

Abstract
T cell exhaustion is a major factor in failed pathogen clearance during chronic viral infections. Immunoregulatory pathways, such as PD-1 and IL-10, are upregulated upon this ongoing antigen exposure and contribute to loss of proliferation, reduced cytolytic function, and impaired cytokine production by CD4 and CD8 T cells. In the murine model of LCMV infection, administration of blocking antibodies against these two pathways augmented T cell responses. However, there is currently no in vitro assay to measure the impact of such blockade on cytokine secretion in cells from human samples. Our protocol and experimental approach enable us to accurately and efficiently quantify the restoration of cytokine production by HIV-specific CD4 T cells from HIV infected subjects.
Here, we depict an in vitro experimental design that enables measurements of cytokine secretion by HIV-specific CD4 T cells and their impact on other cell subsets. CD8 T cells were depleted from whole blood and remaining PBMCs were isolated via Ficoll separation method. CD8-depleted PBMCs were then incubated with blocking antibodies against PD-L1 and/or IL-10Rα and, after stimulation with an HIV-1 Gag peptide pool, cells were incubated at 37 °C, 5% CO2. After 48 hr, supernatant was collected for cytokine analysis by beads arrays and cell pellets were collected for either phenotypic analysis using flow cytometry or transcriptional analysis using qRT-PCR. For more detailed analysis, different cell populations were obtained by selective subset depletion from PBMCs or by sorting using flow cytometry before being assessed in the same assays. These methods provide a highly sensitive and specific approach to determine the modulation of cytokine production by antigen-specific T-helper cells and to determine functional interactions between different populations of immune cells.
Keywords: Immunology, Issue 80, Virus Diseases, Immune System Diseases, HIV, CD4 T cell, CD8 T cell, antigen-presenting cell, Cytokines, immunoregulatory networks, PD-1: IL-10, exhaustion, monocytes
Introduction
During persistent viral infections, virus specific-T cells acquire functional defects in a process known as T cell exhaustion. Early in vivo studies in the murine LCMV model of chronic viral infection indicated that exhausted virus-specific T cells have reduced cytolytic function against virally infected cells, lose their ability to proliferate and have reduced capacity to produce cytokines such as IL-2, TNF-α and IFN-γ1,2. A complex network of immunoregulatory pathways, such as PD-1 and IL-10, are upregulated during chronic infections and contribute to T cell dysfunction (reviewed in3,4). In vivo administration of blocking antibodies against these inhibitory pathways in the LCMV mouse model restored function of exhausted virus-specific T cells and enhanced viral clearance, indicating that T cell exhaustion is a partially reversible phenomenon (reviewed in 5).
Findings on the LCMV model were quickly extended to human chronic viral infections such as HBV, HCV and HIV5. In chronic HIV-1 infection, PD-1 and IL-10 pathways are upregulated in infected subjects and correlate with parameters of disease progression, directly with the viral load and inversely with the CD4 count6-8. Antibody blockade of the PD-1 or IL-10 pathways in vitro restored proliferation of HIV-specific CD4 and CD8 T cells indicating that, similar to the LCMV mouse model, T cell exhaustion in humans is a partially reversible phenomenon. However, due to the delicate nature of experiments on human samples as well as limitations in the sensitivity of in vitro assays, thorough investigation of the functional restoration profiles achieved by these interventions is strikingly absent. Proliferation assays have been, so far, the only reliable in vitro assay tested in most studies, yet there is a remarkable lack of evidence on the impact of these interventions on: 1) the killing capacity of cytotoxic T cells, 2) the antiviral effect of T cells on viral replication, 3) the cytokine secretion profile, and 4) the effect on CD4 T cell help on other cell subsets.
