

Abstract
Purpose
Tumor cells from approximately 40% of patients with Hodgkin or non-Hodgkin lymphoma express the type II latency Epstein-Barr virus (EBV) antigens latent membrane protein 1 (LMP1) and LMP2, which represent attractive targets for immunotherapy. Because T cells specific for these antigens are present with low frequency and may be rendered anergic by the tumors that express them, we expanded LMP–cytotoxic T lymphocytes (CTLs) from patients with lymphoma using autologous dendritic cells and EBV-transformed B–lymphoblastoid cell lines transduced with an adenoviral vector expressing either LMP2 alone (n = 17) or both LMP2 and ΔLMP1 (n = 33).
Patients and Methods
These genetically modified antigen-presenting cells expanded CTLs that were enriched for specificity against type II latency LMP antigens. When infused into 50 patients with EBV-associated lymphoma, the expanded CTLs did not produce infusional toxicities.
Results
Twenty-eight of 29 high-risk or multiple-relapse patients receiving LMP-CTLs as adjuvant therapy remained in remission at a median of 3.1 years after CTL infusion. None subsequently died as a result of lymphoma, but nine succumbed to complications associated with extensive prior chemoradiotherapy, including myocardial infarction and secondary malignancies. Of 21 patients with relapsed or resistant disease at the time of CTL infusion, 13 had clinical responses, including 11 complete responses. T cells specific for LMP as well as nonviral tumor-associated antigens (epitope spreading) could be detected in the peripheral blood within 2 months after CTL infusion, but this evidence for epitope spreading was seen only in patients achieving clinical responses.
Conclusion
Autologous T cells directed to the LMP2 or LMP1 and LMP2 antigens can induce durable complete responses without significant toxicity. Their earlier use in the disease course may reduce delayed treatment-related mortality.
INTRODUCTION
Antigen-specific T cells targeting immunodominant viral antigens from cytomegalovirus and Epstein-Barr virus (EBV) have been used with dramatic success to treat viral reactivation after bone marrow transplantation.1–4 In particular, donor-derived EBV-specific T cells produced complete responses (CRs) resulting in durable remissions in more than 70% of patients with EBV-associated post-transplantation lymphoproliferative disease (PTLD) with minimal infusional toxicity.5,6 However, PTLD, an EBV type III latency tumor expressing highly immunogenic EBV-derived antigens, can only develop in an immunocompromised host. By contrast, EBV-associated tumors of the immunocompetent host with Hodgkin lymphoma (HL; 40% of tumors) or non-Hodgkin lymphoma (NHL; 20% of diffuse large B-cell lymphomas [DLBCLs] and > 90% natural killer [NK]/T-cell NHL nasal type) are associated with type II EBV latency, where only restricted, weakly immunogenic (subdominant) EBV antigens (latent membrane protein 1 [LMP1], LMP2, and EBNA1) are expressed.7–9
The frequency of T cells specific for type II latency antigens in patients with type II latency tumors is low, and such T cells may be rendered anergic in the tumor microenvironment.10,11 Nonetheless, some immunocompetent patients with relapsed EBV-associated HL enter CR after treatment with autologous EBV-specific cytotoxic T lymphocytes (CTLs), even when these lines contain only low frequencies of LMP-specific T cells LMP-CTLs.12,13
To enhance activity against EBV type II latency lymphomas, we developed approaches that increase the frequency of relevant EBV-latency antigen-specific T cells and conserve the memory T-cell populations likely needed for long-term persistence and sustained antitumor responses. We used adenoviral vector (AdV) –transduced dendritic cells (DCs) and EBV-transformed B–lymphoblastoid cell lines (LCLs) as antigen-presenting cells to activate and expand LMP-specific T cells.14 We initially used an AdV-encoding LMP2 antigen alone15 and subsequently used AdV-encoding LMP2 and LMP1, the latter in truncated form to reduce toxicity and potential oncogenicity.15,16 We infused LMP-CTLs into 50 patients who had either relapsed/resistant EBV-positive HL or NHL (n = 21) or were in remission from high-risk or multiple-relapse disease (n = 29). We now report the clinical responses to CTL treatment; the phenotype, fate, and antitumor function of the infused CTLs; and the development of epitope spreading beyond the initially targeted EBV antigens, which may promote and sustain the antitumor response.
PATIENTS AND METHODS
Patients and LMP Status of the Tumors
The protocol for the use of LMP-CTLs as therapy for lymphoma was approved by the US Food and Drug Administration, Recombinant DNA Advisory Committee, and Baylor College of Medicine Institutional Review Board and Institutional Biosafety Committee. Patients were eligible for this study if they had EBV-associated type II or III latency HL or NHL detected by immunohistochemistry for LMP1 and/or in situ hybridization for EBER.17
Patients—who either had relapsed after receiving standard therapy (Table 1) or were considered at high risk for relapse (Table 2)—received two infusions of T cells 2 weeks apart in the General Clinical Research Center of Texas Children's Hospital or the Methodist Hospital, where their vital signs were monitored before and immediately after infusion. If patients had a partial response (PR) or stable disease 8 weeks after receiving CTLs, they were eligible to receive eight additional CTL infusions, consisting of the same number of cells as their second injection. After completing the dose-escalation component for the LMP1/2 study and finding no difference in outcome with dose, we amended the study so additional patients could be treated at the first dose level. Total doses of CTLs received are shown in Tables 1 and 2. Analysis of disease response to CTL therapy was performed using International Working Group response criteria, and scans were reviewed by an independent radiologist.18,19
Table 1.
