

Abstract
In vitro three-dimensional models of cancer have the ability to recapitulate many features of tumors found in vivo, including cell-cell and cell-matrix interactions, microenvironments that become hypoxic and acidic, and other barriers to effective therapy. These model tumors can be large, highly complex, heterogeneous, and undergo time-dependent growth and treatment response processes that are difficult to track and quantify using standard imaging tools. Optical coherence tomography is an optical ranging technique that is ideally suited for visualizing, monitoring, and quantifying the growth and treatment response dynamics occurring in these informative model systems. By optimizing both optical coherence tomography and 3D culture systems, it is possible to continuously and non-perturbatively monitor advanced in vitro models without the use of labels over the course of hours and days. In this article, we describe approaches and methods for creating and carrying out quantitative therapeutic screens with in vitro 3D cultures using optical coherence tomography to gain insights into therapeutic mechanisms and build more effective treatment regimens.
Keywords: 3D in vitro models, optical coherence tomography, time-lapse, overlay culture, image analysis, therapeutic response
1. Introduction
Therapeutics have traditionally been chosen through a process of trial and error, where the selection of potential candidates can be time consuming, laborious, and expensive. While trial and error methods can be acceptable in the fight against some slow-growing cancers, in many cases there is a pressing need to select optimal therapeutics; ineffective therapies can lead to the loss of precious time in fighting growing cancers, while treatments with only moderate success can drive the evolution of therapeutic resistance in patients. For many patients diagnosed with fast growing or advanced stage cancers, such as metastatic ovarian cancer, the use of emergent technologies to build personalized therapies on a case-by-case basis are sorely needed.
Live-cell assays utilizing patient-derived samples can provide important insights into how an individual will respond to therapy. Cultured cells have been extensively used for therapeutic screening, with readouts including traditional metrics of cell death (necrosis, apoptosis), gene expression changes, and morphologic and phenotypic cellular alterations. These approaches have revealed insights into different stages of treatment response, and can be used to identify windows of opportunity for targeted therapy [1]. The downside to single-cell approaches is that the artificial testing environment insufficiently recapitulates the disease in vivo. Therapeutic response in monolayer cellular cultures often overestimates treatment efficacy, and can miss the effects of key parameters affecting treatment outcome including microenvironmental variables such as perfusion, hypoxia, and acidosis.
Three-dimensional (3D) cultures are far more biologically relevant in that they provide many of the important cell-cell and cell-matrix interactions that occur within tumors and can be engineered to replicate crucial microenvironmental features found in vivo. These complex culture systems come in many varieties, including suspension, overlay, and embedded models [2–6], as well as natural and synthetic scaffold-based cultures [7]. Cancer cells grown into 3D cultures can be curated to develop into nodules that replicate morphological parameters of clinical disease, such as the formation of hollow lumens [4] and hypoxic microenvironments in the cores of large tumors [8, 9]. With the ability to provide important therapeutically relevant features and parameters of disease in vivo, 3D culture systems have the ability to more closely replicate the dose-dependent therapeutic responses observed in patients [10].
As 3D cancer culture systems are typically heterogeneous, optical imaging has been found effective in monitoring treatment response and enabling analysis of the complex information 3D culture systems provides. Many approaches, such as histology and fluorescence staining, however, can miss critical dynamic treatment response information, as they require either fixation or the introduction of cytotoxic compounds. Live-cell stains and autofluorescence imaging can achieve non-perturbative imaging, but the limited penetration depth of both fluorescent agents and light into 3D cultures makes examining large (> 500 μm diameter) nodules difficult.
Optical coherence tomography (OCT) [11, 12] offers considerable advantages for monitoring therapeutic response in 3D tissue culture systems. As an optical ranging technique, OCT is analogous to ultrasound and is used to rapidly generate 3D image volumes of samples. OCT systems can be made to probe millimeters deep into samples with subcellular spatial resolution, allowing for detailed cellular analysis throughout entire in vitro cultures. The contrast in OCT derives from the reflection of light from samples, and as such, does not require the use of exogenous labels. Using only a few hundred microwatts of infrared light, time-lapse OCT (TL-OCT) can continuously capture treatment response dynamics in 3D cultures over the course of days and even weeks. Moreover, OCT data can be readily mined and quantified, allowing for comprehensive, 3D mapping of treatment response over time.
In this article, we will introduce methods and provide guidance in demonstrating how to create and optimize OCT systems for use with 3D cultures, integrate such a system into a commercial microscopy platform, and carry out high-content treatment response studies using biologically-relevant 3D culture models. In addition, we will demonstrate an approach for analyzing the generated data sets to reveal insights into therapeutic response mechanisms.
2) Selecting and Building OCT Systems for In Vitro Screening
OCT is based on low-coherence interferometry, where depth information is collected by interfering light reflected from two separate but equal path lengths. Most OCT systems are configured in a Michelson interferometer design: broadband input light is divided by a beam splitting optic into two orthogonal paths, here called the sample arm and the reference arm. Light reflected from the ends of these arms travels back to the beam splitter; if the paths are equal, a strong interference signal is observed. OCT takes advantage of the fact that light can be reflected off of many different surfaces in a sample, each of which creates a contribution to the interferometric signal. Early OCT systems operated in the so-called time-domain: a movable mirror at the end of the reference arm would oscillate over the depth scan range to generate interferometric fringes corresponding to each reflecting surface in the sample [13, 14]. While these time-domain OCT systems (TD-OCT) had much success, they have been superseded by faster spectral (SD-OCT) or frequency (FD-OCT) OCT systems that offer lower noise operation [15]. In these spectral domain systems, the reference arm length is held constant, and instead the entire spectrum of the interferogram is collected on a camera. The depth scan is recovered by simply Fourier transforming the spectral interferogram. Swept-source spectral domain OCT systems have recently been created, including optical frequency domain imaging (OFDI) [16] and frequency domain mode-locked OCT (FDML-OCT) [17], that offer significant speed, depth and sensitivity enhancements.
Like any optical system, OCT imaging platforms can be designed for different goals and end uses. OCT systems developed for in vitro screening have different requirements than those typically built for high-speed in vivo imaging. For most screening requirements, in vitro imaging systems rarely require high data acquisition rates or multiple millimeters of imaging depth for 3D culture systems. In vitro culture systems are typically no more than 1–2 mm in thickness, with experiments generally focused on monitoring growth, migration, and cellular death. For such processes, high resolution is paramount, necessitating the use of broadband light sources. In OCT, the spectral interferogram and depth scan are related by a Fourier transform. Thus, the light source spectral bandwidth is inversely related to system axial resolution: the broader the light source, the more axial resolution afforded. Commercial light sources for OCT are available in three main wavelength regimes: 750–960 nm, 1000–1100 nm, and 1250–1400 nm ranges. Of these three regimes, light sources centered about 800 nm generally offer the greatest bandwidth, and thus axial resolution. For example, a recent μOCT system utilizing extreme broadband light from a supercontinuum source [18] was capable of visualizing cilia in vitro with < 1 μm of axial resolution.
Another important design criteria for in vitro OCT systems is imaging depth, for which there are two considerations. First, the OCT imaging range is limited via the Niquist sampling theorem by the spectral interference frequency resolution. As interferometric modulation frequency increases with distance from time-zero, higher interference frequency resolutions allow for greater ranging depths in samples. In SD-OCT systems the sampling frequency is simply the spectral bandwidth divided by the number of lateral elements in the detection camera. For swept source systems, the sampling frequency is the instantaneous linewidth of the wavelength-tunable laser. While the spectral sampling frequency sets the maximum theoretical imaging range of the system, the penetration depth itself is governed by light scattering in the turbid 3D cultures. Longer wavelengths encounter the least amount of scattering, with systems operating at 1300 nm offering more than 2 mm of potential imaging depth. Unfortunately, current long wavelength systems do not offer large bandwidths, with many 1300 nm-centered systems offering about 7 μm of axial resolution.
For most in vitro time-lapse or longitudinal imaging experiments, the most important metric is stability: system alignment as well as the light source power and spectra need to be stable over days and even weeks for quantitative imaging. In general, the most stable systems are those with as few moving parts as possible. For this reason, time-domain OCT systems may not be ideal, and swept-source OCT systems (OFDI or FDML) need to be rigorously vetted for long term stability. For our in vitro therapeutic screens, we chose an SD-OCT system based on a superluminescent diode light source and grating/camera pair, as similar devices have been found to offer excellent stability over periods of a week or more [19].
For the following protocols described here, we used an overlay culture system [4, 10, 19, 20] to model the studding micrometastatic lesions found in ovarian cancer. These model tumors can grow to sizes of 500–1000 μm in diameter, requiring optical ranging of about 1 mm. 3D tumor nodules are moderately light scattering, which can occasionally lead to difficulties imaging with 800 nm centered OCT systems at depths exceeding 900 μm. Typical growth and treatment response changes occur over the course of hours and days, requiring a highly stable system that can be left to run continuously if desired. Treatment response in model tumors encompasses both large-scale changes, such as fragmentation and matrix invasion, and cellular-level alterations, such as apoptosis and senescence, requiring resolutions better than 3–4 μm. To achieve these goals, we chose to build an SD-OCT system (Fig. 1) using a superluminescent diode source (Superlum, HP855) that provided a stable high bandwidth spectrum (130 nm FWHM) at an output power of 15 mW [21]. The high output power of this source enabled strong reflection signals to be collected from the sample for high signal-to-noise imaging (80 dB) at moderate acquisition rates (5000 frame/s). At these acquisition rates, an entire 96-well plate containing many thousands of model tumors could be screened in less than 30 minutes. As only 1 mm of penetration depth is required for the ovarian tumor culture model, spectral interferograms could be collected on a single 2048 element, single-tap CCD linescan chip (L104k, Basler). The interferometer itself was based off a simple fiber Michelson interferometer (AC Photonics) that provided stable, low-drift operation.
