

Abstract
We present a microfluidic device that enables the quantitative determination of intracellular biomolecules in multiple single cells in parallel. For this purpose, the cells are passively trapped in the middle of a microchamber. Upon activation of the control layer, the cell is isolated from the surrounding volume in a small chamber. The surrounding volume can then be exchanged without affecting the isolated cell. However, upon short opening and closing of the chamber, the solution in the chamber can be replaced within a few hundred milliseconds. Due to the reversibility of the chambers, the cells can be exposed to different solutions sequentially in a highly controllable fashion, e.g. for incubation, washing, and finally, cell lysis. The tightly sealed microchambers enable the retention of the lysate, minimize and control the dilution after cell lysis. Since lysis and analysis occur at the same location, high sensitivity is retained because no further dilution or loss of the analytes occurs during transport. The microchamber design therefore enables the reliable and reproducible analysis of very small copy numbers of intracellular molecules (attomoles, zeptomoles) released from individual cells. Furthermore, many microchambers can be arranged in an array format, allowing the analysis of many cells at once, given that suitable optical instruments are used for monitoring. We have already used the platform for proof-of-concept studies to analyze intracellular proteins, enzymes, cofactors and second messengers in either relative or absolute quantifiable manner.
Keywords: Immunology, Issue 80, Microfluidics, proteomics, systems biology, single-cell analysis, Immunoassays, Lab on a chip, chemical analysis
Introduction
Many studies in the past have revealed cell-to-cell differences within a large cell population1-3, in particular signaling processes4, or the amounts of intracellular biomolecules such as proteins5,6, metabolites, and cofactors7,8. These heterogeneities are considered to be fundamentally important for cell adaptation and evolution9, but also play a key role in the emergence and treatment of diseases such as cancer10-13. Therefore, studies on the single-cell level are of high interest in biological and pharmacological research, particularly if these studies reveal the different cell responses after treatment with bioactive chemical substances.
In recent years, many analytical platforms have been developed that facilitate the analysis of single living cells or the chemical composition of the cell content. While fluorescent activated cell sorting (FACS) is the gold standard for very high-throughput analysis of single living cells, the method cannot be employed for the quantification of intracellular or secreted compounds. The emergence of microfluidic platforms has promised novel analytical strategies for positioning, treatment and observation of single cells. A milestone in microfluidics was reached with the integration of flexible PDMS valves realized by Quake and coworkers14,15. These valves are useful since they can isolate regions on chip, e.g. separate two cultures 16. Furthermore, they are especially applicable for single cell analysis and therefore help to reduce analyte dilution problems. The power of this approach for single-cell analysis has been recently demonstrated by Hansen and coworkers, who analyzed the gene expression from hundreds of single cells in parallel17.
When targeting proteins and metabolites, the analysis is very difficult due to the lack of suitable amplification methods, the large number of different compounds present, and their variations in chemical nature. Furthermore, most intracellular biomolecules are expected to be present in low copy numbers in the order of a few ten thousands18, hence the analytical method used must have a high sensitivity. More powerful assays such as immunoassays and enzyme-linked immunoassays (ELISA) are difficult to integrate into microfluidic devices since they require several washing and incubation steps as well as surface immobilization.
Due to these challenges, it is not surprising that only a few examples have been reported where proteins or metabolites were quantified on the single-cell level. For example, studies on the secretion of fluorescent compounds have been reported19,20. Recently, the implementation with ELISA was presented for the analysis of secreted (nonfluorescent) proteins from a cell culture (THP-1 cells)21 and single (immune) cells10. Targeting intracellular proteins, Shi et al. developed a microfluidic device that facilitated the identification of intracellular proteins for the analysis of signaling pathways in tumor cells by means of an immunoassay11. However, only relative amounts of proteins were determined and no enzymatic amplification was used to increase the signal for low abundance proteins.
Recently, we were able to combine a single-cell trapping microdevice with fluorescence assays8 and immunoassays22 (Figure 1). Cells are passively trapped in microsized hurdle structures, which allow supply and (rapid) exchange of medium and other chemical agents without any movement of the cells. A ring-shaped valve around each trap enables isolation of the cell in a very small volume (“the microchamber”). This valve is actuated immediately after introducing a cell-lysing (hypoosmolar) buffer, hence preventing intracellular molecules or secreted molecules to diffuse away. Most importantly, due to the small size of the volume (625 pl) large dilution of the molecules is avoided. Furthermore, since lysis and analysis are performed in at the same position in the chip, there is no loss of analytes due to transportation. The chip design described here comprises 8 alternating rows of either 7 or 8 microchambers, totaling 60 microchambers. The chambers are actuated in rows, so that cross-contamination along a line is precluded.
The platform can be used in combination with fluorescence assays as well as immunological assays (Figure 1d). For the latter, we established protocols for immobilization of the antibodies, which are compatible with the chip production and assembly process. Hence the platform opens the way for sensitive, reliable and quantifiable assays at the single cell level. Up to now, we have used the device for the analysis of intracellular and secreted enzymes (relative quantification by enzymatic assays), intracellular cofactors, proteins and small molecules (absolute quantification by endpoint assays or ELISA). In the following, we describe the process of chip fabrication by means of multilayer soft lithography and the protocols for patterning of the antibodies by means of microcontact printing and surface chemistry. Additionally, some examples of chip use and operations are given.
Protocol
1. SU-8 Master Fabrication
Prepare both master molds for the channels (fluidic and control, for schematics and dimensions see Figure 2) with the following protocol but with different mask patterns. The process is shown in Figure 3a.
- Start by heating a 4 inch silicon wafer for 10 min at 180 °C. Load the dehydrated wafer on a spin-coater and use the following protocol for spin-coating SU-8 2015:
- Spin wafer at 100 rpm for 20 sec (open lid of spin-coater, dispense SU-8 during this step, close lid after dispensing).
- Spin wafer at 500 rpm for 10 sec (this will spread the resist over the whole wafer).
- Spin wafer at 1,750 rpm for 30 sec (defines the height of the resist to 20 µm).