Intracellular cytokine staining (ICS) is a very useful and widely used flow cytometry based assay that is used to detect production of cytokines in various cell types in both mice and humans. It has also been used to investigate the effect of antibody blockade of inhibitory pathways. In ICS assays, cytokine secretion is blocked by the addition of Brefeldin and/or Monensin. Cytokines trapped inside the cells are then detected with fluorescent antibodies using polychromatic flow cytometry. The duration of the assay is usually limited to 6 or 12 hr after antigen stimulation since both Brefeldin and Monensin, which are typically added within 2 hr of antigen addition, are toxic to the cells. The ICS assay is a powerful technique that has been useful in the investigation of the effect of in vivo administration of blocking antibody intervention in mice where cells were extracted after a certain period of time after antibody exposure9. However, there are several limitations for the application of this experimental approach to evaluate the impact of blockade of inhibitory receptors in human samples. When performing antibody interventions of inhibitory pathways in a 6 or 12 hr stimulation in vitro (typically the case with human samples), blockade of inhibitory receptors in most cases does not alter the frequency of responding antigen-specific T cells as measured by standard ICS assays6, 7 . Measurements of per-cell cytokine production as measured by mean or median fluorescence intensity have given inconsistent results6, 7, 10. On the other hand, when ICS is performed at a late time point after stimulation (i.e. six days), changes in the number of cytokine-secreting cells can be observed. The differences are a combined effect of altered proliferation, survival, and changes in effector function that accumulate over the specified period of time6. One way to partially overcome these limitations is to incubate for longer periods of time (e.g. 36 hr, 60 hr, etc.) with the antigen before the addition of the cytokine secretion blocker without waiting for proliferation to occur. We have successfully used this method to determine the kinetics of cytokine secretion by HIV-specific CD4 and CD8 T cells11,12; however, this approach is not able to detect small responses and will not give an integrated result of the total cytokine secretion occurring since stimulation. Therefore, ICS is not a sensitive enough method to detect the impact of blockade of inhibitory pathways on the quantity of cytokines produced by activated T cells compared with cytokine mRNA quantitation or measurements of cytokine secretion in the supernatant at the same time points. Additionally, most of the cytokines produced by activated T cells act in autocrine fashion by contributing to either positive or negative feedback loops through interaction with antigen presenting cells. Therefore, addition of Brefeldin or Monensin during the ICS assay prevents cytokine secretion and thus stimulation of T cells is not optimal. Finally, detection of multiple cytokines with ICS is limited by the availability and sensitivity of antibodies for detection of cytokines with flow cytometry as well as by constraints in the number of fluorochromes that can be used simultaneously in any given panel.
We developed a highly sensitive in vitro experimental approach to evaluate the impact of immunoregulatory pathways in regulating cytokine secretion by HIV-specific CD4 T cells and subsequently CD4 help to antigen presenting cells (APCs) and natural killer cells (NK cells). This method can also be used to evaluate the impact of blockade of inhibitory molecules on CD8 T cell function by depleting the CD4 T cells from PBMCs or by stimulating with optimal peptides that are recognized only by CD8 T cells. In contrast to intracellular cytokine staining assays, we decided not to interfere with the cytokine secretion by HIV-specific CD4 T cells in order to be able to: 1) perform a more accurate quantification of the cytokine levels produced; 2) investigate a broader panel of cytokines or effector molecules; and 3) evaluate the impact of cytokines produced by HIV-specific CD4 T cells on other cell subsets. We stimulate CD8 depleted PBMCs with an HIV-1 Gag peptide pool in the presence of blocking antibodies for the immunoregulatory pathway of interest. After the desired time of incubation, usually 48 hr, we collect the supernatants to measure cytokine secretion with bead arrays and collect the cell pellets either for phenotypic analysis of the different cell subsets or for transcriptional analysis. Of note, at this 48 hr time point, we do not detect a significant population of proliferating T cells12. This is a flexible approach and several adaptations of this design can be applied to address different hypotheses. For example, the individual cell subsets (such as HIV-specific CD4 T cells or PD-1 high CD4 T cells, etc.) can be sorted after 12 hr of incubation before further incubation for 36 hr followed by collection of supernatants and cell pellets to investigate more specifically the impact of blockade interventions on cytokine secretion by defined subpopulations of PBMCs.
Protocol
1. Depletion and Isolation of PBMCs via Ficoll Separation
Deplete CD8+ T cells by adding Human CD8+ Depletion Cocktail at 50 μl/ml of whole blood.
Mix well and incubate for 20 min at room temperature (18-25 °C).
After incubation, mix whole blood with HBSS (Hank's Balanced Salt Solution without Ca2+ or Mg2+) and layer over Histopaque. Spin at 340 rcf for 30 min (no brake, slow acceleration).