Fig. 1.

Schematic of an example SD-OCT system combined with simple confocal microscope. Light from a broadband light source is coupled into a fiber Michelson interferometer with four arms. The sample arm provides light to the microscope imaging system, while the reference arm is designed to have the same optical path length as the sample arm. Interference between the sample and reference arms is detected on a custom spectrometer. OL: objective lens, M: mirror, DM: dichroic mirror, L: lens, TS: translation stage, PH: pinhole, PMT: photomultiplier tube, FC: fiber coupler, FS: 80/20 fiber splitter, LC: line-scan camera, ST: 5-axis stage, G: transmission grating.
For time-lapse experiments that last days and even weeks, additional steps must be taken to ensure stability of the imaging system. All components must be secured to either a thick breadboard or laser table, and covered by housing material to protect them from room air flow. We have preferred clear acrylic housing materials as they allow both isolation of equipment and the ability to check beam alignment with an infrared viewer. All fiber components should be secured to prevent polarization drift, and a polarization compensator is recommended to help adjust for any long-term drift. A common source of mechanical drift is spring-based mechanical stages. For example, five-axis mechanical stages used to mount and adjust linescan cameras, can lose mechanical stability through constant use, leading to long-term drift that can alter the collected spectral interferogram. Another simple, but difficult to trace source of drift can be room temperature changes. For example, many buildings will often cycle their daytime, nighttime, and weekend ambient temperatures. Such temperature changes can cause periodic drift over long-term experiments that can influence quantitative analysis. In these situations, small heating units or heating tape can be placed around acrylic housings on a control loop to stabilize interferometer temperatures.
It is worth noting that continuous operation of OCT systems over the course of days requires considerable data storage space. A high-resolution (e.g. 2000 × 250 × 2048 element) array acquired every half hour over the course of several days can generate hundreds of gigabytes of data for a single sample position. Screening multiple positions across multiple wells to track treatment response over the course of days can easily generate terabytes of data. It is important to keep these data storage requirements in mind when building and setting up OCT therapeutic screening experiments.
2.1) Integration with Fluorescence Microscopy Systems
A major benefit of OCT is that it can be readily combined with other optical imaging modalities, including wide-field fluorescence, confocal microscopy, and multiphoton imaging technologies. Such a multimodal approach offers significant advantages: OCT can be used for continuous, non-perturbative imaging over long time periods, but does not offer molecular sensitivity. By combining OCT with fluorescence contrast, for example, specific cellular components or processes can be detected within the structural context provided by optical ranging. The addition of confocal fluorescence to a home-built OCT system is relatively straightforward: a dichroic mirror can be introduced into the OCT system allowing the fluorescence excitation and emission light to pass through the scanning and focusing optics (Fig. 1). Multiphoton capabilities can also be added to OCT systems either by ensuring the wavelengths of the two light sources are separate, or by combining the light sources at orthogonal polarizations.
Adding OCT capabilities to existing confocal and multiphoton fluorescence microscopes can be more difficult, especially commercial systems, as the scanning optics are typically controlled by proprietary hardware and software that offer few, if any, integration capabilities. Fortunately, some commercial microscope vendors have recently made efforts to allow for add-on capabilities. One such vendor is Olympus, Inc, whose FV1000 confocal microscope system offers a hardware input/output component that provides key signals required for integration of an OCT system.
2.1.1) General Protocol for Integrating an Existing SD-OCT System with Olympus FV1000 Hardware
This protocol will make use of the SD-OCT system described in the above section [19]. To illustrate the individual components of the imaging system, Figure 2 shows the OCT beam light path into and through the FV1000 scanhead and inverted microscope base. While standard Olympus confocal microscopes will have a reflector in the turret for confocal scanning, this reflector is a dichroic mirror that does not efficiently transmit near infrared light. A protected silver mirror (Fig. 2, M1) should be mounted in a cube in the turret to allow for near infrared light delivery to the objective lens before beginning the setup.
Fig. 2.
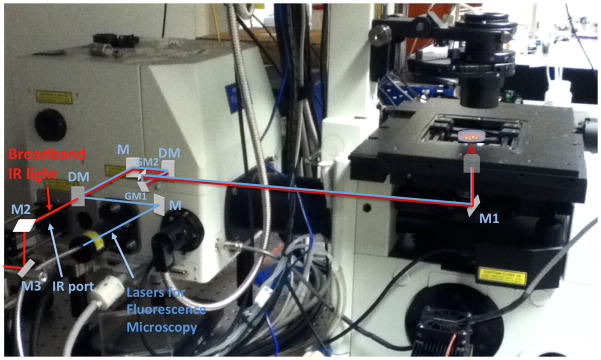
Image of an Olympus FW1000 system overlaid with both the OCT sample arm and fluorescence excitation beam paths. The broadband infrared light for OCT and the lasers for the fluorescence microscopy are shown as red and blue, respectively. M, M1–3: mirror, DM: dichroic mirror, GM1,2: galvanometric mirror, PMT: photomultiplier tube, F: optical band path filter.
To begin the alignment procedure, place a fiber port on the laser table close to the near infrared side port entrance of the FV1000 scanhead. This side port allows infrared light coupling via a dichroic mirror that reflects wavelengths below approximately 700 nm, but allows through longer wavelengths. To provide the degrees of freedom needed to properly align the input beam, be sure to use at least two relay mirrors (Fig. 2, M2, M3) between the fiber port and the side port entrance, with the final mirror placed in line with the side port entrance. Ensure that the propagating beam is level with the laser table and that it enters the side port at exactly the center of the aperture. Additionally, it is recommended to place a neutral density filter wheel into the beam path to control the sample arm light level. At this point, set the filter wheel to allow through the maximum intensity of light.
-
Power up the microscope and set the system to scan at the highest possible zoom level exactly in the middle of the scan area. Alternatively, carry out a scan, and then set the microscope to “point” scan mode at a position directly in the image center. This step is important to ensure that the scanning mirrors are positioned at their central scanning position. Rotate the nosepiece to an empty position or remove an objective lens to observe the near infrared beam. Place a piece of lens cleaning tissue over the objective aperture, and use an infrared viewer to see the beam. Using the second-to-last external coupling mirror, center the beam in the aperture. Then, adjust the last coupling mirror to optimize the brightness of the beam. Iterate the adjustment of these two mirrors until the brightest beam is centered in the aperture.
It is likely that the near infrared beam may not be visible on the first try. If this is the case, open the two back panels on the FV1000 microscope to expose the beam path. Do not attempt this if the microscope is under warranty or a service contract without an Olympus representative present. The two safety shutters can be bypassed by inserting short rods into the two open safety ports. Take extreme care and wear appropriate laser eyewear if bypassing the safety shutters. Position the laser beam using the two coupling mirrors (Fig. 2, M2, M3) such that the last coupling mirror (Fig. 2, M3) centers the beam on the first safety aperture, with the second-to-last mirror (Fig. 2, M2) being used to align the beam on the first galvanometric mirror (Fig. 2, GM1)
Once aligned, approximate the length of the light path from the fiber splitter (Fig. 1, FS) to the objective focus, keeping in mind that lenses add additional optical path length due to their higher index of refraction. Set up the reference arm as described previously [19] to match the estimated path length. A neutral density filter wheel should be inserted into the reference arm beam path.
-
It is now time to find the zero time delay between the sample and reference arms. Time zero is achieved when the sample and reference arm have the same optical path lengths, and is thus the distance difference at which the OCT system experiences maximum interference intensity. Place a mirror in the objective focus to reflect a strong near infrared beam backwards; a 4X objective is recommended. Block the reference arm and use the neutral density wheel to adjust the sample arm spectrum to 40 % of the maximum. Then, block the sample arm beam, and set the reference arm spectrum to 40 % of the maximum. Unblock both arms, and begin adjusting the reference arm delay. As the path lengths become equal and interference is observed, it will be necessary to adjust the sample and reference arm neutral density filters to lower power settings.
Finding time zero can be a time-consuming process, as the initial path estimate is not always easy to accurately calculate, especially for a closed imaging system. It is recommended to move the reference arm delay stage to several positions and carefully adjust the time delay to find time zero.
The electronics of the OCT system now must be configured to trigger off the Olympus input/output box. In our SD-OCT system, the electronics were designed to be driven by a set of internally generated line and frame triggers that act to synchronize scanning and camera acquisition rates. The input/output box provides pixel, line, and frame triggers that can be used to perform the same function and drive the OCT hardware in synchrony with the confocal scanhead. These outputs only occur when the microscope is actively scanning, allowing the entire OCT system to be “slaved” to the Olympus trigger signals. Unfortunately, there does not seem to be a way to communicate the imaging parameters from the Olympus FV1000 software; values such as pixel dwell time, image size in X by Y pixels, the number of line or frame averages, and the number of unique positions must currently be manually entered into the OCT software and camera control settings.