Remove resist from the edge of the wafer with acetone (use a swab) as this will otherwise stick to the hotplate in the next step. Transfer the wafer to a hotplate at 95 °C and heat for 4 min. After the softbake, expose the resist through a photomask taped to sodalime glass in a mask aligner with UV light (150 mJ/cm2, measured at 365 nm).
After the exposure, heat the wafer again at 95 °C for 5 min on a hotplate. Cool down the wafer to RT and then immerse it into a bath of SU-8 developer for 4 min. Shake gently to remove unexposed SU-8. Wash the wafer with clean-room grade isopropanol and blow dry with nitrogen. Check under a microscope if the development was successful (especially the cell traps). If the cell traps are completely developed, the reflection of the silicon wafer should be visible. If necessary, develop the wafer again for a few minutes.
Bake the wafer in an oven for 2 hr at 180 °C to remove all residual solvent. Check the height of the channels with a step profiler. If the height differs from the desired height, repeat this protocol starting at step 1.1 with a new wafer and change the spin-speed (step 1.1.3). Finalize the fabrication of the master mold by silanizing the wafer with 1H,1H,2H,2H-Perfluorodecyl-dimethylchloro-silane in a desiccator overnight.
2. Master Fabrication for Microcontact Printing
- In order to prepare master molds for microcontact printing, dehydrate a silicon wafer for 10 min at 180 °C. Spin-coat 1 ml HDMS onto this wafer at 7,500 rpm for 30 sec (the resist adheres better to a silanized surface). Spin-coat 2 ml of AZ1518 positive resist with this protocol:
- Static dispense of the resist on to the wafer.
- Spin wafer for 5 sec at 500 rpm.
- Spin wafer for 60 sec at 4000 rpm.
After a 50 sec softbake at 100 °C on a hotplate, expose the resist to UV-light (21 mJ/cm2 at 365 nm) through a photomask and on a mask aligner. Develop the resist in a bath of AZ 726 for 75 sec. Rinse the developed wafer with DI-water and blow dry with nitrogen. Remove residual solvent by heating the wafer at 115 °C for 50 sec. Silanize the wafer under vacuum overnight.
3. Fabrication of the Microfluidic Chip
A two-layer device design is used for these experiments. The two layers are prepared separately and are bonded together, before the chip is finally bonded onto a glass slide (Figure 3b). This step describes the production of the PDMS layers and the PDMS stamp for microcontact printing.
Prepare a 10:1 mixture of PDMS and curing agent. Prepare approximately 80 g PDMS in total (using our designs, this will result in 10 chips). Mix both parts vigorously and degas the PDMS until the mixture is bubble free (~30 min).
Place the wafer with the control layer inside a Petri dish and tape it onto the bottom. Next, pour ~50 g of PDMS on top and put the wafer for at least 2 hr in an oven at 80 °C to assure complete curing of the PDMS. Repeat the same procedure for the microcontact printing wafer, for this ~20 g PDMS is needed.
Spin-coat PDMS on the wafer with the fluidic layer at a rotational speed of 2,000 rpm to form an approximately 40 µm high PDMS layer. Cure the PDMS in an oven for at least 1 hr at 80 °C.
When both parts are cured, remove the control layer from the wafer and cut into pieces with a razor blade. Punch pressure connection holes by using a 1 mm biopsy puncher. For storage, it is advisable to put a piece of tape over the channels.
To bond the two layers, take the spin-coated wafer and the top part and place them in a plasma cleaner23. After plasma treatment, quickly align both parts under a microscope with large working distance. Add some spare PDMS around the placed top parts. This step is not mandatory, however it facilitates the removal of the PDMS from the wafer. Place the wafer in an oven at 80 °C for at least 1 hr.
Use a scalpel to carefully remove the PDMS from the wafer and to cut the microchips. Punch access holes for the fluidic connections with a 1.5 mm biopsy puncher.
The PDMS chip is now finished and can be stored for months. Optional: If the chip is used with a reservoir, cut the upper part of a 200 µl pipette tip and use some spare, semi-cured PDMS to glue it on top. Put the chip in the oven again at 80 °C for at least 1 hr.
4. Bonding to Glass Slide
To use the device for direct enzymatic assays, the only step required after bonding is the blocking of surfaces. For this protocol, proceed to A. However, to use the microfluidic chip for immunoassays or ELISAs, please refer to protocol B to restrict the binding sites only to the glass slide (i.e. for TIRF microscopy or SPR). If this restriction is not important, refer to A, but use biotinylated conjugates in step 4.3 and continue with step 4.8 in protocol B to create a fully functional surface.
Prior to performing protocol A and B: It is advisable to filter all of the used protein solutions prior to introducing them into the chip. Debris and protein aggregates may otherwise block cell traps, thereby reducing the number of chambers that can be used for analysis. For this purpose, use a nonprotein adsorbing filter unit to filter all solutions before introducing them into the channels.
Protocol A (enzymatic assays)
To finalize the microfluidic device, bond the PDMS parts onto a glass slide. To do this, first clean a glass slide with soap, distilled water and ethanol. Dry the slide using a nitrogen stream. Clean the PDMS part with scotch tape.
Put the PDMS part and the cleaned glass slide into the plasma cleaner for 45 sec at 18 W. To assure a tight bonding, put the chip on a hot plate (100 °C) for 5 min.
After bonding, fill the chip with fluids. An easy way to achieve this is to use centrifugal force. Cut the lower part of 200 µl pipette tip. For the fluidic channels, fill them with the blocking solution (bovine serum albumin (4% w/v) or poly-L-lysine) grafted polyethylene glycol (0.05% w/v) and place the tips into the inlets. Fill the control layer inlets with either water or with an isosmotic solution, e.g. PBS. Place the chip into the centrifuge (800 x g) for 5 min. The channels should be now filled with fluid completely. If not, repeat this step.
Incubate the blocking solution for at least 30 min at room temperature. Wash the solution out of the fluid layer with PBS at a flow rate of 10 µl/min using a syringe pump. The device is now ready for cell experiments.
