

Summary
Achieving the goal of malaria elimination will depend on targeting Plasmodium pathways essential across all life stages. Here, we identify a lipid kinase, phosphatidylinositol 4-kinase (PI4K), as the target of imidazopyrazines, a novel antimalarial compound class that inhibits the intracellular development of multiple Plasmodium species at each stage of infection in the vertebrate host. Imidazopyrazines demonstrate potent preventive, therapeutic, and transmission-blocking activity in rodent malaria models, are active against blood-stage field isolates of the major human pathogens, P. falciparum and P. vivax, and inhibit liver stage hypnozoites in the simian parasite P. cynomolgi. We show that imidazopyrazines exert their effect through inhibitory interaction with the ATP-binding pocket of PI4K, altering the intracellular distribution of phosphatidylinositol 4-phosphate. Collectively, our data define PI4K as a key Plasmodium vulnerability, opening up new avenues of target-based discovery to identify drugs with an ideal activity profile for the prevention, treatment and elimination of malaria.
Introduction
To eliminate malaria, broadly acting medicines must be developed that cure the symptomatic asexual blood stage, clear the preceding liver stage infection that can cause relapses and block parasite transmission to mosquitoes1. Relapse prevention is especially important for P. vivax, which forms intra-hepatic hypnozoites that can persist for years before reinitiating development and triggering blood stage infection. Primaquine is the only licensed antimalarial capable of eliminating the hypnozoite reservoir and delivering a radical cure. However, side-effects and weak activity against blood stages preclude widespread use of primaquine2. Since primaquine’s target and mechanism of action are not known, the search for novel radical cure drugs has been limited to related analogs, such as Tafenoquine3. There is a clear need for druggable and chemically validated targets that are essential in all lifecycle stages of the malaria parasite.
Here we report that a parasite phosphatidylinositol 4-kinase type III beta (PI4KIIIβ), a ubiquitous eukaryotic enzyme that phosphorylates lipids to regulate intracellular signaling and trafficking, is inhibited by imidazopyrazines. In blood stages, imidazopyrazines block a late step in parasite development by disrupting plasma membrane ingression around developing daughter merozoites. This likely stems from altered phosphatidylinositol 4-phosphate (PI4P) pools and disrupted Rab11A-mediated membrane trafficking. Our findings validate PI4K as the first known drug target required across all Plasmodium lifecycle stages.
Results
Imidazopyrazines target multiple life stages
A cell-based screen against P. falciparum asexual blood-stage parasites4 revealed a new class of antimalarials with an imidazopyrazine core (Fig. 1a). This compound was distinct from known antimalarials and was active against multiple drug-resistant strains (IC50 27–70 nM; Extended Data Table 1 and Extended Data Fig. 1a), suggesting a novel mechanism of action. Synthetic derivatives with potency and pharmacokinetic properties suitable for testing in animal models were designed and showed activity in vivo (data shown for KDU691; Extended Data Table 2 and Extended Data Fig. 1b).
Figure 1. Imidazopyrazines demonstrate potent activity across Plasmodium species and parasite stages.

The central schematic illustrates the Plasmodium lifecycle in mosquitoes and the vertebrate host. Injected sporozoites infect the liver, proliferate and emerge into the bloodstream as merozoites; in some species hypnozoites may remain dormant in the liver. Merozoites undergo multiple cycles of asexual proliferation in RBCs, or at low frequency, differentiate into sexual-stage gametocytes. Mature gametocytes ingested by an Anopheles mosquito become gametes that mate and form oocysts, within which sporozoites develop that migrate to the salivary glands. a, Chemical structures of the imidazopyrazine analogs KAI407 (R=trifluoromethyl, R’=cyano), KDU691 (R=methylamide, R’=chloro), KAI715 and the quinoxaline BQR695. b, In vitro activity of imidazopyrazines against liver-stage schizonts of P. yoelii (means±s.d.; n=4). c, In vivo efficacy of KDU691 against luciferase-expressing P. berghei. Mice were either untreated (+) or given a single oral dose of 7.5 mg/kg KDU691 prior to sporozoite inoculation (t = 0 hr) or after a liver-stage infection was established (t=24, 36 or 48 hr post-infection), then monitored by bioluminescence (one representative mouse per group shown; n=8). d, In vitro inhibition of P. cynomolgi hypnozoites by imidazopyrazines (means±s.d.; n=4). e, Ex vivo analysis of imidazopyrazine activity against asexual blood-stages of P. falciparum (n=8) and P. vivax field isolates (n=6), shown as a boxplot (mean; interquartile range 25–75%) with whiskers (min-max). f, KDU691 inhibition of P. falciparum gametocyte conversion to female gametes after 24 hr incubation with compound, expressed as the percentage of Pfs25-positive female gametes (means; n=2). g, Transmission-blocking effect of KDU691, measured by the number of P. falciparum oocysts in Anopheles stephensi midguts (means±s.d.; n=20) infected with parasites exposed to either 0.1% DMSO, 1 μM DHA or 1 μM KDU691. Abbreviations: ATQ, atovaquone; PQ, primaquine; DHA, dihydroartemisinin.
We evaluated imidazopyrazines (Fig. 1a), against all parasite lifecycle stages in the vertebrate host. First, a cell-based assay showed potent inhibition of liver-stage development of the rodent parasite, P. yoelii, with IC50 values less than 160 nM and as low as 9 nM (Fig. 1b) for KDU691, KAI407 and KAI715. In vivo, KDU691 administered orally at the time of infection prophylactically protected mice from colonization with transgenic P. berghei sporozoites expressing luciferase. Just a single dose (7.5 mg/kg; Fig. 1c and Extended Data Fig. 1c) prevented what would normally be a fatal outcome. To determine whether KDU691 could eliminate an existing infection, animals were dosed 24, 36 or 48 hr post-sporozoite inoculation, well after the onset of liver-stage development (which lasts 48 hr in P. berghei). This delayed treatment rapidly eliminated the luciferase-expressing parasites with a single dose (Fig. 1c). KDU691 was also active at low concentrations (IC50~196 nM; Fig. 1d) against in vitro cultured liver-resident hypnozoites of the simian parasite P. cynomolgi5. Collectively these data demonstrate that KDU691 is active against liver stages in multiple Plasmodium species, including hypnozoites associated with malaria relapse. To assess imidazopyrazine activity against a human parasite that forms hypnozoites, we tested blood-stage P. vivax field isolates for sensitivity. KDU691 potency against P. vivax (mean IC50~69 nM) was comparable to that observed in parallel studies conducted with field isolates of the more prevalent human pathogen, P. falciparum (mean IC50~118 nM; Fig. 1e).
A small proportion of asexual blood-stage parasites sexually differentiate into gametocytes that transmit to the mosquito vector. Gametocytes are often not killed by drugs that provide symptomatic relief6. We sought to determine the transmission-blocking potential by assessing compound activity on gametocyte viability and the mosquito-specific stages of gamete and oocyst formation. An enriched gametocyte population was treated with either KDU691 or dihydroartemisinin (DHA), a licensed antimalarial with gametocytocidal activity7. Both compounds reduced gametocyte viability in a dose-dependent manner, with KDU691 displaying an IC50 value of 220 nM (Extended Data Fig. 1d). Furthermore, 200 nM KDU691 reduced gamete formation by 60%, comparable to that achieved with 1 μM DHA (Fig. 1f). Lastly, we tested compounds in the ‘Standard Membrane Feeding Assay’ that directly monitors transmission to the mosquito8. Transmission was completely inhibited at 1 μM KDU691 (Fig. 1g), with dose-dependent suppression of oocyst densities (IC50~316 nM; Extended Data Fig. 1e) and oocyst prevalence (i.e. the number of infected mosquitoes; IC50~370 nM; Extended Data Fig. 1f). Altogether, these data indicate that KDU691 is effective at blocking transmission and suggest it targets a conserved function throughout the Plasmodium lifecycle.
Inhibition of blood-stage cytokinesis
Imidazopyrazine-treated parasites accumulated multinucleated schizonts (Fig. 2a), leading us to test compound effects when administered at distinct stages during development in red blood cells (RBCs). Parasite reinvasion was dramatically inhibited even when KAI407 was added late in the intraerythrocytic cycle, up to 44 hr post-invasion (Fig. 2b). Conversely, early ring-stage parasites exposed continuously to KAI407 for up to 40 hr followed by drug-washout demonstrated no appreciable loss of parasite viability (Extended Data Fig. 2a). These data suggest that the imidazopyrazine target is most critical at a very late stage in the 48 hr intraerythrocytic cycle.
Figure 2. PfPI4K is required for the completion of cytokinesis in asexual blood-stage schizonts.

a, Microscopy of highly-synchronized parasites continuously treated with ~5×IC50 drug (125 nM KAI407, 15 nM KAI715 or 150 nM KDU691) or DMSO vehicle. Representative images from a single experimental replicate are shown (n=3). b, Drug-free media was supplemented with 125 nM KAI407 in 2 hr intervals starting at the mid/late trophozoite stage (t=34 hr post-invasion). Parasitemia in the next lifecycle (t=72 hr) was normalized to untreated parasites (means±s.d.; n=4). Statistical significance was determined by Mann-Whitney U test: *P<0.05, **P< 0.01, ***P<0.001 (also for panel c). c, Percent reinvasion of RBCs after mechanical rupture of schizonts treated with 125 nM KAI407, 15 nM KAI715, 150 nM KDU691 or 1 μM E-64. (means±s.d.; n=4). d, PfATP4-GFP was used to visualize plasma membrane ingression around developing daughter merozoites, with nuclei stained by Hoechst 33342. Representative images from a single experimental replicate are shown (n=2). Scale bar, 5 μm. Parasites treated with 500 nM KAI407 for 4 hr had a disorganized membrane structure, without clearly defined daughter cells.
We next tested whether drug-treated parasites arrest during maturation of daughter merozoites or form daughter cells that fail to egress from the host cell9,10. Synchronized imidazopyrazine-treated parasites were mechanically ruptured, yielding a reinvasion parasitemia near 0.01% (Fig. 2c), suggesting a defect in merozoite formation. As a control, merozoites that were artificially retained within a mature parasite by treatment with E-64, a protease inhibitor that blocks egress11, and then mechanically released remained viable and capable of reinfecting RBCs, yielding 2–3% parasitemia (Fig. 2c)9,10. Mock-treated parasites that had been allowed to reinvade naturally yielded a parasitemia of 8–10%.
To visualize drug-induced cellular defects, we examined a transgenic line expressing PfATP4-GFP, a plasma-membrane protein12 that reveals merozoite envelopment during biogenesis. Control E-64-treated parasites showed complete plasma membrane ingression around each daughter cell (Fig. 2d and Extended Data Fig. 2b). In contrast, KAI407-treated parasites showed incomplete and disorganized segmentation, lacking merozoites that were fully enclosed by membranes with PfATP4-GFP, suggesting perturbation of plasma membrane ingression (Fig. 2d and Extended Data Fig. 2c). Based on the timing of growth inhibition and the observed cellular phenotype, we conclude that imidazopyrazines impair cytokinesis by interfering with membrane biogenesis around the developing merozoite.
