

Abstract
Homologous recombination (HR), a mechanism to accurately repair DNA in normal cells, is deregulated in cancer. Elevated/deregulated HR is implicated in genomic instability and telomere maintenance, which are critical lifelines of cancer cells. We have previously shown that HR activity is elevated and significantly contributes to genomic instability in BAC. The purpose of this study was to evaluate therapeutic potential of HR inhibition, alone and in combination with telomerase inhibition, in BAC. We demonstrate that telomerase inhibition in BAC cells increases HR activity, RAD51 expression, and association of RAD51 to telomeres. Suppression of HR leads to shorter telomeres as well as markedly reduced genomic instability in BAC cells over time. Combination of HR suppression (whether transgenic or chemical) with telomerase inhibition, causes a significant increase in telomere attrition and apoptotic death in all BAC cell lines tested, relative to either treatment alone. A subset of treated cells also stain positive for β-galactosidase, indicating senescence. The combined treatment is also associated with decline in S-phase and a strong G2/M arrest, indicating massive telomere attrition. In a subcutaneous tumor model, the combined treatment resulted in the smallest tumors, which were even smaller (P=0.001) than those resulted from either treatment alone. Even the tumors removed from these mice had significantly reduced telomeres and evidence of apoptosis. We therefore conclude that although telomeres are elongated by telomerase, elevated RAD51/HR assist in their maintenance/stabilization in BAC cells. Telomerase inhibitor prevents telomere elongation but induces RAD51/HR, which contribute to telomere maintenance/stabilization and prevention of apoptosis, reducing the efficacy of treatment. Combining HR inhibition with telomerase, makes telomeres more vulnerable to degradation and significantly increases/expedites their attrition, leading to apoptosis. We therefore demonstrate that a therapy, targeting HR and telomerase, has potential to prevent both the tumor growth and genomic evolution in BAC.
Keywords: Homologous Recombination, Telomerase, Telomeres, Barrett’s Adenocarcinoma
INTRODUCTION
DNA at the end of each chromosome in eukaryotic cells is comprised of multiple copies of TTAGGG sequence1–3 which fold into a looped conformation4 and associate with specific proteins including TRF2 and TRF14, 5 to form a protective structure called, Telomere. Telomeres prevent chromosomal ends from undergoing unnecessary repair and/or recombination, interchromosomal fusion through end-joining, and protect them from degradation by nucleases6. Thus, telomeres play an important role in maintaining genomic integrity in a cell7–10. The DNA replication system, including that in human, is unable to replicate the portion of telomeric DNA occupied by RNA primers. This leads to loss of a small portion of telomeric DNA with each cell division. Rate of telomere attrition, which may vary with age and lifestyle factors11, has been estimated to be in the range of 50–100 base pairs per replicative cycle12. When the median length of telomeric DNA is reduced below a certain limit, the cell undergoes replicative senescence and/or apoptotic death13–17. Telomere length may, therefore, indicate the lifespan of cells in culture18. It has also been proposed that leucocytes may be used to estimate rate of telomere shortening and pace of aging in human subjects11.
Although the length of telomeric DNA is reduced with cell division in most somatic cells, it is maintained in germline and certain other cell types including hematopoietic stem and gastrointestinal epithelial cells. The length of telomeric DNA in these cells is maintained by addition of TTAGGG repeats to telomeres by an enzyme, telomerase or also known as human telomerase reverse transcriptase (hTERT)19. Telomerase is inactive in most normal somatic cells but active in the cells mentioned above. Telomerase is also activated during oncogenesis in >90% of cases20–22. Since activation of telomerase is believed to occur at or near crisis, the telomeres in most cancer cells are significantly shorter than those in normal cells but their further attrition is prevented by telomerase-mediated addition of telomeres23, 24, which confers unlimited proliferation to these cells25–27. Since telomerase activity is elevated in most cancers but repressed in most normal cells, whereas telomere length is significantly shorter in cancer relative to normal cells28, 29, it has been proposed that inhibitors of telomerase may inhibit proliferation of cancer cells while having little or no effect on normal cells. Consistently, a variety of agents with ability to inhibit telomerase activity have been evaluated in vitro and in animal models against a number of cancer types30–34. We also evaluated and demonstrated the efficacy of a variety of agents in human multiple myeloma and BAC cells; these agents included small molecules interacting with G-quadruplex structures of DNA14, 29, 35, DNA or PNA (peptide nucleic acid) oligonucleotides targeting hTR16, 36, 37, and siRNAs designed against hTERT13. GRN163L, a lipid-attached oligonucleotide targeting RNA component of telomerase (hTR) is the first telomerase inhibitor suited for in vivo delivery and is currently in clinical trial.
Although telomerase inhibition seems to be a promising approach in fighting cancer, it is associated with some limitations. First, the therapy starts to work only after a lag period, which is required for telomere shortening in cancer cells. As soon as the telomeres in cancer cells reach below critical limit, they undergo apoptotic death or replicative senescence. The lag period depends on initial telomere length in cancer cells and probably also on other factors such as levels of nuclease and other activities. Secondly, the presence of an alternate pathway of telomere maintenance, also known as ALT, has also been reported. Infact, certain immortal cell lines and a subset of cancers do not have any detectable telomerase activity but still maintain their telomeres through ALT pathway38, 39, involving homologous recombination (HR)-mediated telomere elongation40. The existence of both telomerase- and HR-dependent telomere maintenance within the same cell has also been reported41.
Telomere maintenance, by telomerase and/or HR, is a lifeline of cancer cells. Effective and relatively expeditious telomere erosion leading to replicative arrest/apoptosis of cancer cells may require rational combinations such as those targeting telomerase and HR. We have shown that HR is elevated in BAC42 as well as other cancer cells43 and this deregulated HR plays a significant role in genomic instability and disease progression. Here we report a novel and critical finding that following telomerase inhibition, HR is further elevated in BAC cell lines, and combining inhibitors of HR (whether chemical or shRNA based) with telomerase inhibition, significantly increases telomere attrition and apoptosis in BAC cell lines both in vitro and in vivo. We therefore propose that inhibitors of homologous recombination have potential to be the most rational combination for telomerase-directed therapy, and together these drugs, would target tumor growth as well as genomic evolution in BAC.
RESULTS
Telomerase inhibition leads to a significant increase in homologous recombination in BAC cells
BAC cells were treated with GRN163L (2.0 µM) for 48 hrs and evaluated for HR activity, using the luminescence-based HR assay, developed in our laboratory42. The HR activity was increased by 71±8%, 143±21%, and 100±23% in OE33, OE19, and FLO-1 cell lines, respectively (P=0.001–0.012; Figure 1A). To confirm the induction of HR by GRN163L, FLO-1 cells treated with GRN163L (2.0 µM) for 48 hrs were also evaluated for HR activity, using an alternate (fluorescence-based) HR assay (Addgene, Cambridge, MA)44as described in Methods. Consistent with the data derived by our luminescence-based assay, the evaluation by fluorescence-based HR assay also showed 92±3% (p=0.0089) increase in HR activity in FLO-1 cells, following treatment with telomerase inhibitor GRN163L (Figure 1B). To further confirm the increase in HR activity following telomerase inhibition, telomerase in FLO-1 cells was suppressed by shRNA-mediated knockdown of catalytic subunit of telomerase (hTERT), inhibition of telomerase activity confirmed as described in Methods (not shown), and cells evaluated for HR. Loss of telomerase by shRNA was also associated with significant (150%) increase in HR activity (Figure 1C). Thus suppression of telomerase by oligonucleotide or shRNA and evaluation of its impact on HR by two different methods, indicate that telomerase inhibition in BAC cells significantly induces HR, which may contribute to telomere maintenance.
Figure 1. Telomerase inhibition induces whereas nilotinib and RAD51-suppression reduce HR activity in BAC cells.