Patient Characteristics: Active Disease Cohort
| UPN | Age (years) | Sex | Disease/EBV Latency Type | No. of Relapses | Most Recent Treatment | ALC at Time of Infusion | Total CTL Dose | Toxicity Attributed to CTLs | Response to CTLs | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| LMP2-specific CTL protocol | ||||||||||
| 824 | 8 | M | T-cell CAEBV, latency II | 2 | AlloSCT (busulfan/cyclophosphamide/alemtuzumab) | 728 | 4 × 107/m2 | None | PR | Relapse at 4 years |
| 652 | 39 | M | DLBCL, latency II | 5 | Brentuximab | 649 | 8 × 107/m2 | None | CR | Relapse at 10 months |
| 909 | 30 | F | HL/CVID, latency III | 2 | ABVD, rituximab | 4,998 | 8 × 107/m2 | None | CR | Durable CR (9+ years) |
| 1316 | 54 | M | NK/T-cell NHL, latency II | 0 | RT, CHOP | 802 | 1.2 × 108/m2 | None | CR | Durable CR (5+ years) |
| 1187 | 17 | M | HL, latency II | 3 | AutoSCT (BEAM) | 334 | 1.2 × 108/m2 | None | NR | NR |
| 1006 | 19 | M | HL, latency II | 6 | ESHAP | 390 | 1.2 × 108/m2 | None | NR | NR |
| 1160 | 15 | F | HL/CVID, latency III | 0 | VP16, doxorubicin, rituximab | 1,483 | 3 × 108/m2 | None | CR | Durable CR (7+ years) |
| 1054 | 64 | F | NK/T-cell NHL, latency II | 3 | R-CHOP | 479 | 3.2 × 108/m2 | None | CR | Relapse at 9 months |
| LMP1/2-specific CTL protocol | ||||||||||
| 1372 | 69 | M | NK/T-cell NHL, latency II | 1 | CHOP, RT | 671 | 4 × 107/m2 | None | CR | Durable CR (5+ years) |
| 1371 | 21 | M | HL, post-SOT, latency III | 1 | Ifosfamide, vinorelbine, Ara-C, VP16, cisplatin, RT | 476 | 4 × 107/m2 | None | CR | Durable CR (5+ years) |
| 2051 | 30 | F | T-cell CAEBV, latency II | 0 | CHOP, VP16, dexamethasone, acyclovir | 1,308 | 4 × 107/m2 | None | NR | NR |
| 2336 | 52 | M | DLBCL/PTLD, latency III | 0 | No treatment | 444 | 4 × 107/m2 | None | CR | Durable CR (2+ years) |
| 2266 | 27 | F | HL, latency II | 3 | SGN-35, bendamustine | 446 | 4 × 107/m2 | None | NR | NR |
| 2457 | 16 | M | HL, latency II | 0 | COG protocol (AHOD0031) | 1,332 | 4 × 107/m2 | None | NR | NR |
| 1811 | 60 | M | NK/T-cell NHL, latency II | 0 | RT, hyperCVAD, high-dose MTX | 411 | 1 × 108/m2 | Inflammatory response? | NR | NR |
| 1409 | 18 | M | NK/T-cell NHL, latency II | 0 | RT, CHOP | 413 | 1.2 × 108/m2 | None | NR | NR |
| 1545 | 79 | M | LYG, latency III | 0 | No treatment | 659 | 1.2 × 108/m2 | None | CR | Durable CR (4+ years) |
| 1351 | 46 | F | HL, latency II | 3 | AutoSCT (BEAM) | 1,889 | 2 × 108/m2 | None | PR | Received T cells on second protocol → durable CR (5+ years) |
| 1356 | 28 | F | DLBCL, latency II | 1 | No treatment | 1,730 | 3 × 108/m2 | None | NR | NR |
| 1990 | 65 | M | CLL/DLBCL, latency III | 0 | FCR, alemtuzumab | 732 | 3 × 108/m2 | Inflammatory response? | CR | Durable CR (1+ year); died as result of infection |
| 1656 | 55 | M | NK/T-cell NHL, latency II | 0 | RT | 808 | 5 × 108/m2 | None | PR → CR | Durable CR (4+ years) |
Abbreviations: ABVD, doxorubicin, bleomycin, vinblastine, dacarbazine; ALC, absolute lymphocyte count; alloSCT, allogeneic stem-cell transplantation; Ara-C, cytarabine; autoSCT, autologous SCT; BEAM, carmustine, etoposide, cytarabine, melphalan; CAEBV, chronic active EBV infection; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; CLL, chronic lymphocytic leukemia; COG, Children's Oncology Group; CR, complete response; CTL, cytotoxic T lymphocyte; CVID, common variable immunodeficiency disease; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; ESHAP, etoposide, methylprednisolone, cytarabine, cisplatin; FCR, fludarabine, cyclophosphamide, rituximab; HL, Hodgkin lymphoma; hyperCVAD, cyclophosphamide, doxorubicin, vincristine, dexamethasone, cytarabine, methotrexate; LMP, latent membrane protein; LYG, lymphoid granulomatosis; MTX, methotrexate; NHL, non-Hodgkin lymphoma; NK, natural killer; NR, no response; PR, partial response; PTLD, post-transplantation lymphoproliferative disease; R-CHOP, rituximab plus CHOP; RT, radiation therapy; SGN-35, brentuximab vedotin; SOT, solid organ transplantation; UPN, unique patient number; VP16, etoposide.
Table 2.
Patient Characteristics: First or Later Remission Cohort
| UPN | Age (years) | Sex | Disease | No. of Relapses | Most Recent Treatment | ALC at Time of Infusion | Total CTL Dose | Toxicity Attributed to CTLs | Outcome (time post-CTLs) |
|---|---|---|---|---|---|---|---|---|---|
| LMP2-specific CTL protocol | |||||||||
| 959 | 57 | F | DLBCL, latency II | 2 | AutoSCT (BEAM-R) | 706 | 4 × 107/m2 | None | Relapse (2 months); died as result of complications related to alloHSCT (2 years) |
| 871 | 66 | F | DLBCL, latency II | 3 | AutoSCT (BEAM-R) | 2,321 | 4 × 107/m2 | None | CCR; died as result of secondary malignancy (6 years) |
| 964 | 50 | F | HL, latency II | 4 | Liposomal vincristine | 819 | 4 × 107/m2 | None | CCR; died as result of second cancer (2 years) |
| 1053 | 30 | M | HL, latency II | 4 | ABVD | 740 | 1.2 × 108/m2 | None | CCR; died as result of secondary MDS (7 years) |
| 1263 | 52 | M | NK/T-cell NHL, latency II | 0 | RT, CHOP | 2,259 | 1.2 × 108/m2 | None | CCR (6+ years) |
| 1286 | 69 | F | Peripheral T-cell NHL, latency III | Primary refractory | RT, EPOCH, intrathecal MTX | 1,184 | 1.2 × 108/m2 | None | CCR; died as result of complications related to lung transplantation (2 years) |
| 1007 | 25 | F | HL, latency II | 1 | AutoSCT (busulfan/melphalan/thiotepa) | 1,462 | 1.2 × 108/m2 | None | CCR (8+ years) |
| 1272 | 59 | M | HL, latency II | 1 | AutoSCT (BEAM) | 1,338 | 1.2 × 108/m2 | None | CCR; died as result of infection (3 years) |
| 1057 | 7 | M | HL, immune suppressed, latency III | 0 | Rituximab | 3,494 | 3 × 108/m2 | None | CCR (7+ years) |
| LMP1/2-specific CTL protocol | |||||||||
| 2053 | 48 | M | NK/T-cell NHL, latency II | 0 | VP16, dexamethasone, ifosfamide, cisplatin | 1,033 | 4 × 107/m2 | None | CCR (2+ years) |
| 2056 | 56 | F | DLBCL, latency II | 6 | R-CHOP | 672 | 4 × 107/m2 | None | Not evaluable; died as result of cardiac disease (< 8 weeks) |
| 2313 | 66 | M | HL, latency II | 1 | ABVD | 410 | 4 × 107/m2 | None | CCR; died as result of infection (4 years) |
| 2433 | 17 | M | HL, latency II | 4 | SGN-35 | 760 | 4 × 107/m2 | None | CCR (1+ years) |
| 1369 | 23 | M | HL, latency II | 1 | AutoSCT (BEAM) | 968 | 4 × 107/m2 | None | CCR (5+ years) |
| 1420 | 34 | M | HL, latency II | 1 | AutoSCT (BEAM) | 999 | 4 × 107/m2 | None | CCR (5+ years) |
| 1595 | 18 | M | HL, latency II | 1 | AutoSCT (BEAM), RT | 285 | 4 × 107/m2 | None | CCR (4+ years) |
| 2493 | 17 | M | HL and NPC, latency II | 0 | ABVE-PC | 1,035 | 4 × 107/m2 | None | CCR (1+ years) |
| 1984 | 65 | F | HL and melanoma, latency III | 1 | Rituximab, AVD ×4 | 530 | 6 × 107/m2 | None | CCR; died as result of CNS hemorrhage (1 year) |
| 1806 | 59 | F | NK/T-cell NHL, latency II | 0 | AutoSCT (BEAM) | 1,145 | 1 × 108/m2 | None | CCR (2+ years) |