Collect PBMCs and transfer to a new 50ml conical. Wash 2x with 45 ml RPMI 1640 supplemented with 50 IU Penicillin, 50 μg/ml Streptomycin, 2 mM L-glutamine, and 1% HEPES by spinning for 10 min at 340 rcf.
2. Antibody Blockade and Antigen-Specific Stimulation of Cells
Resuspend cells at 2x106 per condition in RPMI 1640 containing 10% Human Serum supplemented with 50 IU Penicillin, 50 μg/ml Streptomycin, 2 mM L-glutamine, and 1% HEPES, accounting for 500 μl per condition.
Aliquot 500 μl of cell suspension per FACS tube.
Depending on the pathway investigated, add PD-L1, IL-10Rα blocking antibodies and/or isotype controls at 10 μg/ml and incubate for 15 min at 37 °C, 5% CO2.
Stimulate cells with an HIV-1 Gag peptide pool at 1 μg/ml/peptide final concentration. Also include a "no stimulation" control for each blocking condition. Incubate at 37 °C, 5% CO2 for 48 hr.
3. Collection and Analysis of Supernatant
After 48 hr, spin down FACS tubes for 7 min at 340 rcf.
Carefully collect 2 x 225 μl supernatant in Eppendorf tubes for Luminex bead arrays without disturbing the pellet.
Inactivate virus in the collected supernatant with 25 μl 0.5% PBS-Tween to give a final concentration of 0.05% PBS-Tween.
Store collected supernatants that are not immediately used in a -80 °C freezer.
Measure desired cytokine concentrations using Millipore Luminex kit according to manufacturer's protocol.
4. Collection and Phenotypic Analysis of Cells via Flow Cytometry
After collecting supernatant, wash cells in 3ml PBS. Spin cells for 7 min at 340 rcf and decant excess wash.
Stain for viability using a LIVE/DEAD Fixable Dead Cell Stain Kit according to manufacturer's protocol.
Wash cells in PBS for 7 min at 340 rcf and at 4 °C. Resuspend in 100 μl PBS 1% FBS.
If staining for monocytes or other antigen presenting cells, block Fc receptors by adding 1.4 μl of FcR blocking reagent and vortex to mix.
Incubate for 10 min at 4 °C.
Add surface antibodies, vortex, and incubate for 20 min at 4 °C in the dark.
Wash cells with PBS 1% FBS, decant excess wash, and add 200 μl 4% paraformaldehyde. Incubate at room temperature for 20 min in the dark.
Wash cells with PBS 1% FBS, resuspend in 250 μl PBS 1% FBS, and acquire on a multilaser flow cytometer (e.g. BD LSRII or Fortessa).
5. Analysis of Cells via qRT-PCR
For cells not analyzed via flow cytometry: after supernatant collection, lyse cells in 300 μl Qiagen Buffer RLT containing 1% beta-mercaptoethanol. If not continuing with protocol, cells can be frozen at -80 °C after this point.
Isolate RNA using an RNA isolation kit following the manufacturer's protocol.
Synthesize cDNA from RNA using a cDNA synthesis kit following the manufacturer's protocol.
Perform quantitative PCR using SYBR Green technology.
Create a primer mix for each primer used including housekeeping genes (e.g. IL-13, IL-10, IFN-γ, GAPDH) by adding primers to nuclease-free water to give a final concentration of 10 μM. Keep on ice.
Create a master mix for each primer by adding 10.5 μl nuclease-free water, 12.5 μl SYBR green, and 1 μl primer mix per plate well. Keep on ice.
Pipette 24 μl master mix into each well of the plate that will be used.
Add 1 μl cDNA to each well according to template.
Cap wells and centrifuge plate for 3 min at 800 rcf.
Run plate on a qRT-PCR machine, e.g. a Stratagene MX3005P instrument.
6. Adaptation for Intracellular Cytokine Staining at 48 hr
Add Brefeldin and Monensin 6 hr prior to intracellular cytokine staining. Brefeldin is added at a concentration of 10 μg/ml while Monensin is added according to manufacturer's instructions. For convenience, Brefeldin can be added 12 hr prior to stain at a concentration of 5 μg/ml.
After surface stain, wash cells with PBS 1% FBS and stain for intracellular cytokines such as IL-12, IFN-γ, and TNF-α using a Fixation/Permeabilization Solution kit and protocol.