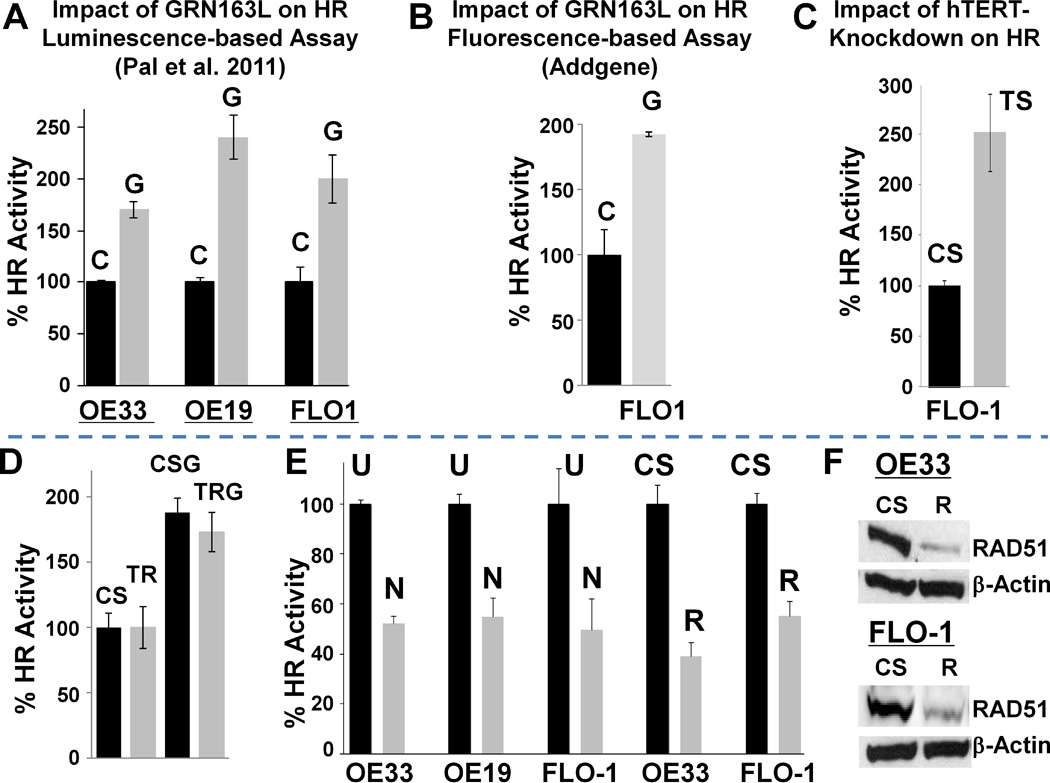
(A) GRN163L, an oligonucleotide targeting RNA component of telomerase, induces HR. BAC cell lines (OE33, OE19, FLO-1) were treated with mismatch control oligonucleotide (C; 2 µM) or GRN163L (G; 2 µM) for 48 hrs, and evaluated for HR activity using the luminescence-based HR assay described in Methods. Error bars represent SEMs of triplicate assays. (B) Confirmation of increase in HR by alternate method. FLO-1 cells treated as above (in panel A) were evaluated for HR activity, using a fluorescence-based HR assay (Addgene) as described in Methods. (C) shRNA, targeting catalytic subunit of telomerase (hTERT), also induces HR. FLO-1 cells were transduced with lentivirus particles, producing control (CS) or telomerase-targeting (TS) shRNAs, and following selection evaluated for HR, using luminescence-based HR assay described in Methods. (D) Impact of TRF2 knockdown on HR. FLO-1 cells, transfected with control (CS) or TRF2 targeting (TR) siRNA, were cultured untreated or treated for 48 hrs with GRN163L (G; 2 µM), and evaluated for HR activity using the luminescence-based HR assay. (E) Inhibition of HR by nilotinib and RAD51-suppression. BAC cell lines were exposed to nilotinib (5 µM) or transduced with lentivirus-based shRNAs (CS, control; R, RAD51-targeting). Untreated cells (U), those treated with nilotinib for 48 hrs (N), and shRNA-transduced cells at day six after transduction, were evaluated for HR activity using a plasmid based assay as described in Methods. Relative HR activity in nilotinib- and shRNA-treated cells is shown as percent of activity in untreated and control shRNA-treated cells, respectively: error bar indicates SEMs of triplicate assays. (F) RAD51-suppression by lentivirus-based shRNAs. BAC cell lines, transduced with lentivirus-based shRNAs (CS, control; R, RAD51-targeting) as described in panel B, were evaluated for RAD51 expression by Western blotting.
To investigate if telomere deprotection leads to increased HR, TRF2 in FLO-1 cells was suppressed by siRNA and suppression confirmed by Western blotting (Supplementary Figures 1B–C). TRF2-suppressed cells were evaluated for HR by luminescence-based assay. As shown in Figure 1D, no difference in HR activity was observed following suppression of TRF2.
Nilotinib and RAD51-suppression significantly inhibit HR in BAC cells
To monitor the role of elevated HR in BAC, HR was suppressed either chemically by nilotinib or transgenically by RAD51-knockdown, as reported previously42. BAC cells were treated with nilotinib (5 µM) for 48 hrs or transduced with lentivirus-based control or RAD51-targeting shRNAs, and evaluated for HR activity, using the luminescence-based HR assay described above. Nilotinib inhibited HR activity by 48±7%, 46±7%, and 53±3% in OE33, OE19 and FLO-1 cells, respectively (P; <.001–0.002; Figure 1E). Consistent with our previous observations, RAD51-knockdown led to 47±1% reduction in HR activity in FLO-1 cells (P=0.005; Figure 1E). Similarly, the HR activity in OE33 cells was also significantly reduced (by 60%±3%; P=<0.001) following RAD51-suppression (Figure 1E); shRNA-mediated knockdown of RAD51 was confirmed by Western blotting (Figure 1F). Thus nilotinib at (5 µM) and RAD51-suppression by the specific shRNAs used in this study, caused a significant and similar (close to 50%) reduction of HR activity in BAC cell lines tested.
Telomerase inhibition increases association of RAD51 with telomeres
To evaluate the impact of telomerase inhibitor GRN163L on RAD51 expression and its binding to telomeres, we conducted chromatin immunoprecipitation assay. OE33 cells were treated with telomerase and/or HR inhibitors for 48 hrs, protein-DNA complexes cross-linked, and immunoprecipitated using anti-RAD51 antibody. The DNA from these complexes was purified and evaluated by PCR, using primers specific for telomeres and One gene used as internal control. As shown in Figure 2A, the exposure to GRN163L was associated with 3-fold increase in the amount of RAD51-bound telomeric DNA. This increased binding of RAD51 to telomeres was reduced by 50% when HR inhibitor nilotinib was added along with GRN163L (Figure 2A). In transduced cells, exposure to GRN163L led to 89% increase in the RAD51-bound telomeric DNA (Figure 2B, column 4). Moderate suppression of RAD51 by “R” and strong suppression by “R4” shRNAs reduced the GRN163L-induced binding of RAD51 to telomeres by 50% and 80% respectively (Figure 2B, columns 5 and 6). These data indicate that a substantial fraction of RAD51, induced following telomerase inhibition, associated with telomeric sequences.
Figure 2. Telomerase inhibition induces RAD51 expression and its binding to telomeres.
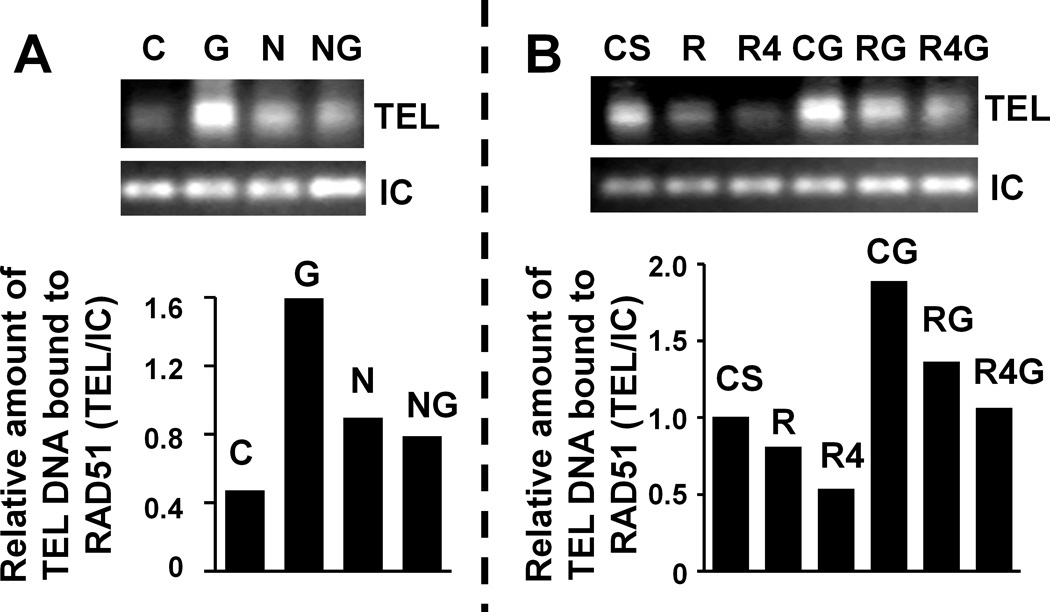
(A) OE33 cells, untreated or treated for 48 hrs with mismatch control (C; 2 µM), GRN163L (G; 2 µM), HR inhibitor nilotinib (N; 5 µM), and combination of N and G (NG) were processed as described above. Top panel is the gel image showing RAD51-bound telomere (TEL) and internal control (IC) bands, whereas bottom is the bar graph showing relative amount of telomeric DNA bound to RAD51 following normalization with internal control DNA. (B) OE33 cells treated with control shRNA (CS), RAD51 shRNA (R), RAD51 shRNA mediating stronger suppression of RAD51 (R4), CS cells treated with GRN163L (CG), R cells treated drug (RG), or R4 cells treated with drug (R4G) were processed as described above. Top panel shows gel image of telomere (TEL) and internal control (IC) bands, whereas bottom is the bar graph showing relative amount of telomeric DNA bound to RAD51 following normalization with internal control DNA.