| 1370 | 61 | M | HL, latency II | 1 | ABVD, RT | 1,220 | 1.2 × 108/m2 | None | CCR (5+ years) |
| 1842 | 38 | M | HL, latency II | 1 | AutoSCT (BEAM-R) | 347 | 1.2 × 108/m2 | None | CCR (3+ years) |
| 1884 | 14 | M | HL, latency II | Primary refractory | AutoSCT (BEAM), mantle RT | 1,114 | 1.2 × 108/m2 | None | CCR (3+ years) |
| 1905 | 18 | M | HL, latency II | Primary refractory | AutoSCT (BEAM) | 1,694 | 1.2 × 108/m2 | None | CCR (3+ years) |
| 1455 | 47 | M | NK/T-cell NHL, latency II | 0 | RT, CHOP | 512 | 1.2 × 108/m2 | None | CCR (4+ years) |
| 1511 | 62 | F | LYG, immune suppressed, latency III | 0 | Surgery, HDMTX, rituximab, cyclophosphamide | 540 | 3 × 108/m2 | None | CCR (3+ years) |
| 1888 | 52 | M | DLBCL, latency II | 0 | CHOP-R | 472 | 3 × 108/m2 | None | CCR (3+ years) |
| 2135 | 43 | M | NK/T-cell NHL, latency II | Primary refractory | AutoSCT (busulfan/melphalan/gemcitabine) | 769 | 3 × 108/m2 | None | CCR (2+ years) |
| 2368 | 64 | F | HL, latency II | 1 | AutoSCT (BEAM-R) | 563 | 3 × 108/m2 | None | CCR (1+ years) |
| 2095 | 24 | M | PTLD, immune suppressed, latency III | 1 | AutoSCT (high-dose melphalan) | 3,171 | 3 × 108/m2 | None | CCR (1+ years) |
Abbreviations: ABVD, doxorubicin, bleomycin, vinblastine, dacarbazine; ABVE-PC: doxorubicin, bleomycin, vincristine, etoposide, prednisone, cyclophosphamide; ALC, absolute lymphocyte count; AVD, adriamycin, vinblastine, dacarbazine; alloHSCT, allogeneic hematopoietic stem-cell transplantation; autoSCT, autologous stem-cell transplantation; BEAM, carmustine, etoposide, cytarabine, melphalan; BEAM-R, BEAM plus rituximab; CCR, continued complete response; CTL, cytotoxic T lymphocyte; DLBCL, diffuse large B-cell lymphoma; EPOCH, etoposide, doxorubicin, vincristine, prednisone, cyclophosphamide; HD, high dose; HL, Hodgkin lymphoma; LMP, latent membrane protein; LYG, lymphoid granulomatosis; MDS, myelodysplastic syndrome; MTX, methotrexate; NHL, non-Hodgkin lymphoma; NK, natural killer; NPC, nasopharyngeal carcinoma; PTLD, post-transplantation lymphoproliferative disease; R-CHOP, rituximab plus CHOP; RT, radiation therapy; SGN-35, brentuximab vedotin; VP16, etoposide.
Generation of LMP-Specific CTLs
The generation of Good Manufacturing Practice (GMP) –grade LMP-CTLs was performed as previously published.13,20 Immature DCs were transduced with either the Ad5f35LMP214 or Ad5f35ΔLMP1-I-LMP2 vector15,21 and matured. Before coculture with peripheral blood mononuclear cells (PBMCs) with or without interleukin-15, DCs were gamma irradiated (30 Gy). From day 10, responder T cells were restimulated weekly with irradiated LCLs transduced with the same LMP vector. At the time of final cryopreservation, the patient-derived CTLs contained both effector-memory populations (CD62L−, CD45RA−) and central memory populations (CD62L+, CD45RA−) and comprised both CD4+ and CD8+ T cells.22,23 Fewer than 1% of cells expressed monocyte or B-cell markers.
Cytotoxicity Assays
The cytotoxic specificity of each CTL line was analyzed in a standard 4-hour chromium-51 release assay as described.24 Details are provided in the Appendix (online only).
Immunophenotyping
Details are provided in the Appendix (online only).
LMP Multimers and Peptides
To detect LMP T cells in the CTL products and in PBMCs, we used pentamers (Proimmune, Springfield, VA) as previously described.17 Panels of 15-mer peptides (overlapping by 11 amino acids) covering the entire amino acid sequence of LMP1 and LMP2 from the white prototype EBV strain B95-8 were synthesized as previously described.25–28 For LMP1, 10 peptide pools were prepared using a strategy similar to that used for LMP2.29
Enzyme-Linked Immunospot Assay
Enzyme-linked immunospot (ELISPOT) analysis was used to determine the frequency of T cells secreting interferon gamma (IFN-γ) in response to EBV- and tumor-associated antigen (TAA) pepmixes (JPT Peptide Technologies, Berlin, Germany) or LCLs as previously described.26 Details are provided in the Appendix (online only).
Statistical Analysis
Survival data were analyzed using the Kaplan-Meier method, and comparisons between groups were performed with the log-rank test. Overall survival (OS) was calculated from the time of first CTL infusion to death resulting from any cause; observations were censored at the date of last follow-up. Event-free survival (EFS) was calculated from the time of first CTL infusion to the date of relapse, death, or last follow-up, whichever occurred first. Cumulative incidence was estimated using the competing risk method described by Gray.30 P values less than .05 were considered statistically significant. Additional details are provided in the Appendix (online only).
RESULTS
Patient Characteristics
Ninety-five patients had EBV-positive tumors and elected to proceed with LCL and CTL generation. LMP1/2-specific CTLs were generated from 52 patients. Of the remaining 43 patients, 26 patients were ineligible. Nine patients (9.5%) died before completion and release of the CTL line, and in eight patients (9%), we were unable to generate the LCL or CTL line. Fifty patients received LMP-CTLs. All 50 patients had type II or III EBV-positive lymphoma as evaluated by EBER and/or LMP1 positivity and the presence or absence of known immune deficiency (Tables 1 and 2; Appendix Table A1, online only). Ages ranged from 7 to 79 years (median, 44.5 years), and initial disease presentation ranged from stage IA to IVB. Patients were recruited from 18 centers in the United States and internationally. We analyzed EFS and assessed the influence of underlying disease and other variables on outcome by stratifying patients into disease and treatment groups, as outlined:
Histologic classification.
Twenty-five patients had HL, 11 had NK/T-cell NHL, seven had DLBCL, two had PTLD, one had peripheral T-cell NHL, and four had other lymphomas, including chronic active EBV infection and lymphomatoid granulomatosis (Appendix Table A1, online only).
Classification by disease stage.
Twelve patients received CTLs as adjuvant therapy after entering initial remission of disease, which was considered to pose a high risk of relapse (eg, NK/T-cell lymphomas, primary refractory lymphomas, and lymphomas developing in immunocompromised host). The remaining 17 patients were in subsequent remissions after one to six relapses, thus generating a first- or later-remission cohort (Table 2). An additional 21 patients received CTLs as treatment for relapsed or resistant disease refractory to standard treatment (active disease cohort; Table 1).
Classification by treatment.
Treatment included T cells enriched for LMP2 versus T cells enriched for LMP1 and LMP2. Seventeen patients received LMP2-enriched T cells, including 16 previously described,17 whereas 33 received LMP1- and LMP2-enriched T cells (Tables 1 and 2). Overall, 22 CTL lines were derived from patients at diagnosis and 28 after first or subsequent relapse.
