86; P = .006; Fig 1A). However, among those without a detectable IDH mutation, there were no differences in survival associated with treatment assignment (PCV plus RT, 1.3 years v RT, 1.8 years; HR, 1.14; 95% CI, 0.63 to 2.04; P = .67; Fig 1B). Also, there were no differences in 10-year survival rates (PCV plus RT, 6% [two of 31] v RT, 4% [one of 23]). For those with wild-type IDH tumors, the likelihood of long-term survival was low irrespective of initial treatment.
Fig 1.
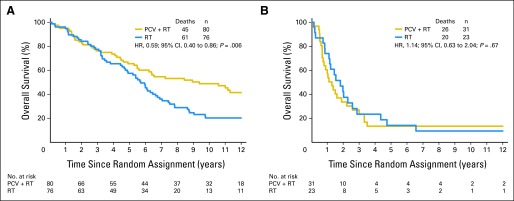
Kaplan-Meier estimates of overall survival (OS) by treatment (procarbazine, lomustine, and vincristine [PCV] plus radiotherapy [RT] or RT) for patients with (A) IDH-mutated and (B) nonmutated tumors. Hazard ratio (HR) ratio for OS for those with mutated tumors was 0.59 (95% CI, 0.40 to 0.86; P = .006); HR for those with nonmutated tumors was 1.14 (95% CI, 0.63 to 2.04; P = .67).
Early Somatic Alterations, Survival, and Treatment Effects
AO and AOA are often screened for 1p/19q codeletion and IDH mutations to obtain prognostic information. Hence, we examined the interaction of 1p/19q codeletion and IDH mutation with survival and treatment in patients in whom both codeletion and mutation status were known. Data on both markers were available in 208 (71%) of 291 patients: 88 (42%) had a codeleted mutated tumor, 66 (32%) had a noncodeleted mutated tumor, and 44 (21%) had a noncodeleted nonmutated tumor. There were 10 codeleted nonmutated patient cases (5%). Ninety percent of codeleted tumors had a detectable mutation, but only 57% of mutated AO/AOA had a visible codeletion. Among the informative patient cases, 111 received PCV plus RT (53%) and 97 received RT (47%). ATRX was undetectable in 41 (72%) of 57 patients with noncodeleted, mutated tumors and present in 16 (28%); it was also present in all codeleted patient cases tested.
Patients with codeleted mutated tumors (AO, 78%) had the longest survival, those with noncodeleted mutated tumors (AOA, 64%) had an intermediate survival, and those with neither had the shortest survival (PCV plus RT: 14.7 years; 95% CI, 6.4 to not reached v 5.5 years; 95% CI, 2.6 to 11.0 v 1.0 years; 95% CI, 0.6 to 1.9; P < .001; Fig 2A; RT alone: 6.8 years; 95% CI, 5.4 to 8.6 v 3.3 years; 95% CI, 2.5 to 4.9 v 1.3 years; 95% CI, 0.8 to 1.9; P < .001; Fig 2B; not described here are ten patients with codeleted tumors with intact IDH whose survival was intermediate). Patients with codeleted mutated tumors lived longer after CRT than RT (14.7 v 6.8 years; HR, 0.49; 95% CI, 0.28 to 0.85; P = .01; Fig 3A), and remarkably, those with noncodeleted mutated tumors also lived longer after CRT (5.5 v 3.3 years; HR, 0.56; 95% CI, 0.32 to 0.99; P = .045; Fig 3B). In exploratory analyses of small subsets of patients with noncodeleted mutated tumors with ATRX results, survival was longer in patients who were ATRX negative (4.4 v 3.5 years; AOA, 75%) and longer still in those who were ATRX positive (11.0 v 2.7 years; AO, 75%). Patients with noncodeleted nonmutated AO/AOA experienced no discernible benefit from the addition of PCV to RT (1.0 v 1.3 years; HR, 0.99; 95% CI, 0.53 to 1.86; P = .97).
Fig 2.