HR suppression, by RAD51-knockdown, increases the efficacy of GRN163L in OE33 cells
To study the impact of HR-suppression on efficacy of telomerase inhibitor, HR in OE33 cells was first inhibited by lentivirus-based shRNAs targeting RAD51. RAD51-knockdown was confirmed by Western blotting and HR suppression by plasmid based assay (Figure 1). The shRNA-transduced cells (control, CS; RAD51, R) were cultured in the presence of mismatch control or GRN163L oligonucleotides for 10 days and evaluated for following characteristics. Impact on telomerase activity: Although RAD51 suppression alone led to a small (19±2%) decline in telomerase activity, the exposure to GRN163L led to near complete loss of activity in both the control and RAD51-suppressed cells (Figure 3A). Cell growth: Relative to control (CS) cells, the viability of RAD51-suppressed (R), GRN163L-treated CS, and GRN163L-treated R cells was reduced by 34±5%, 24±6%, and 65±5%, respectively (P<0.001–0.003; Figure 3B). Thus, inhibition of HR or telomerase, significantly reduced proliferation rate in OE33 cells and combined treatment was associated with a significant 41% (P=0.001) increase in cell death, relative to GRN163L used alone (Figure 3B). Impact on telomeres: Telomere length in RAD51-suppressed (R) cells, GRN163L-treated CS cells, and GRN163L-treated R cells was reduced by 23±4%, 69±5% and 86±1%, respectively (P<.001–0.037) (Figure 3C), indicating that simultaneous suppression of telomerase and HR has a significantly greater impact on telomere maintenance (P=0.02). Impact on apoptosis: Apoptosis was detected by evaluating the ability of cells to bind lactadherin, which interacts with phosphatidylserine exposed during apoptosis. Control-shRNA-transduced (CS) cells, RAD51-suppressed (R) cells, GRN163L-treated CS, and GRN163L-treated R cells had 2±1%, 11±3%, 3±3%, and 39±5% cells undergoing apoptosis, respectively (Figure 3D). Thus, simultaneous suppression of telomerase and HR led to a significant 36% (12-fold) increase in apoptosis (P=0.001), relative to telomerase inhibition alone (Figure 3D). Evidence of senescence. Cells were also evaluated for β-galactosidase staining, a marker for senescence. A subset of cells subjected to both telomerase and HR suppression stained positive for β-galactosidase, indicating senescence (Figure 3E). In summary these data show that when telomerase inhibition is combined with RAD51/HR suppression, there is a significant increase in telomere attrition leading to expedited and stronger growth arrest and senescence/apoptosis in OE33 cells.
Figure 3. Impact of HR inhibition (by RAD51 knockdown) on efficacy of telomerase inhibitor in OE33 cells.
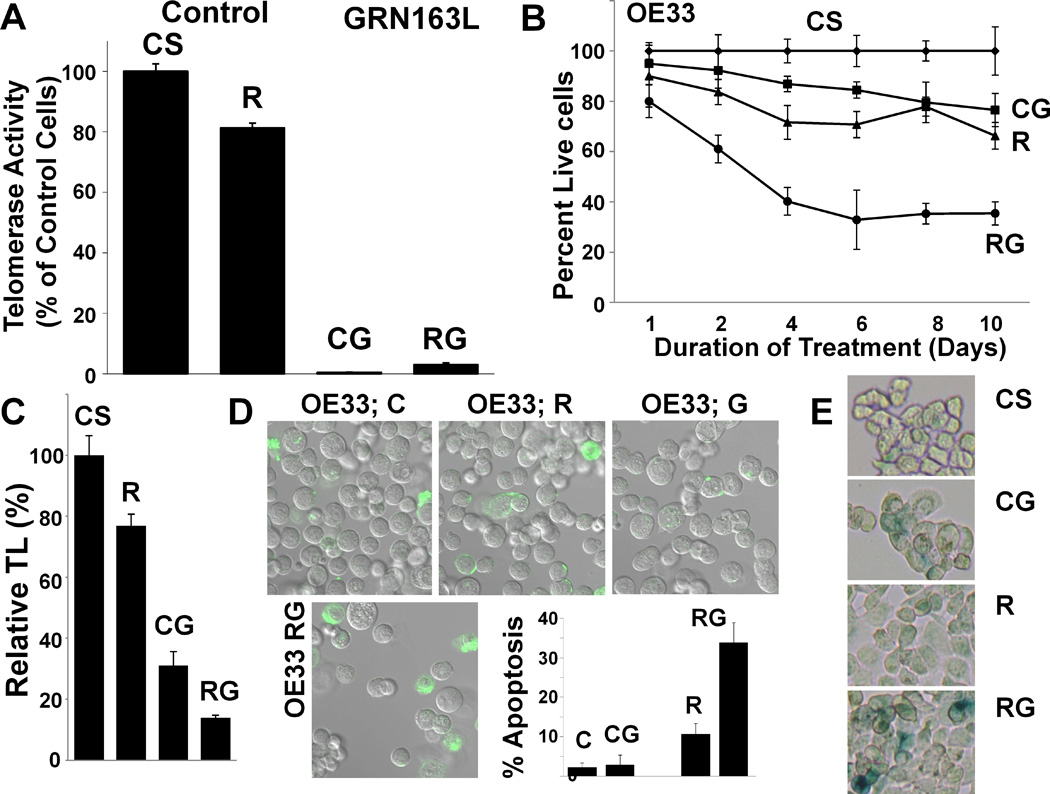
OE33 cells, transduced with lentivirus particles producing control (CS) or RAD51-specific (R) shRNAs, were cultured in the presence or absence of telomerase inhibitor GRN163L (G; 2 µM) and evaluated for impact on growth, apoptosis, and telomere maintenance. (A) Impact on telomerase activity. Cells treated as above for 10 days were evaluated for telomerase activity using TRAPeze Telomerase Detection kit. CS, control shRNA treated cells; R, RAD51 suppressed cells; CG, CS cells treated with GRN163L; RG, R cells treated with GRN163L. (B) Impact on growth. The control (CS) and RAD51-suppressed (R) cells were cultured in the presence or absence of GRN163L and cell viability determined at different time points as indicated by counting the substrate attached cell number, and confirmed by trypan blue exclusion. The growth curve shows the mean of triplicate values, with S.E.M. (C) Impact on telomere length. The control and RAD51-suppressed OE33 cells were cultured in the presence or absence of GRN163L for 10 days and telomere length determined by real time PCR as described; error bars represent SEMs of triplicate assays. (D) Impact on apoptosis. Transduced OE33 cells, treated as above for 10 days, were examined for apoptosis by evaluating their ability to bind lactadherin. Extent of green fluorescence indicates early or late apoptosis. (E) Evidence of senescence. Cells treated as above were evaluated for β-galactosidase staining, a marker for senescence.
Consistent with RAD51-knockdown, HR inhibitor “nilotinib” also increases efficacy of GRN163L
OE33 cells, treated with mismatch control oligonucleotide, telomerase inhibitor GRN163L, nilotinib, or combination of drugs for 19 days were evaluated for various activities. Impact on growth and apoptosis: Whereas control cells continued to proliferate, the treatment with nilotinib and GRN163L led to a gradual decline in live cell number leading to 59±4% and 68±3% cell death in 19 days, respectively (P<.001). However, the combination of drugs was responsible for 90±4% cell death, significantly increased relative to control cells (P<.001) and those treated with GRN163L alone (P=0.007) (Figure 4A). Treatment with nilotinib, GRN163L, and combination of drugs led to apoptotic death in 27±2%, 38±1%, and 74±3% cells, respectively (P<.001; Figure 4B). Thus, simultaneous suppression of HR and telomerase was associated with 36% increase in apoptosis in OE33 cells, relative to GRN163L used alone (P<.001) (Figure 4B). Impact on telomere length. Telomeres in untreated OE33 cells are really short (~2.00 kbp) (Supplementary Figure 1D). Telomere length in cells treated with nilotinib and GRN163L was reduced to 70±5% and 40±5% of control cells, respectively (P=0.023 and 0.002) (Figure 4C). However, the combination of drugs reduced telomere length to only 6±1% of control cells (P<.001), i.e., a significant (35%) increase in telomere attrition relative to GRN163L used alone (P<.001; Figure 4C). Impact on cell cycle. Evaluation of cell cycle by flow cytometry indicated that S-phase was significantly reduced (P<.001) only by combination treatment, whereas G2/M arrest was induced by all three treatments (Figure 4D). The increase in G2-arrest by combination treatment was 5.0±0.2- and 2.4±0.1-fold higher, relative to control and GRN163L-treated cells (P<.001), indicating that severe telomere shortening (in cells treated with both drugs) was associated with prevention of entry into S-phase and a strong arrest in G2/M-phase of cell cycle (Figure 4D). Impact on HR. Figure 1 shows the impact of 48 hr treatment of BAC cells with nilotinib and GRN163L on HR. To evaluate the impact of these treatments on HR beyond 48 hrs, the cells treated with drugs separately or together for two weeks were evaluated for HR activity, using the plasmid based assay described previously42. As shown in Figure 4E, nilotinib was associated with a significant suppression of HR (34±11% of control; P=0.001), even at day 14 of treatment. Although induction of HR by GRN163L at day 14 was not as strong as observed at 48 hrs of treatment, it was still 30±4% higher, relative to control cells (P=0.007). Addition of nilotinib along with GRN163L, kept the level of HR as low as in the cells treated with nilotinib alone (Figure 4E).