Specificity and Clonality of Ex Vivo–Expanded T Cells
At the time of cryopreservation, CTLs comprised CD8+ T cells (median, 72%; range, 6% to 99%), CD4+ T cells (median, 9%; range, 1% to 94%), and NK cells (CD3−/CD56+; median, 1%; range, 1% to 27%). Although the T-cell phenotype was predominantly effector and effector memory (CD45RA−/CD62L−; median, 31%; range, 2% to 92%), a median 25% (range, 2% to 94%) of the infused T cells were CD45RA−/CD62L+ (Fig 1A). No T regulatory cells (CD4+/CD25+/FoxP3+), B cells, or DCs were detected in the final product. The specificity of the LMP-CTLs was determined with IFN-γ ELISPOT assays after stimulation with LMP peptides. Cytotoxicity was tested against LMP-expressing target cells. When available, HLA peptide pentamers were used (data not shown). Of lines generated with Ad5f35LMP2, 53% had LMP2-specific activity, but none had LMP1 activity (Fig 1B; Appendix Table A2, online only). By contrast, 66% lines generated with Ad5f35ΔLMP1-LMP2 had LMP1 and/or LMP2 activity (Fig 1C; Appendix Table A2, online only). Many of the remaining 18 CTL lines were predominantly CD4+ HLA class II restricted, and LMP-specific activity could not be confirmed. Nevertheless, these CD4+ CTLs were cytotoxic and had antitumor activity in vivo. The LMP-directed T cells were also cytolytic to target cells pulsed with peptides derived from LMP2 or LMP1/2 and against EBV LCLs, which also express LMP1 and LMP2 (Fig 1D). As shown in Figures 1E and IF, there was a hierarchy of killing by LMP1/2-specific CTLs: greatest against autologous LCLs, intermediate against targets pulsed with LMP2 peptides, and lowest against targets expressing LMP1. We saw no killing of unpulsed or irrelevant peptide–pulsed targets. The LMP-responding cells were polyspecific because relevant HLA multimers demonstrated the presence of T cells enriched for multiple specificities of LMP1 and LMP2 (Appendix Tables A2 and A3, online only). Flow cytometric analysis confirmed that LMP-specific lines were polyclonal and that a majority of Vβ families were represented (data not shown).
Fig 1.

Characteristics of latent membrane protein (LMP) –specific cytotoxic T lymphocyte (CTL) lines derived from patients with Epstein-Barr virus (EBV) –positive lymphoma. Bars indicate median values. (A) Phenotype of LMP-specific CTL lines at time of freezing, showing predominance of CD3+ and CD8+ T cells. Recognition of LMP1 and LMP2 in interferon gamma (IFN-γ) enzyme-linked immunospot (ELISPOT) assay by CTLs, generated with APCs transduced with (B) Ad5f35LMP2 or (C) Ad5f35ΔLMP1-1-LMP2 vector. Spot counts in response to target antigen stimulation by IFN-γ ELISPOT assay of LMP-CTL lines are shown as solid circles. (D) LMP2-specific CTL lines demonstrate cytotoxicity against autologous lymphoblastoid cell lines (LCLs) and LMP2 pepmix–pulsed phytohemagglutinin (PHA) blasts but not against unpulsed or LMP1-pulsed PHA blasts, at effector cell to target cell (E:T) ratio of 20:1. (E) LMP1/2-specific CTL lines demonstrate similar cytotoxicity against autologous LCL and LMP2 pepmix–pulsed PHA blasts but less against LMP1-pulsed PHA blasts. Unpulsed PHA blasts were not killed. (F) LMP1- and LMP2-specific activity in CTL line generated from patient with relapsed Hodgkin lymphoma. Results represent patients whose CTLs recognized both LMP1 and LMP2. LMP-specific CTL line from this patient showed killing of autologous (auto) LCL and PHA blasts only if pulsed with LMP1 or LMP2 pepmix. There was no killing of PHA blasts alone. Allo, allogeneic; SFC, spot-forming cell.
LMP-Specific CTLs As Adjuvant Therapy
Of the 29 patients in first or later remissions, nine received CTLs generated against LMP2, and 20 received CTLs generated against LMP1/2. One patient died as a result of complications from preexisting cardiac disease before the 8-week disease evaluation, but 27 of the remaining 28 evaluable patients remained in CR (Table 2; Figs 2A and 2B). However, there were nine deaths resulting from nonrelapse causes (Fig 2C), for a 2-year EFS of 82%. Of the 12 high-risk patients treated with CTLs as first-line therapy, one patient (8%) died as a result of complications related to lung transplantation. In contrast, of the 17 patients in remission after receiving T cells for multiple-relapse disease, eight (47%) died (Fig 2C), all of nonrelapse causes associated with extensive prior chemoradiotherapy (Table 2). Univariate analysis found no differences in EFS by LMP2 compared with LMP1/2-specific T cells or lymphoma subtype (P = .22; Fig 2B).
Fig 2.

Outcomes in 29 patients in first or later remission who received latent membrane protein (LMP) –cytotoxic T lymphocyte (CTL) as adjuvant therapy. (A) Remissions were sustained in all but one patient. (B) Two-year event-free survival for 29 patients treated with either LMP2- (n = 9) or LMP1/2-specific CTLs (n = 20; P = .22). (C) Cumulative incidence of death resulting from lymphoma or nonlymphoma causes (eg, cardiac disease, secondary malignancy, infection).
Outcome of CTL Therapy for Relapsed Disease
No immediate or delayed infusional toxicities were attributable to CTL infusion, although one patient had CNS deterioration 2 weeks after infusion. Although this was attributed to disease progression, we cannot exclude an inflammatory response at a site of CNS disease. A second patient developed respiratory complications approximately 4 weeks after the second CTL infusion, coincident with achieving CR. Although this event was attributed to an intercurrent infection, and the patient completely recovered, a systemic inflammatory response syndrome related to CTLs could not be excluded. Overall, 11 of the 21 patients in the active disease cohort achieved CR. Two more achieved PRs, one of whom entered CR after additional CTL therapy (Fig 3A). The probability of response trended higher in recipients with CTL lines containing LMP1 specificity (five of seven responded) versus those receiving lines lacking LMP1 activity (one of six responded), but this trend did not reach significance (P = .103; Fig 3B). The presence of LMP2 specificity had no discernible impact on response rates (Fig 3C). This lack of correlation may be related to the relative insensitivity of the ELISPOT assay, which underestimates the actual frequency of antigen-specific T cells by 10- to 100-fold. Univariate analysis showed no difference in response rates among patients with HL versus NHL, and responses were independent of the total CTL dose.
Fig 3.
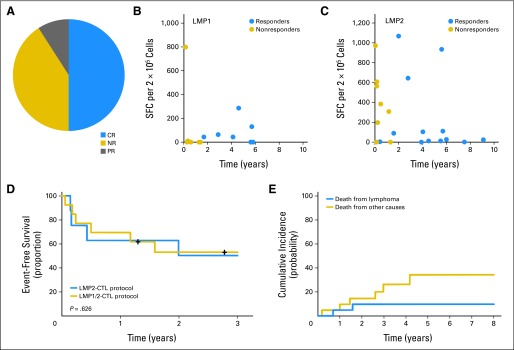
Outcomes in patients who received latent membrane protein (LMP) –cytotoxic T lymphocytes (CTLs) as treatment for relapsed or refractory disease. (A) Distribution of clinical responses among 21 patients. (B) Increased proportion of responding versus nonresponding patients had CTLs with specific activity against LMP1 by interferon gamma enzyme-linked immunospot assay. (C) This relationship was not apparent when analysis was based on CTLs with LMP2-specific activity. (D) Two-year event-free survival for patients treated in LMP2-CTL protocol (n = 8) versus LMP1/2-CTL protocol (n = 13; P = .626). (E) Cumulative risk of death resulting from lymphoma or other causes. CR, complete response; NR, no response; PR, partial response; SFC, spot-forming cell.
Overall, the 2-year EFS rate among patients treated for resistant/recurrent disease was approximately 50% for both the LMP2 and LMP1/2 T-cell groups (P = .626; Fig 3D), with deaths evenly distributed between relapse and nonrelapse causes in both groups (Fig 3E).