Protocol B (immunoassays)
For a complete overview of the surface modification from step 4.7 onwards, please refer to Table 1.
Incubate PDMS stamps for microcontact printing with a bBSA/BSA solution (e.g. 1-100). After 30 min, clean the stamps thoroughly with distilled water and dry the stamp under a stream of nitrogen. Quickly place the stamps on a cleaned glass slide (Figure 4).
Expose the glass slide with the stamp together with the top part of the microfluidic chip to oxygen plasma for 45 sec at maximum power (18 W) in a plasma cleaner. After plasma treatment, remove the stamp and align the printed surface underneath the microchambers of the device. Place the chip for 30 min on a hot plate at 50 °C.
To block the remaining surface, introduce sterile-filtered BSA solution (4% (w/v) in PBS) into the chip with a centrifuge (see step 4.2). Incubate for 1 hr and wash the solution out of the fluid layer with PBS (see step 4.3). The chip can then be used directly or stored for up to 2 weeks at 4 °C in a humid box.
For a fully functional binding surface, introduce avidin (0.025% (w/v) in PBS) into the reservoir. Use a flow rate of 5 µl/min for 30 min to withdraw the avidin solution through the fluid channels. Afterwards, flush PBS for 10 min. Rinse the reservoir thoroughly.
Afterwards, add Protein G (0.0025% (w/v) in PBS) and flush for another 30 min. Wash with PBS for another 10 min afterwards. Next, add the antibody of interest (0.0001% (w/v) in PBS) for 10 min. After 10 min, stop the flow for 15 min to allow binding. Next, wash with PBS for 10 min.
5. Cell Experiments
The protocol is written in general terms because of the variety of possible assays. As an example the reagents needed for the G6PDH toxicity assay are given. The initial cell trapping efficiency of the device is around 2.5%, i.e. 5 out of 200 cells will be trapped. The final occupancy of the cell traps strongly depends on the used cell line, the protocol to suspend the cell line and the time the cells are flushed through the channel. For cells naturally growing in suspension (such as U937), single cells can be found in about 75% of the chambers after a few minutes. The other chambers are either not filled, or occupied by two or more cells. In general, the single-cell occupancy is smaller for adherent cell lines. When using the rather mild suspending protocol presented here, the percentage decreases to about 30% (HEK, MLT cells). The single-cell occupancy can be improved by trypsin treatment of the cells, but this may also alter the results of the experiments.
Depending on the target molecule, adsorption or absorption to PDMS can influence the results. Adsorption can be reduced by blocking the surfaces with BSA or PLL-g-PEG. Absorption is unlikely for most hydrophilic biomolecules, but it should be checked for (small) lipophilic cell components.
Prepare the cells according to the specific protocol (e.g. drug addition). For suspension cells, add cells directly to the chip. For adhesive cell lines, use enzyme-free dissociation buffer to produce a cell suspension. Filter the cells before introducing them to the chip to reduce the amount of cell clusters.
Load the cells onto the chip using a syringe pump and forward flow, rather than to withdraw the cell suspension. Use a flow rate of 5 µl/min for suspension cell lines and higher flow rates of 10-20 µl/min for adhesive cells since to reduce nonspecific attachment of the cells onto the channel walls.
When the traps are filled, close the chambers. If many cells adhere nonspecifically to the surface, try to wash them away with cell dissociation buffer at a flow rate of 10-30 µl/min.
Protocol A (enzymatic assays)
Withdraw lysis buffer through the chip. For this example, this buffer is a hypoosmolar buffer with added Tween 20 (10 mM Tris-HCl, 10 mM KCl, 1.5 mM MgCl2, 1% (v/v) Tween 20). For lysis, quickly open and close chambers (i.e. 500 msec for flow rates of 10-50 µl/min 7,21). For the G6PDH assay, add 2 mM glucose-6-phosphate, 0.5 mM NADP, 0.5 U/ml diaphorase and 0.3 mM resazurin to the lysis buffer. Start monitoring the kinetic reaction immediately after lysis.
Note: Choose any buffer which is suitable for the assay, however bare in mind that strong detergents (like Triton, SDS) lyse cells immediately and will lead to a loss of analyte during chamber opening.
Protocol B (immunoassays)
For a short description of the ELISA cell experiments (flow rates, chamber status, etc.), please refer to Table 1.
For ELISA, add the required detection reagent (e.g. secondary antibody, labeled antigen) directly to the lysis buffer. The herein added Tween 20 reduces the nonspecific adsorption. Lyse the cells according to step 5.4. Incubate the mixture for 10-15 minutes to allow binding (incubation time depending on the used antigen and antibody).
Antibody enzyme conjugates that adsorbed nonspecifically to the channel walls can lead to a conversion of the detection reagent already inside the reservoir and channels. To reduce issues from this adsorption, flush a permanent inhibitor of the used detection enzyme through the chip during the incubation time (chambers closed, before adding the detection reagent). For HRP, use 20 mM HCl in water to permanently inactivate the nonspecifically adsorbed enzymes.
After incubation time, introduce the detection reagent (e.g. Amplex Red and hydrogen peroxide for HRP) to the chip. Open the chambers shortly again to wash the nonbound secondary antibody away and to add the detection reagent. Start monitoring the kinetic reaction immediately.
Calibration: Immobilize the antibody against the target of interest. Close chambers. Prepare different concentrations of analyte in buffer or in cell lysate off-chip. Apply a concentration to the chip and quickly exchange the volume inside the chambers (500 msec opening time). This opening time is sufficiently short to ensure that no additional accumulation occurs from analyte flushing, and only a specific number of molecules from the volume of the microchamber are detected. From the known concentration of the antigen inside the solution and the volume of the chamber (625 pl), calculate the amount inside the chamber. Afterwards, continue with step 5.5 and introduce the detection moiety.