Figure 4. Impact of HR inhibition by nilotinib on efficacy of telomerase inhibitor in OE33 cells.
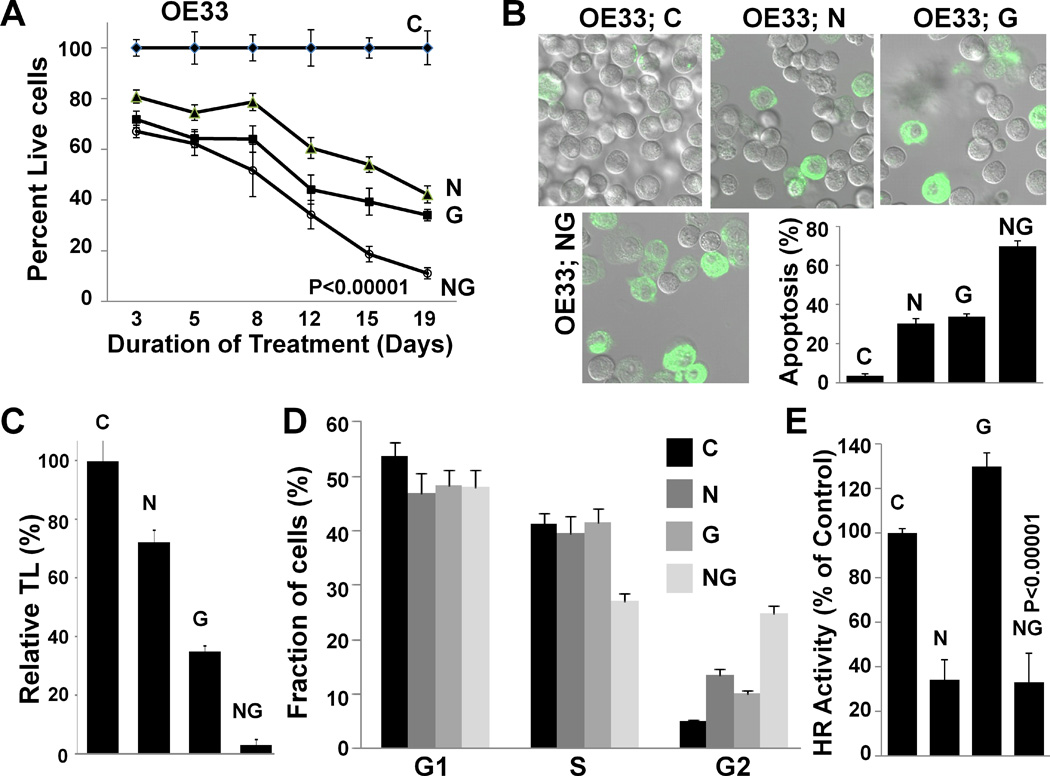
(A) Nilotinib significantly enhances anti-proliferative activity of GRN163L. The BAC (OE33) cells were treated with control oligonucleotide (C), oligonucleotide GRN163L targeting telomerase (G; 2 µM), nilotinib (N; 5 µM), or combination (NG), and viable cell number determined at different time points as indicated. The growth curve shows the mean of triplicate assays, with S.E.M. (B) Impact on apoptosis. OE33 cells, treated as described above for 19 days, were examined for apoptosis by evaluating their ability to bind lactadherin. Extent of green fluorescence indicates early or late apoptosis. (C) Impact on telomere length. OE33 cells, treated as described above for 19 days, were harvested, genomic DNA purified and telomere length determined by real time PCR as described in Methods. Error bars represent SEMs of triplicate assays. (D) Impact on cell cycle. OE33 cells, treated as described above for 19 days, were examined for cell cycle by flow cytometry. (E) Impact on HR activity. OE33 cells, treated as described above for 19 days, were evaluated for HR activity which was assessed using a plasmid based assay as described in Methods. Error bars represent SEMs of triplicate assays.
To extend the observations made in OE33 cells to other cell lines, HR was also suppressed (chemically or transgenically) in OE19 and FLO-1 cell lines (Supplementary Figure 2). Suppression of HR in all three cell lines (OE33, OE19, and FLO-1) led to shorter telomeres, whereas combination of HR and telomerase suppression, caused a significant increase in telomere attrition and apoptotic death, relative to either treatment alone. Overall these data indicate that although telomerase inhibition disrupts telomere maintenance and inhibits cell cycle progression and proliferation, it induces recombinase (RAD51) and HR activity, which contribute to telomere maintenance and prevention of apoptosis. Inhibition of HR, by transgenic or chemical manipulations, also affects above oncogenic activities and when combined with GRN163L, keeps HR downregulated, leading to significantly increased telomere attrition, cell cycle arrest, and apoptosis in OE33 cells.
We have previously shown that transgenically-mediated suppression of HR leads to significant reduction in the acquisition of genomic changes over time in BAC cells42. Here we show that chemical suppression of HR (by nilotinib) also reduces genomic instability in BAC cells. DNA from OE33 cells, untreated or treated with nilotinib (5 µM) for three weeks, was purified and evaluated for acquisition of genomic changes. DNA from cells harvested and frozen at the beginning of experiment (Day 0) was used as baseline genome. Accrual of copy number changes in cultured vs. baseline control cells throughout genome was monitored by investigating single nucleotide polymorphism (SNP) and CNV loci of SNP6.0 arrays (Affymetrix), using Bioconductor and dChip softwares. Copy number changes were reduced both in number and extent by HR inhibitor, nilotinib, throughout genome (Supplementary Figure 3). These observations are consistent with and serve to corroborate our data demonstrating reduction of genomic instability following transgenic-suppression of HR42.
Impact of HR and telomerase suppression on tumor growth and telomere maintenance in vivo
To investigate the effect of HR and/or telomerase suppression in a subcutaneous tumor model of BAC, HR was inhibited either chemically by nilotinib or transgenically by RAD51 knockdown. SCID mice were divided into two subgroups. In first group, the FLO-1 cells were injected subcutaneously and following appearance of tumors, mice treated with either PBS, nilotinib, GRN163L, or the combination. In second group of mice, the tumors were developed by injecting control and RAD51-suppressed FLO-1 cells, and following appearance of tumors, mice treated with PBS or GRN163L. Relative to control mice, the tumor size in mice treated with nilotinib, GRN163L, and the combination was reduced to 47±3%, 48±4%, and 27±6%, respectively (P<0.001–0.002; Figure 5A). In another group of mice, tumors developed by RAD51-suppressed cells without and with GRN163L treatment were 41±5% and 21±6% of control mice, respectively (Figure 5B). Relative to corresponding controls, the tumor sizes resulted from nilotinib, GRN163L, and RAD51-suppression were not significantly different, whereas simultaneous suppression of telomerase and HR (whether mediated chemically or transgenically) resulted in the smallest tumor sizes, significantly smaller than control mice and those treated with GRN163L alone (Figure 5C). Relative to control, treatment with nilotinib, GRN163L, and combination reduced telomeres in tumor cells to 53±8%, 42±6%, and 13±3% of relative telomere length in tumor cells derived from control mice, respectively (P=0.001–0.045; Figure 5D). Thus simultaneous suppression of telomerase and HR led to a significant (29%; P=0.006) increase in telomere shortening, compared to GRN163L alone, in vivo. Simultaneous suppression of telomerase and HR was also associated with ~40% of tumor cells undergoing apoptosis, whereas only 10% of cells had evidence of apoptosis in tumors derived from control mice (Figure 5E).
Figure 5. Impact of HR suppression on antitumor activity of telomerase inhibitor GRN163L.