Tumor Responses to Adoptively Transferred LMP-Specific CTL Lines Associated With Increases in Frequency of Circulating LMP-Specific T Cells
We measured changes in the frequency of LMP1- and LMP2-specific CTLs in the blood before and after LMP-CTL infusion using IFN-γ ELISPOT assays. Figures 4A and 4B shows that most patients who achieved a clinical response or remained in a durable remission (responders) had circulating LMP1- and/or LMP2-specific T cells. Few nonresponding patients showed this pattern (Figs 4C and 4D). Of note, neither responders nor nonresponders had a concomitant rise in cytomegalovirus-specific T cells. Thus, changes in LMP1/2 T-cell frequency were not simply a marker of a generalized increase in virus reactivity (data not shown). We also evaluated suppressive T regulatory cells (CD4+/CD25+/CD69−) immune reconstitution in patients who received LMP1/2 CTLs. The mean change from 0 to 2 weeks was −50.1% in the nonresponders and 9.7% in the responders; from 2 to 8 weeks, it was 25.8% in the nonresponders and 11.2% in the responders (data not shown).
Fig 4.
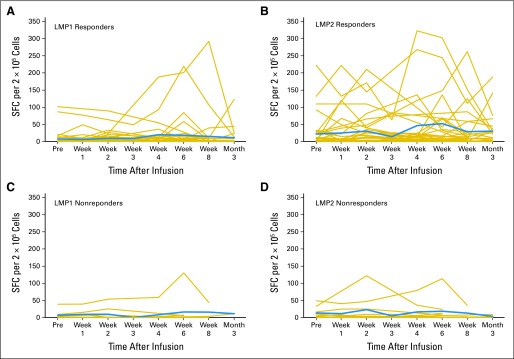

Frequency of latent membrane protein (LMP) –specific and tumor antigen–specific T cells in responding versus nonresponding patients. Immune reconstitution was evaluated in individual patients who received LMP–cytotoxic T lymphocytes. (A-D) Peripheral blood T cells were incubated with LMP1 or LMP2 pepmixes. Number of interferon gamma (IFN-γ) spot-forming cells (SFCs) per 2 × 105 mononuclear cells was measured in enzyme-linked immunospot (ELISPOT) assays. Gold lines represent individual LMP-specific T cells; blue lines represent mean of LMP-specific T cells over time. Note greater frequencies of reactivity for responding patients. (A) LMP1 responders. (B) LMP2 responders. (C) LMP1 nonresponders. (D) LMP2 nonresponders. (E) Evidence for epitope spreading in 12 patients with non-Hodgkin or Hodgkin lymphoma. Peripheral blood T cells were incubated with lymphoma antigen pepmixes (MAGE A4, survivin, PRAME); number of IFN-γ SFCs per 2 × 105 mononuclear cells was measured in ELISPOT assays.
To determine if an LMP-CTL–mediated attack on tumor cells elicited broader immune reactivity against tumor cells, we investigated 12 cases of NHL or HL in which patients received LMP-CTLs as treatment. We evaluated tumor-specific T-cell populations from seven responding patients and five nonresponding patients to identify epitope spreading. In four of the seven responders, infusion of LMP-directed T cells was followed over an 8-week period by a striking increase in T cells specific for the lymphoma-associated antigens MAGE A4, survivin, and PRAME. In contrast, none of the five nonresponders demonstrated such epitope spreading (Fig 4C).
DISCUSSION
We administered LMP2- or LMP1/2-specific CTLs to 50 patients with EBV-associated HL or NHL, showing that administration is safe and that 29 patients treated in remission from high-risk or multiple-relapse disease had an 82% EFS rate at 2 years (Fig 2B). Among 21 patients with active disease, 11 entered sustained CR with CTL therapy alone, and two more achieved PRs. Responses were associated with effector and central memory LMP1-specific T cells in the infused population but not with type of disease treated or recipient's lymphopenic status. Strikingly, CRs were seen even in patients with limited apparent in vivo expansion of LMP-directed T cells, and this effect was associated with epitope spreading, as evidenced by the emergence of fresh/endogenous nonviral tumor antigen–directed T cells targeting non-EBV antigens.
Although high cure rates are achievable with conventional therapeutics for patients with HL and NHL, such treatment may induce unacceptable organ toxicities and immune suppression and can lead to secondary cancers and cardiac disease. These problems are accentuated in patients receiving salvage therapy after relapse and have led to increasing interest in more targeted immunotherapies such as monoclonal antibodies and more recently T lymphocytes genetically modified with CAR-CD19 constructs.31–37 However, monoclonal antibodies have a limited half-life and require repeated infusions, and when targeted to a pan–B-cell antigen, they deplete the normal B-cell pool for at least 6 months. CAR-CD19–modified T cells require prior lymphodepleting chemotherapy, may be associated with a systemic inflammatory response syndrome, and deplete normal B cells indefinitely. By contrast, T cells targeting viral antigens via their native T-cell receptors persist long-term, do not require prior chemotherapy to potentiate their action, have minimal toxicity, and do not eliminate healthy tissues.
EBV-directed T-cell therapy for PTLD, which displays a type III latency motif, is a robust model for immunotherapy because of the highly immunogenic nature of these tumors. Here, we predominantly focused on EBV latency type II lymphomas. Compared with type III latency tumors, type II latency lymphomas are substantially less immunogenic, because viral gene expression is limited to the immune subdominant latent membrane proteins. When developing our studies, we chose to target LMP1 and/or LMP2, because EBNA1 is not well processed by the major histocompatibility complex class I processing machinery. CD4-restricted EBNA1 epitopes have been described, but although healthy donor-derived EBNA1-specific T cells have shown efficacy in patients with PTLD after allogeneic stem-cell transplantation,38 there is only a single case report of activity of EBNA1-specific T cells in the autologous setting or against type II latency tumors.39 However, we cannot rule out the activity of T cells specific for other viral proteins presented by LCLs from the second simulation. Nevertheless, this is the first large study to our knowledge to demonstrate effective control of both type II and III latency EBV lymphomas using patients' own LMP1- and LMP2-specific T cells.
It is a challenge in heavily pretreated patients with relapsed lymphoma to expand sufficient autologous CTLs for treatment.14 In fact, 120 mL of blood sufficed for T-cell expansion, obviating the need for an apheresis procedure. The ease of blood collection enabled blood samples to be shipped to our GMP facility, allowing recruitment of patients both nationally and internationally. Because pretreatment with chemotherapy was not required,40 T-cell infusions could be delivered with a 1- to 4-hour postinfusion outpatient monitoring period, thus simplifying the treatment approach. Furthermore, clinical responses were achieved with few major toxicities.33,37,41,42
A limitation of adoptive immunotherapy outside the setting of hematopoietic stem-cell transplantation is poor lymphocyte persistence in vivo and a lack of durable antitumor responses. Naive T cells have the most aggressive antitumor effects in murine models, but they are also associated with significant toxicities.33,37,41,42 The LMP-specific T-cell lines we used contained a combination of CD45RA−/CD62L− and CD45RA−/CD62L+ T cells (Fig 1A), the progeny of which is responsible for long-term persistence in nonhuman primates.43 The persistence of LMP-specific T cells is also facilitated by the continued presence of EBV in memory B-cell reservoirs, where the relevant antigens are constantly available for effective imumune responses.