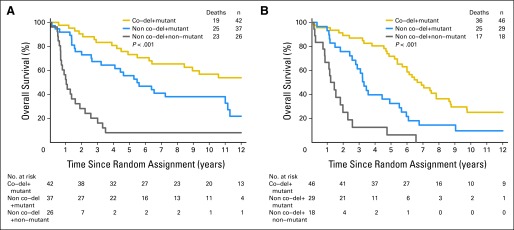
Kaplan-Meier estimates of overall survival for patients whose tumors were IDH mutated and 1p/19q codeleted (co-del; gold), mutated (mut) and noncodeleted (blue), and nonmutated and noncodeleted (gray) after (A) procarbazine, lomustine, and vincristine (PCV) plus radiotherapy (RT) and (B) RT alone. Median survivals after (A) PCV plus RT were 14.7 (95% CI, 6.4 to not reached), 5.5 (95% CI, 2.6 to 11.0), and 1.0 years (95% CI, 0.6 to 1.9; P < .001), respectively. Median survivals after (B) RT alone were 6.8 (95% CI, 5.4 to 8.6), 3.3 (95% CI, 2.5 to 4.9), and 1.3 years (95% CI, 0.8 to 1.9; P < .001), respectively.
Fig 3.
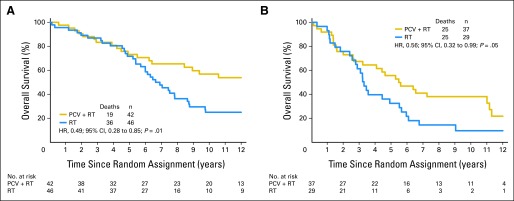
Kaplan-Meier estimates of overall survival (OS) by treatment (procarbazine, lomustine, and vincristine [PCV] plus radiotherapy [RT] or RT) for those with (A) IDH mutated and 1p/19q codeleted and (B) IDH mutated noncodeleted tumors. Hazard ratio (HR) for OS for those with (A) mutated codeleted tumors was 0.49 (95% CI, 0.28 to 0.85; P = .01); HR for those with (B) mutated noncodeleted tumors was 0.56 (95% CI, 0.32 to 0.99; P = .05).
Status of the rs55705857 Allele, Survival, and Treatment Effects
Risk status was assessable in 245 (84%) of 291 patients. Seventy-six patients (31%) were carriers of the G risk allele: 75 were heterozygous carriers (ie, GA), and one patient was a homozygous carrier (ie, GG). The G allele frequency for the entire RTOG 9402 cohort was 0.16. The allele frequency was 0.22 for the subset of patients with 1p/19q codeleted tumors, not significantly different from the frequency in an independent cohort at the Mayo Clinic with codeleted oligodendroglial tumors.9 G allele status was associated with good function, steroid-free condition, and AO pathology. Moreover, patients who were carriers of the G allele lived significantly longer than those who were homozygous for the A allele (PCV plus RT: 8.9 v 3.3 years; HR, 0.51; 95% CI, 0.31 to 0.83; P = .006; RT: 5.3 v 3.8 years; HR, 0.67; 95% CI, 0.42 to 1.06; P = .08; survival curves not shown), and treatment assignment may have had a differential effect on the survival of carriers versus noncarriers.
The G allele was present in the germ-line DNA of 40 (32%) of 124 patients who were randomly assigned to PCV plus RT and 36 (30%) of 121 randomly assigned to RT alone. For G allele carriers, progression-free survival (PFS) was significantly longer after CRT than RT (6.8 v 1.7 years; HR, 0.42; 95% CI, 0.24 to 0.71; P = .001; Fig 4A). Also, in G allele carriers, there was a trend toward longer OS after PCV plus RT than RT (8.9 v 5.3 years; HR, 0.65; 95% CI, 0.36 to 1.16; P = .14; Fig 4B). In contrast, there were no survival differences related to treatment among patients who did not have the G risk allele in their germ-line (OS: PCV plus RT, 3.3 years v RT, 3.8 years; HR, 0.87; 95% CI, 0.62 to 1.22; P = .41; PFS: PCV plus RT, 1.4 years v RT, 1.3 years; HR, 0.80; 95% CI, 0.58 to 1.12; P = .19; survival curves not shown).