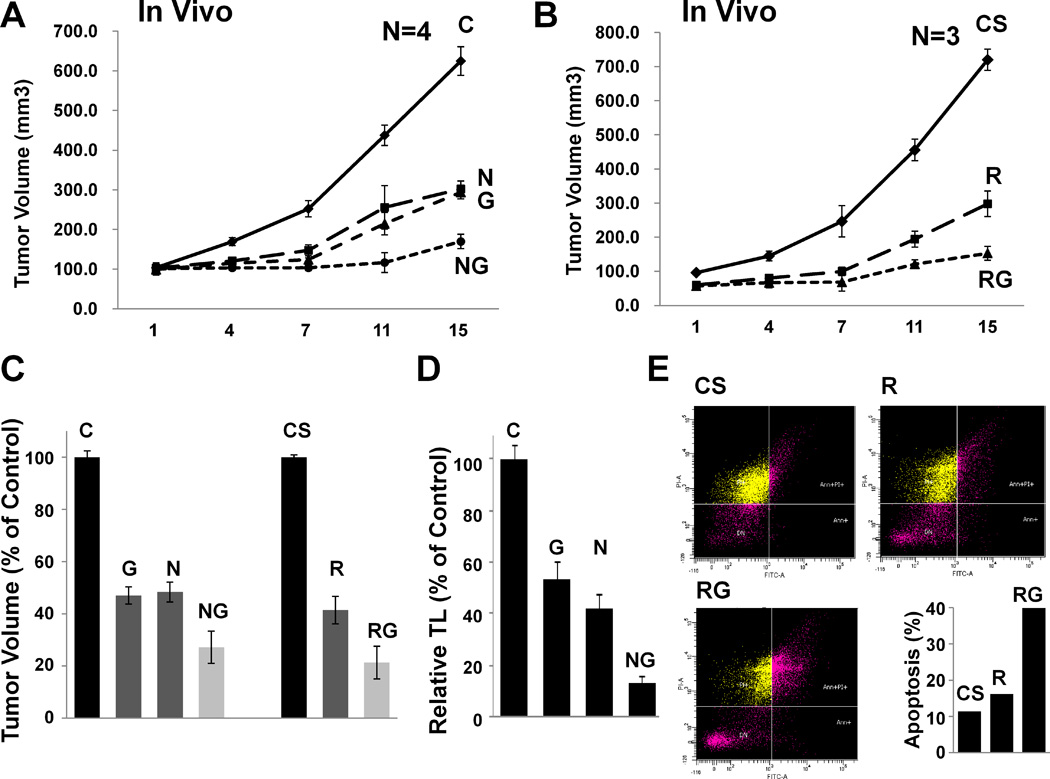
Homologous recombination (HR) activity was suppressed either chemically (by nilotinib) or transgenically (by RAD51 suppression). (A) Impact of nilotinib on efficacy of GRN163L in a subcutaneous tumor model of BAC. FLO-1 cells were injected subcutaneously in the interscapular area of SCID mice. Following appearance of tumors, mice were treated with normal saline containing working dilution of DMSO (control mice), nilotinib (N; 100 mg/kg), GRN163L (G; 45 mg/kg), or combination of nilotinib and GRN163L, injecting four times per week, intraperitoneally. C, control mice; N, mice treated with nilotinib, G, mice treated with GRN163L; NG, mice treated with combination. (B) Impact of RAD51-suppression on efficacy of GRN163L in a subcutaneous tumor model of BAC. FLO-1 cells, transduced with lentivirus particles producing control (CS) or RAD51-targeting (R) shRNAs were cultured for two weeks and injected subcutaneously in the interscapular area of SCID mice. Following appearance of tumors, the mice were treated with normal saline or GRN163L (G; 45 mg/kg), injecting four times per week, intraperitoneally. CS, mice in which tumors were developed by injecting control shRNA-transduced FLO-1 cells; R, mice in which tumors were developed by injecting RAD51 shRNA-transduced FLO-1 cells; RG, R mice treated with GRN163L. (C) Summary of in vivo data. Bar graph summarizes all in vivo data and shows average tumor size in mice subjected to telomerase and/or HR inhibitors. (D) Telomere length in vivo. Tumors from group of mice treated with nilotinib and/or GRN163L were removed and telomere length evaluated by Q-PCR as described in Methods. (E) Evidence of apoptosis in vivo. To evaluate if reduced tumor size was due to apoptotic cell death, tumors from second group of mice were removed and evaluated for apoptosis using flow cytometry. CS, mice in which tumors were developed by injecting control shRNA-transduced FLO-1 cells; R, mice in which tumors were developed by injecting RAD51 shRNA-transduced FLO-1 cells; RG, R mice treated with GRN163L.
DISCUSSION
We have previously shown that telomerase activity is elevated and inhibition of telomerase by GRN163L induces growth arrest and apoptosis in BAC cells17. Telomerase is believed to get activated at or near crisis. Telomeres in most cancer cells are therefore significantly shorter than those in normal cells, but their further shortening is prevented by telomerase-mediated elongation of telomeres23, 24. The fact that telomeres are shorter28, 29 whereas telomerase activity is elevated in most cancer cells20–22 and confers unlimited proliferation potential25–27, makes telomerase a relatively specific and ideal target to inhibit proliferation of cancer cells. Consistent with this, telomerase inhibition achieved by various methods/agents, has been shown to induce growth arrest and senescence and/or apoptosis in a number cancer cell types13, 14, 30–34, 37, 45, 46. However, cell death following telomerase inhibition mostly occurs after a lag period of few weeks, required for telomere shortening below critical length13, 14, 35–37, 46. The requirement of this lag period makes telomerase a somewhat less favorable target, especially for aggressively proliferating cancer cells. Moreover, there is always a possibility that cancer cells exposed to telomerase inhibitor for relatively longer time periods, may activate alternate pathway of telomere elongation, involving homologous recombination (HR)40. Although rarely, but exposure to telomerase inhibitor has led to development of resistance to therapy in certain cancer cell lines (unpublished data from our laboratory). Identification of agents/strategies which may significantly expedite telomere erosion by telomerase inhibitors, can provide novel cancer treatments which may prove to be more effective and less toxic than currently used chemotherapeutics.
We have previously shown that HR activity is spontaneously elevated and contributes to genomic instability in BAC42. Here, we report a novel and important finding that inhibition of telomerase leads to a further increase in HR activity, in all BAC cell lines tested. The induction of HR, following suppression of telomerase by GRN163L, was confirmed by two different methods. Additionally, the suppression of telomerase by shRNA was also associated with increased HR activity in BAC cells. The mechanism of increased HR in BAC cells, following inhibition of telomerease activity, is currently under investigation in our laboratory (manuscript in preparation). Our previous data also show that suppression of elevated HR by RAD51-knockdown, significantly reduces acquisition of genomic changes in BAC cells42. In an effort to search for a chemical inhibitor of HR, we have identified that nilotinib which targets ABL tyrosine kinase, one of the key signaling molecules implicated in HR47, significantly reduces HR activity in a variety of cancer cell lines. Consistent with transgenic suppression of HR, we show that HR suppression by nilotinib also reduces genomic instability in BAC cells.
Elevated HR has also been implicated in telomere maintenance through ALT mechanism40. Telomerase inhibitor, GRN163L, binds to template region of RNA subunit of telomerase and acts as a competitive substrate inhibitor, preventing both the binding to telomerase and the extension process. If, however, telomeric substrate is pre-bound to telomerase, GRN163L may not replace it from the enzyme, at least during one cycle of elongation48. We therefore hypothesized that telomeric ends in BAC cells are mostly occupied by telomerase and relatively inaccessible to RAD51, whereas treatment with GRN163L exposes them for RAD51 to bind. This is consistent with our chip data demonstrating that telomerase inhibition increases RAD51 expression and its association with telomeres.
To evaluate the impact of HR suppression, alone or in combination with GRN163L, HR was inhibited either chemically or transgenically (by RAD51-knockdown). Consistent with our previous observations42, lentiviral shRNA-mediated suppression of RAD51 led to significant reduction in HR activity. We also demonstrated for the first time that nilotinib, a BCR/ABL tyrosine kinase inhibitor, consistently reduces HR activity in BAC (Figure 1) and other cancer cell lines tested by us (not shown). The reduction in HR activity by nilotinib at the dose used in this study was similar as that observed by RAD51-suppression. Suppression of HR, by either method, was associated with telomere shortening (by 23–58%) in all BAC cell lines tested, indicating that besides telomerase, elevated RAD51 and HR also participate in telomere maintenance in these cells, probably by repair/stabilization of eroding telomeres. The increased association of RAD51 to telomeres following telomerase inhibition, could also contribute to telomere stabilization by making them relatively inaccessible to degradation machinery. Since induction of HR following telomerase inhibition was not associated with lengthening of telomeres, it is clear that maintenance of telomeres by RAD51/HR in BAC cells is not associated with any significant increase in their length. Although recombination is usually associated with production of long heterogenous telomeres49, 50, it has been reported that in yeast, the maintenance of telomeres by recombination can also be accomplished through pathways which do not lead to a significant increase in telomere length51. Our data therefore indicate that elevated HR/RAD51 in BAC cells contribute to telomere maintenance/stabilization, providing support to telomerase. In the presence of telomerase inhibitor, HR is further induced to stabilize telomeres, but the absence of continued elongation (by telomerase) leads to gradual telomere shortening. Occasionally the induced HR succeeds in elongating telomeres, leading to development of resistance to telomerase-inhibition (unpublished data from our laboratory). Consistent with this, the suppression of HR significantly increased the efficacy of telomerase inhibitor (GRN163L), leading to a stronger telomere reduction and an expeditious growth arrest and apoptotic death in BAC cells. Combined treatment was also associated with a strong G2/M arrest and senescence, as detected by β-galactosidase staining. Simultaneous suppression of HR and telomerase, in different experiments and cell lines, was associated with 82–94% reduction in median telomere length relative to control cells; the telomeres were significantly smaller than even the cells treated with GRN163L alone (P value range; 0.02–<0.001). The production of smallest telomeres and induction of G2/M arrest by combined treatment in this study are consistent with the observations in yeast showing that extensive telomere shortening induces G2/M arrest52. Moreover, it has been shown that key HR protein RAD51, loaded onto telomeres by BRCA2, significantly contributes to the integrity of telomeric DNA in mouse embryonic fibroblasts53. This further supports our data demonstrating a significant increase in telomere attrition and apoptosis in BAC cells by simultaneous suppression of telomerase and RAD51/HR.