Wash cells, resuspend in 250 μl PBS 1% FBS, and run on a multilaser flow cytometer (e.g. BD LSR II or Fortessa).
7. Adaptation for Sorting Cells
16 hr after stimulation, stain for viability using LIVE/DEAD Fixable Dead Cell Stain Kit according to manufacturer's protocol. Keep cells on ice during entire procedure.
Wash cells with PBS for 7 min at 340 rcf and at 4 °C. Resuspend in 100 μl PBS 1% FBS.
Add surface antibodies, vortex to mix, and incubate cells on ice in the dark for 20 min.
Wash cells with PBS 1% FBS at 340 rcf and at 4 °C and resuspend in 500 μl cold RPMI 1640 containing 10% Fetal Bovine Serum supplemented with 50 IU Penicillin, 50 μg/ml Streptomycin, 2 mM L-glutamine, and 1% HEPES.
Filter cells using 5 ml Polystyrene Round-Bottom Tube with Cell-Strainer cap.
Prepare collection tubes for sort by adding 200 μl RPMI 1640 containing 10% Fetal Bovine Serum supplemented with 50 IU Penicillin, 50 μg/ml Streptomycin, 2 mM L-glutamine, and 1% HEPES to each tube. Keep all tubes on ice during sort.
Live sort the cell subsets of interest on an instrument (e.g. BD FACS Aria II) located in a facility equipped for biohazardous material.
After cells have been sorted, place cells back in the incubator for desired amount of time and follow with supernatant collection/Luminex analysis and cell pellet collection/PCR analysis.
Representative Results
When performing cytokine measurements using this in vitro system, it is essential to be able to distinguish the antigen-specific responses compared to the no stimulation control. In general, we considered positive responses as any values of the antigenic stimulation that give at least three-fold increase in cytokine levels compared to the no stimulation control and which were within the linear range of the assay. We are using high sensitivity bead array assays that can detect concentrations of cytokines as low as 0.16 pg/ml, enabling us to observe weak antigen-specific CD4 T cell responses. Samples are run in duplicate in order to ensure that accurate values are obtained. Figure 2A shows that for IFN-γ production, the assay can detect a median of 800-fold increase compared to no stimulation. These values depend on the cytokines tested because for some of them, such as IL-13 and IL-2, lower values compared to the no stimulation will be observed. A similar approach applies to transcriptional profiling of the mRNA of various cytokines tests. Figure 2B shows the level of mRNA for IFN-γ that is increased significantly (more than three-fold) after stimulation. With this assay, there is a strong correlation between IFN-γ protein secretion and IFN-γ mRNA levels (Figure 2C). Even though we developed this in vitro approach to study HIV-specific CD4 T cell function, we have also successfully used this approach to evaluate the impact of PD-L1 and/or IL-10Rα blockade on CD4 T cell responses after stimulation with SEB, anti-CD3/CD28, CMV lysate and RD-1 peptides from mycobacterium tuberculosis (data not shown). This method can be used to assess responses to bacterial antigens, including vaccine-induced responses, equally well. Figure 2D shows that stimulation with HIV-Gag results in greater IFN-γ secretion while stimulation with Tetanus toxoid results in higher IL-13 secretion, demonstrating the different cytokine profile of HIV and Tetanus-specific CD4 T cell responses.
A major strength of this assay is the ability to detect differences in cytokine secretion after antibody manipulation of inhibitory pathways that are not detected by other in vitro assays like ICS. It should be noted that some cytokines, such as IL-2, may be consumed by cells so measurements in the supernatant may potentially underestimate the cytokine levels produced. For this reason, any differences in cytokine secretion observed after antibody blockade should be also confirmed at the mRNA level to evaluate whether observed differences are due to changes in a per cell production. Figure 3A shows the impact of a PD-L1 blocking antibody on IFN-γ and IL-2 secretion by HIV-specific CD4 T cells after 48 hr of stimulation with the cognate antigen. Blockade of the PD-1 pathway generally does not have a significant impact on cytokine secretion without any antigen stimulation but enhances IFN-γ and IL-2 secretion when cells are stimulated with HIV-1 Gag peptide pools. When CD3 cells are depleted from the PBMCs, there is no IFN-γ or IL-2 produced in response to antigen stimulation (Figure 3B) indicating that secretion of IFN-γ and IL-2 is induced in response to HIV-specific CD4 T cell antigen-specific stimulation. For some cytokines, such as IL-21, the sensitivity of bead arrays does not allow detection of protein levels after weak antigen responses such as HIV. For those cytokines, quantification of mRNA can be performed instead12.