Fig 4.
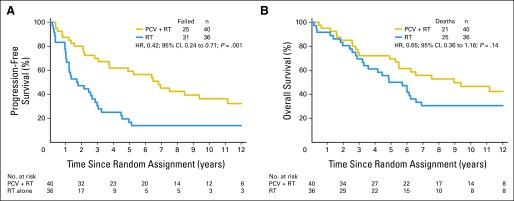
Kaplan-Meier estimates of (A) progression-free survival (PFS) and (B) overall survival (OS) by treatment (procarbazine, lomustine, and vincristine [PCV] plus radiotherapy [RT] or RT) for G allele carriers. Hazard ratio (HR) for (A) PFS was 0.42 (95% CI, 0.24 to 0.71; P = .001); HR for (B) OS was 0.65 (95% CI, 0.36 to 1.16; P = .14).
Genetic Associations and Multivariable Analysis
The odds ratios for IDH mutation and 1p/19q codeletion, IDH mutation and G allele carrier status, and 1p/19q codeletion and G allele status were 5.87 (95% CI, 2.75 to 12.5; P < .001), 2.27 (95% CI, 1.04 to 4.96; P = .038), and 2.79 (95% CI, 1.55 to 5.02; P < .001), respectively. The features listed in Table 2 were significant, with P values less than .1 in the univariable Cox hazards model. In the stepwise multivariable Cox model that incorporated these variables, survival was strongly associated with IDH mutation (HR, 0.41; 95% CI, 0.27 to 0.63; P < .001) and 1p/19q codeletion (HR, 0.39; 95% CI, 0.27 to 0.58; P < .001); it was also associated with CRT at diagnosis (HR, 0.65; 95% CI, 0.46 to 0.92; P = .02; Table 2). The global tests for treatment interactions with IDH mutation, 1p/19q codeletion, and G allele carrier status were not significant (P > .05), nor were any pairwise tests of interaction. The c-index for the model with treatment plus the clinical and genetic features listed in Table 2 was 0.68. The indices after inclusion of mutation only, codeletion only, and both were 0.71, 0.72, and 0.73, respectively. G allele carrier status did not contribute additional effect.
Table 2.
Cox Proportional Hazards Model for OS
| Variable | HR* | 95% CI | Multivariable P | Univariable P |
|---|---|---|---|---|
| Treatment (PCV plus RT v RT) | 0.65 | 0.46 to 0.92 | .0157 | .099 |
| Surgery (resection v biopsy) | 0.50 | 0.28 to 0.89 | .0195 | .008 |
| Disability (none/minor v other) | 0.45 | 0.25 to 0.81 | .0073 | < .001 |
| Multifocal disease (no v yes) | 0.39 | 0.20 to 0.76 | .0056 | < .001 |
| 1p/19q (codeleted v other) | 0.39 | 0.27 to 0.58 | < .001 | < .001 |
| IDH mutation (positive v negative) | 0.41 | 0.27 to 0.63 | < .001 | < .001 |
| Sex (female v male) | — | — | .07 | |
| Age, years (< 50 v ≥ 50) | — | — | < .001 | |
| KPS (80-100 v 60-70) | — | — | < .001 | |
| Anaplastic features (2-3 v 4-5) | — | — | .002 | |
| Steroid use (no v yes) | — | — | < .001 | |
| Tumor type (pure v mixed) | — | — | < .001 | |
| G allele status (with v without) | — | — | < .001 |
NOTE. Variables dropped during stepwise selection are found at the bottom; all were significant at P < .10 in univariable models. Those at the top were significant at P < .05 in multivariable model. Interaction terms were not statistically significant. Bold font indicates favorable status.