As this system utilizes 800–950 nm light and separate sample and reference arms, it will be finally necessary to determine the phase compensation for high-resolution imaging. As light travels through any material, the wavelength-dependent index of refraction can cause temporal delays between colors. For broadband light sources in the 800–900 nm regime, this leads to a phase delay between different wavelengths that must be compensated for to achieve high-resolution imaging. The phase compensation algorithm described by Mujat et al. [22] uses an iterative fitting routine and is highly recommended.
3) Optimizing In Vitro 3D Cultures for OCT Imaging
The recent increase in the use of 3D cultures in research settings has largely been driven by the development of commercially available scaffolds and matrices, as well as the advancement of novel culture methods and models. In combination with the wealth of primary, immortalized and genetically engineered cell lines, researchers are now able to use 3D culture systems modeling various aspects or stages of clinical disease to investigate morphogenesis, disease progression, and the factors underlying treatment response and resistance in a wide range of diseases. 3D culture systems can be highly cost-effective compared to animal experiments, and offer the ability to carry out parallel high-content imaging studies across tens of thousands of model tumors. The individual characteristics of these models can be exploited to develop targeted therapies, such as those focused on fighting resistance subpopulations that reside in hypoxic tumor microenvironments [23], or to evaluate the value of a compound as a broad spectrum agent against a full panel of different disease models.
OCT is a natural partner for 3D culture studies as it enables continuous monitoring of spatiotemporal dynamics throughout cultures non-perturbatively and non-destructively. Though it does not directly provide molecular-level information, OCT can enable insight into molecular effects by capturing alterations in structural properties such as architecture, phenotype, and cellular organization. For example, OCT can be used to quantitatively monitor tumor burden in vitro throughout the course of therapy by mapping the volumes of individual 3D tumors. As the contrast in OCT arises from backscattering, it is also possible to visualize the progression of apoptosis in vitro without labels via the elevated light scattering caused by chromatin condensation. Moreover, the wealth of 4-dimensional data (X, Y, Z and time) can be mined long after experiments have concluded to reveal insights that would be difficult or impossible to achieve using conventional imaging techniques.
3.1) Creating 3D Cultures for OCT Imaging
There are several characteristics of 3D culture models that should be optimized for visualization and monitoring with OCT. In order to address these parameters in sufficient detail, this discussion will be limited to overlay models, though the methods described here can likely be applied to other culture models and systems to improve imaging quality. In an overlay model, cells are typically seeded atop a layer of extracellular matrix (ECM), such as commercially available Matrigel (BD Biosciences, 354230) or a synthetic substitute, after which they grow and develop to form 3D multicellular structures that are partially embedded in the surface of the matrix. This model makes imaging tasks simple as all spheroids essentially grow in the same plane. As such, the consistency (i.e. the evenness or flatness) of the basement membrane surface is an important parameter for optimized OCT monitoring of overlay culture models. Potential pitfalls to avoid are the creation of bubbles in the matrix layer and not completely spreading the matrix to the well edges during plating. These errors can cause breakdown or even detachment of the gel from the glass, leading to loss of 3D nodules or cellular invasion through the gel, both of which may compromise the integrity of the experiment. Protocols using Matrigel [4] have previously discussed that simply pre-cooling culture plates and tips while using a freezer pack during plating can be used to help avoid these problems. It should be noted that Matrigel can vary greatly between different lots, in terms of its protein and endotoxin levels, which may affect 3D culture growth as well as its structural integrity. Consequently, using different lots of Matrigel within a given experiment or set of experiments within a screen will introduce a great deal of variability in the collected data and should be avoided at all costs. It is strongly advised to test out several lots for plating consistency and effects on nodule growth and morphogenesis prior to beginning any series of experiments. Moreover, it is recommended to always purchase extra Matrigel for projects so that all experiments can be carried out using a single lot even in the case of experimental failures or setbacks.
Another major consideration for OCT experiments is the choice of culture plate type and format. Maintaining sterility is crucial for long-term imaging of in vitro 3D cultures, as bacteria and fungi can significantly affect growth and molecular response even at mild or latent infection levels. Therefore, it is important that the cover of culture plates never be removed, particularly under non-sterile conditions. While upright imaging geometries can be used to visualize 3D cultures with OCT, experience has shown that imaging through the plastic plate cover can introduce unwanted reflections, distortions and other imaging artifacts. Imaging through standard plastic culture plates using an inverted geometry suffers from similar drawbacks, but fortunately culture plates are available with glass coverslip bottoms that enable distortion free, long-term sterile imaging. Although slightly more expensive, dishes incorporating a glass bottom within a circular cutout are preferable to simple glass-bottom wells or plates. The inset or microwell provided by the cutout in these types of dishes or wells allows for the easy creation of flat, consistent growth areas when using extracellular matrices such as Matrigel [10, 19, 20].
Another non-trivial consideration is to ensure that backscattering from the coverslip bottom does not interfere with imaging. Backreflection of near infrared light from the coverslip can far exceed the intensity of backscattered light from biological samples, leading to poor image quality if samples are created too close to the coverslip. A straightforward way to reduce this effect is to simply tilt the plate holder mount a few degrees: light reflected from the coverslip/air interface will be deflected, thus reducing its detected intensity. However, even with reduced reflections from plate tilting, there is still a minimum required distance between the biological sample of interest and the coverslip that is largely determined by the depth of focus of the system’s objective lens. If both the coverslip and biological target of interest are within 2–3 focal depths of each other, the signal from the coverslip can easily dominate. For low numerical aperture (NA) objectives, such as a .15 NA lens, the minimum recommended coverslip-sample distance is approximately 200 μm. This space needs to be filled with transparent, low-scattering gel or matrix to provide a buffer between the coverslip and biological sample. A series of trial and error tests to determine the optimal distance (i.e. Matrigel or matrix thickness) is recommended when starting to work with OCT systems.
There is a great deal of variation between different tumor cell lines, as some will form individual spherical nodules that are consistent in their shape and form (e.g. PC-3, H460) while others (e.g. A2780) form thick sheets of cells resembling multicellular layer models [24, 25] (Fig. 3). Depending on the characteristics of the model, these tumors may be homogeneous or may display a wide range of heterogeneity in size, extent of implantation in the basement membrane matrix, and shape among other features. Even after selecting a particular cell line, parameters such as seeding density, growth duration, and maintenance and growth conditions can significantly influence culture phenotype and development. In order to demonstrate the optimization of 3D culture parameters for OCT therapeutic monitoring in sufficient detail, the OVCAR5 overlay 3D tumor model will be used as a specific example below.
Fig. 3.

OCT b-scans (xz cross-section images) of 3D in vitro cultures. 3D cultures grown from the PC-3 prostate cancer cells form loosely packed spheroids after 16 days. Similarly, OVCAR5 ovarian cancer cells grown for a period of 13 days form into spherical nodules that display smooth surface features. A2780 ovarian cancer cells, on the other hand, rapidly proliferate and spread to form large sprawling “carpet-like” masses after 14 days in culture.
3.2) Manipulating Overlay 3D Culture Conditions for Optimal OCT Therapeutic Experiments
The seeding density for a given culture can influence a host of parameters, including growth rate, cellular and nodular migration, sample density, and the architectural organization of individual culture models. This is dependent on the biological mechanisms of nodule formation and growth, which can vary considerably between different cell lines and 3D culture systems. For example, at plating densities close to 18,000 cells/mL, OVCAR5 cells deposited on top of the Matrigel surface will migrate to generate “polyclonal” nodules that are formed by the proliferation of several distinct seeded cells, and nodules, that have congregated. At higher densities (30–40,000 cell/mL), OVCAR5 cells and semi-mature multicellular complexes migrate and merge to form large irregularly shaped aggregates than can blanket large areas of the Matrigel surface. On the other hand, low plating densities (e.g. 1,000 cells/mL) do not seem to allow for cellular congregation, and cultures will not properly form nodules of multicellular origin. For OCT treatment response experiments, we have found it ideal to have a controlled size distribution of spherical polyclonal nodules to map out therapeutic uptake and penetration with measured treatment response. Thus, in our experiments with OVCAR5 cells, we utilize a seeding density of approximately 18,600 cell/mL. It should be noted that other cell lines and cell types, primary cells in particular, can require higher seeding densities and the resulting cell-cell contacts to stimulate proliferation, migration, and tumor nodule formation. This can be achieved by seeding single-cell suspensions at higher densities or alternatively, as is often true with primary cell lines, by seeding cultures with multicellular clusters [26].
Another cell culture parameter that can dramatically influence therapeutic experimental conditions such as hypoxia, acidosis, and ECM formation is the growth rate and culture growth time. The rate at which the growth medium is depleted and must be refreshed is specific to the model or cell line being used, as is the composition of the culture medium with respect to essential amino acids, sugars and/or serum. The dependence on exogenous growth factors for the formation and growth of 3D models is another important consideration. To form the spherical nodules optimal for our OCT imaging studies, complete OVCAR5 culture media [23] (CellGro, Cat #10 040 CV) is supplemented with 2% Matrigel to provide the exogenous stimulation that drives nodule formation and growth [4]. The growth factors present in Matrigel which stimulate 3D formation and morphogenesis can be individually added at specific concentrations to create highly specialized media formulations optimized for a given cell type or model [26]. However, simply adding Matrigel to the growth medium at a certain concentration is sufficient to induce 3D formation and growth in the OVCAR5 culture system as well as many other 3D models [4], and we believe it to be a far simpler and less error-prone approach for media supplementation. In other cell lines, for example the H460 lung carcinoma line, cells will spontaneously form nodules or multicellular complexes even without stimulation from contact with a basement membrane matrix or by the addition of exogenous growth factors to the media.