Plasmodium PI4K target identification
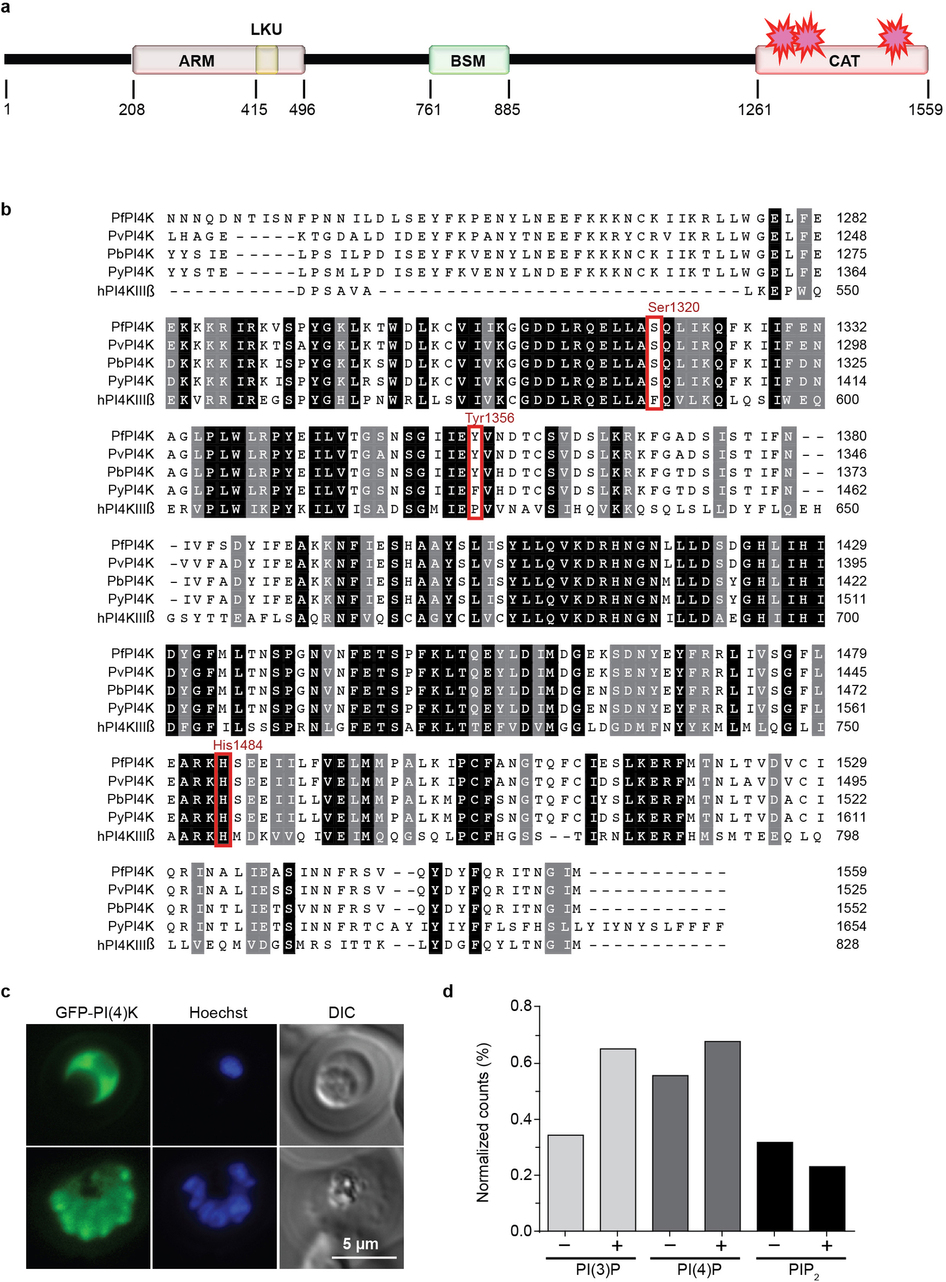
To determine the mechanism of broad antimalarial activity by imidazopyrazines, we next sought to elucidate their target using forward genetics. We selected imidazopyrazine-resistant blood-stage P. falciparum parasites in vitro using either KAI407 or KAI715. Resistant lines were evolved in three independent flasks for each compound, with multiple clones isolated from each flask for IC50 characterization (Fig. 3a). For each clone, we identified genomic differences relative to the parent using a genome-tiling microarray13. All five KAI407-resistant clones shared alterations to a single gene, pfpi4k (PF3D7_0509800; PI4KIIIβ), one of two annotated phosphatidylinositol 4-kinases. Type III β phosphatidylinositide kinases (Extended Data Fig. 3a,b) are conserved amongst eukaryotes and function to convert phosphatidylinositol (PI) to phosphatidylinositol 4-phosphate (PI4P). Within the cell, PI4P regulates effector protein recruitment and lipid-sorting events at the Golgi, and is a precursor for the generation of PI4,5P2 at the plasma membrane14,15.
Figure 3. Resistance to imidazopyrazine and quinoxaline compounds is mediated by gene variations in PfPI4K and PfRab11A.

a, Fold-change in IC50 values between 11 drug-evolved clones and the drug-sensitive parent, for the imidazopyrazines, KAI407, KAI715, KDU691, and the quinoxaline BQR695 (means±s.d.; n=8). Statistically significant mean IC50 values for each drug-resistant line were identified using the Mann-Whitney U test (also for panels c, d, e). CNVs and SNVs are noted. b, Schematic overview of the pfpi4k gene editing strategy to introduce putative resistance mutations into a wild type parasite. ZFNs target a 34-bp site on pfpi4k (thunderbolt); following cleavage, homology-dependent repair from a 1.7 kb donor sequence resulted in incorporation of the specific SNV (shown in red), as well as additional silent mutations at the ZFN cut site. c, Fold-change in IC50 values of ZFN-edited lines H1484Y-, S1320L-and Y1356F-PfPI4K against KAI407, KDU691, KAI715 and BQR695 (means±s.d.; n=12). IC50 values for other known antimalarials are provided in Extended Data Fig. 5a. d, Chromosomal integration of GFP fusions of wild type Rab11A, mutant Rab11A (D139Y) or wild type PI4K conferred resistance to imidazopyrazine and quinoxaline compounds. A control line expressing GFP alone was used to normalize the IC50 shift (means±s.d.; n=6). e, Fold-change in IC50 values of P. berghei in vitro liver-stage schizonts expressing PbPI4K-H1477Y (equivalent to PfPI4K-H1484Y). Data shown as means±s.d. (n=12).
Three KAI407-resistant clones possessed copy number variations (CNVs) that included pfpi4k (Extended Data Fig. 4a). In the remaining two clones, a single nucleotide variant (SNV) was detected in the second exon of pfpi4k (Extended Data Fig. 4b). Sequencing of pfpi4k in these two independently evolved clones revealed an identical non-synonymous SNV that led to a H1484Y coding change. Removal of drug pressure for three months resulted in resensitization of one clone (KAI407-R3-REV) to KAI407, with a corresponding reversion in pfpi4k to the wild type sequence, supporting a role for this variant in KAI407 resistance (Fig. 3a). Selections with KAI715 yielded two resistant clones, one of which also implicated pfpi4k amplification in resistance (KAI715-R4; Extended Data Fig. 4c). The second clone (KAI715-R5) possessed a solitary D139Y mutation in a second gene product, the small GTPase PfRab11A (PF3D7_1320600) (Extended Data Fig. 4d). Notably, PI4K binds and regulates Rab11A in Drosophila spermatocytes16, consistent with a related function in membrane trafficking.
Our imidazopyrazine-resistant lines provide compelling evidence for PI4KIIIβ as the plasmodial drug target. Orthologs of PI4KIIIβ are found in all Plasmodium species and are extensively conserved at the amino acid level, including 97% identity in the catalytic domain between the falciparum and vivax orthologs (Extended Data Fig. 3b). Conservation with the catalytic region of human PI4KIIIβ is less significant, with only 43% identity. The partial sequence conservation between the human and Plasmodium orthologs of PI4KIIIβ (Extended Data Fig. 3b) led us to test whether some inhibitors of the human kinase might also exert antimalarial activity.
Subsequent screening of a Novartis compound library led us to identify BQR695 (Fig. 1a), a quinoxaline that displayed sub-micromolar potency against both human PI4KIIIβ (IC50~90 nM; Extended Data Table 3) and P. falciparum asexual blood stages (IC50~71 nM). BQR695, which showed no evidence of toxicity against mature RBCs, induced a schizont-stage arrest indistinguishable from that observed in imidazopyrazine-treated parasites (Extended Data Fig. 2d,e) and exhibited cross-resistance with the imidazopyrazine-resistant lines (Fig. 3a). These data confirm a shared mechanism of action and of resistance with the imidazopyrazines. To further test this association, we evolved BQR695-resistant parasites. Whole-genome scanning of three independently selected lines (BQR695-R) showed two instances of CNVs involving pfpi4k (Extended Data Fig. 4e), and a novel mutation (S1320L) in pfpi4k (Extended Data Fig. 4f). Sequencing of pfpi4k in the CNV-containing lines revealed that BQR695-R7 had at least one pfpi4k copy with a Y1356F coding change, which was associated with a higher-degree of resistance to BQR695 (Fig. 3a). All three resistance-associated SNVs, S1320L, Y1356F and H1484Y, mapped to the catalytic domain of PfPI4K (Extended Data Fig. 3a,b). Importantly, the three imidazopyrazines (KAI407, KAI715, and KDU691) and the quinoxaline BQR695 demonstrated comparable cross-resistance against all drug-resistant lines (Fig. 3a). Collectively these data provide compelling chemical and genetic evidence that PI4K is the direct drug target of imidazopyrazine and quinoxaline compounds.