Abbreviations: HR, hazard ratio; KPS, Karnofsky performance score; OS, overall survival; PCV, procarbazine, lomustine, and vincristine; RT, radiotherapy.
HRs expressed as λfavorable/λunfavorable.
DISCUSSION
AO and AOA share histologic similarities and IDH mutations.7 In most AOs, IDH mutations coexist with loss of 1p/19q15,16 and seem pathognomonic.17 Mutations of CIC on 19q, FUBP1 on 1p, and the TERT promoter complete the mutational spectrum of AO.18–20 In most AOAs, ATRX and TP53 mutations accompany those of IDH.21 AO/AOA also share the methylator (G-CIMP) phenotype and proneural expression profile related to IDH mutations and 2-hydroxyglutarate production.22–27 Moreover, the MGMT promoter is often hypermethylated in AO/AOA.28 Furthermore, as a consequence of DNA hypermethylation and 1p allelic loss, NHE-1 is frequently silenced in 1p/19q codeleted AO/AOA, perhaps contributing to the remarkably slow rate of growth.29 As highlighted here, a recent postgenome-wide association fine mapping study has identified a single-nucleotide polymorphism at 8q24.21 that is associated with a six-fold risk of developing an IDH-mutant glioma, including both AOs and AOAs. This degree of risk is similar to that for breast cancer associated with BRCA1 and BRCA2 variants30,31 and greater than the CHEK2 variant associated with breast cancer.32
This unique constellation of genetic, epigenetic, germ-line, and somatic events define a cancer family that is sensitive to drugs that alkylate DNA.33–41 Within this family, AOs, most of which are codeleted, are especially chemosensitive; they often shrink dramatically when treated with chemotherapy alone; furthermore, the survival of patients with codeleted AO/AOA doubled after early treatment with chemotherapy and RT.4,5 AOAs, most of which lack codeletion of 1p/19q, are also sensitive to alkylating agents.35 Their radiographic responses are less complete and durable, but the results of RCTs show that a substantial (but currently unknown) subset of noncodeleted AOs/AOAs benefit from CRT.4,5 Here, we inquired whether IDH mutation or susceptibility to a mutation might be a key determinant of the behaviors of AO/AOA, including sensitivity to DNA-damaging therapies. This question is important and timely because benefit from CRT is not predicted precisely by codeletion status, and IDH mutations alter the expression of genes that may affect growth and treatment effects.42
In this expanded molecular analysis of RTOG 9402, genetic background, tumor histology, cancer genotype, and survival after treatment were closely related. Like codeletion status, there were significant associations between the presence of the rs55705857 G allele in normal DNA, detection of an IDH mutation in tumor DNA, diagnosis of AO, and better prognosis after either CRT or RT alone. These findings reinforce that some individuals are susceptible to gliomas and may be susceptible to subtypes that harbor particular genetic alterations.8,9 In turn, these alterations are accompanied by a predictable set of behaviors, including prognosis after DNA-damaging therapies.43 Looking forward, one can imagine that presurgical testing for germ-line features might be used to predict tumor type and survival.
Perhaps more intriguing is the suggestion that benefit from neoadjuvant PCV might be related to IDH mutation or risk of an IDH mutation, characteristics of AO/AOA that predate overt tumorigenesis. Presence of the risk allele at rs55705857 was associated with longer PFS after CRT, and IDH mutation with longer OS after CRT. Moreover, in the absence of an IDH mutation, equally low rates of long-term survival were seen after CRT and RT, findings that stand in contrast to the data on survival in relation to codeletion status, where more patients with noncodeleted tumors had long survival after CRT than RT alone.4
Our results point to an association between IDH mutation and benefit from pre-RT PCV in AO/AOA. In RTOG 9402, benefit from CRT was seen in patients with codeleted tumors, virtually all with IDH mutations, but also in patients with noncodeleted tumors with mutations; among the latter, those who were ATRX positive might have benefitted more than those who were negative. Whether these noncodeleted ATRX-positive tumors are biologically identical to codeleted tumors, save for absence of visible losses of 1p/19q, or represent laboratory errors in codeletion testing is unknown. Likewise, whether the association between IDH mutation and survival benefit from neoadjuvant PCV is causal through epigenetic silencing of MGMT2 or other mechanisms,44 is unresolved here. However, our findings are consistent with other correlative data. Analyses of EORTC (European Organisation for Research and Treatment of Cancer) 26951 suggest that IDH mutations and the hypermethylated phenotype in AO/AOA are associated with survival benefit from adjuvant PCV. Although unequivocal statistical significance was not reached, there was a trend toward benefit from CRT.45,46 Readers are cautioned, however, that these important secondary analyses of RTOG 9402 and EORTC 26951 were unplanned and underpowered and had incomplete molecular data.