Since telomeres on individual chromosomes are heterogeneous in size and the heterogeneity may be more extensive in cancer cells, the chromosomes with extremely small telomeres are eroded and lost much faster than those with very long telomeres, and erosion of even one or more critical chromosomes may be enough to inhibit cell growth. Infact the length of the smallest telomere is a major determining factor for initiation of senescence in a cell54. Therefore, additional reduction in median telomere length, as seen by combination treatment, is expected to significantly expedite senescence and/or apoptosis. A large telomere reduction in BAC cells following suppression of telomerase and HR activities, over a shorter period of time, could be attributed to several factors, including proliferation rate (doubling time ranging from 18–24 hrs), initial telomere length, and intracellular levels of nucleases etc. For example, mean TRF in OE33, being ~2.0 kbp (Supplementary Figure 1D), is very close to the minimum telomere length required for continued survival of human cells12; thus any additional reduction is expected to cause a rapid loss of telomeres. However, our data does not rule out the possibility that besides impact on telomere shortening, RAD51-suppression and nilotinib may also have other anticancer activities, contributing to increased and expeditious growth arrest.
In summary, this manuscript is the first illustration that: 1) Inhibition of telomerase induces homologous recombination (HR) activity and recombinase (RAD51) in BAC cells; 2) HR contributes to telomere maintenance in telomerase positive human cells without significantly increasing their length; 3) Identifies nilotinib as HR inhibitor; 4) and demonstrates a marked inhibition of genomic instability by nilotinib, a chemical inhibitor of HR, in BAC cells. We also show that inhibition of telomerase increases the association of RAD51 with telomeres in BAC cells. The induced RAD51/HR and association of RAD51 with telomeres contribute to prevention of telomere attrition and apoptosis. Simultaneous suppression of HR and telomerase, significantly increases telomere attrition, leading to G2M arrest, senescence and/or apoptosis both in vitro and in vivo. Combination of GRN163L with transgenic and chemical suppression of HR was associated with the smallest tumor sizes, which were 73% and 79% smaller relative to tumors in control mice, respectively (P=0.001). Since HR is implicated in telomere maintenance and genomic instability, it may contribute to survival of cancer cells through these and probably other roles as well. We are currently using a high throughput assay to screen a library of 300,000 small molecules to identify more specific and potent inhibitors of HR, to target genomic instability and telomere maintenance. A therapeutic strategy, targeting HR and telomerase, has potential to prevent both the tumor growth and genomic evolution in BAC and probably other cancers as well, and our data provide the preclinical rationale for clinical evaluation of HR inhibitors in BAC, in combination with GRN163L.
MATERIALS AND METHODS
Cell lines, drugs, and shRNAs
Barrett’s esophageal adenocarcinoma (BAC) cell lines OE33 and OE1942, 55, 56, purchased from Sigma-Aldrich (St. Louis, MO, USA), were cultured in RPMI-1640 medium, supplemented with 2 mM L-glutamine and 10% fetal bovine serum (FBS). BAC cell line FLO-142, 56, 57, provided by Dr. David G. Beer, University of Michigan, Ann Arbor, MI, USA, was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma Chemical CO., St. Louis, MO, USA) supplemented with 10% FBS. Cells were maintained in a state of logarithmic growth at 37°C in a humidified incubator with 5% CO2. Nilotinib (which targets BCR/ABL tyrosine kinase), siRNA targeting TRF2, and lentivirus particles producing either non-targeted control (CS) shRNAs and those targeting human recombinase RAD51 (RS and RS4) or telomerase (hTERT) were purchased from Sigma-Aldrich. RS mediates a moderate suppression of RAD5142 whereas R4 a strong or near complete (80–100%) suppression56. Telomerase inhibitor GRN163L, a palmitoyl (C16) lipid – attached N3’-P5’ thio-phosphoramidate oligonucleotide (5’-Palm-TAGGGTTAGACAA 3’), targeting RNA component of telomerase is synthesized at Geron Inc., Menlo Park, CA).
Cell treatments, growth kinetics, and apoptosis assays
Telomerase activity in BAC cells was suppressed by oligonucleotide GRN163L or lentivirus based shRNA, targeting telomerase; mismatch oligonucleotide or control shRNA were used as control. HR was reduced either chemically (by treatment with nilotinib) or transgenically (using lentivirus-based RAD51 targeting shRNAs). For transgenic suppression of HR, lentivirus particles, producing non-targeted control (CS) or RAD51-targeting (RS and RS4) shRNAs were transduced into BAC cells as described previously42. The transduced cells were selected in puromycin at concentration of 1 µg/ml for seven days and evaluated for RAD51 suppression by Western blotting. To evaluate the impact on growth, the transduced cells were cultured in the presence of GRN163L or mismatch control oligonucleotide. In another set of experiments, BAC cells were treated with nilotinib and GRN163L, either alone or in combination. For all growth kinetics experiments, the untreated and treated cells were plated in triplicate at constant number and cultured for various durations. Cells were harvested at indicated time points, viability assessed by counting the number of attached cells, and confirmed by trypan blue exclusion and MTT assays. Aliquots of cells were collected for various analyses and the remaining cells replated in new flasks at the same cell number and at drug concentration/s.
Apoptotic cells were detected by their ability to bind lactadherin, which interacts with phosphatidylserine exposed during early apoptosis, as reported previously58. Briefly, the cell samples were incubated in dark with FITC-labeled lactadherin (Haematologic Technologies, Inc.) at RT for 10 minutes. Cells were then centrifuged at RT and the pellets resuspended gently in Tyrode solution (140 mM NaCl, 2.7 mM KCl, 12 mM NaHCO3, 0.42 mM NaH2PO4, 1 mM MgCl2, 2.5 mM CaCl2, 5.5 mM glucose, 5 mM HEPES, 0.35% bovine serum albumin; pH adjusted to 7.4 with 1.5 mM CaCl2). Cells were immediately viewed and photographed under a confocal microscope (LSM 710; Zeiss) as described previously. Approximately 100–200 cells, representing at least five different microscopic fields, were examined to assess the fraction of apoptotic (FITC-labeled) cells for each sample.
Western blot analyses
Protein extracts from untreated or treated BAC cells were made and processed as described previously42,56,59. Expression of RAD51 was detected using an enhanced chemiluminescence system, according to the instructions provided by the manufacturer (Amersham Life Sciences Inc., Arlington Heights, IL, USA).
Chromatin immunoprecipitation (chip) assays
The chip assays were performed using Chromatin Immunoprecipitation Assay kit (USB corporation, Cleveland, Ohio, USA) as described by the manufacturer. Briefly, the cells treated with telomerase and/or HR inhibitors for 48 hrs were harvested at 70–80% confluence and fixed in 1% formaldehyde. After rinsing with PBS, the cell pellets were resuspended in lysis buffer and sonicated to fragment chromatin DNA. Pre-blocked Protein A agarose beads were added to the supernatant, incubated on a shaker for 1 hour and centrifuged. Anti-RAD51 antibody was added to the supernatant, samples incubated on the shaker overnight, and then mixed with pre-blocked Protein A agarose beads. The complex was washed sequentially with lysis buffer, high salt buffer, lithium salt buffer, and TE buffer (pH 7.5). Elution buffer was then added to the washed beads and following 20 min incubation, NaCl was added and beads incubated at 65°C overnight. DNA complex was extracted using phenol/chloroform/isoamyl alcohol and resuspended in TE buffer. PCR, using primers specific to telomeres and single-copy (One gene; used as internal control) were conducted to assess the fraction of telomeric DNA associated with RAD51.
Assay of Telomerase Activity
Telomerase activity was measured using the TRAPeze XL Telomerase Detection Kit (Chemicon International, Inc.Temecula, CA) as described by the manufacturer and reported by us previously13, 14, 35. Briefly, the cells were lysed in CHAPS buffer (Chemicon International, Inc) in triplicate, mixed with TRAPeze XL reaction mix (containing fuorescent primers), and incubated for 30 minutes at 30°C. Telomerase products were amplified by PCR and quantitated with a fluorescence plate reader. Telomerase activity, for each lysate, was calculated from the ratio of telomerase products to an internal standard, as instructed by the manufacturer.