PI4K variation confers drug resistance
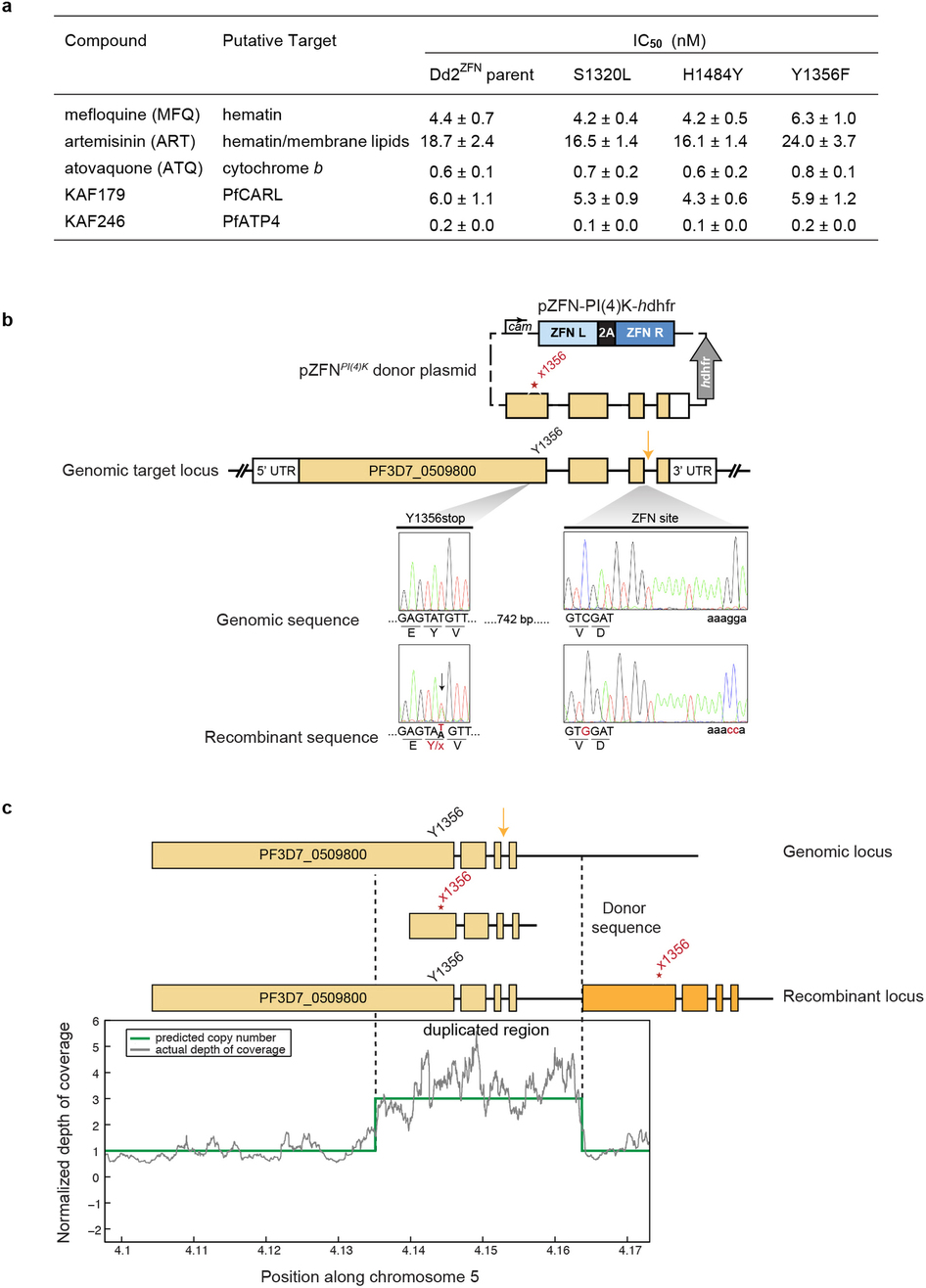
To confirm the role of pfpi4k and pfrab11a mutations in resistance, we engineered these variants into drug-sensitive parasites. We generated recombinant parasites bearing individual PfPI4K SNVs using zinc-finger nuclease (ZFN) technology17. Genome editing was confirmed by direct sequencing (Fig. 3b) and whole-genome analysis (>20× coverage across 96.4% of the genome; Extended Data Table 4). Genome-edited Dd2 parasites containing either the S1320L or H1484Y mutations were resistant to both imidazopyrazine and quinoxaline compounds (Fig. 3c and Extended Data Fig. 5a) and yielded resistance comparable to the original drug-evolved lines. In contrast, the Y1356F mutation conferred resistance specifically to the quinoxaline BQR695 but not to the imidazopyrazines (Fig. 3c). In comparison, the evolved line BQR695-R7, which possessed both the Y1356F mutation as well as a CNV, showed resistance to both imidazopyrazine and quinoxaline compounds (Fig. 3a).
Because several resistant lines contained CNV events, we sought to determine whether increased copy numbers of wild type PI4K could alone mediate protection. We generated a stable parasite line over-expressing GFP-tagged PI4K18. GFP-PI4K was diffusely localized in trophozoite-stage parasites and enriched at the apical ends of developing daughter merozoites (Extended Data Fig. 3c). Expression of this additional wild type copy of PfPI4K resulted in decreased susceptibility to both imidazopyrazine and quinoxaline compounds, confirming that CNVs can contribute to resistance and providing evidence that the GFP-tagged protein is functional (Fig. 3d). Collectively, these data demonstrate that PfPI4K can mediate resistance to the imidazopyrazines in asexual blood stages either through increased copy number or point mutations.
We next investigated whether PI4K SNV variants could confer resistance in liver-stage parasites. Introduction of a mutation analogous to H1484Y into P. berghei (H1477Y) conferred a significant IC50 shift for the imidazopyrazine and quinoxaline compounds against in vitro liver-stage parasites (Fig. 3e). Importantly, these data show that the mechanism of action for these inhibitors is conserved between lifecycle stages.
Finally, we tested the validity of the Rab11A D139Y variation, which conferred a modest, but significant, resistance phenotype (KAI715-R5; Fig. 3a). The D139Y SNV lies on the α4 helix of Rab11A, distant from the GTP binding site (Extended Data Fig. 6a,b). GFP-tagged versions of both wild type and D139Y Rab11A showed a predominantly cytosolic localization that became apically punctate in late schizonts (Extended Data Fig. 6c,d), consistent with a previous report19. As expected, the D139Y mutant conferred modest resistance against KAI407 (Fig. 3d).
If PI4K is the cellular target of the imidazopyrazines, then its function should be essential in most stages of the Plasmodium lifecycle. We therefore tested the essentiality of pfpi4k in the asexual blood stages by using ZFNs to introduce a stop codon at position Y1356, which lies within the catalytic site of the kinase domain (see Fig. 4a,b). This Y1356stop editing experiment was attempted in three independent transfections. Unlike the Y1356F mutation, which was readily isolated in ~18% of clones, two transfection reactions produced 0/35 clones with a stop codon. From a third transfection, one clone yielded a duplication on chromosome 5 that partially encompassed pfpi4k and placed the edited stop codon within a truncated copy of the gene (Extended Data Fig. 5b,c). These data strongly suggest that gene editing could not be achieved without a duplication event that preserved one copy of the gene, confirming pfpi4k essentiality to blood-stage growth.
Figure 4. Imidazopyrazines and quinoxalines bind the ATP-binding site of PI4K and alter PI4P intracellular distribution.

a, In silico docking of a homology model of the PfPI4K catalytic domain showing KAI407 (green) and BQR695 (orange) binding to the ATP-binding pocket of PfPI4K (cartoon representation; grey). Resistance SNVs (yellow) are labeled. b, Zoomed view of the ATP-binding pocket rotated 90° along the y-axis, showing the predicted hydrogen bond (green dash) between the imidazole nitrogen in KAI407 and the amide bond from the V1357 backbone (grey sticks). c, d, Recombinant full-length P. vivax PI4K (PvPI4K) activity in the presence of (c) KDU691 or (d) BQR695 monitored across a range of ATP concentrations (1.25–40 μM). Data shown as means±s.d. (n=9). e, Recombinant PvPI4K mutants S1286L, Y1322F or H1450Y (equivalent to PfPI4K-S1320L, -Y1356F or -H1484Y respectively) were assayed against imidazopyrazine and quinoxaline compounds. Fold-change in IC50 value is shown relative to wild type enzyme (means±s.d.; n=9). Statistical significance was determined by the Mann–Whitney U test: *P<0.05; **P<0.01; ***P<0.001 (also for panel h). f, Illustration of the PI4P-dependent localization of GFP-PHOsh2. g, In vivo distribution of PI4P was detected in parasites expressing GFP-PHOsh2. In untreated parasites, GFP-PHOsh2 localized to intracellular foci (blue arrow) and the plasma membrane (white arrow). Treatment with 500 nM KAI407 or BQR695 for 4 hr depleted the intracellular pool and redistributed the probe to the plasma membrane. Representative images from a single experimental replicate are shown (n=3). Nuclei were stained with Hoechst 33342 (blue); DIC, differential interference contrast. Scale bar, 5 μm. h, Quantification of intracellular GFP-PHOsh2-labeled foci after drug treatment (means±s.d.; n=3).
Biochemical validation of PfPI4K
We performed in silico modeling on a PfPI4K homology model based on related PI3-kinases (Fig. 4a), finding that both KAI407 and BQR695 were accommodated in the ATP-binding pocket. The nitrogen at position 1 of the imidazole ring of KAI407 is predicted to make a critical hydrogen bond contact with the main chain amide from V1357 that lies adjacent to the Y1356F SNV (Fig. 4b). This ‘hinge-binding’ contact is common among protein kinase inhibitors and mimics the hydrogen bonds made by the adenine of ATP20.
To confirm that imidazopyrazines directly inhibit PI4K function we expressed full-length recombinant P. vivax PI4K (PvPI4K). KDU691 and BQR695 showed dose-dependent inhibition of PvPI4K activity (Fig. 4c,d), with an IC50 of 1.5 and 3.5 nM, respectively, in the presence of 10 μM ATP. Furthermore, compound potency decreased with increasing concentrations of ATP, indicative of ATP-competitive inhibition, providing further evidence that imidazopyrazines and quinoxalines bind to the ATP-binding pocket of parasite PI4K. Mutations homologous to the PfPI4K resistance-conferring variants (Extended Data Fig. 3b) generated resistance profiles (Fig. 4e) similar to those observed with the respective drug-resistant parasite lines (Fig. 3a,c). In contrast to the low nanomolar activity observed against parasite PI4K, KDU691 and KAI407 did not significantly inhibit any of the >40 enzymes in a panel of human kinases, and both were at least 1000× more potent against the parasite enzyme than against any human lipid kinase (Extended Data Table 3), demonstrating remarkable selectivity. This specificity precludes the possibility that imidazopyrazine antimalarial activity derives from inhibition of host kinases.
Perturbed cellular distribution of PI4P
Finally, we examined the consequences of PfPI4K inhibition on intracellular PI4P. Somewhat unexpectedly, HPLC analysis of radiolabeled phospholipids did not reveal a decrease in total PI4P after imidazopyrazine treatment (Extended Data Fig. 3d). To determine if imidazopyrazines might instead change the intracellular distribution of PI4P, we generated a transgenic parasite expressing a validated PI4P-specific probe, GFP-PHOsh2, which comprises GFP fused to two tandem PI4P-binding pleckstrin homology (PH) domains from yeast Osh2 (Fig. 4f)21. In yeast, this marker defines two populations of PI4P – one at the plasma membrane and one intracellular, with the latter diminished by inhibition of the Golgi-localized PI4K homolog, Pik121. Similarly, in P falciparum, GFP-PHOsh2 localized to intracellular foci and, to a lesser degree, the parasite plasma membrane (Fig. 4g). Treatment with 0.5 μM of either KAI407 or BQR695 caused GFP-PHOsh2 to redistribute to the parasite plasma membrane (Fig. 4g,h), consistent with depletion of intracellular PI4P upon inhibition of PfPI4K function.