OVCAR5 3D cultures seeded at approximately 18,000 cells/mL require refreshment of Matrigel-supplemented media every three days, which must be continuously maintained throughout therapy cycles and subsequent monitoring of treatment response to avoid artifacts resulting from the removal of growth stimuli. This will vary depending on the metabolic activity of the cell line being used, the rate of tumor growth and proliferation, and the requirement of the cultures for maintenance of levels of their endogenous secreted growth factors. For example, 3D cultures utilizing patient-derived cells often require more frequent media changes on the order of every two days to remove dead cells, and require conditioned media in order to facilitate continued growth. The removal of all growth medium from such cultures can deprive cells of crucial secreted growth factors, and therefore refreshment using conditioned media is strongly recommended by re-administering at least 50% of the existing culture media [26]. Regularity in the maintenance of cultures is a significant factor ensuring that they display the desired sizes and phenotypes at the expected intervals for uniformity between experiments.
The selection of a 3D culture model depends on the needs of the experiment or OCT screen; different cell lines or culture models will exhibit a range of phenotypes, morphogenic stages, and/or tumor microenvironmental factors which can be exploited to guide drug selection [23]. This heterogeneity of culture systems also extends to the size and structure of 3D cultures in a given sample that can either exist with great uniformity or within a defined distribution. All of these factors, and the variability of their reproducibility, exist on a spectrum that is specific to the model. For example, OVCAR5 3D cultures naturally develop a reproducible range of heterogeneous tumor nodules that display not only different sizes, but different degrees of therapeutically relevant factors such as hypoxia and acidity (Table 1). Generally, OVCAR5 nodules display hypoxic and acidic tumor core microenvironments after ten days of growth, and reach peak levels of hypoxia and acidosis across the majority of tumors in the sample after thirteen days.
Table 1.
Timeline of OVCAR5 model Treatment-relevant Stages of Morphogenesis
| Day 0 – Day 3 |
|
| Day 7 |
|
| Day 10 |
|
| Day 13 |
|
| Day 17 |
|
| Day 21 |
|
3.2.1) Plating Overlay OVCAR5 3D Nodules on Growth Factor Reduced (GFR) Matrigel Basement Membrane Matrix
-
1
Thaw enough GFR Matrigel on ice to make 2% Matrigel-supplemented culture medium and coat all of the glass well inserts as described below. Allow 1–2 hours for Matrigel to thaw completely, depending on the volume in which it is stored (1 mL aliquots are recommended).
-
2
Place all plates in a −20°C freezer for at least 20 minutes prior to plating. Additionally, ensure that a freezer block has been chilled to −20°C. As Matrigel rapidly solidifies above 8°C, it is extremely helpful to pre-cool and maintain all culture plates at or below 4°C during Matrigel application. Under sterile conditions, place the pre-chilled culture plates on top of the freezer block for the duration of the application process to allow more time to correct any errors in Matrigel application (see steps below).
-
3
Dispense the appropriate volume of thawed GFR Matrigel in the center of the glass coverslip insert and expand the diameter of the Matrigel with the pipette tip by continuously spreading using a circular motion until the Matrigel and pipette tip contact the edge of the glass insert. For both glass bottom 35 mm dishes (MatTek, P35G-0-14-C) and individual wells of 24-well glass bottom plates (MatTek, P24G-0-13-F), a volume of 115 μL of thawed GFR Matrigel is recommended.
After spreading the Matrigel, trace the periphery of the well cutout and glass insert several times until all regions of the glass/plastic interface are coated. Then, continue to spread the Matrigel in a circular motion back throughout the entirety of the well until it is evenly layered to a uniform thickness.
-
3
Avoid leaving bubbles in the Matrigel and if necessary, direct them to the edge of the inset with the pipette tip to minimize their impact on tumor growth.
Holding the dishes and gently rotating or tilting them in all directions may also be helpful in spreading the Matrigel uniformly across the glass surface, particularly in the case of multiwell plates. Even while cooled below 4°C, we have found that it is crucial to fix any Matrigel spreading errors within the first one or two minutes. Attempting to correct defects in the Matrigel once it has begun to set can result in tears or other degradation issues days or weeks later.
-
4
Incubate the freshly coated dishes or plates in a sterile, humidified 37°C incubator for 30 minutes to allow the Matrigel to solidify.
This process is time-sensitive and after 45 minutes of incubation at 37°C the Matrigel can dry out and crack, rendering plates unusable. Alternatively, incubation at 37°C for less than 20 minutes may prevent Matrigel from sufficiently solidifying, altering the cellular response after seeding.
-
5
Create a single cell suspension of OVCAR5 cells at a density of 18,600 cells/mL using normal cell splitting protocols [19, 23].
Break apart cellular aggregates following trypsinization by forcing cells through a serological pipet pressed down into the bottom of a 50 mL conical tube at a rate of approximately 1 mL per second. Repeat this process until clusters of cells are no longer visible (typically 10 times is sufficient).
After thoroughly resuspending the single-cell suspension to ensure uniformity in the density of cells seeded across all samples, dispense the appropriate volume of the 18,600 cells/mL suspension onto the solidified Matrigel bed according to the size of the glass coverslip inset (200 μL and 170 μL for 35 mm and 24-well plates, respectively). Surface tension will hold the dispensed cell suspension in place on top of the Matrigel bed.
Be careful to apply the cell suspension only to the Matrigel bed and to avoid spilling any fluid beyond the Matrigel boundary, as this will result in the growth of monolayer cells on the surrounding plastic which can accelerate the depletion of the culture media and alter the growth conditions over the two week culture period.
If plating multiple dishes or wells, it is highly recommended to resuspend the single-cell suspension periodically, as cells can settle to the bottom of the conical tube causing variation in the density of cells seeded across different wells.
-
6
Allow cells to adhere to the Matrigel surface for a period of forty minutes in a 37°C incubator under 5% CO2.
Cellular adherence can be confirmed under a phase-contrast microscope by gently moving the plate and confirming that cells remain stationary and are not simply settled on the surface of the Matrigel. While 40 minutes is sufficient to allow OVCAR5 cells to adhere to the Matrigel basement membrane matrix surface, this parameter is specific to the cell line being used. Consequently, when adapting a culture model to use a new cell line or cell type it is essential to ensure that cells have completely adhered before proceeding to the next step using the method outlined above.
-
7
Add a volume of Matrigel-supplemented complete cell culture media to bring the media to a final volume of 2 mL or 1 mL at 2% Matrigel-supplemented media, for 35 mm and 24-well plates respectively (1.8 mL of 2.2% Matrigel-supplemented media for 35 mm dishes, and 830 μL of 2.4% Matrigel-supplemented media for 24-well plates).
-
8
Return cultures to the sterile 37°C incubator and maintain them under 5% CO2 until they develop the desired phenotype (Table 1).
-
9
Refresh the growth medium by replacing it with fresh 2% Matrigel-supplemented complete culture media every three days throughout therapeutic cycles and subsequent visualization experiments with OCT.
Continuous morphogenesis of cultures is stimulated by the presence of Matrigel in the culture media, and its absence will cause tumor growth arrest, and profound changes in their therapeutic response.
4) Carrying Out OCT Screens on In Vitro 3D Cultures
The last few decades have seen the rise of vast libraries of biologically active compounds and synthetic derivatives available for therapeutic screening. While high-throughput in vitro screening approaches have been of great use to the drug development community, a large body of evidence points to major differences between the activity of drug candidates in monolayer culture experiments and their in vivo counterparts, requiring novel methods and techniques focused on 3D culture screening systems. Chemotherapeutic interventions are a prime example of a front-line treatment modality whose pitfalls and limitations can be addressed by research taking advantage of molecular screening techniques in pre-clinical settings. For example, the role of membrane transport proteins and extracellular acidity on the efflux of chemotherapy agents [27, 28] can be investigated in a highly controlled manner using 3D tumor models that replicate interstitial acidity [23], an investigation which would be difficult or impossible to achieve in animal models. Similarly, the limited penetration of chemotherapeutics into tumors, as a factor contributing to reduced therapeutic efficacy, has been demonstrated using 3D tumor cultures [29]. Similar models that develop a thick layer of secreted ECM on the tumor periphery can be used to investigate the role of ECM as a barrier to drug penetration.
Photodynamic therapy (PDT) is another example of a treatment modality for which the application of 3D cultures is extremely well suited. The cytotoxic effects of PDT are mediated by the oxidative damage generated directly or indirectly by the excited-state photosensitizer (PS) following absorption of photons. PDT can mediate cellular death through necrosis in addition to apoptotic mechanisms, the ultimate balance of which can be partially controlled by optimization of treatment parameters such as PS dose, incubation duration, drug-light interval, fluence rate or power level of the light source and total light dose delivered. PDT requires optimization and fine-tuning, making 3D culture systems a natural partner for screening studies, as carrying out these experiments in vivo would be both costly and highly time-consuming.