With these limitations in mind, a diagnostic approach to AO/AOA that includes assessment of IDH and codeletion might be helpful clinically.46,47 Patients with mutated codeleted tumors will benefit from CRT; those with mutated noncodeleted tumors may also benefit but have shorter survival, whereas those with AO/AOA with neither alteration are unlikely to benefit from the addition of PCV chemotherapy to RT. Moreover, two simple, reliable, and inexpensive tests, well suited to biopsy samples, provide complementary information and might help oncologists decide when to use CRT versus RT alone for AO/AOA.
Two final issues merit comment. First, it is unclear whether our findings with regard to IDH and PCV apply to TMZ or to astrocytomas or GBMs with mutant IDH. Second, it is also unclear why patients with codeleted tumors had long survival after CRT. Does codeletion synergize with mutant IDH or the G allele to enhance sensitivity to PCV or slow tumor regrowth? The answers to these questions await new clinical trials and a thorough understanding of the biologic consequences 1p/19q codeletion and IDH mutation.
Acknowledgment
We thank the staff of the Anatomic Pathology Immunohistochemistry Laboratory and Molecular Genomics Shared Resource at the Mayo Clinic, who performed the genetic studies, and Andrew Lassman, MD, for reviewing the manuscript.
Appendix
Table A1.
Pretreatment Characteristics by IDH Availability
| Characteristic | With IDH (n = 210) |
Without IDH (n = 81) |
||
|---|---|---|---|---|
| No. | % | No. | % | |
| 1p 19q deletion | ||||
| Codeleted | 98 | 46.7 | 28 | 34.6 |
| Noncodeleted | 110 | 52.4 | 27 | 33.3 |
| Missing | 2 | 1.0 | 26 | 32.1 |
| Age, years | ||||
| < 50 | 140 | 66.7 | 61 | 75.3 |
| ≥ 50 | 70 | 33.3 | 20 | 24.7 |
| Sex | ||||
| Male | 126 | 60.0 | 48 | 59.3 |
| Female | 84 | 40.0 | 33 | 40.7 |
| KPS | ||||
| 60-70 | 22 | 10.5 | 8 | 9.9 |
| 80-100 | 188 | 89.5 | 73 | 90.1 |
| Prior surgery | ||||
| Biopsy | 24 | 11.4 | 11 | 13.6 |
| Partial resection | 120 | 57.1 | 40 | 49.4 |
| Total resection | 64 | 30.5 | 29 | 35.8 |
| Surgery (no details) | 2 | 1.0 | 1 | 1.2 |
| Neurologic function | ||||
| No symptoms | 65 | 31.0 | 29 | 35.8 |
| Minor symptoms | 108 | 51.4 | 34 | 42.0 |
| Moderate (fully active) | 20 | 9.5 | 9 | 11.1 |
| Moderate (not fully active) | 17 | 8.1 | 8 | 9.9 |
| Unknown | 0 | 0.0 | 1 | 1.2 |
| Histology | ||||
| AO | 111 | 52.9 | 39 | 48.1 |
| AOA (oligo dominant) | 41 | 19.5 | 24 | 29.6 |
| AOA (oligo equal to astro) | 33 | 15.7 | 6 | 7.4 |
| AOA (astro dominant) | 25 | 11.9 | 12 | 14.8 |
| Grade | ||||
| Moderately anaplastic | 113 | 53.8 | 48 | 59.3 |
| Very anaplastic | 97 | 46.2 | 33 | 40.7 |
| Multifocal disease | ||||
| No | 192 | 91.4 | 71 | 87.7 |
| Yes | 16 | 7.6 | 9 | 11.1 |
| Unknown | 2 | 1.0 | 1 | 1.2 |
| Steroid use at baseline | ||||
| No | 84 | 40.0 | 36 | 44.4 |
| Yes | 126 | 60.0 | 45 | 55.6 |
| Treatment assigned | ||||
| PCV plus RT | 111 | 52.9 | 37 | 45.7 |
| RT alone | 99 | 47.1 | 44 | 54.3 |
Abbreviations: AO, anaplastic oligodendroglioma; AOA, anaplastic oligoastrocytoma; KPS, Karnofsky performance score; PCV, procarbazine, lomustine, and vincristine; RT, radiotherapy.