Discussion
Here, we report the identification of PI4K as an essential and druggable target of imidazopyrazines and quinoxaline compounds that block development at multiple stages of the Plasmodium lifecycle, implicating this phosphatidylinositol kinase as a critical cellular regulatory molecule. PfPI4K functionally complements yeast Pik1, an essential Golgi-localized PI4KIIIβ22. Our data suggest that, like Pik123-25, PfPI4K regulates essential membrane trafficking events, with the plasmodial protein required for the late-stage membrane ingression that drives completion of the asexual erythrocytic stages. We propose that Plasmodium PI4K acts at the Golgi to locally generate PI4P, which in turn recruits effectors that generate transport vesicles destined for the ingressing plasma membrane (Fig. 5). One obvious candidate effector is Rab11A, which has also been linked to PI4K in Drosophila and mammalian cells16,26. Drosophila PI4K and Rab11 act during cytokinesis, regulating membrane delivery to complete cell division16, which mirrors our findings of failed membrane ingression upon PfPI4K inhibition. Rab11A may bind directly to PI4K and/or PI4P or may instead be recruited indirectly by an intermediate protein. In either case, Rab11A likely marks these Golgi-derived vesicles to modulate delivery to the target compartment, a role consistent with previous findings implicating Plasmodium Rab11A in cytokinesis19.
Figure 5. Proposed role of PfPI4K within asexual blood-stage parasites.

Model depicting daughter merozoite biogenesis in a late-stage schizont, showing plasma membrane ingression and development of the underlying inner membrane complex (IMC). Delivery of post-Golgi secretory vesicles to the invaginating plasma membrane requires PfPI4K-driven production of PI4P, which likely recruits lipid-binding effector proteins in a Rab11A-dependent process. PVM, parasitophorous vacuolar membrane; N, nucleus.
Our results provide genetic, chemical and biochemical validation of Plasmodium PI4K as a target suitable for antimalarial drug discovery. Although the imidazopyrazine compounds described here may serve primarily as tool compounds, further optimization could lead to clinical candidates with desirable drug-like characteristics. Maintaining their remarkable selectivity for Plasmodium PI4K will be an important feature for an ultimate antimalarial drug because chronic PI4KIIIβ inhibition in humans causes immunosuppressive effects27 that could preclude long-term malaria prophylaxis. Because the emergence of resistant parasites has diminished the effectiveness of all licensed antimalarial therapies, most of which were introduced as monotherapies, clinical candidates will necessarily require combination with mechanistically distinct drug partner(s) to mitigate the potential for drug resistance and to complement the PI4K inhibitors’ narrow and late stage of action in the asexual blood stage.
Collectively, our data identify Plasmodium PI4K as a novel drug target that operates in all lifecycle stages of the parasite and is therefore suitable for the development of next-generation antimalarial drugs capable of curing, preventing and blocking the transmission of the disease. We anticipate that our findings will rapidly yield clinical candidates compatible with single exposure, radical cure and prophylaxis (SERCAP), a profile widely heralded as critical for the success of worldwide malaria elimination efforts28.
Methods
Statistical analysis
Data are shown as mean ± s.d., and represent at least two independent experiments performed in duplicate unless stated otherwise. Statistical significance between groups was determined using an unpaired, nonparametric analysis via the Mann–Whitney U test (with Prism, GraphPad Software, Inc). The Mann-Whitney test does not require the assumption that the differences between the two samples are normally distributed. P-values were considered significant if P<0.05.
P. berghei and P. yoelii sporozoite generation
The New York University Insectary provided Anopheles stephensi mosquitoes infected with rodent malaria: Plasmodium yoelii (17XNL), a transgenic P. berghei expressing luciferase (ANKA) or an engineered P. berghei ANKA line with H1477Y-PbPI4K (see below). Plasmodium-infected mosquitoes were dissected 22–25 days after an infective blood meal and sporozoites purified as described by Meister et al.29.
P. yoelii liver schizont assay
A previously described29 in vitro high-content imaging assay was used to measure the proliferation of P. yoelii sporozoites after invasion of a transgenic hepatoma line (HepG2-A16-CD81-EGFP; obtained from the laboratory of Dominique Mazier (INSERM, France); tested monthly for mycoplasma contamination; used for up to 50 passages). Anti-PyHSP70-stained exoerythrocytic forms (EEFs) were quantified using the Opera Confocal High Content Screening System (PerkinElmer). Atovaquone and DMSO-treated wells were used as controls on each assay plate. IC50 values were obtained using mean parasite area and a custom curve fitting model, and a standard logistic regression model was applied for curve fitting.
In vivo pharmacokinetic studies and causal prophylaxis efficacy
All animal-related procedures at GNF were non-blinded and conducted under an IACUC approved protocol in compliance with Animal Welfare Act regulations and the Guide for the Care and Use of Laboratory Animals. To determine PK properties, non-randomized mice (n=3; naïve male Balb/C; 7–9 weeks old; 23–25 g) were dosed with KDU691 (HCl salt; formulated in 75% polyethylene glycol 300 (PEG300) and 25% of dextrose (5 mg/ml) in distilled water (D5W)). Sample size and analysis protocol followed a standardized protocol 36. The causal prophylaxis model was carried out in two independent experiments with non-randomized mice (n=4 per experimental group; female CD1; 7–9 weeks old; 25–32 g) inoculated with 1×105 P. berghei sporozoites (i.v.) and orally administered 7.5 mg/kg KDU691 (suspension formulation of 0.5% (w/v) methylcellulose and 0.5% (v/v) Tween 80). Sample size was determined by a power analysis using binary outcome data (infected or cured mice) and showed that the use of 4 mice per experimental group had 90% power to detect a difference of 75% (e.g. 3 mice). The timing of drug administration occurred either immediately after parasite inoculation (Day 0), 24 hr post-inoculation (Day 1), 36 hr post-inoculation or 48 hr post-inoculation (Day 2). Parasitemia was monitored by two methods. The In Vivo Imaging System (IVIS 100, Xenogen; Calipar Life Science) was used to acquire the bioluminescence signal (5 minute acquisitions) as previously described29. Alternatively, blood smear samples were obtained from each mouse on days 4, 5, 6, 7, 10, 15, 21 and 30 post-inoculation, stained with Giemsa, and viewed under a microscope for visual detection of blood parasitemia. Animals with parasitemia exceeding 20% were euthanized.
P. cynomolgi liver assay: sporozoite generation
All Rhesus macaques (Macaca mulatta) used in this study were bred in captivity for research purposes and were housed at the Biomedical Primate Research Centre (BPRC; AAALAC-certified institute) facilities under compliance with the Dutch law on animal experiments, European directive 86/609/EEC and with the ‘Standard for Humane Care and Use of Laboratory Animals by Foreign institutions’ identification number A5539-01, provided by the Department of Health and Human Services of the USA National Institutes of Health (NIH). The local independent ethical committee first approved all protocols. Non-randomized rhesus macaques (male or female; 5–14 years old; one animal per month) were infected with 1×106 P. cynomolgi M strain blood stage parasites, and bled at peak parasitemia. Approximately 300 female Anopheles stephensi mosquitoes (Sind-Kasur strain, Nijmegen University Medical Centre St Radboud) were fed with this blood as described previously37.
P. cynomolgi liver assay: sporozoite infection of primary rhesus hepatocytes
Rhesus monkey hepatocytes were isolated from liver lobes as described by Guguen-Guillouzo et al.38. Sporozoite infections were performed within three days after hepatocyte isolation. Sporozoite inoculation of primary Rhesus hepatocytes was performed according to Mazier et al.39 and Dembele et al.5. On day 6, intracellular P. cynomolgi malaria parasites were fixed, stained with purified rabbit antiserum reactive against P. cynomolgi Hsp70.15, and visualized with FITC-labeled goat anti-rabbit IgG antibodies. Quantification of small “hypnozoite” EEFs (1 nucleus, a small round shape, maximally 7 μm in diameter) or large “developing parasite” EEFs (more than 1 nucleus, larger than 7 μm and round or irregular shape) was determined for each well using a high-content imaging system (Operetta®, Perkin-Elmer).
P. falciparum asexual blood stage culture and assay
P. falciparum isolates were maintained by standard methods40 in an atmosphere of 93% N2, 4% CO2, 3% O2 at 37°C in complete culturing medium (CM: 10.4 g/L RPMI 1640 (without phenol red, with 2.1 mM glutamine), 5.94 g/L Hepes, 5 g/L Albumax II, 50 mg/L hypoxanthine, 2.1 g/L sodium bicarbonate, 10% human serum and 43 mg/L gentamicin). Human erythrocytes served as host cells. In vitro antimalarial activity was measured using a modified SYBR Green cell proliferation assay4.
In vivo mouse therapeutic efficacy assay
All in vivo efficacy studies were approved by the veterinary authorities of the Canton Basel-Stadt. In vivo antimalarial activity was assessed in non-randomized, non-blinded mice (n=3 (qd regimen) or n=5 (bid regimen) per experimental group; female NMRI; 6 weeks old; 20–22 g) intravenously infected on day 0 with 2×107 erythrocytes infected with P. berghei GFP parasites (PbGFPCON, kindly donated by Drs. AP Waters and CJ Janse, Glasgow and Leiden Universities)40. Power analysis using a binary outcome data (infected or cured mice) showed that the use of 3 mice had >80% power to detect a difference of 67% (e.g. 2 mice), whereas the power increased to >90% to detect a difference of 40% (e.g. 2 mice) when the sample size increased to 5. Compounds formulated in 0.5% methylcellulose/0.5% Tween 80 were administered orally in a volume of 10 mL/kg as a single dose (24 hr post-infection) or as four consecutive doses (6, 24, 48 and 72 hr post-infection). With the single-dose regimen at 96 hr post-infection, parasitemia was measured using standard flow cytometry techniques or standard microscopy41. Activity was calculated as the difference between the mean percent parasitemia for the control and treated groups, expressed as a percentage of the control group. The survival time in days was also recorded up to 30 days after infection, at which time parasite-free animals were considered cured.
Ex vivo schizont assay of P. falciparum and vivax isolates
Clinical isolates of P. vivax and P. falciparum (8 samples each) were collected from the Mae Sot region of Tak Province (Thailand) in 2011. All samples were collected from patients with acute malaria (with a mono species parasitemia of 5,000–8,000 parasites/μl, >80% of the parasites at ring stage and no antimalarials or antibiotics taken by the patient in the past 30 days) attending the clinics of Shoklo Malaria Research Unit. After obtaining written consent, the sensitivity of the parasite isolates was tested, as previously described12. The clinical samples were collected and tested in accordance to protocols approved by The Center for Clinical Vaccinology and Tropical Medicine at University of Oxford (OXTREC 58-09), which had been ratified by the Ethics Committee of the Faculty of Tropical Medicine at Mahidol University.