Telomere length analysis
Genomic DNA from untreated and treated cells was purified using DNeasy Tissue Kit (Qiagen Inc.), as described by the manufacturer. A slight modification of a previously described real-time PCR based assay was performed, using Applied Biosystems 7900HT Thermocycler60. Average relative telomere length was presented as telomere repeat and single-copy (One gene) copy-number (T/S) ratio, as described previously17. Briefly, the genomic DNA in 96-well plate was resuspended in either the telomere or One gene qPCR reaction mixtures containing corresponding primers60, in triplicate. Since PCR product is approximately doubled in each cycle of the PCR, the T/S ratio is [2Ct(Telomeres)/2Ct(One gene)]−1 = 2−ΔCt. The relative TS ratio (T/S of one sample relative to the T/S of another sample) is 2−(ΔCt1−ΔCt2) = 2−ΔΔCt. Using this formula, a relative T/S ratio for each of the 96 wells was determined, as the T/S of the well relative to the mean T/S for all 96 wells. Standard curve was made by plotting T/S ratio of four cell lines (HTB, ARP, RPMI, U266) for which median telomere length in kbp has been determined by gel-based analyses; TL of samples in kbp was derived from this standard curve.
Homologous Recombination Assays
Homologous recombination (HR) activity was assessed by two different methods, the luminescence-based HR assay established in our laboratory42 and fluorescence-based HR assay, available commercially (Addgene, Cambridge, MA)44. The HR substrate plasmid developed by us contains two incomplete fragments of a firefly luciferase (Fluc) gene, sharing a small region of homology. The Fluc fragments are separated by an AmpR gene, serving as spacer. HR between homologous sequences of two Fluc fragments generates a functional gene, resulting in the excision of the AmpR gene. The plasmid also has a Gaussia luciferase (Gluc) gene which serves as an internal control and is not affected by recombination. This plasmid is introduced into BAC cells, the cells are incubated for an appropriate duration, harvested, and the HR is assessed from the ratio of two luciferase activities. Fluorescence-based HR assay substrate (pDRGFP; Addgene44) is comprised of two defective copies of GFP, separated by a drug resistance marker. One of the GFP contains the restriction site for I-Sce I enzyme; the introduction of break at this site promotes homology-based recombination between two mutated genes, generating a functional GFP. FLO-1 cells stably transfected with HR substrate (Addgene), were transfected with a plasmid expressing I-Sce I enzyme. The cells were then plated in two different flasks, one treated with control oligonucleotide and the other with GRN163L as treated for other HR assay and subsequently evaluated for confocal microscopy. HR was assessed from fluorescence intensity of each microscopic field divided by total number of cells in the corresponding field. Average background fluorescence/cells, determined from untransfected cells, was subtracted from values of transfected cells.
Assessment of genomic instability
BAC cells were cultured in the presence or absence of nilotinib. An aliquot of cells was harvested and frozen at the beginning of the experiment (Day 0), to be used as a reference. The acquisition of genomic changes, relative to reference cells, was monitored by genome-wide screens for copy-number alterations, using single nucleotide polymorphism (SNP) arrays (Affymetrix) and dchip software as described previously42.
Senescence
One day before this assay, the treated cells were plated on Lab-Tek slides in the presence of mismatch or match (GRN163L) oligonucleotide and the attached cells were stained for β-galactosidase expression, a marker for cellular senescence61H. Briefly, the cells were rinsed three times with PBS and fixed in 2% formaldehyde and 0.2% glutaraldehyde solution in PBS. The cells were then washed again as described above and stained overnight in solution containing 1 mg/ml X-gal, 40 mM citric acid/sodium phosphate (pH 6), 5 mM potassium ferrocyanide, 150 mM NaCl, and 2 mM MgCl2. The stain was then removed, cells were rinsed with PBS, and staining was viewed under a fluorescence microscope (Olympus).
Impact of HR/Telomerase suppression in a subcutaneous tumor model
Homologous recombination (HR) activity was suppressed either chemically (by nilotinib) or transgenically (by RAD51-knockdown). Five week old male CB-17 SCID mice were purchased from Charles River Laboratories (Wilmington, MA) and maintained following guidelines of the Institutional Animal Care and Use Committee (IACUC); all experimental procedures were approved by the IACUC and the Occupational Health and Safety Department of Dana Farber Cancer Institute, Boston, MA, USA. FLO-1 cells (3.0×106 in 100 µl saline) were injected subcutaneously into the interscapular area of each mouse. Following appearance of palpable tumors (~7–10 days), the mice were treated with normal saline containing working dilution of solvent DMSO, nilotinib (100 mg/kg dissolved in DMSO, diluted in normal saline), GRN163L (40 mg/kg dissolved in normal saline), or combination of nilotinib and GRN163L, injecting four times per week, intraperitoneally. To evaluate if efficacy of GRN163L can be enhanced by RAD51 suppression, FLO-1 cells transduced with control (CS) or RAD51-specific (R) shRNAs were cultured for two weeks and then injected subcutaneously into the mice as described above. After appearance of tumors, the mice were treated with either normal saline or GRN163L (40 mg/kg dissolved in normal saline), injecting four times a week. Tumor sizes were measured twice a week and animals sacrifized when tumors reached 2 cm3 in volume or when paralysis or major compromise in their quality of life occurred.
Supplementary Material
Acknowledgements
We are grateful to Dr. David G. Beer, Department of Surgery, University of Michigan, Ann Arbor, MI for providing FLO-1 cells.
FUNDING: This work was supported in part by grants from National Cancer Institute R01CA125711 to MAS, from the Dept. of Veterans Affairs Merit Review Awards (to NCM) and from the National Institutes of Health Grants RO1-1375555, P50-100007 and PO1-78378 to NCM.
ABBREVIATIONS
- HR
Homologous recombination
- BAC
Barrett’s esophageal adenocarcinoma
Footnotes
Conflict of interest
The authors declare no conflict of interest.
REFERENCES
- 1.Allshire RC, Gosden JR, Cross SH, et al. Telomeric repeat from T. thermophila cross hybridizes with human telomeres. Nature. 1988;332:656–659. doi: 10.1038/332656a0. [DOI] [PubMed] [Google Scholar]
- 2.de Lange T, Shiue L, Myers RM, et al. Structure and variability of human chromosome ends. Mol Cell Biol. 1990;10:518–527. doi: 10.1128/mcb.10.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moyzis RK, Buckingham JM, Cram LS, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith JD, Comeau L, Rosenfield S, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 5.van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 6.Day JP, Marder BA, Morgan WF. Telomeres and their possible role in chromosome stabilization. Environ Mol Mutagen. 1993;22:245–249. doi: 10.1002/em.2850220411. [DOI] [PubMed] [Google Scholar]
- 7.Blasco MA, Lee HW, Hande MP, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA [see comments] Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 8.Collins K. Mammalian telomeres and telomerase. Curr Opin Cell Biol. 2000;12:378–383. doi: 10.1016/s0955-0674(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 9.Ducray C, Pommier JP, Martins L, et al. Telomere dynamics, end-to-end fusions and telomerase activation during the human fibroblast immortalization process. Oncogene. 1999;18:4211–4223. doi: 10.1038/sj.onc.1202797. [DOI] [PubMed] [Google Scholar]
- 10.McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet. 2000;34:331–358. doi: 10.1146/annurev.genet.34.1.331. [DOI] [PubMed] [Google Scholar]
- 11.Shammas MA, Rao MY. Purification of diseased cells from Barrett's esophagus and related lesions by laser capture microdissection. Methods Mol Biol. 2011;755:181–187. doi: 10.1007/978-1-61779-163-5_14. [DOI] [PubMed] [Google Scholar]
- 12.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 13.Shammas MA, Koley H, Batchu RB, et al. Telomerase inhibition by siRNA causes senescence and apoptosis in Barrett's adenocarcinoma cells: mechanism and therapeutic potential. Mol Cancer. 2005;4:24. doi: 10.1186/1476-4598-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shammas MA, Koley H, Beer DG, et al. Growth arrest, apoptosis, and telomere shortening of Barrett's-associated adenocarcinoma cells by a telomerase inhibitor. Gastroenterology. 2004;126:1337–1346. doi: 10.1053/j.gastro.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 15.Shammas MA, Koley H, Bertheau RC, et al. Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia. 2008;22:1410–1418. doi: 10.1038/leu.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shammas MA, Liu X, Gavory G, et al. Targeting the single-strand G-rich overhang of telomeres with PNA inhibits cell growth and induces apoptosis of human immortal cells. Experimental Cell Research. 2004;295:204–214. doi: 10.1016/j.yexcr.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Shammas MA, Qazi A, Batchu RB, et al. Telomere maintenance in laser capture microdissection-purified Barrett's adenocarcinoma cells and effect of telomerase inhibition in vivo. Clin Cancer Res. 2008;14:4971–4980. doi: 10.1158/1078-0432.CCR-08-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harley CB. Telomere loss: mitotic clock or genetic time bomb? Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- 19.Blackburn EH, Greider CW, Henderson E, et al. Recognition and elongation of telomeres by telomerase. Genome. 1989;31:553–560. doi: 10.1139/g89-104. [DOI] [PubMed] [Google Scholar]