Representative Results
Our platform is able to analyze a variety of intracellular as well as secreted molecules present in or produced by single cells. Here, we would like to present different example studies to underline the variety of possible assays. We will give an example for a secreted enzyme (Figure 5a) as well as an intracellular enzyme (Figure 5b) and protein (Figures 5c and d). For more examples, such as cofactors or small molecules, please refer to Eyer et al. (2012) and Eyer et al. (2013).
First, we present an assay for a secreted enzyme (Figure 5a). Upon activation with phorbol myristate acetate (PMA), human monocytes induce secretion of lysozyme, an enzyme that induces bacterial cell lysis. For this assay, the cell buffer contains 4-Methylumbelliferyl β-D-N,N′-diacetylchitobioside, a weakly fluorescent substrate for lysozyme. Upon cleavage of the sugar residues by the enzyme, the fluorophore is liberated and can be detected inside the chamber. Here, the advantage of the microchamber is the minimized dilution of the secreted enzyme, allowing the detection of low enzyme concentrations within a short time period. In Figure 5a, several example curves are shown representing the enzymatic turnover of lysozyme secreted from single stimulated U937 cells. The U937 cell line derives from a patient with histiocytic lymphoma, and can be induced to terminal monocytic differentiation. The cell line produces TNFα, lysozyme and β2-microglobulin after stimulation with PMA. No turnover is detectable in open chambers or cell-free chambers (dashed line).
Figure 5b shows the relative quantification of an intracellular enzyme, here glucose 6-phosphate dehydrogenase (G6PDH). G6PDH is a housekeeping protein and cells from the same tissue origin should not vary largely in their G6PDH concentration24. Here, U937 cells were trapped in the chamber, and we supplied a fluorescence assay for the detection of G6PDH together with the lysis buffer. It consists of glucose-6-phosphate, NADPH, the enzyme diaphorase and the substrate resazurin, which is converted to fluorescent resorufin when G6PDH is present. The rate of increase in fluorescence over time corresponds to the amount of G6PDH. The microchip further facilitates studies on the effect of toxic compounds, e.g. by incubation of the cells with a toxin (here camptothecin) for a defined time (60 min). After this treatment, the released enzyme was flushed away and the cells were lysed, and the level of G6PDH was measured. Indeed, a significant loss of intracellular enzyme content was visible due to a slower increase of fluorescence (red lines and columns in Figure 5b). Details of this study can be found in Eyer et al. 8
Furthermore, we show the results from an immunoassay for the analysis of intracellular GFP amounts. For induced expression, HEK293 cells where transfected with the T-REx expression system. In this system, GFP expression can be induced by the addition of tetracycline. After 8 hr of incubation, the cells were suspended, flushed onto the chip, trapped, and subsequently lysed. Capturing anti-GFP antibodies were immobilized on the glass surface following the protocol presented here to bind the released GFP from the cell. Figure 5c shows the binding kinetics of GFP to the antibody. In this example, GFP could be directly measured by total internal fluorescence microscopy due to the fluorescence of the protein. We want to emphasize that nonfluorescent proteins can also be detected by using a sandwich ELISA or similar formats. For this, the required assay components can be introduced after the washing steps. The advantage of ELISA is the signal amplification due to the enzyme, yielding a fluorescent product that is accumulated inside the microchamber.
Finally, we would like to present a single-cell sandwich ELISA with GAPDH as the target protein. We determined GAPDH in U937 suspension cells as well as adherent HEK293 cells (Figure 5d). For this, we immobilized anti-GAPDH antibody (capture antibody). After cell lysis, the second enzyme-labeled antibody (detection antibody) was introduced. The chambers were opened and due to the flow, unbound detection antibodies were washed away and Amplex Red was introduced to the chamber. The conversion of Amplex Red to fluorescent resorufin by HRP (detection antibody) was monitored over time in all 60 microchambers at the same time. The slope from the linear part of the reaction was determined. This slope is directly correlated to the concentration of the enzyme and hence, to the concentration of analyte. The results from single U937 cells showed an average GAPDH amount of 2.6 attomol, with minimal and maximal values of 2.0 and 3.2 attomol, respectively. The data analysis of 46 HEK cells revealed that those cells contained in average 2.5 attomol GAPDH (minimal 0.9 attomol; maximal 4.1 attomol).
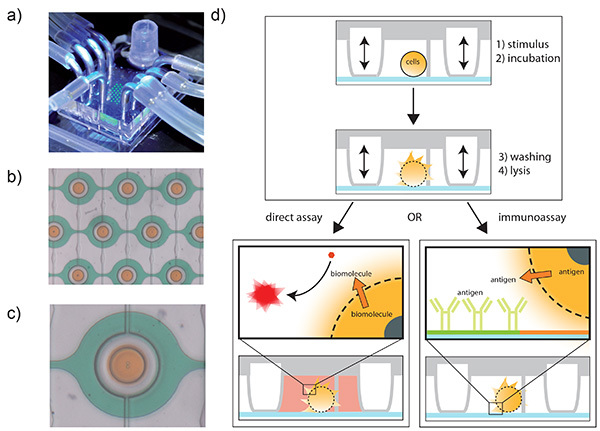 Figure 1. The microdevice and schematics of the different assays presented. a) The assembled microdevice. Here, the chambers are filled with fluorescein for visualization (green fluorescence). Also visible is the reservoir, the connections for the control layer (pressure, 2x4 connectors in the back left and front right) and the connection to the syringe pump (front). b) Zoom in on the microchamber area. Many of these chambers can be arranged in an array. For visualization, orange food dye was isolated inside the chambers, whereas green food dye is flushed through the channels. The pressure control layer is filled with water. c) Zoom on a single microchamber. d) Schematic of the device operation. Within a microchamber (side view), a single cell is trapped and isolated. The cell can be stimulated, incubated, washed and finally lysed within the chip. The cellular content can be analyzed directly with assays for enzymes or coenzymes where a fluorescent moiety is generated upon reaction (left). For other proteins, the surface can be modified with antibodies that bind specifically to the target protein (right). Since the antigen is bound to the surface, the lysate can be washed away and detection molecules such as secondary antibodies can be added to quantify the target protein. Click here to view larger image.