Tumors frequently modulate target antigen expression to prevent T-cell recognition; indeed, treatment failures resulting from loss of a single targeted antigen are already becoming evident.42 Hence, after initially targeting only LMP2, we added LMP1. Although the numbers were small, the number of responding patients was greatest among those who received an infused product with abundant LMP1-specific T cells, compared with those with little or no LMP1-specific activity. The success of the LMP-CTL approach in eliciting clinical responses in 13 of 21 patients may also be related to epitope spreading (as previously observed in vaccine trials),44–46 which implies a beneficial change in the immunosuppressive tumor microenvironment.47 In contrast to the apparent lack of epitope spreading in nonresponding patients, more than 50% of patients achieving durable clinical responses produced T cells specific for the nonviral TAA within 2 months of T-cell therapy. We and others have previously demonstrated upregulation of TAAs by hematologic malignancies using hypomethylating agents, decitabine, or 5-azacytidine.48,49 Therefore, given the potential importance of eliciting a T-cell response to TAAs, epigenetic modifiers to increase TAA expression by tumor cells could be incorporated into LMP-CTL therapy.
In conclusion, we have shown that it is possible to resurrect powerful immunity to subdominant tumor-associated viral antigens in heavily pretreated patients with lymphoma. Our results suggest that such targeted therapies have a place not only in eliminating chemoradiotherapy-resistant malignant cell populations in relapsed patients but also in preventing relapse and achieving durable remissions without off-target adverse effects or long-term toxicities when administered early in the disease process.
Acknowledgment
We thank A. Durett for expert technical assistance and staff in the Good Manufacturing Practice facilities for assisting in cytotoxic T lymphocyte preparation and quality assurance. We thank all the clinicians who referred patients to this study: Babis Andreadis, Kelty Baker, Jeff Cohen, James Essell, Michelle Fanalae, Roger Giller, Branden Hsu, Roy Jones, Michael Keating, Sharon Lockhart, Don Mahoney, Vera Malkovska, Ken McClain, Peter McLaughlin, Phil McMahill, Richard T. McMahon, Rene McNall, Monika Metzger, Martha Mims, Sattva Neelapu, Pamela New, Cesar Nuñez, Owen O'Connor, Naomi Runnegar, Roger Strair, Raymond Thertulien, and Anas Younes.
Appendix
Cytotoxicity Assays
The cytotoxic specificity of each cytotoxic T lymphocyte (CTL) line was analyzed in a standard 4-hour chromium-51 release assay. The target cells tested were: autologous lymphoblastoid cell lines (LCLs), HLA class I and II mismatched LCLs, or phytohemagglutinin (PHA) -stimulated peripheral blood mononuclear cells (PBMCs; ie, PHA blasts) pulsed with latent membrane protein 1 (LMP1) or LMP2 pepmix (JPT Peptide Technologies, Berlin, Germany). As additional controls, we used LMP-negative target cells or autologous PHA blasts either alone or pulsed with irrelevant peptides from a CMVpp65 pepmix.
Immunophenotyping
CTL lines were stained with CD3, CD4, CD8, CD16, CD56, TCRαβ, TCRγδ, CD19, CD28, CD62L, CCR7, CD45RA, and CD45RO (Becton Dickinson, San Jose, CA). For each sample, 10,000 cells were analyzed by FACSCalibur using Cell Quest software (Becton Dickinson).
Enzyme-Linked Immunospot Assay
Enzyme-linked immunospot (ELISPOT) assay analysis was used to determine the frequency and function of T cells secreting interferon gamma in response to Epstein-Barr virus– and tumor-associated antigen pepmixes (JPT Peptide Technologies) or LCLs. To reduce interassay variability, patient PBMC samples were cryopreserved and batched for ELISPOT analysis. Spots were quantified by Zellnet Consulting (New York, NY), and the frequency of spot-forming cells was calculated based on the input cell numbers.
Statistical Analysis
Descriptive statistics were calculated to summarize CTL line characteristics and immune reconstitution data. Comparisons were made between groups using the nonparametric Wilcoxon rank sum test for continuous variables and Fisher's exact test for categoric variables. Survival data were analyzed using the Kaplan-Meier method, and comparisons between groups were performed with the log-rank test. Overall survival was calculated from the time of first CTL infusion to death resulting from any cause; observations were censored at the date of last follow-up. Event-free survival was calculated from the time of first CTL infusion to the date of relapse, death, or last follow-up, whichever occurred first. Cumulative incidence was estimated using the competing risk method. P values less than .05 were considered statistically significant.
Table A1.
Patient Characteristics
| Treatment Protocol | UPNs |
|
|---|---|---|
| HL | NHL/Other* | |
| Patients treated as adjuvant therapy | ||
| LMP2-specific CTLs | 964, 1053, 1007, 1272, 1057 | 959, 871, 1263, 1286 |
| LMP1/2-specific CTLs | 2313, 2433, 1369, 1420, 1595, 1984, 1370, 1842, 1884, 1905, 2368, 2493 | 2053, 2056, 1806, 1455, 1511, 1888, 2135, 2095 |
| Patients with active disease | ||
| LMP2-specific CTLs | 909, 1187, 1006, 1160 | 0824, 652, 1316, 1054 |
| LMP1/2-specific CTLs | 1371, 2266, 2457, 1351 | 1372, 2051, 2336, 1811, 1409, 1545, 1356, 1990, 1656 |
NOTE. Bold font indicates patients who did not respond to CTL therapy.
Abbreviations: CTL, cytotoxic T lymphocyte; HL, Hodgkin lymphoma; LMP, latent membrane protein; NHL, non-Hodgkin lymphoma; UPN, unique patient number.
NHL/other includes: natural killer/T-cell NHL, diffuse large B-cell lymphoma, post-transplantation lymphoproliferative disease, chronic active Epstein-Barr virus infection, and lymphoid granulomatosis.
Table A2.
LMP2 Specificity of CTL Lines Generated for LMP2-Specific CTL Protocols
| UPN | HLA Type | Diagnosis | Strength of LMP2-Specific Response in ELISPOT Assay* | No. of Epitopes Recognized by CTL Line |
|---|---|---|---|---|
| 1006 | A23,24/B35,55 | HL | +4 | 3 |
| 1187 | A1,3/B7,8 | HL | 0 | None identified |
| 964 | A2/B8,51 | HL | +4 | 2 |
| 1007 | A3,68/B7,1402(65) | HL | +4 | 2 |
| 1053 | A1,68/B27,37 | HL | +1 | 1 |
| 1272 | A3/B7,44 | HL | 0 | None identified |
| 1057 | A1,3/B14,37 | HL, immune suppressed | 0 | None identified |
| 909 | A3,24/B18 | HL, CVID | +1 | 1† |
| 1160 | A3,24/B41,52 | HL, CVID | 0 | None identified |
| 1286 | A1,2/B7,39 | Peripheral T-cell NHL, immune suppressed | 0 | None identified |
| 1263 | A66/B15(63),58 | NK/T-cell NHL | 0 | None identified |
| 1316 | A3,36/B15(71),53 | NK/T-cell NHL | +2 | None identified |
| 1054 | A1,32/B1401(64) | NK/T-cell NHL | 0 | None identified† |
| 652 | A3,24/B35 | DLBCL | +4 | 2 |
| 871 | A2,29/B13,27 | DLBCL | +4 | 4 |
| 959 | A2,68/B27,51 | DLBCL | 0 | 2 |
| 824 | A2,3/B51,57 | T-cell CAEBV | 0 | None identified |
Abbreviations: CAEBV, chronic active Epstein-Barr virus infection; CTL, cytotoxic T lymphocyte; CVID, common variable immunodeficiency disease; DLBCL, diffuse large B-cell lymphoma; ELISPOT, enzyme-linked immunospot; HL, Hodgkin lymphoma; LMP, latent membrane protein; NHL, non-Hodgkin lymphoma; NK, natural killer; SFC, spot-forming cell; UPN, unique patient number.