Gametocyte viability assay
P. falciparum NF54 stage III–V gametocytes were isolated by discontinuous Percoll gradient centrifugation of parasite cultures treated with 50 mM N-acetyl-D-glucosamine for three days to kill asexual parasites. Gametocytes were seeded in 96-well plates and viability was measured by a modified pLDH assay using resazurin as a substrate following a 72-hr compound exposure42. Compounds were tested in 9-point dose response series (0.1 nM – 1 μM in 0.5 log10 dilutions) in quadruplicate (2 different plates with 2 different parasite batches). Controls MIN (1 μM dihydroartemisinin; DHA), MAX (vehicle) and full dose response of reference compound (DHA) were run in a standardized plate format. Data were expressed as % effect relative to the MIN and MAX wells of the assay plate. Data were analyzed with GraphPad Prism 5 and IC50 values were calculated by applying a four-parameter non-linear regression model.
Gametogenesis assay
Cultures containing P. falciparum NF54 mature (stage V) gametocytes were pre-incubated with compound for 24 hr. KDU691 was tested at 0.2 μM (1×IC50) and 20 μM (100×IC50), and controls included 0.1% DMSO (vehicle) and 1 μM DHA. Subsequently, gametogenesis was induced by adding xanthurenic acid to a final concentration of 50 μM. An additional control included a sample treated with 0.02% w/v NaN3 to block gametogenesis (sample t=0 hr). Parasites were incubated for eight hr at 20°C and emerged gametes were immunostained with an Alexa Fluor® 488-labeled anti-Pfs25 antibody as previously described33. Gametes were quantified by flow cytometry and data were expressed as the fraction of Pfs25-positive cells in the population of uninfected and infected red blood cells.
Standard Membrane Feeding Assay
Cultures containing mature (stage V) P. falciparum NF54 gametocytes were pre-incubated with compound for 24 hr. Cells were spun down (10,000×g, 20 sec) and resuspended in human serum and red blood cells to a final hematocrit of 50%. Fresh compound was added and the blood meal was fed to Anopheles stephensi mosquitoes. Seven days after the blood meal the number of midgut oocysts was determined by microscopy. Compounds were tested in a 9-point dose response series (0.1 nM – 1 μM in 0.5 log10 dilutions) in duplicate. Controls included 2 vehicle controls (0.1% DMSO) and 2 positive controls (1 μM dihydroartemisinin; DHA). Per sample, a total of 20 mosquitoes were dissected to determine oocysts loads. Representative data are shown from one of two independent experiments and expressed as % effect relative to vehicle control and analyzed with the GraphPad Prism 5 software package by applying a four-parameter non-linear regression model.
Parasite synchronization
In brief, a Dd2 culture was treated consecutively with 5% sorbitol (6 hr spacing) and 36 hr after the first sorbitol treatment, the cultures were captured on MACS® LD columns (Miltenyi Biotec). Purified schizonts were eluted with complete culturing media (CM) and combined into a fresh culture. The flask was incubated at 37°C with gentle shaking (110 rpm) for 2.5 hr to promote optimal re-invasion. After this incubation, the culture was filtered over a fresh MACS® LD column to re-capture any remaining schizonts, and to allow the uninfected and newly parasitized red blood cells with ring-stage parasites to flow through. The entirety of reinvasion occurred over a 3 hr period, so the midpoint was used to establish t=0 hr in the parasite lifecycle.
Onset of inhibition studies
A synchronized culture was divided into 100 μL volumes (~2% parasitemia in 2.5% hematocrit) within a 96-well tissue culture plate (BD Biosciences). Stage of action studies were carried out by immediately adding 3×IC50 inhibitor and parasites were visualized at t= 24, 32, 40, 48 and 60 hr post-synchronization. Giemsa-stained parasites were visualized (>1000 cells per sample) and recorded by a light microscope (Carl Zeiss Axiostar Plus microscope, 100× objective lens) equipped with a PowerShot G6 digital camera (Canon). To test the effect of KAI407 exposure duration on parasite maturation throughout a single (~48 hr) asexual blood stage cycle, 125 nM KAI407 was added either immediately following synchronization or every two hr starting at lifecycle t=34 hr (trophozoite stage). Alternatively, 125 nM KAI407 was present in the media immediately following synchronization, and then washed out (3×1 ml CM, then resuspended to the original volume) at these same time points. In both experiments, the parasitemia of ring-stage parasites was determined by microscopy 60 hr post-final synchronization. Drug-treated wells were compared to a DMSO-treated control to determine the % inhibition.
Physical disruption/merozoite release assay
Synchronized parasites were cultured to schizogony (t=42 hr) and then treated with either DMSO, 1 μM E-64, or ~5×IC50 drug: 125 nM KAI407, 15 nM KAI715 or 150 nM KDU691. At=51 hr, parasites were sheared by 20 strokes through a 28.5 gauge needle as previously described10. The cultures were incubated for an additional 8 hr and ring stage parasitemias determined by microscopy (>1000 cells counted per sample).
Imaging of drug-treated PfATP4-GFP
Generation of the PfATP4-GFP line and subsequent fluorescence imaging was previously described by Rottmann et al.12. The drug-arrested phenotype induced by KAI407 treatment of this line was investigated with sorbitol synchronized populations. Either 500 nM KAI407 or DMSO (control) was added to early schizonts (t=40 hr) and incubated for 4 hr. Cells were then immediately fixed with paraformaldehyde and glutaraldehyde for imaging. Images are representative of two biological replicates.
In vitro resistance selection and microarray analysis
A clonal population of P. falciparum Dd2 parasites was used to initiate two or three independent parasite cultures under the initial selection pressure of 12 nM KAI407, 1 nM KAI715 or 40 nM BQR695. Stepwise drug evolution continued until the final concentration was at least 3-fold higher than the initial concentration (typically 80–120 days). For each of the ten resistant strains, copy number variations (CNVs) and single nucleotide variations (SNVs) were detected using a whole-genome tiling array and analyzed with PfGenominator (freely available at http://winzeler.ucsd.edu/software.html)13. The susceptibility of each resistant strain to KAI407, KAI715, KDU691 and BQR695 was determined by the 72-hr SYBR Green cell proliferation assay4 with four independent experiments assayed in duplicate.
PI4K variation in a transgenic P. berghei line
Introduction of the PI4K resistance-conferring mutation H1477Y-PbPI4K (equivalent to H1484Y in PfPI4K) into a transgenic P. berghei ANKA line expressing luciferase (PbANKA; obtained from MR4, MRA-868) was generated using an established protocol35. In brief, an 1857 base pair (bp) fragment of PbPI4K (PBANKA_110940) was PCR amplified using primer pair p1/p2 (Supplementary Table 1) and cloned into a Topo TA vector (Invitrogen). Sequence fidelity was verified using primers p5 to p9. Subsequently, site-directed mutagenesis (Stratagene) was performed with primers p3/p4 to introduce the non-synonymous coding mutation C4519T (encodes 1477Y; mutation located 556 bp upstream of the stop codon). The KpnI /PspM01-digested fragment was cloned into pl0001-TgDHFR-KO (obtained from MR4, MRA-770), in which the pycrt-3′UTR (terminating sequence; primers p12/p13) and PbPI4K 3′UTR (second homology site; primers p10/p11) were also included. In parallel, a second plasmid was constructed with the native pbpi4k gene fragment to serve as recombinant control. Following transfection, recombinant PbANKA parasites were selected in vivo with non-randomized mice (female Swiss Webster mice, 6 weeks old, ~25 g) drinking water supplemented with pyrimethamine. Animal protocols were approved and performed in accordance with rules and regulations of Institution Animal Care and Use Committee (IACUC) of Columbia University. Presence of transfected parasites was verified with direct sequencing using primers p14 to p20. Clonal parasite lines were isolated by in vivo limiting dilution and the PI4K variation verified by direct sequencing. All primers for this study are listed in Supplementary Table 1. Drug activity was measured in three independent experiments performed in quadruplicate using the in vitro P. yoelii liver schizont assay.
Zinc-finger nuclease engineering
ZFNs targeting the genomic region encoding the kinase domain of pfpi4k (PF3D7_0509900) were designed by Sigma-Aldrich, and a ZFN pair recognizing the sequence (5′-CTGACAGTCGATGTG gtaaaaataaaaggaaata) at the exon 3 – intron 3 boundary was selected based on activity in a yeast proxy assay43. The ZFN pair was subcloned into a pDC2-based expression vector downstream of a calmodulin (PF14_0323) promoter essentially as described17, yielding the vector pZFNPI4K-hDHFR. A ~1.7 kb donor template, encompassing 1217 bp of the pfpi4k coding sequence and 466 bp of the 3′ untranslated region, was amplified with primers p3480 and p3498 and inserted into the BstAPI and ZraI sites of pZFNPI4K-hDHFR to yield the editing plasmid pZFN-PI4K-hDHFR. To prevent recleavage of an edited genome, three silent mutations were introduced by site-directed mutagenesis into the ZFN-binding site using primers p3496 and p3497 (see Figure 3b). Additional modifications were overlaid on this donor sequence to generate transfection plasmids bearing mutations at S1320L (pZFN-PI4KS1320L-hDHFR; primers p3506 and p3507), Y1356F (pZFN-PI4KY1356F-hDHFR; primers p3508 and p3509), Y1356stop (pZFN-PI4KY1356X-hDHFR; primers p3510 and p3511) and H1484Y (pZFN-PI4KH1484Y-hDHFR; primers p3512 and p3513). The ZFN primers are listed in Supplementary Table 1.
A clonal Dd2 line was transfected with the pZFN-PI4K-hDHFR editing plasmids and selected with 2.5 nM WR99210 the day following electroporation. On day 6 post-electroporation, WR99210 pressure was removed, and parasites were observed microscopically at day 14. Clonal parasites were obtained by limiting dilution, and successful editing was determined by direct sequencing. The susceptibility of each edited line to KAI407, KAI715, KDU691 and BQR695 was determined by three independent experiments performed in triplicate using the 72-hr SYBR Green cell proliferation assay4.
Whole-genome sequencing analysis
Genomic DNA libraries were prepared for whole-genome sequencing (WGS) using the Illumina NexTera protocol of simultaneous fragmentation and adapter ligation and indexed using the TruSeq Dual Index Sequencing primers (v. 2013, Illumina.). DNA libraries were clustered and run on an Illumina HiSeq 2000, according to manufacturer’s instructions. An internally developed WGS pipeline, named PlaTypUS (Manary et al., submitted), was used to align and analyze all WGS data. The program integrates many community-developed tools into its processing pipeline. To generate alignments, PlaTypUS follows a multi-step alignment process to execute quality-control measures on an alignment map. FASTQ files obtained from sequencing were aligned to the Pf3D7 reference (PlasmoDB v9.1) for SNV and CNV analysis.