Conventional fluorescence-based viability assays operate by using a combination of live cell and dead cell markers that perturb cultures, making their use a terminal event in any screening study [10, 20]. As a non-perturbative technique, OCT imaging overcomes this limitation by capturing many therapeutic time points in the same cultures over the course of hours and days. For example, chemotherapeutic agents operate by targeting highly proliferative cancer cells to slow or halt tumor growth, making OCT a natural tool for monitoring response as longitudinal evaluation of tumor growth arrest and subsequent shrinkage are well within its capabilities. Chemotherapeutic response can occur on long timescales and, at the same time, can be punctuated by rapid events whose timing can be difficult to predict, requiring longitudinal imaging to capture the full time course of treatment dynamics. For example, treatment of the OVCAR5 model with cisplatin results in growth arrest during the first 36 hours [19]. Cellular death and tumor degradation begin only two days post-administration, but then quickly evolve over the course of several hours. The cytotoxic effects of PDT manifest on far shorter timetables than chemotherapy. The PDT-induced therapeutic effects typically manifest within a 4–6 hour period, and residual and/or indirect therapeutic effects such as cell death mediated by “the bystander effect” [30] are completed within a 24 hour period. Rapid changes in tumor architecture induced by PDT as well as secondary cytotoxic effects can be captured by continuous OCT monitoring in a far more informative fashion than the brief snapshots provided by terminal fluorescence-based approaches which can lack 3D information. It should also be noted that cellular senescence, which is often induced by treatments such as chemotherapy, renders markers of cell death and apoptosis uninformative, making post-experiment, quantitative volumetric measurements essential in understanding therapeutic outcome.
OCT time-course imaging can also be invaluable for combination therapeutic studies where it is critical to observe the effects of not only different therapeutic doses, but also the timing of therapeutic administration. For example, the therapeutic effects of PDT have been shown to trigger the breakdown of tumor architecture, resulting in a time-dependent synergistic enhancement of chemotherapy, which may be at least partially attributable to PDT-induced structural degradation, enabling the influx of drugs deep into normally impenetrable tumors [10].
4.1) Preparing for and Running OCT Imaging Sessions for 3D Cultures
As in any experiment, there are a number of considerations that should be addressed when carrying out OCT imaging studies. It is important to set the acquisition rate (and thus integration time) such that the signal-to-noise ratio (SNR) during imaging is as high as possible. While imaging processing can be carried out with as little as 60 dB of contrast, it is recommended to have at least 80 dB of SNR for automated image analysis algorithms. As a point of reference, the time-lapse OCT system described in Evans et al. [19] carried out OCT imaging at rates of approximately 1,000 frames/s, and was capable of capturing growth and apoptotic events over the course of four days.
When setting the acquisition parameters for longitudinal imaging, it is also crucial to ensure that the collected interferogram intensity always falls within the detector dynamic range. Biological processes that result in enhanced scattering, for example the condensation of nuclear chromatin that occurs during apoptosis, can significantly elevate the backreflected sample signal, potentially saturating cameras or other detectors during a long-term experiment. Saturation of the spectral interferogram results in a loss of information and degradation of OCT images. It is strongly suggested to run a pilot experiment to collect images at relevant time points before an formal experiment to determine the range of signal allowed on the detector.
Another consideration is the objective lens: selection of an objective lens is generally a balance between lateral resolution and axial depth of field. This choice largely depends on the physical size of the 3D model being studied; in order to capture information throughout large samples, it is necessary to select low numerical aperture lenses to maximize depth of focus. For smaller models, mid to high NA lenses with reduced depth of focus can be used to provide increased lateral resolution. Apodization can be used to provide extended depth of focus even with high NA objectives. While apodization itself can lead to an increase in the lateral point spread function, Liu et al [18] have found that combining apodization with high NA objective lenses provides both an improvement in the overall lateral resolution with an extended of depth of focus suitable for micrometer scale resolution OCT imaging.
An important, but routinely overlooked, parameter is the selection of regions of interest during OCT imaging experiments. Consistently visualizing the same regions in every dish or well prevents observer bias introduced by visually identifying regions ideally suited for imaging. As 3D models can have region-dependent densities, which are caused by sample plating methods, imaging the same areas across all samples ensures that such variations in sample density are accounted for. These variations can introduce measurement errors if regions of samples for imaging are randomly selected.
As a representative example, the protocol below will focus on a time-lapse experiment carried out on an SD-OCT system.
4.1.1) Carrying out 3D Model OCT Experiments
Allow the OCT system to fully stabilize prior to beginning an experiment. This includes detectors, light sources, microscopes, and even incubator stages. While many system components can be ready for use in 45 minutes, we have found that extending the warm up phase is particularly helpful for long-term and time-lapse experiments. Even after one hour of warm up, components still may not have completely reached equilibrium. For example, line scan cameras can continue to experience thermal drift as much as two hours after power up.
As in a normal startup procedure, block the sample arm, and carefully adjust the line scan camera to find the maximum spectral bandwidth. Importantly, to avoid mechanical drift, always be sure to engage the threads in the camera mount in one single direction at a time; adjusting the mount back and forth can introduce “backlash” that can manifest as mechanical drift in the hours following alignment.
To achieve the best signal-to-noise ratio, it is optimal to have the highest reference to sample arm power possible; this will ensure that intensity changes in the sample do not appear as modulations in the spectral interferogram. For the 3D OVCAR5 model introduced above, it is rare that the backscattered intensity exceeds a few percent of the reference arm intensity, but different models will exhibit different scattering intensities. To avoid such problems, be sure to adjust the sample arm to less than 5% of the reference arm intensity. Keep in mind that the backscattering intensity can increase during any given long-term experiment, so be sure to limit the maximum signal intensity as determined in pilot experiments so as to avoid detector saturation.
Set the OCT system to collect xz depth scans, also know as b-scans, and bring the 3D model tumors into focus. If using an OCT system based off a microscope, it is most helpful to first find and bring into focus the 3D cultures using transmitted light with the microscope eyepiece. While imaging with OCT, next adjust the microscope focus to optimize the OCT imaging intensity over the 3D cultures. It has been found helpful to place the focus closer to the upper limit of the cultures (i.e. the “top” of a 3D nodule) as this has been found to provide the most even signal intensity distribution throughout the in vitro models. Additionally, if any axial drift occurs, this will ensure that the 3D nodules remain in focus as long as possible.
To bring the 3D cultures into the optimal imaging depth range for the OCT system, adjust the delay stage that controls the reference/sample arm time delay. As detailed above, biological cultures of interest should be positioned within the Niquist sampling roll-off point to avoid signal loss. It is usually optimal to adjust the timing delay such that the 3D model tumors are as close to time-zero (that is, the zero ranging depth) as possible.
-
Use the sample stage, whether manual or automatic, to locate regions of interest for OCT imaging. Based on our experience, we recommend imaging 3D cultures at the most level regions of the sample. For the OVCAR5 model described above, the most level region of the Matrigel surface is at the center of the well; the edges of the gel are typically curved due to the formation of a meniscus when the liquid gel is first set. To remove confounding errors and observer bias, it is recommended to always image the same physical areas in all wells and all samples. When plating the OVCAR5 tumor model in 24-well plates, for example, we typically collect a minimum of four regions of interest at the center of the well, which depending on growth conditions and/or sample variability, can capture the complex volumetric details of twenty to fifty nodules.
If using multiwell plates on systems with automated stages, it is recommended to save the stage protocol for future imaging sessions so that one can easily follow the same 3D nodules over time. Using the same stage protocol for all multiwell plates in a study can standardize the imaging positions to further reduce observer bias. It should be noted that the gel thickness is known to change over the time course of therapy, which may require re-registering the axial focus at the start of each subsequent imaging session.
With the state positions set, now adjust the imaging system to acquire image volumes, also known as c-scans. It is highly recommended to save the raw interferometric data, not the display data, as the interferogram can be analyzed during post-processing to achieve high signal-to-noise ratio images.
As OCT is a quantitative imaging tool, it is critical to ensure that a consistent set of imaging parameters are used across all imaging sessions. This set of parameters includes the camera speed, integration time, light power, pixel dwell time, and warm up time. It is highly recommended to explore these settings in an initial test experiment.
4.2) Therapeutic Administration to OVCAR5 In Vitro Model Tumors
Initial OCT imaging of treatment response in 3D models should be carried out regularly, if not continuously with a time-lapse system, to capture and map out the timeline of tumor growth and cellular death dynamics. For chemotherapy and biological therapies, it may be important to maintain the agent at uniform levels in the treatment media over time. Cisplatin or carboplatin, for example, should be refreshed every two or three days in fresh Matrigel-supplemented media, as the therapeutic acts against, and is consumed by, the 3D model nodules.