Figure 1. The microdevice and schematics of the different assays presented. a) The assembled microdevice. Here, the chambers are filled with fluorescein for visualization (green fluorescence). Also visible is the reservoir, the connections for the control layer (pressure, 2x4 connectors in the back left and front right) and the connection to the syringe pump (front). b) Zoom in on the microchamber area. Many of these chambers can be arranged in an array. For visualization, orange food dye was isolated inside the chambers, whereas green food dye is flushed through the channels. The pressure control layer is filled with water. c) Zoom on a single microchamber. d) Schematic of the device operation. Within a microchamber (side view), a single cell is trapped and isolated. The cell can be stimulated, incubated, washed and finally lysed within the chip. The cellular content can be analyzed directly with assays for enzymes or coenzymes where a fluorescent moiety is generated upon reaction (left). For other proteins, the surface can be modified with antibodies that bind specifically to the target protein (right). Since the antigen is bound to the surface, the lysate can be washed away and detection molecules such as secondary antibodies can be added to quantify the target protein. Click here to view larger image.
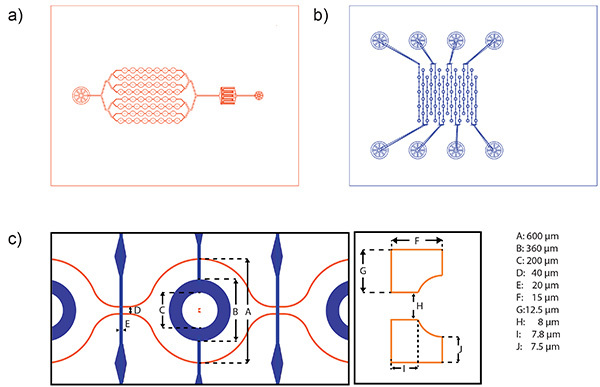 Figure 2. Schematic of channel systems and dimensions of the microchamber. a) Mask design of the fluidic layer. The fluidic layer consists of (from left to right) an outlet, the chamber region, a filter to retain particles and cell clusters and an inlet. A 4 in wafer can harbor the structures for the production of 10 chips. b) Mask design of the corresponding control layer. Due to the needed space for alignment, a 4 in wafer contains the structures for the production of 5 chips. c) Schematic of aligned layers (left) and enlargement showing a cell trap (right), both with length scales. Click here to view larger image.
Figure 2. Schematic of channel systems and dimensions of the microchamber. a) Mask design of the fluidic layer. The fluidic layer consists of (from left to right) an outlet, the chamber region, a filter to retain particles and cell clusters and an inlet. A 4 in wafer can harbor the structures for the production of 10 chips. b) Mask design of the corresponding control layer. Due to the needed space for alignment, a 4 in wafer contains the structures for the production of 5 chips. c) Schematic of aligned layers (left) and enlargement showing a cell trap (right), both with length scales. Click here to view larger image.
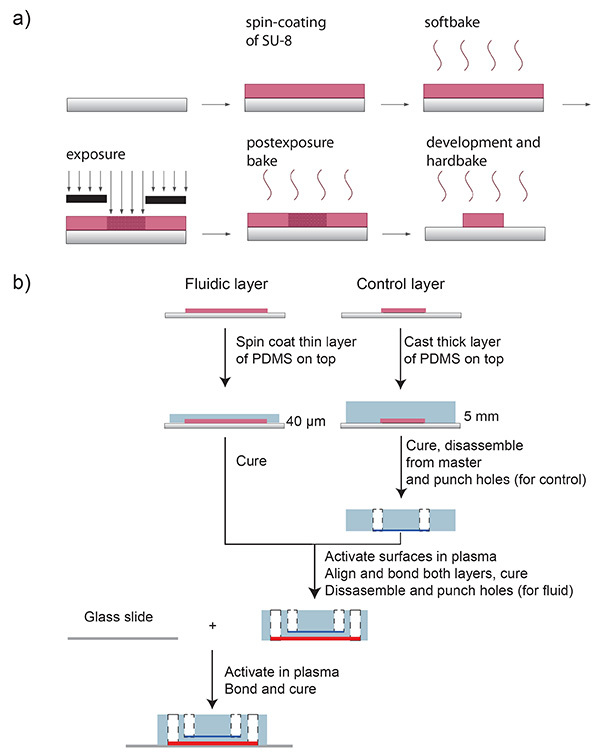 Figure 3. Schematic of of SU-8 processing and PDMS production. a) Schematic of the SU-8 processing protocol. A silicon wafer is spin-coated with SU-8 and soft-baked at 95 °C for 4 min. The desired microfluidic channel design is transferred onto the SU-8 using a photomask and UV-light exposure. After a post-exposure bake (95 °C, 5 min), SU-8 is developed and hard-baked at 180 °C for 2 hr. b) Schematic of the microfluidic chip fabrication. A mixture of PDMS oligomer and curing agent is spin-coated onto the master form for the fluidic channels, and poured onto the master form for the control layer. The channel negatives are depicted in pink. Both layers are cured in an oven. The control layer chip part is disassembled from the wafer (control channel in blue) and holes for the pressure connections are punched (white). Both parts –control and fluid layer- are placed in oxygen plasma, and afterwards, they are aligned and bonded. The whole chip is then disassembled from the fluidic master form (fluid channel in red) and fluidic access holes are punched (white). Finally, the chip and a glass slide are bonded after surface activation in oxygen plasma. Click here to view larger image.