SFCs per 105: 0-24 → 0; 25-49 → +1; 50-99 → +2; 100-499 → +3; and > 500 → +4.
CD4+ CTL line.
Table A3.
LMP1 and LMP2 Specificity of CTL Lines Generated for LMP1/2-Specific CTL Protocol
| UPN | HLA Type | Diagnosis | Strength of Response in ELISPOT Assay |
No. of Epitopes Recognized by CTL Line | |
|---|---|---|---|---|---|
| LMP1 Specific | LMP2 Specific | ||||
| Patients with active disease | |||||
| 2266 | A3,26/B7,38 | HL | 0 | +3 | 1* |
| 2457 | A2,23/B44,51 | HL | 0 | 0 | None identified |
| 1351 | A24/B7;52 | HL | +2 | +4 | 3 |
| 1371 | A26,68/B15(62),49 | HL, immune suppressed | 0 | 0 | 1* |
| 2336 | A2,31/B35,50 | DLBCL/PTLD | +2 | +4 | 4 |
| 1356 | A2,24/B51 | DLBCL | 0 | +3 | 3 |
| 1990 | A2,30/B27,38 | DLBCL (Richter's transformation) | +1 | +1 | 2 |
| 1372 | A11/B46;51 | NK/T-cell NHL | 0 | +3 | 1 |
| 1811 | A2,3/B7,39 | NK/T-cell NHL | +3 | +3 | 5* |
| 1409 | A23,31/B38,52 | NK/T-cell NHL | 0 | +4 | 1 |
| 1656 | A2,68/B15,51 | NK/T-cell NHL | +1 | +3 | 2 |
| 2051 | A24,29/B40,44 | T-cell CAEBV | 0 | 0 | None identified |
| 1545 | A3,31/B35 | LYG | +3 | 0 | 2 |
| Patients treated as adjuvant therapy | |||||
| 2313 | A3/B7 | HL | 0 | 0 | None identified |
| 2433 | A3,26/B38,47 | HL | 0 | 0 | None identified |
| 1369 | A2,29/B7,15(62) | HL | +3 | +4 | 5 |
| 1420 | A1,24/B37,51 | HL | +2 | +2 | 2 |
| 1595 | A2,31/B15,27 | HL | +1 | 0 | 2 |
| 1984 | A1,68/B15(72),57 | HL and melanoma | +1 | 0 | 1 |
| 1370 | A29,68/B15(71),49 | HL | 0 | 0 | None identified |
| 1842 | A1,33/B14(65),37 | HL | +1 | +1 | 3 |
| 1884 | A2,32/B44,51 | HL | 0 | +1 | 2 |
| 1905 | A1,31/B15(62),51 | HL | 0 | 0 | None identified |
| 2368 | A30,33/B15(71),18 | HL | 0 | 0 | None identified |
| 2493 | A2,3/B42,45 | HL and NPC | 0 | 0 | None identified |
| 2056 | A1,26/B35,38 | DLBCL | +1 | +1 | 1 |
| 1888 | A1,29/B8,44 | DLBCL | +3 | +2 | 2 |
| 2053 | A24,32/B7,27 | NK/T-cell NHL | +3 | +4 | 3 |
| 2135 | A24,31/B40(61),44 | NK/T-cell NHL | +3 | +4 | 2 |
| 1806 | A3,24/B18,35 | NK/T-cell NHL | 0 | 0 | None identified |
| 1455 | A2,24/B39 | NK/T-cell NHL | +3 | +4 | 4 |
| 1511 | A2,11/B8,14 | LYG, immune suppressed | 0 | 0 | None identified |
| 2095 | A2/B40(60),58 | PTLD, immune suppressed | +4 | +3 | 2 |
Abbreviations: CAEBV, chronic active Epstein-Barr virus infection; CTL, cytotoxic T lymphocyte; DLBCL, diffuse large B-cell lymphoma; ELISPOT, enzyme-linked immunospot; HL, Hodgkin lymphoma; LMP, latent membrane protein; LYG, lymphoid granulomatosis; NHL, non-Hodgkin lymphoma; NK, natural killer; NPC, nasopharyngeal carcinoma; PTLD,post-transplantation lymphoproliferative disease; UPN, unique patient number.
CD4+ CTL line.
Footnotes
See accompanying article on page 830 and Listen to the podcast by Dr Timmerman at www.jco.org/podcasts
Supported in part by National Institutes of Health (NIH) Grants No. RO1 CA74126 (National Cancer Institute), P50CA126752, and PO1 CA94237; Specialized Center of Research Award from Leukemia Lymphoma Society; Dan L. Duncan Chair (H.E.H.); Fayez Sarofim Chair (M.K.B.); Production Assistance for Cellular Therapies (PACT) program (National Heart, Lung, and Blood Institute Contract No. HHSN268201000007C); Clinical Research Center at Texas Children's Hospital; Methodist Hospital; Dan L. Duncan Institute for Clinical and Translational Research at Baylor College of Medicine; and shared resources from Dan L. Duncan Cancer Center Support Grant No. P30CA125123. Latent membrane protein 2 vector provided by grant from National Gene Vector Laboratories (NIH National Center for Research Resources Grant No. U42 RR16578).