Expression of GFP-tagged PfPI4K and PfRab11A
The full-length pfpi4k and pfrab11a genes were PCR amplified from genomic DNA using primers p3727/p3772 and p3730/p3731, respectively, and inserted into a pDC2-based vector behind GFP to generate the expression plasmids pDC2-PbEF1alphapro-GFP-PfPI4K-attP-BSD and pDC2-PbEF1alphapro-GFP-PfRab11A-attP-BSD. The D139Y mutation was introduced by site-directed mutagenesis into the pfrab11a coding sequence using primers p3736/p3737, yielding the plasmid pDC2-PbEF1alphapro-GFP-PfRab11A-D139Y-attP-BSD. The expression plasmids were then transfected into the Dd2-attB parasite line using attP×attB integrase-mediated recombination18. Drug activity against each transgenic line was determined by three independent experiments performed in duplicate using the 72-hr SYBR Green cell proliferation assay4.
Human lipid kinase panel
The biochemical inhibitory activities of human lipid kinases were measured with either Kinase-Glo® (Promega) for PI4KIIIβ and the α and β isoforms of PI3K or Adapta® (Invitrogen) for the δ and γ isoforms of PI3K. A detailed description of the Kinase-Glo® assays is available from Furet et al.44. The Adapta® assays were run as follows: 50 nL of compound dilutions were dispensed into white 384-well low-volume polystyrene plates (Greiner). Then 5 μL of purified recombinant PI3K and lipid substrate (PI or PIP2:PS) were dispensed and followed by a subsequent dispense of 5 μL ATP (final assay volume 10 μL). The enzymatic reaction was incubated at room temperature. The standard reaction buffer for the Adapta® TR-FRET assay contained 10 mM Tris–HCl pH 7.5, 3 mM MgCl2, 50 mM NaCl, 1 mM DTT, 0.05% CHAPS (v/v). Reactions were stopped with 5 μL of a mixture of EDTA containing the Eu3+-labeled anti-ADP antibody and the Alexa Fluor® 647-labeled ADP tracer in a proprietary TR-FRET dilution buffer. Plates were read 15–60 min later in a Synergy2 reader using an integration time of 0.4 s and a delay of 0.05 s. Controls for 100% inhibition of the kinase reaction were performed by replacing PI3K with the equivalent volume of the standard reaction buffer. The controls for the 0% inhibition were performed with the solvent vehicle of the compounds (90% DMSO in H2O (v/v)). A reference compound was included in all assay plates and each compound was assayed in a 16-point, 1:3 dilution series.
Human protein kinase panel
The human protein kinase selectivity profile for KAI407 and KDU691 was carried out against a panel of 40 recombinant kinases. Inhibition of kinase-dependent substrate phosphorylation was measured by the Caliper mobility shift assay technology using microfluidic capillary electrophoresis as previously described45.
Homology modeling
The P. falciparum phosphatidylinositol 4-kinase model was built by homology modeling using the X-ray crystallographic coordinates for human PI3K gamma in complex with an inhibitor (PDB ID 3ENE) as a template: The kinase domain of PfPI4K (amino acid boundaries: N1261-M1559) were aligned to that of PI3K gamma (kinase domain: A557-A1102). Fully flexible ligand docking was performed with Glide (version 5.5; Schrödinger). The PyMOL Molecular Graphics System (version 1.2r2, Schrödinger) was used to render the models and prepare figures.
Cloning and expression of PvPI4K
The full-length coding sequence of PvPI4K (PVX098_050) was codon-optimized for baculovirus expression, synthesized and cloned into pFastBac-HTa (Invitrogen) in frame with the N-terminal polyhistidine tag using the EcoRI and HindIII restriction sites. Recombinant bacmid clones were generated by site-specific transposition in E.coli DH10Bac (Invitrogen). The pFastBacHTa-PvPI4K plasmid was used as a template for site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The primers used for mutagenesis are listed in Supplementary Table 1. All mutation sites, as well as absence of errors across the whole gene, were confirmed by direct DNA sequencing. The subsequent steps for bacmid isolation, transfection, and selection of the recombinant viruses were performed according to the vendor’s protocol (Bac-to-Bac system, Invitrogen).
SF9 cells, cultured in SF-900 III serum-free medium, were transfected with recombinant baculovirus at 1/200 (v/v) and incubated at 27°C for 72 hr. The pellets were collected after centrifugation and re-suspended in cell lysis buffer (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 5% glycerol, 0.01% Triton-X 100, 0.5 mM TCEP and 1× complete protease inhibitor cocktail without EDTA (Roche Diagnostics). The cell suspension was lysed by sonication and the clarified supernatant was loaded onto a 1 mL HisTrap affinity column (GE Healthcare) pre-equilibrated with Buffer A (20 mM Tris, pH 7.5, 500 mM NaCl, 5% glycerol, 0.5 mM TCEP, 20 mM imidazole and 1× complete protease inhibitor cocktail without EDTA). The protein was eluted with Buffer B (Buffer A with 300 mM imidazole) and fractions containing PvPI4K were pooled and purified by a gel filtration column (Hi-Load 16/60 Superdex 200, GE Healthcare) equilibrated with 20mM Tris pH 7.5, 5% glycerol, 500 mM NaCl, 5 mM DTT and 1× protease inhibitor cocktail without EDTA. Protein concentrations were determined by the calculated protein extinction coefficient (ε280=125875 M−1 cm−1). Aliquots were flash frozen in liquid nitrogen and immediately stored at −80°C. All PvPI4K constructs were purified with this same protocol.
PI4K enzymatic assay
L-alpha-phosphatidylinositol (PI; Avanti Polar Lipid), dissolved in 3% n-Octylglucoside (OG; Roche Diagnostics), was used as the lipid substrate for the phosphatidylinositol 4-kinase activity assay. PvPI4K was assayed using Transcreener™ ADP2 FP detection kit (BellBrook) in a black, solid 384-well plate (Corning). The final assay volume was 10 μL and contained 1 nM of the respective PvPI4K construct in 10 mM Tris pH 7.5, 1 mM DTT, 10 μM ATP, 5 mM Mn2+, 0.2 % Triton-X 100, and 11 μM PI/OG. The enzyme reaction was performed for 1 hr at room temperature and was stopped by adding 10 μL of detection mix containing 1× Stop Buffer, 4 nM AMP Alexa Fluor® 633 tracer, and 20 μg/ml ADP antibody. Fluorescence polarization measurements were performed on the Infinite M1000 plate reader (Tecan) with λex= 635 nm and λem= 680 nm (20 nm bandwidth). IC50 values were calculated from three independent experiments performed in triplicate using Graphpad PRISM software.
In vivo detection of PI4P pools
To detect PI4P within the parasite, the PH domain of yeast Osh2 was amplified from pREP4146,21 (kindly provided by Mark Ellis and Fred Chang, Columbia University Medical Center, New York) using primers p2987b/p2988. The Osh2 PH domain was inserted as a dimer behind GFP to generate the expression vector pDC2-PbEF1alphapro-GFP-PHOsh2-attP-BSD, and transfected into the Dd2-attB parasite line using attP×attB integrase-mediated recombination. Sorbitol synchronized parasites followed the above methodology to image drug-treated parasites expressing PfATP4-GFP. Parasites (100 cells per treatment) were imaged from three independent experiments.
Phosphatidylinositol phosphate (PIP) quantification
The sorbitol/MACS method described above was used to generate a highly synchronized parasite culture (10% parasitemia in 2.5% hematocrit). At t=41.5 hr in the parasite lifecycle, the culture was washed three times with pre-warmed complete media prepared from phosphate-free RPMI (US Biological; PF-CM). The final culture was resuspended in 4 mL PF-CM and split into two equal volumes. One volume was treated with 500 nM KAI407 and the remaining volume with DMSO and incubated at 37°C for 15 min. PIP radiolabeling was initiated by the addition of 800 μCi/mL Na2[32P]O4 (Perkin Elmer) and incubated 1–4 hr at 37°C (t=42–46 hr in lifecycle). The samples were then washed five times with chilled PBS with 0.15% saponin. The final supernatant was aspirated and to each parasite pellet the following were successively added with mixing in between: 0.4 mL 1 N HCl, and 0.4 mL MeOH, 0.4 mL CHCl3. Samples were vortexed vigorously and then centrifuged. The lower organic layer was transfer to a new tube and extracted once with 400 uL (1.0:0.9, (v/v)) MeOH:0.1 M EDTA, pH 8.0. The lower organic layer was transferred to a new tube and dried under a nitrogen stream. A previously described method by Mandl et al.47 was used to quantify PIPs. In brief, phosphatidylinositides were separated by anion-exchange HPLC (Agilent) using an ammonium phosphate gradient, detected by a flow scintillation analyzer (Perkin-Elmer), and quantified using ProFSA software (Perkin-Elmer). The [32P]-labeled PIP peaks were identified using internal standards. The counts in each peak were normalized against total phosphatidylinositol counts in each individual sample.
Supplementary Material
Extended Data Table 1 | Activity of imidazopyrazines against drug-resistant lines of P. falciparum.
*CG, cycloguanil; CQ, chloroquine; MEF, mefloquine; PYR, pyrimethamine; QN, quinine; SFX, sulfadoxine.
†IC50 values are represented as means±s.d. (N=2, n=2).
{kind=link}
Extended Data Table 2 | Pharmacokinetic properties of KDU691.
Cmax, Maximum concentration achieved; *AUC, Area Under Curve; t1/2, half life; F, % oral bioavailability. Vss, steady state volume of distribution; CL, clearance.
†p.o.: oral gavage of 20 mg/kg KDU691 (n=3) formulated in PEG300/D5W (3:1, v/v).
‡i.v.: intravenous injection (via tail vein) of 5 mg/kg KDU691 (n=3) formulated in PEG300/D5W (3:1, v/v).
{kind=link}
Extended Data Table 3 | In vitro activities of KAI407, KDU691 and BQR695 against recombinant human lipid and protein kinases.
*IC50 values are represented as means±s.d. (N=2, n=2). ND, not determined.
{kind=link}
Extended Data Table 4 | Whole-genome sequencing results of zinc finger nuclease-edited lines.
*Chr, chromosome; Pos, base pair (bp) position within chromosome; Ref, bp in reference genome; Alt, bp call in sequenced sample
†IG, intergenic; INT, intron; NSC, nonsynonymous coding; SC, synonymous coding
‡AA: amino acid change
§Comparison of whole-genome sequencing of two populations, the parental clone (par.) and the child zinc-finger-edited clone. Ref: number of reads matching to the reference (PlasmoDB version 9.1); Alt: number of read differences from the reference in the test clone. Desired alleles are shaded in grey.