- 20.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 21.Shay JW. Telomerase in human development and cancer. J Cell Physiol. 1997;173:266–270. doi: 10.1002/(SICI)1097-4652(199711)173:2<266::AID-JCP33>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 22.Shay JW, Wright WE. The reactivation of telomerase activity in cancer progression. Trends Genet. 1996;12:129–131. doi: 10.1016/0168-9525(96)30018-8. [DOI] [PubMed] [Google Scholar]
- 23.Engelhardt M, Martens UM. The implication of telomerase activity and telomere stability for replicative aging and cellular immortality (Review) Oncol Rep. 1998;5:1043–1052. doi: 10.3892/or.5.5.1043. [DOI] [PubMed] [Google Scholar]
- 24.Wai LK. Telomeres, telomerase, and tumorigenesis--a review. Med Gen Med. 2004;6:19. [PMC free article] [PubMed] [Google Scholar]
- 25.Counter CM, Avilion AA, LeFeuvre CE, et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. Embo J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer [see comments] Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 27.Lord RV, Salonga D, Danenberg KD, et al. Telomerase reverse transcriptase expression is increased early in the Barrett's metaplasia, dysplasia, adenocarcinoma sequence. J Gastrointest Surg. 2000;4:135–142. doi: 10.1016/s1091-255x(00)80049-9. [DOI] [PubMed] [Google Scholar]
- 28.Bechter OE, Eisterer W, Pall G, et al. Telomere length and telomerase activity predict survival in patients with B cell chronic lymphocytic leukemia. Cancer Res. 1998;58:4918–4922. [PubMed] [Google Scholar]
- 29.Shammas MA, Shmookler Reis RJ, Akiyama M, et al. Telomerase inhibition and cell growth arrest by G-quadruplex interactive agent in multiple myeloma. Molecular Cancer Therapeutics. 2003;2:825–833. [PubMed] [Google Scholar]
- 30.Fu W, Begley JG, Killen MW, et al. Anti-apoptotic role of telomerase in pheochromocytoma cells. J Biol Chem. 1999;274:7264–7271. doi: 10.1074/jbc.274.11.7264. [DOI] [PubMed] [Google Scholar]
- 31.Nakajima A, Tauchi T, Sashida G, et al. Telomerase inhibition enhances apoptosis in human acute leukemia cells: possibility of antitelomerase therapy. Leukemia. 2003;17:560–567. doi: 10.1038/sj.leu.2402825. [DOI] [PubMed] [Google Scholar]
- 32.Sumi M, Tauchi T, Sashida G, et al. A G-quadruplex-interactive agent, telomestatin (SOT-095), induces telomere shortening with apoptosis and enhances chemosensitivity in acute myeloid leukemia. Int J Oncol. 2004;24:1481–1487. [PubMed] [Google Scholar]
- 33.Tauchi T, Shin-Ya K, Sashida G, et al. Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: involvement of ATM-dependent DNA damage response pathways. Oncogene. 2003;22:5338–5347. doi: 10.1038/sj.onc.1206833. [DOI] [PubMed] [Google Scholar]
- 34.Seimiya H, Oh-hara T, Suzuki T, et al. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991. Molecular Cancer Therapeutics. 2002;1:657–665. [PubMed] [Google Scholar]
- 35.Shammas MA, Reis RJ, Li C, et al. Telomerase inhibition and cell growth arrest after telomestatin treatment in multiple myeloma. Clin Cancer Res. 2004;10:770–776. doi: 10.1158/1078-0432.ccr-0793-03. [DOI] [PubMed] [Google Scholar]
- 36.Akiyama M, Hideshima T, Shammas MA, et al. Effects of oligonucleotide N3'-->P5' thio-phosphoramidate (GRN163) targeting telomerase RNA in human multiple myeloma cells. Cancer Research. 2003;63:6187–6194. [PubMed] [Google Scholar]
- 37.Shammas MA, Simmons CG, Corey DR, et al. Telomerase inhibition by peptide nucleic acids reverses 'immortality' of transformed human cells. Oncogene. 1999;18:6191–6200. doi: 10.1038/sj.onc.1203069. [DOI] [PubMed] [Google Scholar]
- 38.Bryan TM, Englezou A, Gupta J, et al. Telomere elongation in immortal human cells without detectable telomerase activity. Embo J. 1995;14:4240–4248. doi: 10.1002/j.1460-2075.1995.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Opitz OG, Suliman Y, Hahn WC, et al. Cyclin D1 overexpression and p53 inactivation immortalize primary oral keratinocytes by a telomerase-independent mechanism. J Clin Invest. 2001;108:725–732. doi: 10.1172/JCI11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunham MA, Neumann AA, Fasching CL, et al. Telomere maintenance by recombination in human cells.[see comment] Nature Genetics. 2000;26:447–450. doi: 10.1038/82586. [DOI] [PubMed] [Google Scholar]
- 41.Cerone MA, Londono-Vallejo JA, Bacchetti S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum Mol Genet. 2001;10:1945–1952. doi: 10.1093/hmg/10.18.1945. [DOI] [PubMed] [Google Scholar]
- 42.Pal J, Bertheau R, Buon L, et al. Genomic evolution in Barrett's adenocarcinoma cells: critical roles of elevated hsRAD51, homologous recombination and Alu sequences in the genome. Oncogene. 2011;30:3585–3598. doi: 10.1038/onc.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shammas MA, Shmookler Reis RJ, Koley H, et al. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood. 2009;113:2290–2297. doi: 10.1182/blood-2007-05-089193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pierce AJ, Johnson RD, Thompson LH, et al. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Munshi NC, Hideshima T, Carrasco D, et al. Identification of genes modulated in multiple myeloma using genetically identical twin samples. Blood. 2004;103:1799–1806. doi: 10.1182/blood-2003-02-0402. [DOI] [PubMed] [Google Scholar]
- 46.Shammas MA, Shmookler Reis RJ, Li C, Koley H, Hurley LH, Anderson KC, Munshi NC. Telomerase Inhibition and Cell Growth Arrest Following Telomestatin Treatment in Multiple Myeloma. Clinical Cancer Research. 2003 doi: 10.1158/1078-0432.ccr-0793-03. In Press. [DOI] [PubMed] [Google Scholar]
- 47.Ganapathipillai SS, Medova M, Aebersold DM, et al. Coupling of mutated Met variants to DNA repair via Abl and Rad51. Cancer Res. 2008;68:5769–5777. doi: 10.1158/0008-5472.CAN-08-1269. [DOI] [PubMed] [Google Scholar]
- 48.Wallweber G, Gryaznov S, Pongracz K, et al. Interaction of human telomerase with its primer substrate. Biochemistry. 2003;42:589–600. doi: 10.1021/bi026914a. [DOI] [PubMed] [Google Scholar]
- 49.Dunham MA, Neumann AA, Fasching CL, et al. Telomere maintenance by recombination in human cells. Nat Genet. 2000;26:447–450. doi: 10.1038/82586. [DOI] [PubMed] [Google Scholar]
- 50.Henson JD, Neumann AA, Yeager TR, et al. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. doi: 10.1038/sj.onc.1205058. [DOI] [PubMed] [Google Scholar]
- 51.Morrish TA, Greider CW. Short telomeres initiate telomere recombination in primary and tumor cells. PLoS Genet. 2009;5:e1000357. doi: 10.1371/journal.pgen.1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.AS IJ, Greider CW. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:987–1001. doi: 10.1091/mbc.02-04-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Badie S, Escandell JM, Bouwman P, et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol. 2010;17:1461–1469. doi: 10.1038/nsmb.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdallah P, Luciano P, Runge KW, et al. A two-step model for senescence triggered by a single critically short telomere. Nat Cell Biol. 2009;11:988–993. doi: 10.1038/ncb1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogunwobi OO, Beales IL. Statins inhibit proliferation and induce apoptosis in Barrett's esophageal adenocarcinoma cells. Am J Gastroenterol. 2008;103:825–837. doi: 10.1111/j.1572-0241.2007.01773.x. [DOI] [PubMed] [Google Scholar]
- 56.Pal J, Fulciniti M, Nanjappa P, et al. Targeting PI3K and RAD51 in Barrett's adenocarcinoma: impact on DNA damage checkpoints, expression profile and tumor growth. Cancer Genomics Proteomics. 2012;9:55–66. [PMC free article] [PubMed] [Google Scholar]
- 57.Aggarwal S, Taneja N, Lin L, et al. Indomethacin-induced apoptosis in esophageal adenocarcinoma cells involves upregulation of Bax and translocation of mitochondrial cytochrome C independent of COX-2 expression. Neoplasia. 2000;2:346–356. doi: 10.1038/sj.neo.7900097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi J, Shi Y, Waehrens LN, et al. Lactadherin detects early phosphatidylserine exposure on immortalized leukemia cells undergoing programmed cell death. Cytometry A. 2006;69:1193–1201. doi: 10.1002/cyto.a.20345. [DOI] [PubMed] [Google Scholar]
- 59.Shammas MA, Neri P, Koley H, et al. Specific killing of multiple myeloma cells by (−)-epigallocatechin-3-gallate extracted from green tea: biologic activity and therapeutic implications. Blood. 2006;108:2804–2810. doi: 10.1182/blood-2006-05-022814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.