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00671164.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Catherine M. Bollard, Cellmedica (U); Daniel Lee, Cellmedica (C); Helen E. Heslop, Cellmedica (U); Cliona M. Rooney, Cellmedica (C) Stock Ownership: None Honoraria: Catherine M. Bollard, Cellmedica; Helen E. Heslop, Cellmedica Research Funding: None Expert Testimony: None Patents: Helen E. Heslop, licensing Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Catherine M. Bollard, Stephen Gottschalk, Malcolm K. Brenner, Helen E. Heslop, Cliona M. Rooney
Provision of study materials or patients: George Carrum, Carlos Ramos, Luis Fayad, Elizabeth J. Shpall, Barbara Pro
Collection and assembly of data: Catherine M. Bollard, Vicky Torrano, Oumar Diouf, Stephanie Ku, Yasmin Hazrat, George Carrum, Carlos Ramos, Luis Fayad, Elizabeth J. Shpall, Barbara Pro, Hao Liu, Meng-fen Wu, Andrea M. Sheehan, Youli Zu, Adrian P. Gee, Helen E. Heslop, Cliona M. Rooney
Data analysis and interpretation: Catherine M. Bollard, Stephanie Ku, Yasmin Hazrat, Hao Liu, Meng-fen Wu, Daniel Lee, Malcolm K. Brenner, Helen E. Heslop, Cliona M. Rooney
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Riddell SR, Watanabe KS, Goodrich JM, et al. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 2.Peggs KS, Verfuerth S, Chow C, et al. Adoptive cellular therapy for cytomegalovirus following allogeneic stem cell transplantation: Toxicity and efficacy. Blood. 2005:104. doi: 10.1016/j.bcmd.2007.07.003. (abstr 191) [DOI] [PubMed] [Google Scholar]
- 3.Heslop HE, Brenner MK, Rooney CM. Donor T cells to treat EBV-associated lymphoma. N Engl J Med. 1994;331:679–680. doi: 10.1056/NEJM199409083311017. [DOI] [PubMed] [Google Scholar]
- 4.Leen AM, Myers GD, Sili U, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12:1160–1166. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- 5.Doubrovina E, Oflaz-Sozmen B, Prockop SE, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119:2644–2656. doi: 10.1182/blood-2011-08-371971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heslop HE, Slobod KS, Pule MA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Küppers R, Engert A, Hansmann ML. Hodgkin lymphoma. J Clin Invest. 2012;122:3439–3447. doi: 10.1172/JCI61245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grogg KL, Miller RF, Dogan A. HIV infection and lymphoma. J Clin Pathol. 2007;60:1365–1372. doi: 10.1136/jcp.2007.051953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox CP, Haigh TA, Taylor GS, et al. A novel latent membrane 2 transcript expressed in Epstein-Barr virus-positive NK- and T-cell lymphoproliferative disease encodes a target for cellular immunotherapy. Blood. 2010;116:3695–3704. doi: 10.1182/blood-2010-06-292268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 11.Tierney RJ, Steven N, Young LS, et al. Epstein-Barr virus latency in blood mononuclear cells: Analysis of viral gene transcription during primary infection and in the carrier state. J Virol. 1994;68:7374–7385. doi: 10.1128/jvi.68.11.7374-7385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roskrow MA, Rooney CM, Heslop HE, et al. Administration of neomycin resistance gene marked EBV specific cytotoxic T-lymphocytes to patients with relapsed EBV-positive Hodgkin disease. Hum Gene Ther. 1998;9:1237–1250. doi: 10.1089/hum.1998.9.8-1237. [DOI] [PubMed] [Google Scholar]
- 13.Bollard CM, Aguilar L, Straathof KC, et al. Cytotoxic T lymphocyte therapy for Epstein-Barr virus+ Hodgkin's disease. J Exp Med. 2004;200:1623–1633. doi: 10.1084/jem.20040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bollard CM, Straathof KC, Huls MH, et al. The generation and characterization of LMP2-specific CTLs for use as adoptive transfer from patients with relapsed EBV-positive Hodgkin disease. J Immunother. 2004;27:317–327. doi: 10.1097/00002371-200407000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Chia WK, Wang WW, Teo M, et al. A phase II study evaluating the safety and efficacy of an adenovirus-DeltaLMP1-LMP2 transduced dendritic cell vaccine in patients with advanced metastatic nasopharyngeal carcinoma. Ann Oncol. 2012;23:997–1005. doi: 10.1093/annonc/mdr341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottschalk S, Ng CY, Perez M, et al. An Epstein-Barr virus deletion mutant associated with fatal lymphoproliferative disease unresponsive to therapy with virus-specific CTLs. Blood. 2001;97:835–843. doi: 10.1182/blood.v97.4.835. [DOI] [PubMed] [Google Scholar]
- 17.Bollard CM, Gottschalk S, Leen AM, et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood. 2007;110:2838–2845. doi: 10.1182/blood-2007-05-091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 19.Cheson BD. New response criteria for lymphomas in clinical trials. Ann Oncol. 2008;19(suppl 4):iv35–iv38. doi: 10.1093/annonc/mdn191. [DOI] [PubMed] [Google Scholar]
- 20.Bollard CM, Gottschalk S, Helen Huls M, et al. Good manufacturing practice-grade cytotoxic T lymphocytes specific for latent membrane proteins (LMP)-1 and LMP2 for patients with Epstein-Barr virus-associated lymphoma. Cytotherapy. 2011;13:518–522. doi: 10.3109/14653249.2011.561983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottschalk S, Edwards OL, Sili U, et al. Generating CTLs against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive immunotherapy of EBV-associated malignancies. Blood. 2003;101:1905–1912. doi: 10.1182/blood-2002-05-1514. [DOI] [PubMed] [Google Scholar]
- 22.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 23.Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol. 2002;2:982–987. doi: 10.1038/nri959. [DOI] [PubMed] [Google Scholar]
- 24.Rooney CM, Roskrow MA, Suzuki N, et al. Treatment of relapsed Hodgkin's disease using EBV-specific cytotoxic T cells. Ann Oncol. 1998;9(suppl 5):S129–S132. doi: 10.1093/annonc/9.suppl_5.s129. [DOI] [PubMed] [Google Scholar]
- 25.Meij P, Leen A, Rickinson AB, et al. Identification and prevalence of CD8(+) T-cell responses directed against Epstein-Barr virus-encoded latent membrane protein 1 and latent membrane protein 2. Int J Cancer. 2002;99:93–99. doi: 10.1002/ijc.10309. [DOI] [PubMed] [Google Scholar]
- 26.Straathof KC, Leen AM, Buza EL, et al. Characterization of latent membrane protein 2 specificity in CTL lines from patients with EBV-positive nasopharyngeal carcinoma and lymphoma. J Immunol. 2005;175:4137–4147. doi: 10.4049/jimmunol.175.6.4137. [DOI] [PubMed] [Google Scholar]
- 27.Miller G, Lipman M. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc Natl Acad Sci U S A. 1973;70:190–194. doi: 10.1073/pnas.70.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kern F, Faulhaber N, Frömmel C, et al. Analysis of CD8 T cell reactivity to cytomegalovirus using protein- spanning pools of overlapping pentadecapeptides. Eur J Immunol. 2000;30:1676–1682. doi: 10.1002/1521-4141(200006)30:6<1676::AID-IMMU1676>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 29.Chang ST, Ghosh D, Kirschner DE, et al. Peptide length-based prediction of peptide-MHC class II binding. Bioinformatics. 2006;22:2761–2767. doi: 10.1093/bioinformatics/btl479. [DOI] [PubMed] [Google Scholar]
- 30.Gray RJ. A class of K-sample tests for comparing the culmulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 31.Reichert JM, Rosensweig CJ, Faden LB, et al. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 32.Gattinoni L, Powell DJ, Jr, Rosenberg SA, et al. Adoptive immunotherapy for cancer: Building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363:1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 35.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Icheva V, Kayser S, Wolff D, et al. Adoptive transfer of EBNA1-specific T cells as a treatment of Epstein-Barr virus reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J Clin Oncol. 2013;31:39–48. doi: 10.1200/JCO.2011.39.8495. [DOI] [PubMed] [Google Scholar]
- 39.Straathof KC, Bollard CM, Popat U, et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus–specific T lymphocytes. Blood. 2005;105:1898–1904. doi: 10.1182/blood-2004-07-2975. [DOI] [PubMed] [Google Scholar]
- 40.Muranski P, Boni A, Wrzesinski C, et al. Increased intensity lymphodepletion and adoptive immunotherapy: How far can we go? Nat Clin Pract Oncol. 2006;3:668–681. doi: 10.1038/ncponc0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger C, Jensen MC, Lansdorp PM, et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Disis ML, Gooley TA, Rinn K, et al. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol. 2002;20:2624–2632. doi: 10.1200/JCO.2002.06.171. [DOI] [PubMed] [Google Scholar]
- 45.Butterfield LH, Ribas A, Dissette VB, et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin Cancer Res. 2003;9:998–1008. [PubMed] [Google Scholar]
- 46.Wierecky J, Müller MR, Wirths S, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 2006;66:5910–5918. doi: 10.1158/0008-5472.CAN-05-3905. [DOI] [PubMed] [Google Scholar]
- 47.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cruz CR, Gerdemann U, Leen AM, et al. Improving T-cell therapy for relapsed EBV-negative Hodgkin lymphoma by targeting upregulated MAGE-A4. Clin Cancer Res. 2011;17:7058–7066. doi: 10.1158/1078-0432.CCR-11-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Craddock C. Epigenetic manipulation of the immune response: A novel treatment strategy in hematologic malignancies. Cytotherapy. 2011;13:516–517. doi: 10.3109/14653249.2011.561652. [DOI] [PubMed] [Google Scholar]