Some cytokines, such as IFN-γ and TNF-α, can be produced by a variety of cell subsets. When performing experiments on CD8-depleted PBMCs, it is sometimes difficult to identfy the source of protein secretion, and, therefore, it is difficult to identify the cell subset that responded to blockade of the inhibitory molecules. For this reason we have developed a variant of this method to perform a more thorough investigation of the impact of blocking inhibitory molecules on cytokine secretion by sorting different cell subsets according to their phenotypic characteristics. Figure 4 shows the impact of PD-1 blockade on IFN-γ and IL-2 secreted by sorted CD4 T cells. For this experiment, CD8-depleted PBMCs were stimulated for 12 hr in the presence of a blocking PD-L1 antibody or an isotype control. After 12 hr, cells were surface stained with fluorescent antibodies and CD4 T cells were sorted using a flow cytometer cell sorter. Sorted CD4 T cells were then incubated further for 36 hr and cytokines were measured in the supernatant using bead arrays as described above. We have successfully used this approach in the past to sort CD4 T cells according to their levels of PD-1 expression and have shown that PD-L1 blockade can restore function of HIV-specific CD4 T cells that express high levels of PD-112.
The number of immunoregulatory pathways identified as governing T cell function in HIV infection is constantly increasing. We have successfully used this assay in the past to show the impact of IL-10 blockade on IFN-γ and IL-2 secretion by HIV-1-specific CD4 T cells8,13 . One of the advantages of this method is the capacity to evaluate how immunoregulatory molecules regulate the interplay of HIV-specific CD4 T cells with antigen presenting cells (APCs). Figure 5A shows the impact of IL-10 blockade on restoring IL-12 secretion from antigen presenting cells. With an adaptation of this technique, by performing ICS at 48 hr after stimulation, we were able to show that monocytes are the primary source of IL-12 after stimulation with HIV-1 Gag peptide pools (Figure 5B).
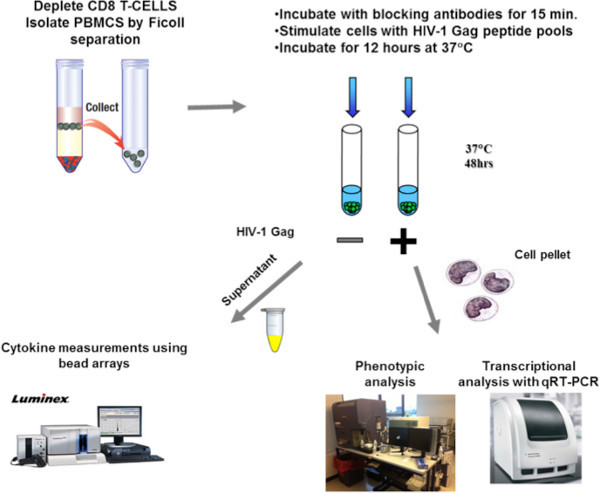 Figure 1. Schematic representation of the methodology. Isolated CD8-depleted PBMCs from HIV infected subjects are incubated for 15 min either with isotype control antibodies or blocking antibodies against PD-L1 or IL-10Rα. After a 48 hr stimulation with an HIV-1 Gag peptide pool, supernatants are collected for measurements of cytokine secretion with bead arrays and cell pellets are collected either for phenotypic analysis using flow cytometry or transcriptional analysis of mRNA levels using qRT-PCR.
Figure 1. Schematic representation of the methodology. Isolated CD8-depleted PBMCs from HIV infected subjects are incubated for 15 min either with isotype control antibodies or blocking antibodies against PD-L1 or IL-10Rα. After a 48 hr stimulation with an HIV-1 Gag peptide pool, supernatants are collected for measurements of cytokine secretion with bead arrays and cell pellets are collected either for phenotypic analysis using flow cytometry or transcriptional analysis of mRNA levels using qRT-PCR.