See accompanying article on page 774
Written on behalf of the Radiation Therapy Oncology Group, North Central Cancer Treatment Group, Southwest Oncology Group, National Cancer Institute of Canada Clinical Trials Group, and Eastern Cooperative Oncology Group.
Support information appears at the end of this article.
The content of this report is the responsibility of the authors and does not necessarily represent the viewpoint of the National Cancer Institute, Canadian Cancer Society, National Institutes of Health, or National Institute of Neurological Disorders and Stroke.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Support
Supported in part by National Cancer Institute Grants No. U10 CA21661 and U10 CA32115 to the Radiation Therapy Oncology Group, No. U10 CA25224 to the North Central Cancer Treatment Group, No. CA17145 and CA21115 to the Eastern Cooperative Oncology Group, No. CA32102 to the Southwest Oncology Group, and No. U10 CA37422 to the Community Clinical Oncology Program; by a Canadian Cancer Society grant to the National Cancer Institute of Canada Clinical Trials Group; by National Institutes of Health Grants No. P50CA108961 and P30 CA15083; by National Institute of Neurological Disorders and Stroke Grant No. RC1NS068222Z; by a generous gift from Bernie and Edith Waterman; and by the Ting Tsung and Wei Fong Chao Family Foundation.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: J. Gregory Cairncross, Robert B. Jenkins, Edward G. Shaw
Financial support: Robert B. Jenkins, Walter J. Curran
Administrative support: Minesh P. Mehta, Walter J. Curran
Provision of study materials or patients: J. Gregory Cairncross, Robert B. Jenkins, Edward G. Shaw, Caterina Giannini, David G. Brachman, Jan C. Buckner, Karen L. Fink, Luis Souhami, Normand J. Laperriere, Minesh P. Mehta
Collection and assembly of data: J. Gregory Cairncross, Meihua Wang, Robert B. Jenkins, Caterina Giannini, David G. Brachman, Jason T. Huse
Data analysis and interpretation: J. Gregory Cairncross, Meihua Wang, Robert B. Jenkins, Caterina Giannini, David G. Brachman, Jan C. Buckner, Karen L. Fink, Luis Souhami, Normand J. Laperriere, Jason T. Huse, Minesh P. Mehta, Walter J. Curran
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for patients with newly diagnosed glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 3.Levin VA, Edwards MS, Wright DC, et al. Modified procarbazine, CCNU, and vincristine (PCV 3) combination chemotherapy in the treatment of malignant brain tumors. Cancer Treat Rep. 1980;64:237–244. [PubMed] [Google Scholar]
- 4.Cairncross G, Wang M, Shaw E, et al. A phase 3 trial of chemo-radiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J Clin Oncol. 2013;31:337–343. doi: 10.1200/JCO.2012.43.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC Brain Tumor Group study 26951. J Clin Oncol. 2013;31:344–350. doi: 10.1200/JCO.2012.43.2229. [DOI] [PubMed] [Google Scholar]
- 6.Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myelogenous leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 7.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenkins RB, Wrensch MR, Johnson D, et al. Distinct germline polymorphisms underlie glioma morphologic heterogeneity. Cancer Genet. 2011;204:13–18. doi: 10.1016/j.cancergencyto.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenkins RB, Xiao Y, Sicotte H, et al. A low frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumours and IDH1 or IDH2 mutated astrocytomas. Nat Genet. 2012;44:1122–1125. doi: 10.1038/ng.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cairncross G, Berkey B, Shaw E, et al. A phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707–2714. doi: 10.1200/JCO.2005.04.3414. [DOI] [PubMed] [Google Scholar]