{kind=link}
Extended Data Figure 1 | Imidazopyrazines are active against all liver- and blood-stage forms within the vertebrate host. The imidazopyrazines KAI407, KDU691, and KAI715 were tested, as available, in a comprehensive array of in vitro assays comprised of diverse Plasmodium species at representative stages of the vertebrate lifecycle. a, The imidazopyrazines retained potency within 1–2 log units of atovaquone (ATQ), a licensed antimalarial with dual-stage activity, in the P. falciparum asexual blood-stage assay. IC50 values for each compound are shown within an inset table above the dose-response curves (n=4). b, Table showing the in vivo efficacy of KDU691 against the blood stages of P. berghei in a malaria mouse model. c, A survival curve for mice (n=8 per group) tested in a causal prophylaxis assay with varying doses of KDU691 (5, 7.5 and 10 mg/kg). A single oral dose of 7.5 mg/kg or greater, administered just prior to intravenous infection with 50,000 sporozoites, protected mice from malaria. Blood parasitemia was monitored for all mice up to 30 days, at which time the experiment was discontinued and surviving mice were deemed cured. Atovaquone was included as a control and is known to provide 100% causal prophylactic protection at 2.5 mg/kg. d, The effect of KDU691 on P. falciparum stage III–IV gametocytes was determined by measuring parasite lactate dehydrogenase activity. Data are expressed as a percent effect relative to the positive (1 μM dihydroartemisinin; DHA) and negative (vehicle) controls (means±s.d.; n=4). DHA and KDU691 were found to have IC50 values of 15.8 nM and 220 nM, respectively. e, f, The results of KDU691 tested in the Standard Membrane Feeding Assay are shown following pre-exposure of P. falciparum mature gametocytes to compound for 24 hr. Oocyst counts from the dissected mosquito midguts (means±s.d.; n=20) are expressed as a percent effect relative to the negative (vehicle) control. An IC50 value of 316 nM on (e) oocyst densities and 370 nM on (f) oocyst prevalence, i.e. the percentage of mosquitos with one or more oocysts, was determined for KDU691.
{kind=link}
Extended Data Figure 2 | Imidazopyrazine and quinoxaline compounds arrest parasites in late schizogony prior to completion of daughter cell formation. a, The onset of action for KAI407 was investigated using a highly synchronized population of blood-stage parasites. Complete culture medium containing 125 nM KAI407 was replaced with drug-free medium at the time indicated in 2 hr intervals. The resultant parasitemia in the next lifecycle (t = 72 hr) was normalized to untreated parasites (means±s.d.; n=4). The time course represents trophozoite-stage parasites (t = 34 hr) through the maturation of schizonts (t = 48 hr). b, c, The plasma membrane marker PfATP4-GFP was used to visualize plasma membrane ingression around developing daughter merozoites, with nuclei stained by Hoechst 33342. Parasites treated with DMSO (control) formed clearly defined daughter cells uniformly surrounded by plasma membrane. Conversely, parasites treated with 500 nM KAI407 for 4 hr (KAI407-treated) had a disorganized membrane structure. Representative images from a single experimental replicate are shown (n=2). Scale bar, 5 μm. d, Microscopy of Giemsa-stained parasites treated with ~5× IC50 drug (125 nM KAI407, 15 nM KAI715, 150 nM KDU691 or 400 nM BQR695) or DMSO vehicle. Representative images from a single experimental replicate are shown (n=3). e, Measurement of merozoite viability via the merozoite release assay. The ability of merozoites to re-invade fresh RBCs after mechanical rupture of drug-arrested schizonts treated with 125 nM KAI407, 15 nM KAI715, 150 nM KDU691 or 400 nM BQR695 was compared to parasite re-invasion of untreated (DMSO) parasites. E-64 (grey), a known inhibitor of merozoite egress, was used as a control at 1 μM (means±s.d.; n=4).
{kind=link}
Extended Data Figure 3 | Domain organization of P. falciparum phosphatidylinositol 4-kinase, amino acid alignment, and expression of GFP-PfPI4K. a, Schematic of the domain organization of PfPI4K: ARM, Armadillo-type fold; LKU, lipid kinase unique domain; BSM, Beta-signature motif; CAT, PI3/4-kinase catalytic domain. Amino acid boundaries are given below the schematic, and the locations of the resistance SNVs starred. The beta-signature motif is exclusive to PI4-kinases. b, Amino acid alignment of PI4KIIIβ from P. falciparum (Pf), P. vivax (Pv), P. berghei (Pb), P. yoelii (Py) and the human ortholog. Amino acids shaded in black are identical between species and those shaded grey are highly conserved. Resistance SNVs are highlighted in red. c, Fluorescence microscopy shows apical enrichment of GFP-PI4K in late schizonts. Representative images from a single experimental replicate are shown (n=3). Scale bar, 5 μm. d, Quantification of phosphatidylinositol phosphate (PIP) species in synchronized asexual blood-stage parasites treated with 500 nM KAI407 (+) or DMSO (−). PIP species were normalized to phosphatidylinositol (PI) and representative data (n=2) are shown as means±s.d.
{kind=link}
Extended Data Figure 4 | Resistance to imidazopyrazine and quinoxaline compounds is mediated by genes encoding PfPI4K or PfRab11A. DNA microarray analysis of the parasite genomic DNA extracted from clonal lines evolved-to-resistance against KAI407, KAI715 or BQR695. Copy number variation (CNV) analysis is shown for all 14 chromosomes (Chr) and organelle-specific plasmids in the mitochondria (mito) and apicoplast (api) for a, KAI407-R lines c, KAI715-R lines, and e, BQR695-R lines. The inset box is a zoomed view of chromosome 5 centered on pfpi4k (indicated by black arrow). Key single nucleotide variations (SNVs) detected in the remaining b, KAI407-R d, KAI715-R, and f, BQR695-R parasite lines lacking significant CNV events. Results of direct DNA sequencing are given next to each detected mutation and the gene model is provided beneath the SNV analysis output for reference.
{kind=link}
Extended Data Figure 5 | IC50 values for ZFN-edited lines and genome editing strategy to introduce a stop codon within the PfPI4K catalytic domain. a, Table summarizing IC50 values for PI4K mutants generated via ZFN editing. *IC50 values are represented as means±s.d. and were calculated from three independent experiments performed in quadruplicate. †Previously referred to as GNF179 (Meister et al.29) ‡Previously referred to as NITD246 (Rottmann et al.12). b, The ZFN plasmid contains an expression cassette for the ZFNs, which target a 34 base pair (bp) site on pfpi4k (thunderbolt), and a 1.7 kb homologous donor sequence with a single SNV introducing a stop codon at amino acid residue 1356 in the protein coding sequence. Although the edited mutation was detected in the bulk culture, analysis of cloned lines continued to show a mixed T/A peak at position 4068 (see chromatogram), suggesting a potential gene duplication. c, Whole-genome sequencing confirmed the direct sequencing results, yielding approximately equivalent numbers of reads encoding the wild type and stop codons. These result from a partial amplification (from 413,512 to 416,372 bp; green line in the normalized coverage) of the 3′ region of pfpi4k, with the edited stop codon within the downstream truncated copy.
{kind=link}
Extended Data Figure 6 | Amino acid alignment and localization of PfRab11A. a, Comparison of amino acid identity (shaded in black) and similarity (shaded in grey) in ClustalW2 between the human (HsRab11a), P. falciparum (PfRab11A), and S. cerevisiae (ScYpt31) Rab11A homologs. The phosphate-binding P-loop and the switch regions that respond conformationally to the nucleotide-bound state of the protein are labeled above the amino acid alignment and consensus residues (100% identity) are indicated with an asterisk. Aspartate residue 139 (D139), which confers imidazopyrazine and quinoxaline resistance when mutated to tyrosine (Y), is labeled with an arrow. A negatively charged residue, either aspartate (D) or glutamate (E), occupies this position in all three species. b, Mapping of the resistance-conferring mutation (D139Y; yellow) within the X-ray crystallographic structure of PfRab11A (slate blue cartoon representation; PDB ID: 3BFK) in complex with GDP (green stick model). The structure was visualized in Pymol. Visualization of c, GFP-PfRab11A-WT and d, GFP-PfRab11A-D139Y by fluorescence microscopy in early (top panel) or segmented (bottom panel) schizonts (n=3). Hoechst 33342 was used to stain the nuclei of the daughter merozoites (blue) that develop during schizogony. A differential interference contrast (DIC) image of the parasitized red blood cell is shown. For each, representative images from a single experimental replicate are shown. Scale bar, 5 μm.
{kind=link}
Acknowledgments
We thank Elizabeth Miller for critical review of the manuscript and figure design. We also thank Ana Rodriguez and the insectary core facility team at New York University for reliable supplies of malaria-infected mosquitoes. We gratefully acknowledge translational research grants (WT078285 and WT096157) from the Wellcome Trust and funding from the Medicines for Malaria Venture (MMV) to the Genomics Institute of the Novartis Research Foundation, the Swiss Tropical and Public Health Institute, Columbia University, the Novartis Institute for Tropical Diseases, the Singapore Immunology Network and Horizontal Programme on Infectious Diseases under the Agency Science Technology and Research (A*STAR, Singapore), and the Wellcome Trust (UK). SMRU is sponsored by The Wellcome Trust of Great Britain, as part of the Oxford Tropical Medicine Research Programme of Wellcome Trust-Mahidol University. E.A.W. and D.A.F. are supported by grants from the Bill and Melinda Gates Foundation, MMV, and the NIH (R01AI090141 to E.A.W. and R01085584 and R01079709 to D.A.F.).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors wish to disclose the following, which may be considered a conflict of interest: C.W.M, C.S.L, S.H.L, J.R., O.S., B.K.S.Y., K.L.K., K.G., D.M.P., B.B., J.T., D.H., D.T., R.J.G., C.B. and T.T.D. are employed by Novartis. C.W.M, J.R., K.L.K., B.B., J.T., D.H., D.T., R.J.G, T.T.D. and E.A.W. own shares of Novartis AG stock. E.A.W. has received grants from Novartis.