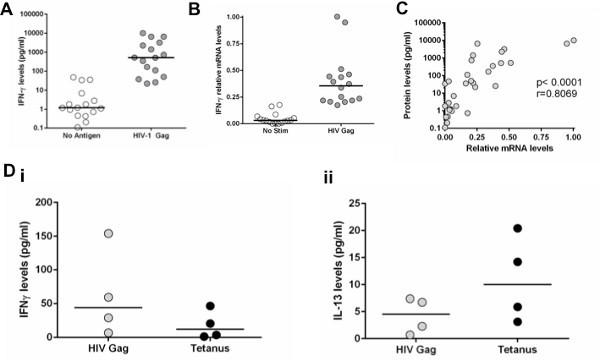 Figure 2. Detection of antigen-specific T cell responses with mRNA or protein secretion. A: CD8-depleted PBMCs stimulated with an HIV-1 Gag peptide pool. IFN-γ secretion was measured at 48 hr after stimulation. B: IFN-γ mRNA levels were measured with qRT-PCR in cell pellets at 48 hr after stimulation. C: Correlation of IFN-γ concentration in supernatants with IFN-γ mRNA levels. D: Comparison of IFN-γ and IL-13 protein levels between CD8-depleted PBMCs stimulated with HIV-1 Gag peptide pool or Tetanus Toxoid. Click here to view larger figure.
Figure 2. Detection of antigen-specific T cell responses with mRNA or protein secretion. A: CD8-depleted PBMCs stimulated with an HIV-1 Gag peptide pool. IFN-γ secretion was measured at 48 hr after stimulation. B: IFN-γ mRNA levels were measured with qRT-PCR in cell pellets at 48 hr after stimulation. C: Correlation of IFN-γ concentration in supernatants with IFN-γ mRNA levels. D: Comparison of IFN-γ and IL-13 protein levels between CD8-depleted PBMCs stimulated with HIV-1 Gag peptide pool or Tetanus Toxoid. Click here to view larger figure.
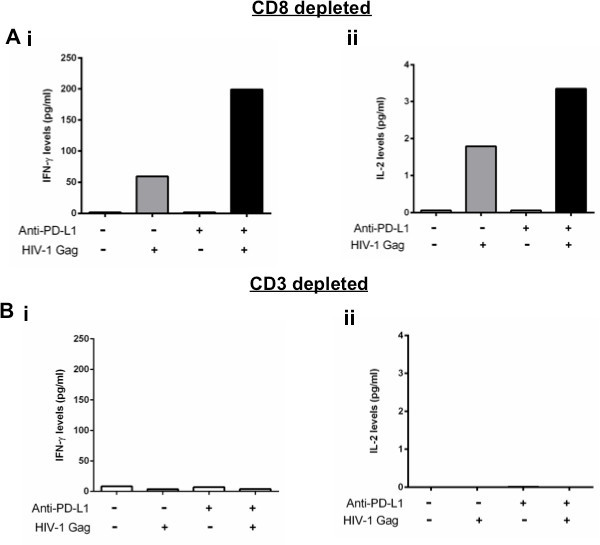 Figure 3. Changes of cytokine secretion after manipulation of inhibitory pathways is CD4 T cell-dependent. A and B: CD8 depleted or CD3 depleted PBMCs stimulated with an HIV-1 Gag peptide pool in the presence of isotype control or PD-L1 blocking antibody. IFN-γ and IL-2 secretion was measured at 48 hr after stimulation.
Figure 3. Changes of cytokine secretion after manipulation of inhibitory pathways is CD4 T cell-dependent. A and B: CD8 depleted or CD3 depleted PBMCs stimulated with an HIV-1 Gag peptide pool in the presence of isotype control or PD-L1 blocking antibody. IFN-γ and IL-2 secretion was measured at 48 hr after stimulation.