- 11.Smith JS, Perry A, Borell TJ, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastro-cytomas. J Clin Oncol. 2000;18:636–645. doi: 10.1200/JCO.2000.18.3.636. [DOI] [PubMed] [Google Scholar]
- 12.Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casella G, Berger R. Statistical Inference. ed 2. Pacific Grove, CA: Duxbury Press; 2001. [Google Scholar]
- 14.Harrell FE., Jr . Regression Modeling Strategies. New York, NY: Springer; 2001. [Google Scholar]
- 15.Reifenberger J, Reifenberger G, Liu L, et al. Molecular analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994;145:1175–1190. [PMC free article] [PubMed] [Google Scholar]
- 16.Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473–1479. doi: 10.1093/jnci/90.19.1473. [DOI] [PubMed] [Google Scholar]
- 17.Labussière M, Idbaih A, Wang XW, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74:1886–1890. doi: 10.1212/WNL.0b013e3181e1cf3a. [DOI] [PubMed] [Google Scholar]
- 18.Bettegowda C, Agrawal N, Jiao N, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yip S, Butterfield YS, Morozova O, et al. Concurrent CIC mutations, IDH mutations and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noushmehr H, Weisenberger D, Diefes K, et al. Identification of a CpG island methyl-ator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christensen BC, Smith AA, Zeng S, et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J Natl Cancer Inst. 2011;103:143–153. doi: 10.1093/jnci/djq497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2010;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ducray F, Idbaih A, de Reyniès A, et al. Anaplastic oligodendrogliomas with 1p19q co-deletion have a proneural gene expression profile. Mol Cancer. 2008;7:41. doi: 10.1186/1476-4598-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper LA, Gutman DA, Long Q, et al. The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PLoS One. 2010;5:e12548. doi: 10.1371/journal.pone.0012548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by anomalies of PDGFRA, IDH1, EGFR and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Möllemann M, Wolter M, Felsberg J, et al. Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer. 2005;113:379–385. doi: 10.1002/ijc.20575. [DOI] [PubMed] [Google Scholar]
- 29.Blough MD, Al-Najjar M, Chesnelong C, et al. DNA hypermethylation and 1p loss silence NHE-1 in oligodendroglioma. Ann Neurol. 2012;71:845–849. doi: 10.1002/ana.23610. [DOI] [PubMed] [Google Scholar]
- 30.Whittemore AS, Gong G, John EM, et al. Prevalence of BRCA1 mutation carriers among US non-Hispanic whites. Cancer Epidemiol Biomarkers Prev. 2004;13:2078–2083. [PubMed] [Google Scholar]
- 31.Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer. 2000;83:1301–1308. doi: 10.1054/bjoc.2000.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.CHEK2 Breast Cancer Case-Control Consortium. CHEK2*1100delC and susceptibility to breast cancer: A collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004;74:1175–1182. doi: 10.1086/421251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cairncross JG, Macdonald DR. Successful chemotherapy for recurrent malignant oligodendroglioma. Ann Neurol. 1988;23:360–364. doi: 10.1002/ana.410230408. [DOI] [PubMed] [Google Scholar]