Figure 3. Schematic of of SU-8 processing and PDMS production. a) Schematic of the SU-8 processing protocol. A silicon wafer is spin-coated with SU-8 and soft-baked at 95 °C for 4 min. The desired microfluidic channel design is transferred onto the SU-8 using a photomask and UV-light exposure. After a post-exposure bake (95 °C, 5 min), SU-8 is developed and hard-baked at 180 °C for 2 hr. b) Schematic of the microfluidic chip fabrication. A mixture of PDMS oligomer and curing agent is spin-coated onto the master form for the fluidic channels, and poured onto the master form for the control layer. The channel negatives are depicted in pink. Both layers are cured in an oven. The control layer chip part is disassembled from the wafer (control channel in blue) and holes for the pressure connections are punched (white). Both parts –control and fluid layer- are placed in oxygen plasma, and afterwards, they are aligned and bonded. The whole chip is then disassembled from the fluidic master form (fluid channel in red) and fluidic access holes are punched (white). Finally, the chip and a glass slide are bonded after surface activation in oxygen plasma. Click here to view larger image.
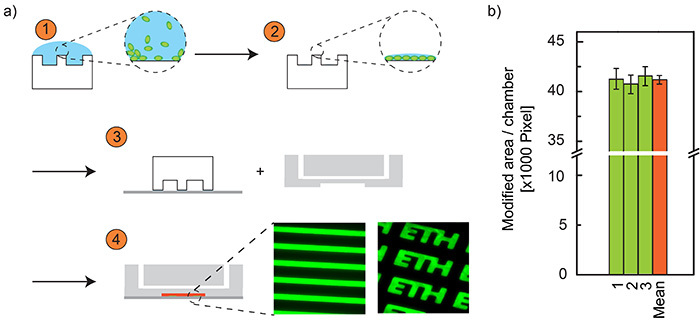 Figure 4. Schematic of the microcontact printing. a) Master fabrication for soft lithography. (1) A PDMS stamp is incubated with protein (bBSA) solution. (2) The solution is removed, the stamp is washed and a monolayer of protein remains on the stamp. (3) The stamp is placed on a glass slide, thereby the proteins are transferred to the glass surface. During this time, the microchip together with the stamp on the glass are exposed to an oxygen plasma. (4) The stamp is removed and the microchip is bonded to the glass slide. For visualization, two example structures are shown, printed with fluorescent BSA-fluorescein conjugate. b) Microcontact printed area per chamber. Since printing and chambers are not aligned, we evaluated the variations on binding spots (printed area) on three different chips (numbers 1-3) and all 60 microchambers. As a result, chamber-to-chamber variability is relatively small (<2.5%) and chip-to-chip variability was negligible (<1%), demonstrating that there is indeed no need to align the printed structures to the chambers. Click here to view larger image.
Figure 4. Schematic of the microcontact printing. a) Master fabrication for soft lithography. (1) A PDMS stamp is incubated with protein (bBSA) solution. (2) The solution is removed, the stamp is washed and a monolayer of protein remains on the stamp. (3) The stamp is placed on a glass slide, thereby the proteins are transferred to the glass surface. During this time, the microchip together with the stamp on the glass are exposed to an oxygen plasma. (4) The stamp is removed and the microchip is bonded to the glass slide. For visualization, two example structures are shown, printed with fluorescent BSA-fluorescein conjugate. b) Microcontact printed area per chamber. Since printing and chambers are not aligned, we evaluated the variations on binding spots (printed area) on three different chips (numbers 1-3) and all 60 microchambers. As a result, chamber-to-chamber variability is relatively small (<2.5%) and chip-to-chip variability was negligible (<1%), demonstrating that there is indeed no need to align the printed structures to the chambers. Click here to view larger image.
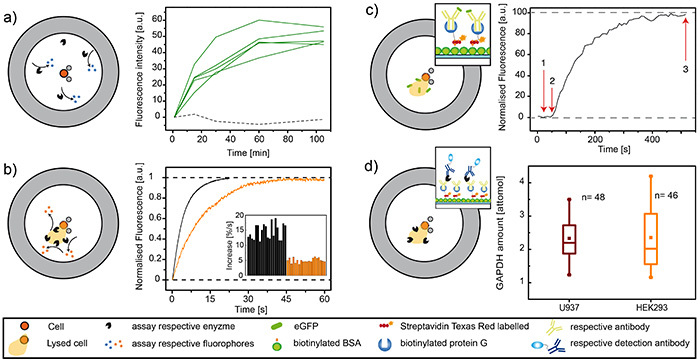 Figure 5. Results from G6PDH, lysozyme, GFP and GAPDH assays. Representative results. a) Enzyme secretion from single cells. Left: schematic of the assay for a secreted enzyme, here lysozyme. Lysozyme is secreted by single, PMA-activated U937 cells and accumulates inside the closed microchambers, where it catalyzes the production of fluorescent 4-methylumbelliferone. Right: Data obtained from a secretion experiment. The green curves show the fluorescence intensity over time for chambers occupied by one cell, also shown is a chamber without cells (black, dotted line) for comparison. b) Detection of intracellular enzymes in single cells. Left: Schematic of the assay for the intracellular enzyme G6PDH. Upon cell lysis, intracellular G6PDH is released into the chamber, where the other components for an enzymatic reaction cascade are present. Right: Example curves and representative data. Single U937 cells were analyzed for their enzyme content (black). Upon the toxic influence of camptothecin, which permeabilizes the membrane, two thirds of the enzyme was lost (orange). c) Immunoassay of intracellular proteins of single cells. Left: Schematic of an immunoassay, here for the intracellular protein GFP. Anti-GFP antibodies were immobilized on the surface according protocol described herein. Cells were then lysed on chip and the binding spots were monitored over time using TIRF microscopy. Right: Graph from such an experiment showing the binding kinetics of GFP to the immobilized antibodies. Time point 1 marks the introduction of the lysis buffer. At time point 2, GFP is starting to accumulate on the surface, meaning that cell lysis has occurred. Binding of GFP to the antibodies is completed at time point 3. d) Sandwich ELISA of intracellular proteins of single cells. Left: Schematic of a sandwich ELISA, here for the intracellular protein GADPH. Anti-GAPDH antibodies were immobilized on the surface according protocol described herein. Cells were then lysed on chip and incubated. Liberated GPADH bound to the antibody, the chamber was washed and detection antibody (HRP-coupled) was introduced. In a last step, nonbound detection antibody was washed away, the detection reagent was introduced and the reaction was monitored over time. Right: Quantification of GAPDH inside U937 and HEK293 cells. Click here to view larger image.