PDT agents are similarly administered in complete cell culture medium, but due to their shorter incubation times, these molecules can be administered without Matrigel supplementation. Due to the inherent light-sensitivity of PS steps should be taken to avoid light exposure, particularly during the early stages of PDT experiments. Working under subdued light is strongly recommended. Consideration should be given to the layout of samples when plating PDT experiments in a multiwell format in order to avoid unintentional treatment light cross talk between adjacent wells. Black-walled multiwell plates can substantially reduce the amount of treatment light that is scattered into adjacent wells during PDT. If clear plastic multiwell dishes must be used, PS-containing samples need to be plated in alternating wells to prevent treatment light from adjacent wells from causing unwanted therapeutic effects.
Great care should be taken when manipulating 3D cultures that are undergoing or recovering from therapy. Cells that may have detached from tumor nodules as well as portions of structurally weakened nodules may be unintentionally removed from the culture well or artificially disrupted. It is strongly recommended that all steps involving the addition and/or removal of media should be done slowly and gently with a large volume, single-channel pipette. Vacuum-assisted aspiration techniques should be avoided at all costs. Aspiration by hand should be carried out with the plate maintained level, and without tilting culture dishes, even if the entirety of the growth media cannot be removed. Though many monolayer protocols may call for washing of cultures after any treatment, investigators are recommended to minimize or entirely avoid washing steps during and following therapeutic administration to prevent causing perturbations and artifacts.
The evaluation and subsequent optimization of combinatorial treatment approaches requires a special set of considerations for the quantification of therapeutic response. Each therapeutic modality must be evaluated individually at the same dose and under the same treatment parameters as they are utilized in combination, in addition to running identical untreated controls to which the sample viability can be compared. In order to evaluate any potential additive or synergistic enhancements in therapeutic efficacy from combination of therapeutic regimens, it is recommended to administer low doses of each therapeutic modality such that the “addition” of the cytotoxic levels of each individual therapy in combination fall below the maximum cytotoxic response measurable in the model. This allows for the identification of synergistic enhancements in therapeutic efficacy resulting from the combination of therapeutic regimens with different therapeutic mechanisms. Furthermore, when screening for time-dependent therapeutic enhancements, it is recommended to carry out all time combinations and controls within a given multiwell plate to normalize for different plating, experimental, and measurement conditions.
4.2.1) Administration of Therapeutics to 3D Cultures
Components of the OCT system should be fully stabilized as described above prior to administration of any therapeutics to 3D cultures to ensure that all components are ready for use. Additionally, be sure to thaw enough GFR Matrigel on ice to make the required volume of 2% Matrigel-supplemented culture medium.
Create therapeutic working treatment media solutions by adding the concentrated stock solution of the therapeutic agent to the appropriate volume of 2% GFR Matrigel-supplemented complete cell culture media. The accuracy of the dose administered can be improved by first adding concentrated drug stock solution to the most concentrated working treatment solution, which should then be diluted in Matrigel-supplemented media to create all other working treatment solutions.
Remove the existing culture media. It is crucial to avoid vacuum-assisted aspiration methods to remove the growth media from samples to minimize disruption of the Matrigel and 3D model tumors. Instead, slowly remove media by hand with a single channel P1000 pipette. Gently administer the appropriate volume of drug-containing working treatment media solution to samples, taking care to add the solution to the side of wells to avoid disrupting the 3D culture. Use 1 mL and 2 mL volumes for individual wells of a 24-well plate and 35 mm dishes, respectively. To avoid cultures drying out during media exchanges, it is recommended to dose samples individually or in small groups.
Return cultures to the incubator for the appropriate period of time. For chemotherapy with carboplatin, for example, cultures are typically incubated for 24 to 48 hours to observe maximum effects.
Specifically for chemotherapy and biological therapies: after several days of treatment, aspirate the treatment medium from samples very gently and slowly from sample dishes/wells using a P1000 pipette. Apply a fresh dose of therapeutic-containing (or therapeutic-free, for untreated control samples) 2% GFR Matrigel-supplemented complete media, administering the solution to each culture dish/well slowly and gently so as not to disrupt treated nodules.
-
Specifically for PDT administration:
Turn on the PDT illumination source and set the output intensity to the appropriate power level. If necessary, allow time for the illumination source to warm up and for the temperature to stabilize to avoid changes in output power during the experiment.
After the incubation period, aspirate the photosensitizer-containing dosing media from culture dishes (or plain media in the case of “no treatment” or “light only” controls).
Add either 2 mL or 1 mL of warmed, 2% Matrigel-supplemented complete cell culture media to 35 mm dishes or individual wells of a 24-well plate, respectively.
Illuminate samples for a period of time calculated to deliver the intended total light dose to individual samples based on the fluence rate of the light source. In our experience, irradiations can be carried out at ambient/room temperature without confounding effects on 3D cultures, provided that they are kept outside the incubator for less than 45–60 minutes. Similarly, the effects on the pH of the culture media from removal of cultures from 5% CO2 environments are also negligible within this time frame. Nonetheless, control samples should be placed under identical conditions to ensure that any potential effects from prolonged exposure to ambient conditions are uniformly experienced by all samples.
Following irradiation, samples should be sterilized before being returned to the 37°C incubator at 5% CO2. This can be achieved by spraying the exterior of dishes with 70% isopropyl or ethyl alcohol and wiping them afterwards to ensure that the entire surface has been sterilized.
It should be noted that combination treatment regimens will require special modification of these protocols. For example, to carry out PDT followed by carboplatin administration, carboplatin working media stocks in 2% Matrigel would be added to cultures at time points following light administration, with great care taken not to disturb the 3D cultures. It is always recommended for combination treatment studies to run a series of test experiments where imaging is carried out either continuously or regularly to discover critical periods of treatment effects to guide follow-up imaging studies.
-
Remove individual or multiwell plates from the incubator at select times for OCT imaging.
Minimize the number of samples removed from the incubator at a time to avoid exposing cultures to room CO2 and temperature conditions for longer than necessary. For experimental consistency, ensure that all samples experience the same ambient environmental conditions to minimize unwanted variations that may introduce experimental error between samples.
Sterilize all samples before placing them back into the incubator to prevent potential sample infections.
4.3) Considerations for Preparing and Running TL-OCT
Proper planning is critical for running successful time-lapse OCT imaging experiments, and investigators are urged to always carry out a full set of mock experiments prior to using real, and potentially precious, samples and drugs. A pilot study that simulates the conditions of the time-lapse experiment should be run to gather image acquisition parameters, such as the dynamic range of scattering intensities experienced during the entire study. Changes in the experimental room ambient temperature and humidity should be prevented, or at least compensated for, if at all possible. All aspects of the imaging system should be tested and monitored, with particular attention paid towards light source power and spectral stability, camera or detector position drift, microscope stage and focal drift, acquisition software errors and stability, and computer hardware stability, especially data storage. Perhaps not surprisingly, automated operating system and software updates can have profound negative effects on time-lapse OCT sessions. For example, automatic updates to the Windows operating system can lead to forced system restarts during the middle of TL-OCT experiments. It is highly recommended to either disconnect computer systems from the network during TL-OCT, or disable the operating system from undergoing automatic restarts.
For long-term or time-lapse experiments, cultures need to be properly protected in an environmental chamber that provides temperature control and maintains the proper CO2 composition of the air. Of utmost importance is to prevent media evaporation. This can be accomplished through the use of humidified weatherstations or stage-top incubators. Many of these environmental chambers have an integrated heating apparatus which maintains a programmed temperature through negative feedback, with a sensor that reports the temperature of either the culture media itself or the surface of the plate and a variable heating element. Additionally, CO2 composition is maintained by a port through which air can be flowed in under slight positive pressure. We prefer to use 5% CO2 balanced air which is pre-mixed, as it eliminates the need for an oxygen mixing system which blends pure CO2 and oxygen to the desired composition. For long-term experiments, the incoming air can be humidified by bubbling through water either inside the environmental chamber or externally. Maintenance of proper CO2 levels is crucial in long-term experiments in order to maintain proper cellular physiology and is even more important in drug screening experiments, as changes in the CO2 levels can influence the pH of the culture media, which can have profound impact on the partitioning and/or uptake of therapeutics. In the absence of a humidified system, or if evaporation still occurs over the duration of the experiment, we recommend covering the top of the culture media with a thin layer of light mineral oil, which will dramatically reduce evaporation while still allowing for gas exchange. Evaporation of the culture media can dramatically impact experimental results by effectively concentrating the media along with any therapeutics being administered. The use of mineral oil can have negative effects if highly hydrophobic compounds are administered, so investigators are urged to consider how to prevent evaporation on an experiment-by-experiment basis.
In spite of the best preparations, drift, misalignment, and other errors are bound to occur even to the most carefully planned TL-OCT experiment. Monitoring of the imaging system throughout the time-lapse experiment is crucial, as many problems can be fixed on-the-fly. Misalignment of the line scan camera, for example, can be compensated for by adjusting the mount, and software or computer problems can be corrected to prevent gaps in data acquisition. Based on our experience, we recommend monitoring the system every six to eight hours at a minimum, especially if the laboratory experiences daily thermal cycling.
Otherwise, the parameters for OCT imaging in TL-OCT experiments are essentially the same as those used in standard OCT imaging experiments as described above. The main differences to consider are the use of pre-programmed image capture routines and to decrease camera acquisition rates to reduce memory usage. The camera acquisition rates we typically use for TL-OCT are between 1.67 and 3.33kHz, which correspond to imaging speeds of 0.81 to 1.63 b-scans/s [21].