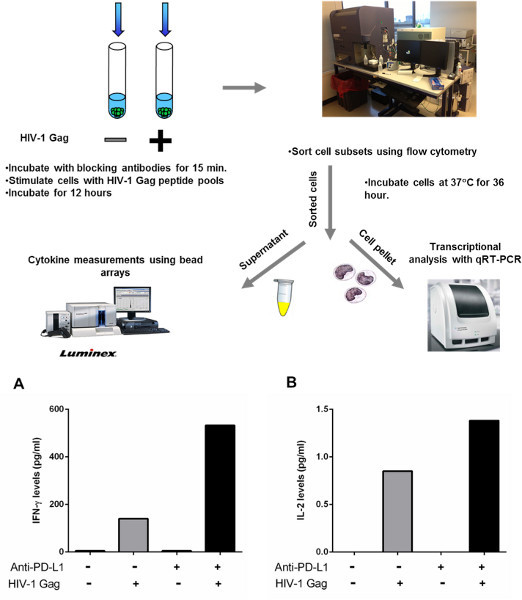 Figure 4. Sorting CD4 T cells to detect cytokine secretion after manipulation of PD-1 blockade. A-B: CD8-depleted PBMCs from three chronically infected, untreated subjects were incubated with an HIV-1 Gag peptide pool or left unstimulated for 12 hr in the presence of isotype control or PD-L1 blocking antibody. CD4 T cells were then negatively selected by lineage exclusion on the lymphocytes and incubated further for 36 hr.
Figure 4. Sorting CD4 T cells to detect cytokine secretion after manipulation of PD-1 blockade. A-B: CD8-depleted PBMCs from three chronically infected, untreated subjects were incubated with an HIV-1 Gag peptide pool or left unstimulated for 12 hr in the presence of isotype control or PD-L1 blocking antibody. CD4 T cells were then negatively selected by lineage exclusion on the lymphocytes and incubated further for 36 hr.
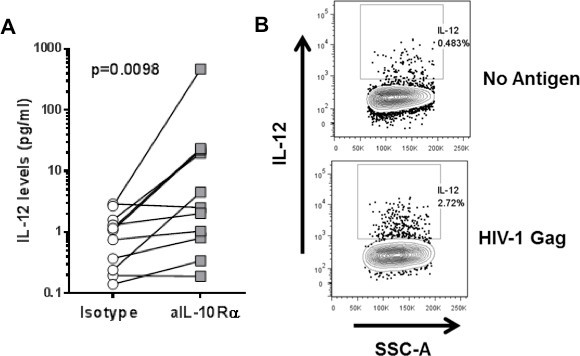 Figure 5. Interplay between CD4 T cells and antigen-presenting cells. A: CD8-depleted PBMCs were stimulated with an HIV-1 Gag peptide pool in the presence of isotype control or IL-10Rα blocking antibody. IL-12 levels in the supernatant were measured at 48 hr after stimulation. B: CD8-depleted PBMCs stimulated with an HIV-1 Gag peptide pool. IL-12 secretion on monocytes was measured with ICS at 48 hr after stimulation.
Figure 5. Interplay between CD4 T cells and antigen-presenting cells. A: CD8-depleted PBMCs were stimulated with an HIV-1 Gag peptide pool in the presence of isotype control or IL-10Rα blocking antibody. IL-12 levels in the supernatant were measured at 48 hr after stimulation. B: CD8-depleted PBMCs stimulated with an HIV-1 Gag peptide pool. IL-12 secretion on monocytes was measured with ICS at 48 hr after stimulation.
Discussion
Supernatant collection is an imperative part of this experimental design. When harvesting supernatants for the Luminex assay, aliquots were made and kept at -80 °C to prevent protein degradation due to freeze-thaw cycles. Freezing and thawing can be harsh processes for proteins, and by minimizing freeze-thaws, signal will be maximized in assays. Therefore, it is easier to obtain repeatable and more accurate data when working from aliquots that have been freeze-thawed the same number of times.
We have previously performed kinetic analysis of cytokine secretion and mRNA levels after stimulation with HIV-Gag peptide pools12 . Based on these kinetics, we chose the 48 hr time point to perform the cytokine secretion measurements. Given the differences observed between mRNA and protein secretion as well as between different stimuli, it is imperative that researchers perform their own kinetic analyses depending on the stimuli they use and whether they are measuring mRNA expression or protein cytokine secretion.
When cell subsets are sorted (using the approach shown in Figure 3), it is important to adjust the volume of the culture according to the cell number sorted. In the standard assay, we typically incubate 1 million CD8-depleted PBMCs in 500 μl of medium. However, when we sort cell subsets, such as CD4 T cells, we usually sort up to 100,000 cells. For this reason, we decrease the volume of the medium to 200 μl in order to keep from diluting the cells and to achieve a cytokine concentration that is detectable with the bead array. For each experiment, the volume of the cell cultures should be experimentally tested, taking into consideration the cell type, the cell numbers sorted, the cytokine of interest, and the sensitivity of the detection kit.