Figure 5. Results from G6PDH, lysozyme, GFP and GAPDH assays. Representative results. a) Enzyme secretion from single cells. Left: schematic of the assay for a secreted enzyme, here lysozyme. Lysozyme is secreted by single, PMA-activated U937 cells and accumulates inside the closed microchambers, where it catalyzes the production of fluorescent 4-methylumbelliferone. Right: Data obtained from a secretion experiment. The green curves show the fluorescence intensity over time for chambers occupied by one cell, also shown is a chamber without cells (black, dotted line) for comparison. b) Detection of intracellular enzymes in single cells. Left: Schematic of the assay for the intracellular enzyme G6PDH. Upon cell lysis, intracellular G6PDH is released into the chamber, where the other components for an enzymatic reaction cascade are present. Right: Example curves and representative data. Single U937 cells were analyzed for their enzyme content (black). Upon the toxic influence of camptothecin, which permeabilizes the membrane, two thirds of the enzyme was lost (orange). c) Immunoassay of intracellular proteins of single cells. Left: Schematic of an immunoassay, here for the intracellular protein GFP. Anti-GFP antibodies were immobilized on the surface according protocol described herein. Cells were then lysed on chip and the binding spots were monitored over time using TIRF microscopy. Right: Graph from such an experiment showing the binding kinetics of GFP to the immobilized antibodies. Time point 1 marks the introduction of the lysis buffer. At time point 2, GFP is starting to accumulate on the surface, meaning that cell lysis has occurred. Binding of GFP to the antibodies is completed at time point 3. d) Sandwich ELISA of intracellular proteins of single cells. Left: Schematic of a sandwich ELISA, here for the intracellular protein GADPH. Anti-GAPDH antibodies were immobilized on the surface according protocol described herein. Cells were then lysed on chip and incubated. Liberated GPADH bound to the antibody, the chamber was washed and detection antibody (HRP-coupled) was introduced. In a last step, nonbound detection antibody was washed away, the detection reagent was introduced and the reaction was monitored over time. Right: Quantification of GAPDH inside U937 and HEK293 cells. Click here to view larger image.
Surface modification protocol for ELISA
| Step | Time | Concentration | Flow rate | Flow-free incubation time | Chamber open/closed | ||
| [min] | (w/v) % | [µl/min] | [min] | ||||
| BSA | 30 | 4 | - | Open | |||
| PBS | 10 | 5 | Open | ||||
| Avidin | 30 | 0.025 | 5 | Open | |||
| PBS | 10 | 5 | Open | ||||
| Protein G, biotin | 30 | 0.0025 | 5 | Open | |||
| PBS | 10 | 5 | Open | ||||
| Antibody | 20 | 0.0001 | 5 | 15 | Open | ||
| PBS | 10 | 5 | Open | ||||
| Cells (300,000 cells/ml) | ~5 | 30 | Close when traps are occupied. | ||||
| Lysis buffer | 5 | 30 | Open shortly | ||||
| Inhibitor (e.g. 20 mM HCl) | 15 | 10 | Closed | ||||
| PBS | 2 | 30 | Closed | ||||
| Detection reagent | 5 | 30 | Open shortly | ||||
| Start monitoring reaction |
Table 1. Surface modification protocol for ELISA. For explanation, see text.
Discussion
Microfluidics technology has opened new and fascinating possibilities for single-cell analysis. In particular, the possibility to trap and immobilize cells individually by microfluidic tools has allowed systematic short and long-term studies on single-cell properties and response. Additionally, encapsulation of cells in high frequency microdroplets, generated on a microchip, has enabled single cell secretion studies, which cannot be performed with conventional cytometry devices. The microdroplet approach, however, has some limitations when incubation and washing steps are required for the analytical protocol. Our approach combines both concepts of cell trapping and isolation and therefore, it facilitates performing more complex assays including immunoassays based on surface-immobilized antibodies. Additionally, the method is very flexible with respect to the application and target analytes.
The design of the microchip enables (i) efficient cell trapping, (ii) supply of various buffers and reagents, and (iii) reliable isolation in a very small volume when required. In this way the dilution of the target molecules is avoided. Sixty (to hundreds) of microchambers are positioned on one single device allowing many experiments in parallel. The round-shaped valves closing the microchambers can be addressed together or separately by rows or columns, which is important when cross-contamination to adjacent chambers should be prevented. With the microchambers closed, the channels can be treated with harmful solutions such as hydrochloric acid for cleaning purposes without affecting the trapped cells. On the other hand, fast exchange of the volume inside the microchamber is possible. Currently a minimal time of 200 msec is needed to fully open the chamber and to introduce new reagents (for complete exchange of the microchamber volume, the flow rate has to be considered).
While our method is very reliable and reproducible, one should keep in mind that there are two main critical points in the protocol of chip fabrication and operation. First, the bonding of the two chip layers after plasma activation has to be done quickly. The alignment is critical, otherwise some chambers or even the whole chip is not usable, since the bonding is irreversible. This step could be difficult for inexperienced users, however, after some training even less experienced laboratory workers obtain usually good results. The second critical point is the cleanness during assembling and use of the microchip. If dust particles are present on the surface of the chip, they can compromise control channels and the valves or even lead to a breaking of the PDMS-PDMS or PDMS-glass bond. We therefore spend a lot of attention to cleaning procedures. It should be noted that besides the master form fabrication (performed in a clean room), we produce and use the microchips in a normal laboratory environment, where dust and particles are present.
Despite all the care, if the chambers are not closing correctly, it could be that either pressure lines are blocked or the spin coating process did not provide the right membrane thickness. Also, from time to time we observed insufficient bonding, resulting in breakage of the bond between the two PDMS layers. If many chips from the same batch are showing this problem, it is possible that the time between plasma activation and alignment is too long. Sometimes, we experienced sticky PDMS on the wafers that could not be properly removed and was a result of insufficient silanization of the wafer.