- 34.Macdonald DR, Gaspar LE, Cairncross JG. Successful chemotherapy for newly diagnosed aggressive oligodendroglioma. Ann Neurol. 1990;27:573–574. doi: 10.1002/ana.410270519. [DOI] [PubMed] [Google Scholar]
- 35.Kim L, Hochberg FH, Thornton AF, et al. Procarbazine, lomustine and vincristine (PCV) chemotherapy for grade III and IV oligoastrocytomas. J Neurosurg. 1996;85:602–607. doi: 10.3171/jns.1996.85.4.0602. [DOI] [PubMed] [Google Scholar]
- 36.Cairncross G, Macdonald D, Ludwin S, et al. Chemotherapy for anaplastic oligodendroglioma. J Clin Oncol. 1994;12:2013–2021. doi: 10.1200/JCO.1994.12.10.2013. [DOI] [PubMed] [Google Scholar]
- 37.van den Bent MJ, Kros JM, Heimens JJ, et al. Response rate and prognostic factors of recurrent oligodendroglioma treated with procarbazine, CCNU and vincristine chemotherapy: Dutch Neuro-oncology Group. Neurology. 1998;51:1140–1145. doi: 10.1212/wnl.51.4.1140. [DOI] [PubMed] [Google Scholar]
- 38.Chinot OL, Honore S, Dufour H, et al. Safety and efficacy of temozolomide in patients with recurrent anaplastic oligodendrogliomas after standard radiotherapy and chemotherapy. J Clin Oncol. 2001;19:2449–2455. doi: 10.1200/JCO.2001.19.9.2449. [DOI] [PubMed] [Google Scholar]
- 39.van den Bent MJ, Taphoorn MJB, Brandes AA, et al. Phase II study of first line chemotherapy with temozolomide in recurrent oligodendroglial tumors: The European Organisation for the Research and Treatment of Cancer Brain Tumor Group Study 26971. J Clin Oncol. 2003;21:2525–2528. doi: 10.1200/JCO.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 40.Hoang-Xuan K, Capelle L, Kujas M, et al. Temozolomide as initial treatment for adults with low-grade oligodendrogliomas and oligoastrocytomas and correlation with chromosome 1p deletions. J Clin Oncol. 2004;22:3133–3138. doi: 10.1200/JCO.2004.10.169. [DOI] [PubMed] [Google Scholar]
- 41.Giannini C, Burger PC, Berkey BA, et al. Anaplastic oligodendroglial tumors: Refining the correlation among histopathology, 1p/19q co-deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol. 2008;18:360–369. doi: 10.1111/j.1750-3639.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van den Bent MJ, Dubbink HJ, Yannick M, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumours: A report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res. 2010;16:1597–1604. doi: 10.1158/1078-0432.CCR-09-2902. [DOI] [PubMed] [Google Scholar]
- 44.Duncan T, Trewick SC, Koivisto P, et al. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci U S A. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van den Bent MJ, Gravendeel LA, Gorlia T, et al. A hypermethylated phenotype is a better predictor of survival than MGMT methylation in anaplastic oligodendroglial tumours: A report from the EORTC study 26951. Clin Cancer Res. 2011;17:7148–7155. doi: 10.1158/1078-0432.CCR-11-1274. [DOI] [PubMed] [Google Scholar]
- 46.Erdem-Eraslan L, Gravendeel L, de Rooi J, et al. Intrinsic molecular subsets of glioma are prognostic and predictive of benefit from adjuvant procarbazine, lomustine and vincristine chemotherapy in combination with other prognostic factors in anaplastic oligodendroglial brain tumors: A report from the EORTC study 26951. J Clin Oncol. 2013;31:328–336. doi: 10.1200/JCO.2012.44.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Theeler BJ, Yung WK, Fuller GN, et al. Moving toward molecular classification of diffuse gliomas in adults. Neurology. 2012;79:1917–1926. doi: 10.1212/WNL.0b013e318271f7cb. [DOI] [PMC free article] [PubMed] [Google Scholar]