5) Discussion of Example Results: Collection and Processing of TL-OCT Therapeutic Screening Data
The data in Figure 4 were collected during a time-lapse experiment spanning a 24 hour period following PDT of OVCAR5 model tumors with EtNBS-OH, with c-scans acquired every fifteen minutes. The total collected data set was comprised of 97 individual interferometric c-scans at 483MB each, accumulating to a total set of data close to 47GB in size. Processing of this raw data into a viewable image set was achieved using an automated routine written in Matlab that Fourier transformed and processed each spectral interferogram across the entire data set to build a 4D OCT image data set. Without using parallel processing, building the 4D image from the raw data takes just under 5 hours. However, this time can be shortened by using multicore-aware programs, including versions of Matlab. Figure 4 and the corresponding time-lapse movie in Supplementary Movie 1 were compiled from individual b-scans using Matlab’s image processing toolbox.
Fig. 4.

A TL-OCT time sequence of OVCAR5 ovarian cancer 3D nodules following photodynamic therapy (PDT) with the PS EtNBS-OH at 20 J/cm2. In the time sequence, nodules are observed to undergo “unpacking” as they undergo structural degradation. The backscattering intensity of individual cells increases in the hours following PDT as cells in the spheroid undergo apoptosis.
Alterations in the nodules are observed within the first hours following PDT, with OVCAR5 nodules seen to undergo rapid structural degradation following an “inside-out” pattern of treatment response. Two major effects can be observed: (1) the formation of highly scattering bodies throughout each tumor nodule and (2) the apparent increase in volume of each nodule throughout the 24 hours following therapy. These effects are caused by PDT-induced oxidative damage, with the pattern of treatment response being consistent with the selective nodular core localization of the photosensitizer [23]. The highly scattering bodies result from chromatin condensation of cells undergoing apoptosis induced by PDT. The therapeutic effects of PDT result in structural damage, resulting in the observed “unpacking” [23]. While there is a strong correlation between the structural degradation and cellular apoptosis induced by PDT, both of which occur in a dose-dependent manner, there is no direct evidence that structural degradation in this model is an effect caused by treatment-induced apoptosis. However, there is a strong likelihood that the hypothesized relationship between the unpacking and cellular apoptosis is at least in part due to the impact of apoptotic events on cell-cell contacts and adhesion and cellular junctions (adherens junctions in particular). This process could be mediated, for example, by caspase-dependent dismantling of cell-cell contacts during apoptosis [31]. This is likely not a therapeutic response specifically induced by PDT, as it has been reported that treatment of cells with the chemotherapeutic agent cisplatin caused the early loss of cell-cell adhesions and cellular interactions in a caspase-independent manner, an effect which has been proposed to play a role in the onset of apoptosis [32]. The use of TL-OCT enables the complete, label-free visualization of the apoptotic and structural degradation processes throughout large tumor nodules over the entire duration of the therapeutic response.
This OCT time-lapse data can be further mined and processed to build quantitative metrics that can reveal insight into the detailed mechanisms of treatment response. One such aspect of treatment response is nodule unpacking, which can be quantified using a segmentation analysis approach that calculates the total surface-area-to-volume ratio of all objects in the image. The surface area of a 3D object increases as it is divided into smaller units. Therefore, if one measures the surface area of 3D nodules and their post-treatment fragments, changes in surface area can be detected as a function of treatment parameters. However, this surface area also depends on the number of nodules and fragments along with their size. Thus, the surface area is normalized to the total volume of the nodules and nodule fragments. Finally, this surface-area-to-volume metric must be normalized to the original volume of the intact sphere to represent the degree of therapeutic disruption. For this type of volumetric analysis, it is important have enough experimental repetitions to account for the inherent variability found in 3D cultures.
Briefly, this segmentation analysis can be carried out by first creating binary images by thresholding the grayscale cross-sectional OCT images, keeping in mind that the image scale is logarithmic. An interferometric imaging technique, OCT images naturally contain what are called “speckles”. These speckles, however, can appear as small fragments or holes in the processed binary image, and should be removed using image processing tools. The “imfill” command in the Matlab image processing toolbox can be used to eliminate such speckle artifacts. Next, the boundaries, and thus volumes, of tumor nodules can be calculated by measuring the perimeter of each object; the “regionprops” command in Matlab is most useful for this task (Fig. 5). Then, the perimeters of all objects are summed to calculate their surface areas. In the same way, summing the area enclosed by the nodular boundary provides the corresponding volume.
Fig. 5.
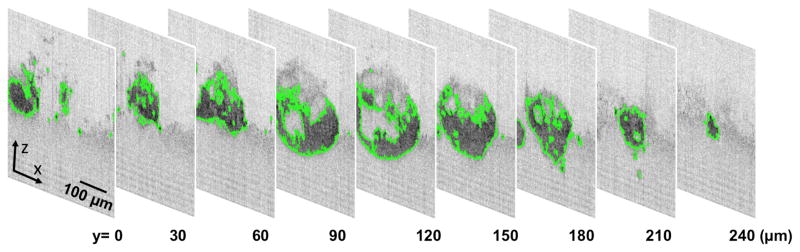
Illustration of the process used to calculate the surface area and volume of 3D nodules. By first determining the boundary (green) of 3D nodules along a series of b-scans (xz 2D cross-sections), the measured perimeters and areas can be tabulated into nodule surface area and volume, respectively.
It is highly recommended to use systematic thresholding for grayscale to binary conversion to avoid observer bias in the analysis process. A high threshold results in only partial recognition of nodules in the binary image, while a low threshold picks up excessive background noise. One approach toward optimizing the threshold is to generate binary images over a range of thresholds and count the number of unique fragments enclosed by a boundary in each image. When the number of fragments is plotted as a function of threshold intensity, there will be an inflection point where a large increase in fragment count occurs. This uptick in fragment number is a result of the background noise being counted as unique fragments. Automating the selection of a threshold below this inflection point not only avoids the observer bias introduced by the individual examiner, but also allows tolerance to variations in signal intensity that occur during the acquisition of OCT images.
Figure 6 demonstrates the potential to identify key mechanistic insights into therapeutic drug candidates using segmentation analysis of TL-OCT data. Comparison of single-area TL-OCT experiments was carried out for the 24 hours immediately following PDT using three benzo[a]phenothiazinium derivatives. The evaluation reveals time-dependent unpacking of nodules following treatment that are linked to key differences in the accumulation and localization of each derivative in the 3D nodules. The reduced unpacking caused by EtNBS-NH2-PDT is a consequence of the limited uptake and penetration of the compound into 3D nodules [23] caused by its net charge and molecular charge and distribution. In contrast, PDT with EtNBS-OH, a more water soluble molecule than its parent compound, is revealed to cause the most rapid rate of structural unpacking, a result uniquely observed by dynamic treatment response monitoring with TL-OCT. This insight, which was discovered by longitudinally tracking less than ten experimental samples, would have taken many more attempts and would have yielded less accurate results using conventional, terminal fluorescence-based microscopy methods.
Fig. 6.

Evaluation of the treatment response of OVCAR5 ovarian cancer nodules to different EtNBS derivatives. OVCAR5 nodules were grown for 13 days, treated with each derivative at 500 nM and 20 J/cm2, and visualized using TL-OCT for 24 hours. Segmentation analysis on the resulting data yields a dynamic treatment response profile for each derivative. The differences between each derivative result from known differences in nodule uptake and accumulation patterns of each compound [23]. (Red: EtNBS, Blue: EtNBS-OH, Green: EtNBS-NH2, Black: No treatment control).
6. Conclusions
As a longitudinal and label-free imaging tool, OCT has the capability to play a large role in future in vitro therapeutic screening and drug development studies. A natural imaging partner for 3D culture systems, OCT is an ideal platform for quantitative, non-perturbative monitoring of therapeutic response. Once the domain of optics experts, the increasing availability of commercial OCT systems now allows for its direct application by biologists to comprehensively study the evolution of therapeutic response and resistance. As has been observed with the current availability of high-throughput fluorescence screening platforms, integration of OCT with automated in vitro culture systems has the ability to increase its capacity for more complex screening studies. This will enable experiments that seek to avoid potential problems such as limited uptake and penetration, prevent drug inactivation in key microenvironments, and overcome other barriers to therapy that are currently difficult or impossible to study in monolayer platforms or in vivo systems. The deep tissue imaging capabilities of OCT will support studies building the next generation of therapeutics targeted against specific tumor subpopulations, such as those present in hypoxic microenvironments. Furthermore, the techniques elaborated here can be readily applied to screening therapeutics against patient-derived samples, an approach that hopefully will lead to the creation of personalized therapeutic regimens.
Supplementary Material
Supplementary Movie 1: Time-lapse OCT movie of 3D OVCAR5 nodules undergoing treatment response following PDT. Cross-sectional images were collected every 15 minutes for 24 hours following PDT with EtNBS-OH (500 nM, 20 J/cm2). Nodules are seen to follow an inside out pattern of structural degradation resulting from the cytotoxic effects of therapy beginning one hour following PDT. The development of dark, highly scattering bodies towards the end of the 24 hour period correlate with the appearances of apoptotic cells undergoing nuclear condensation.
Highlights.