Another important aspect when performing the Luminex assay is virus inactivation. Virus needs to be inactivated to be able to safely run samples on the instrument. Tween-20 was used for this reason. Previously, other detergents had been tested for this purpose. Various dilutions of 0.5% Tween, 5-10% Triton, and wash buffer were added to HIV infected cells and were tested against noninactivated cells. Tween was found to inactivate the virus best while interfering the least with the assays performed.
When performing flow cytometry, an essential step when staining for monocytes is to block the Fc receptor with an Fc blocking reagent to prevent nonspecific binding of monoclonal antibodies. The Fc receptor is present on the surface of antigen presenting cells and binds to the Fc region of antibodies. If this receptor is not blocked before staining, monoclonal antibodies that are added to target specific markers may instead bind to this receptor, therefore giving a false positive result. This is especially important to avoid when analyzing cytokines that derive from a certain cell type, as not blocking this receptor could be detrimental to results. Incubating cells in media containing human serum can also help with this issue, as there are copious amounts of IgG in the serum that bind to the Fc receptor.
Although Luminex assays provide more sensitive detection than ICS assays, certain effector molecules such as IL-21 or perforin are still difficult to detect with this method. This in vitro approach allows for transcriptional analysis using qRT-PCR that provides greater sensitivity in detecting other effector molecules.
One limitation of our experimental approach is that interpretation of its results is difficult when the cytokine of interest is produced by different cell subsets from the heterogeneous mix of leukocytes present in whole PBMCs or CD8-depleted PBMCs. For example, this is the case for the cytokine IL-10 which is upregulated in different cell types in the setting of progressive HIV disease8. Sorting of the population of interest before further incubation and collection of supernatants is an alternative approach we have successfully used in this context.
In summary, the techniques presented in this paper provide a reliable means of measuring restoration of cytokine production by HIV-specific CD4 or CD8 T cells in the presence of blockade of the PD-L1 and IL-10 immunoregulatory pathways. These techniques can be applied in instances where cytokines are not easily detectable by flow cytometry.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Gordon Freeman for providing the anti-PD-L1 blocking antibody. We thank the clinical and laboratory staff at the Massachusetts General Hospital and all study participants for their invaluable role in this project.
This study was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (PO1 AI-080192; D.E.K), the National Heart Lung and Blood Institute of the National Institutes of Health (RO1 HL-092565; D.E.K). D.E.K is supported by a Research Scholar Career Award of the Quebec Health Research Fund (FRQS). FP is supported by a fellowship grant of the Massachusetts General Hospital Executive Committee on Research and the Harvard Global Health Institute (HGHI).
References
- Zajac AJ, et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, vander Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon DS, Kaufmann DE. Protective and detrimental roles of IL-10 in HIV pathogenesis. Eur. Cytokine Netw. 2010;21:208–214. doi: 10.1684/ecn.2010.0201. [DOI] [PubMed] [Google Scholar]
- Porichis F, Kaufmann DE. Role of PD-1 in HIV pathogenesis and as target for therapy. Curr. HIV/AIDS Rep. 2012;9:81–90. doi: 10.1007/s11904-011-0106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ. T cell exhaustion. Nat. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Trautmann L, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- Brockman MA, et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood. 2009;114:346–356. doi: 10.1182/blood-2008-12-191296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Zhang JY, et al. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood. 2007;109:4671–4678. doi: 10.1182/blood-2006-09-044826. [DOI] [PubMed] [Google Scholar]
- Ndhlovu ZM, et al. High-dimensional immunomonitoring models of HIV-1-specific CD8 T-cell responses accurately identify subjects achieving spontaneous viral control. Blood. 2013;121:801–811. doi: 10.1182/blood-2012-06-436295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porichis F, et al. Responsiveness of HIV-specific CD4 T cells to PD-1 blockade. Blood. 2011;118:965–974. doi: 10.1182/blood-2010-12-328070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon DS, et al. CD4+ CD25+ regulatory T cells impair HIV-1-specific CD4 T cell responses by upregulating interleukin-10 production in monocytes. 2012;86:6586–6594. doi: 10.1128/JVI.06251-11. [DOI] [PMC free article] [PubMed] [Google Scholar]