Once the production method is established, the microfluidic platform can be used for many different studies on the single cell level. Here, we showed the detection of intracellular and secreted agents with different assays, but many other assays are possible on the chip. Most proteins are not fluorescent and their detection via immunoassays is not straightforward. Implementation of ELISA into the chip will bring many benefits. Firstly, the assay can be designed very specifically for the desired protein (when suitable antibodies are present). Secondly, information from ELISA can be quantified even at low concentration limits, giving the number of protein or molecule copies present in the cell.
Our current chip is designed for the analysis of single mammalian cells. However, modification of the chip for other cells such as bacteria, algae and yeast, should be possible. Up to now, the only limitation for experiments is the need of an assay based on fluorescence. However, we are currently studying the implementation of mass spectrometry as a detection technique. Once this is established, the range of analytical problems that can be investigated with the platform will be even broader. Furthermore, since not every analysis is possible in the presence of cell lysis buffers, we are currently also evaluating various other lysis techniques on the platform. We are sure that the versatile microfluidic platform presented herein provides the basis for new single-cell studies on the proteome, metabolome and secretome level.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors gratefully acknowledge Tom Robinson for proof reading of the manuscript, C. Bärtschi and H. Benz for the construction of the custom-built pressure control system. We would also like to acknowledge the use of the clean room facility FIRST and the Light Microscopy Center (LMC), both at ETH Zürich. The work was funded by Merck Serono and the European Research Council (ERC) under the 7th Framework Program (ERC Starting Grant, project no. 203428, nμLIPIDs).
References
- Walling MA, Shepard JRE. Cellular heterogeneity and live cell arrays. Chem. Soc. Rev. 2011;40(7):4049–4076. doi: 10.1039/c0cs00212g. [DOI] [PubMed] [Google Scholar]
- Schmid A, Kortmann H, Dittrich PS, Blank LM. Chemical and biological single cell analysis. Curr. Opin. Biotechnol. 2010;20(1):12–20. doi: 10.1016/j.copbio.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Lecault V, White AK, Singhal A, Hansen CL. Microfluidic single cell analysis: from promise to practice. Curr. Opin. Chem. Biol. 2012;16(3-4):381–390. doi: 10.1016/j.cbpa.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Faley S, Seale K, et al. Microfluidic platform for real-time signaling analysis of multiple single T cells in parallel. Lab Chip. 2008;8(10):1700–1712. doi: 10.1039/b719799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wu H, et al. Counting low-copy number proteins in a single cell. Science. 2007;315(5808):81–84. doi: 10.1126/science.1133992. [DOI] [PubMed] [Google Scholar]
- Di Carlo D, Aghdam N, Lee LP. Single-cell enzyme concentrations, kinetics, and inhibition analysis using high-density hydrodynamic cell isolation arrays. Anal. Chem. 2006;78(15):4925–4930. doi: 10.1021/ac060541s. [DOI] [PubMed] [Google Scholar]
- Cecala C, Rubakhin SS, Mitchell JW, Gillette MU, Sweedler JV. A hyphenated optical trap capillary electrophoresis laser induced native fluorescence system for single-cell chemical analysis. Analyst. 2012;137(13):2965–2972. doi: 10.1039/c2an35198f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyer K, Kuhn P, Hanke C, Dittrich PS. A microchamber array for single cell isolation and analysis of intracellular biomolecules. Lab Chip. 2012;12(4):765–772. doi: 10.1039/c2lc20876h. [DOI] [PubMed] [Google Scholar]
- Agresti JJ, Antipov E, et al. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. U.S.A. 2010;107(9):4004–4009. doi: 10.1073/pnas.0910781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Fan R, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat. Methods. 2011;17(6):738–743. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Qin L, et al. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc. Natl. Acad. Sci. U.S.A. 2012;109(2):419–424. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AA, Talasaz AH, et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE. 2012;7(5):e33788. doi: 10.1371/journal.pone.0033788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nature. 2012;12(1):23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288(5463):113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- Thorsen T, Maerkl SJ, Quake SR. Microfluidic large-scale integration. Science. 2002;298(5593):580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]
- Gao Y, Majumdar D, et al. A versatile valve-enabled microfluidic cell co-culture platform and demonstration of its applications to neurobiology and cancer biology. Biomed. Microdevices. 2011;13(3):539–548. doi: 10.1007/s10544-011-9523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AK, VanInsberghe M, et al. High-throughput microfluidic single-cell RT-qPCR. Proc. Natl. Acad. Sci. U.S.A. 2011;108(34):13999–14004. doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7437):337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Kortmann H, Kurth F, Blank LM, Dittrich PS, Schmid A. Towards real time analysis of protein secretion from single cells. Lab Chip. 2009;9(21):3047–3049. doi: 10.1039/b908679j. [DOI] [PubMed] [Google Scholar]
- Huang Y, Cai D, Chen P. Micro- and Nanotechnologies for Study of Cell Secretion. Anal. Chem. 2011;83(12):4393–4406. doi: 10.1021/ac200358b. [DOI] [PubMed] [Google Scholar]
- Huang NT, Chen W, et al. An integrated microfluidic platform for in situ cellular cytokine secretion immunophenotyping. Lab Chip. 2012;12(20):4093–4101. doi: 10.1039/c2lc40619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyer K, Stratz S, Kuhn P, Küster SK, Dittrich PS. Implementing enzyme-linked immunosorbent assays on a microfluidic chip to quantify intracellular molecules in single cells. Anal. Chem. 85(6):3280–3287. doi: 10.1021/ac303628j. [DOI] [PubMed] [Google Scholar]
- Aebi U, Pollard TD. A glow discharge unit to render electron microscope grids and other surfaces hydrophilic. J. Electron Microsc. Tech. 1987;7(1):29–33. doi: 10.1002/jemt.1060070104. [DOI] [PubMed] [Google Scholar]
- Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995;14(21):5209–5215. doi: 10.1002/j.1460-2075.1995.tb00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]