We describe methods for high-content drug screening in 3D culture systems with OCT
The creation of an example time-lapse OCT setup is described
Example results from a set of time-lapse OCT experiments are presented and analyzed
Acknowledgments
The authors would like to acknowledge the members of Evans group for their assistance in editing this manuscript. This research was support by the National Institutes of Heath (NIH) Innovative Molecular Analysis Technologies (IMAT) program, grant number R21 CA155535, and the NIH Director’s New Innovator Award, grant number DP2 OD007096. Information on the New Innovator Award Program is at http://nihroadmap.nih.gov/newinnovator/.
Abbreviations
- 3D
Three-dimensional
- 4D
Four-dimensional
- CCD
Charge-coupled device
- GFR
Growth factor reduced
- FD-OCT
Frequency domain OCT
- FDML-OCT
Frequency domain mode-locked OCT
- FWHM
Full width at half maximum
- ECM
Extracellular matrix
- NA
Numerical aperture
- OCT
Optical coherence tomography
- OFDI
Optical frequency domain imaging
- PDT
Photodynamic therapy
- PS
Photosensitizer
- SNR
Signal-to-noise ratio
- S-OCT
Spectral domain OCT
- TD-OCT
Time-domain OCT
- TL-OCT
Time-lapse OCT
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
O. J. Klein, Email: oklein@partners.org.
Y. K. Jung, Email: jung.yookyung@mgh.harvard.edu.
C. L. Evans, Email: evans.conor@mgh.harvard.edu.
References
- 1.Chang SK, Rizvi I, Solban N, Hasan T. In vivo optical molecular imaging of vascular endothelial growth factor for monitoring cancer treatment. Clin Cancer Res. 2008;14:4146–4153. doi: 10.1158/1078-0432.CCR-07-4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mueller-Klieser W. Three-dimensional cell cultures: from molecular mechanisms to clinical applications. Am J Physiol. 1997;273:C1109–1123. doi: 10.1152/ajpcell.1997.273.4.C1109. [DOI] [PubMed] [Google Scholar]
- 3.Kim JB. Three-dimensional tissue culture models in cancer biology. Semin Cancer Biol. 2005;15:365–377. doi: 10.1016/j.semcancer.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 5.Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smalley KSM, Lioni M, Herlyn M. Life isn’t flat: taking cancer biology to the next dimension. In Vitro Cell Dev Biol Anim. 2006;42:242–247. doi: 10.1290/0604027.1. [DOI] [PubMed] [Google Scholar]
- 7.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 8.Folkman J, Hochberg M. Self-regulation of growth in three dimensions. J Exp Med. 1973;138:745–753. doi: 10.1084/jem.138.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans CL, Abu-Yousif AO, Park YJ, Klein OJ. Killing Hypoxic Cell Populations in a 3D Tumor Model with EtNBS-PDT. PLoS ONE. 2011:e23434. doi: 10.1371/journal.pone.0023434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi I, Celli JP, Evans CL, Abu-Yousif AO, Muzikansky A, Pogue BW, Finkelstein D, Hasan T. Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer. Cancer Res. 2010;70:9319–9328. doi: 10.1158/0008-5472.CAN-10-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujimoto JG. Optical coherence tomography for ultrahigh resolution in vivo imaging. Nat Biotechnol. 2003;21:1361–1367. doi: 10.1038/nbt892. [DOI] [PubMed] [Google Scholar]
- 12.Bouma BE, Tearney GJ. Handbook of optical coherence tomography. Marcel Dekker; New York: 2002. [Google Scholar]
- 13.Huang D, Swanson E, Lin C. Optical Coherence Tomography. Science. 1991;254:1178–1181. doi: 10.1126/science.1957169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tearney GJ, Brezinski ME, Bouma BE, Boppart Sa, Pitris C, Southern JF, Fujimoto JG. In vivo endoscopic optical biopsy with optical coherence tomography. Science. 1997;276:2037–2039. doi: 10.1126/science.276.5321.2037. [DOI] [PubMed] [Google Scholar]
- 15.de Boer JF, Cense B, Park BH, Pierce MC, Tearney GJ, Bouma BE. Improved signal-to-noise ratio in spectral-domain compared with time-domain optical coherence tomography. Opt Lett. 2003;28:2067–2069. doi: 10.1364/ol.28.002067. [DOI] [PubMed] [Google Scholar]
- 16.Yun S, Tearney G, Boer Jd. High-speed optical frequency-domain imaging. Opt Express. 2003;11:2953–2963. doi: 10.1364/oe.11.002953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber R, Wojtkowski M, Fujimoto JG. Fourier Domain Mode Locking (FDML): A new laser operating regime and applications for optical coherence tomography. Opt Express. 2006;14:3225–3237. doi: 10.1364/oe.14.003225. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Chu KK, Houser GH, Diephuis BJ, Li Y, Wilsterman EJ, Shastry S, Dierksen G, Birket SE, Mazur M, Byan-Parker S, Grizzle WE, Sorscher EJ, Rowe SM, Tearney GJ. Method for quantitative study of airway functional microanatomy using micro-optical coherence tomography. PloS one. 2013;8:e54473. doi: 10.1371/journal.pone.0054473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans CL, Rizvi I, Hasan T, de Boer JF. In vitro ovarian tumor growth and treatment response dynamics visualized with time-lapse OCT imaging. Opt Express. 2009;17:8892–8906. doi: 10.1364/oe.17.008892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celli JP, Rizvi I, Evans CL, Abu-Yousif AO, Hasan T. Quantitative imaging reveals heterogeneous growth dynamics and treatment-dependent residual tumor distributions in a three-dimensional ovarian cancer model. J Biomed Opt. 2010;15:051603. doi: 10.1117/1.3483903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung Y, Nichols AJ, Klein OJ, Roussakis E, Evans CL. Label-Free, Longitudinal Visualization of PDT Response In Vitro with Optical Coherence Tomography. Isr J Chem. 2012;52:728–744. doi: 10.1002/ijch.201200009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mujat M, Park BH, Cense B, Chen TC, de Boer JF. Autocalibration of spectral-domain optical coherence tomography spectrometers for in vivo quantitative retinal nerve fiber layer birefringence determination. J Biomed Opt. 2007;12:041205–041205. doi: 10.1117/1.2764460. [DOI] [PubMed] [Google Scholar]
- 23.Klein OJ, Bhayana B, Park YJ, Evans CL. In vitro optimization of EtNBS-PDT against hypoxic tumor environments with a tiered, high-content, 3D model optical screening platform. Mol Pharm. 2012;9:3171–3182. doi: 10.1021/mp300262x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Padrón J, Wilt Cvd. The multilayered postconfluent cell culture as a model for drug screening. Crit Rev Oncol Hematol. 2000;36:141–157. doi: 10.1016/s1040-8428(00)00083-4. [DOI] [PubMed] [Google Scholar]
- 25.Al-abd AM, Lee J-h, Kim SY, Kun N, Kuh H-j. in situ evaluation of cytotoxicity and penetration of. 2008;99:423–431. doi: 10.1111/j.1349-7006.2007.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan Z, Hooley J, Smith DH, Young P, Roberts PE, Mather JP. Establishment of human ovarian serous carcinomas cell lines in serum free media. Methods. 2012;56:432–439. doi: 10.1016/j.ymeth.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Reichert M, Steinbach JP, Supra P, Weller M. Modulation of growth and radiochemosensitivity of human malignant glioma cells by acidosis. Cancer. 2002;95:1113–1119. doi: 10.1002/cncr.10767. [DOI] [PubMed] [Google Scholar]
- 28.Thews O, Gassner B, Kelleher DK, Schwerdt G, Gekle M. Impact of extracellular acidity on the activity of P-glycoprotein and the cytotoxicity of chemotherapeutic drugs. Neoplasia. 2006;8:143–152. doi: 10.1593/neo.05697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alderden RA, Mellor HR, Modok S, Hall MD, Sutton SR, Newville MG, Callaghan R, Hambley TW. Elemental tomography of cancer-cell spheroids reveals incomplete uptake of both platinum(II) and platinum(IV) complexes. JACS. 2007;129:13400–13401. doi: 10.1021/ja076281t. [DOI] [PubMed] [Google Scholar]
- 30.Chakraborty A, Held K, Prise K. Bystander Effects Induced by Diffusing Mediators after Photodynamic Stress. Radiat Res. 2009;172:74–81. doi: 10.1667/RR1669.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brancolini C, Lazarevic D, Rodriguez J, Schneider C. Dismantling cell-cell contacts during apoptosis is coupled to a caspase-dependent proteolytic cleavage of beta-catenin. JCB. 1997;139:759–771. doi: 10.1083/jcb.139.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imamdi R, Graauw MD, Water BVD. Protein kinase C mediates cisplatin-induced loss of adherens junctions followed by apoptosis of renal proximal tubular epithelial cells. JPET. 2004;311:892–903. doi: 10.1124/jpet.104.072678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1: Time-lapse OCT movie of 3D OVCAR5 nodules undergoing treatment response following PDT. Cross-sectional images were collected every 15 minutes for 24 hours following PDT with EtNBS-OH (500 nM, 20 J/cm2). Nodules are seen to follow an inside out pattern of structural degradation resulting from the cytotoxic effects of therapy beginning one hour following PDT. The development of dark, highly scattering bodies towards the end of the 24 hour period correlate with the appearances of apoptotic cells undergoing nuclear condensation.