References
- 1.Greenwood BM, et al. Malaria: progress, perils, and prospects for eradication. J Clin Invest. 2008;118:1266–76. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vale N, Moreira R, Gomes P. Primaquine revisited six decades after its discovery. Eur J Med Chem. 2009;44:937–53. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Wells TN, Burrows JN, Baird JK. Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 2010;26:145–51. doi: 10.1016/j.pt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Plouffe DM, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci USA. 2008;105:9059–64. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dembele L, et al. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PLoS One. 2011;6:e18162. doi: 10.1371/journal.pone.0018162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bousema T, Drakeley C. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin Microbiol Rev. 2011;24:377–410. doi: 10.1128/CMR.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adjalley SH, et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc Natl Acad Sci U S A. 2011;108:E1214–23. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Pelt-Koops JC, et al. The spiroindolone drug candidate NITD609 potently inhibits gametocytogenesis and blocks Plasmodium falciparum transmission to Anopheles mosquito vector. Antimicrob Agents Chemother. 2012;56:3544–8. doi: 10.1128/AAC.06377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyle MJ, et al. Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proceedings of the National Academy of Sciences. 2010 doi: 10.1073/pnas.1009198107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dvorin JD, et al. A Plant-Like Kinase in Plasmodium falciparum Regulates Parasite Egress from Erythrocytes. Science. 2010;328:910–912. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drew ME, et al. Plasmodium food vacuole plasmepsins are activated by falcipains. J Biol Chem. 2008;283:12870–6. doi: 10.1074/jbc.M708949200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rottmann M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–80. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dharia NV, et al. Use of high-density tiling microarrays to identify mutations globally and elucidate mechanisms of drug resistance in Plasmodium falciparum. Genome Biol. 2009;10:R21. doi: 10.1186/gb-2009-10-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balla A, Balla T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 2006;16:351–61. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Mayinger P. Phosphoinositides and vesicular membrane traffic. Biochim Biophys Acta. 2012;1821:1104–13. doi: 10.1016/j.bbalip.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polevoy G, et al. Dual roles for the Drosophila PI 4-kinase four wheel drive in localizing Rab11 during cytokinesis. J Cell Biol. 2009;187:847–58. doi: 10.1083/jcb.200908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Straimer J, et al. Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat Methods. 2012;9:993–8. doi: 10.1038/nmeth.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nkrumah LJ, et al. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods. 2006;3:615–21. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agop-Nersesian C, et al. Rab11A-controlled assembly of the inner membrane complex is required for completion of apicomplexan cytokinesis. PLoS Pathog. 2009;5:e1000270. doi: 10.1371/journal.ppat.1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noble MEM, Endicott JA, Johnson LN. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 21.Roy A, Levine TP. Multiple pools of phosphatidylinositol 4-phosphate detected using the pleckstrin homology domain of Osh2p. J Biol Chem. 2004;279:44683–9. doi: 10.1074/jbc.M401583200. [DOI] [PubMed] [Google Scholar]
- 22.Kruger T, Sanchez CP, Lanzer M. Complementation of Saccharomyces cerevisiae pik1ts by a phosphatidylinositol 4-kinase from Plasmodium falciparum. Mol Biochem Parasitol. 2010;172:149–51. doi: 10.1016/j.molbiopara.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 23.Strahl T, Hama H, DeWald DB, Thorner J. Yeast phosphatidylinositol 4-kinase, Pik1, has essential roles at the Golgi and in the nucleus. The Journal of cell biology. 2005;171:967–979. doi: 10.1083/jcb.200504104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walch-Solimena C, Novick P. The yeast phosphatidylinositol-4-OH kinase Pik1 regulates secretion at the Golgi. Nature cell biology. 1999;1:523–525. doi: 10.1038/70319. [DOI] [PubMed] [Google Scholar]
- 25.Hama H, Schnieders EA, Thorner J, Takemoto JY, DeWald DB. Direct involvement of phosphatidylinositol 4-phosphate in secretion in the yeast Saccharomyces cerevisiae. J Biol Chem. 1999;274:34294–300. doi: 10.1074/jbc.274.48.34294. [DOI] [PubMed] [Google Scholar]
- 26.de Graaf P, et al. Phosphatidylinositol 4-kinaseβ is critical for functional association of rab11 with the Golgi complex. Mol Biol Cell. 2004;15:2038–47. doi: 10.1091/mbc.E03-12-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamarche MJ, et al. Anti-hepatitis C virus activity and toxicity of type III phosphatidylinositol-4-kinase beta inhibitors. Antimicrob Agents Chemother. 2012;56:5149–56. doi: 10.1128/AAC.00946-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.A research agenda for malaria eradication: drugs. PLoS Med. 2011;8:e1000402. doi: 10.1371/journal.pmed.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meister S, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science. 2011;334:1372–7. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell B, et al. Determinants of in vitro drug susceptibility testing of Plasmodium vivax. Antimicrob Agents Chemother. 2008;52:1040–5. doi: 10.1128/AAC.01334-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell BM, et al. Simple in vitro assay for determining the sensitivity of Plasmodium vivax isolates from fresh human blood to antimalarials in areas where P. vivax is endemic. Antimicrob Agents Chemother. 2003;47:170–3. doi: 10.1128/AAC.47.1.170-173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Alessandro S, et al. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J Antimicrob Chemother. 2013;68:2048–58. doi: 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- 33.Dechering KJ, Thompson J, Dodemont HJ, Eling W, Konings RN. Developmentally regulated expression of pfs16, a marker for sexual differentiation of the human malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 1997;89:235–44. doi: 10.1016/s0166-6851(97)00123-0. [DOI] [PubMed] [Google Scholar]
- 34.van der Kolk M, et al. Evaluation of the standard membrane feeding assay (SMFA) for the determination of malaria transmission-reducing activity using empirical data. Parasitology. 2005;130:13–22. doi: 10.1017/s0031182004006067. [DOI] [PubMed] [Google Scholar]
- 35.Janse CJ, Ramesar J, Waters AP. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc. 2006;1:346–56. doi: 10.1038/nprot.2006.53. [DOI] [PubMed] [Google Scholar]
- 36.Li C, et al. A modern in vivo pharmacokinetic paradigm: combining snapshot, rapid and full PK approaches to optimize and expedite early drug discovery. Drug Discov Today. 2013;18:71–78. doi: 10.1016/j.drudis.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Ponnudurai T, et al. Infectivity of cultured Plasmodium falciparum gametocytes to mosquitoes. Parasitology. 1989;98:165–173. doi: 10.1017/s0031182000062065. [DOI] [PubMed] [Google Scholar]
- 38.Guguen-Guillouzo C, et al. High yield preparation of isolated human adult hepatocytes by enzymatic perfusion of the liver. Cell biology international reports. 1982;6:625–628. doi: 10.1016/0309-1651(82)90187-4. [DOI] [PubMed] [Google Scholar]
- 39.Mazier D, et al. Complete development of hepatic stages of Plasmodium falciparum in vitro. Science. 1985;227:440–442. doi: 10.1126/science.3880923. [DOI] [PubMed] [Google Scholar]
- 40.Fidock DA, Nomura T, Wellems TE. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol. Pharmacol. 1998;54:1140–7. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- 41.Franke-Fayard B, et al. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Gamo FJ, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–10. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 43.Doyon Y, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008;26:702–8. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furet P, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013 doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 45.Manley PW, et al. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 2010;1804:445–53. doi: 10.1016/j.bbapap.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 46.Onishi M, et al. Role of septins in the orientation of forespore membrane extension during sporulation in fission yeast. Mol Cell Biol. 2010;30:2057–74. doi: 10.1128/MCB.01529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mandl A, Sarkes D, Carricaburu V, Jung V, Rameh L. Serum withdrawal-induced accumulation of phosphoinositide 3-kinase lipids in differentiating 3T3-L6 myoblasts: distinct roles for Ship2 and PTEN. Mol Cell Biol. 2007;27:8098–112. doi: 10.1128/MCB.00756-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data Table 1 | Activity of imidazopyrazines against drug-resistant lines of P. falciparum.
*CG, cycloguanil; CQ, chloroquine; MEF, mefloquine; PYR, pyrimethamine; QN, quinine; SFX, sulfadoxine.
†IC50 values are represented as means±s.d. (N=2, n=2).
Extended Data Table 2 | Pharmacokinetic properties of KDU691.
Cmax, Maximum concentration achieved; *AUC, Area Under Curve; t1/2, half life; F, % oral bioavailability. Vss, steady state volume of distribution; CL, clearance.
†p.o.: oral gavage of 20 mg/kg KDU691 (n=3) formulated in PEG300/D5W (3:1, v/v).
‡i.v.: intravenous injection (via tail vein) of 5 mg/kg KDU691 (n=3) formulated in PEG300/D5W (3:1, v/v).
Extended Data Table 3 | In vitro activities of KAI407, KDU691 and BQR695 against recombinant human lipid and protein kinases.
*IC50 values are represented as means±s.d. (N=2, n=2). ND, not determined.
Extended Data Table 4 | Whole-genome sequencing results of zinc finger nuclease-edited lines.
*Chr, chromosome; Pos, base pair (bp) position within chromosome; Ref, bp in reference genome; Alt, bp call in sequenced sample
†IG, intergenic; INT, intron; NSC, nonsynonymous coding; SC, synonymous coding
‡AA: amino acid change
§Comparison of whole-genome sequencing of two populations, the parental clone (par.) and the child zinc-finger-edited clone. Ref: number of reads matching to the reference (PlasmoDB version 9.1); Alt: number of read differences from the reference in the test clone. Desired alleles are shaded in grey.
Extended Data Figure 1 | Imidazopyrazines are active against all liver- and blood-stage forms within the vertebrate host. The imidazopyrazines KAI407, KDU691, and KAI715 were tested, as available, in a comprehensive array of in vitro assays comprised of diverse Plasmodium species at representative stages of the vertebrate lifecycle. a, The imidazopyrazines retained potency within 1–2 log units of atovaquone (ATQ), a licensed antimalarial with dual-stage activity, in the P. falciparum asexual blood-stage assay. IC50 values for each compound are shown within an inset table above the dose-response curves (n=4). b, Table showing the in vivo efficacy of KDU691 against the blood stages of P. berghei in a malaria mouse model. c, A survival curve for mice (n=8 per group) tested in a causal prophylaxis assay with varying doses of KDU691 (5, 7.5 and 10 mg/kg). A single oral dose of 7.5 mg/kg or greater, administered just prior to intravenous infection with 50,000 sporozoites, protected mice from malaria. Blood parasitemia was monitored for all mice up to 30 days, at which time the experiment was discontinued and surviving mice were deemed cured. Atovaquone was included as a control and is known to provide 100% causal prophylactic protection at 2.5 mg/kg. d, The effect of KDU691 on P. falciparum stage III–IV gametocytes was determined by measuring parasite lactate dehydrogenase activity. Data are expressed as a percent effect relative to the positive (1 μM dihydroartemisinin; DHA) and negative (vehicle) controls (means±s.d.; n=4). DHA and KDU691 were found to have IC50 values of 15.8 nM and 220 nM, respectively. e, f, The results of KDU691 tested in the Standard Membrane Feeding Assay are shown following pre-exposure of P. falciparum mature gametocytes to compound for 24 hr. Oocyst counts from the dissected mosquito midguts (means±s.d.; n=20) are expressed as a percent effect relative to the negative (vehicle) control. An IC50 value of 316 nM on (e) oocyst densities and 370 nM on (f) oocyst prevalence, i.e. the percentage of mosquitos with one or more oocysts, was determined for KDU691.
Extended Data Figure 2 | Imidazopyrazine and quinoxaline compounds arrest parasites in late schizogony prior to completion of daughter cell formation. a, The onset of action for KAI407 was investigated using a highly synchronized population of blood-stage parasites. Complete culture medium containing 125 nM KAI407 was replaced with drug-free medium at the time indicated in 2 hr intervals. The resultant parasitemia in the next lifecycle (t = 72 hr) was normalized to untreated parasites (means±s.d.; n=4). The time course represents trophozoite-stage parasites (t = 34 hr) through the maturation of schizonts (t = 48 hr). b, c, The plasma membrane marker PfATP4-GFP was used to visualize plasma membrane ingression around developing daughter merozoites, with nuclei stained by Hoechst 33342. Parasites treated with DMSO (control) formed clearly defined daughter cells uniformly surrounded by plasma membrane. Conversely, parasites treated with 500 nM KAI407 for 4 hr (KAI407-treated) had a disorganized membrane structure. Representative images from a single experimental replicate are shown (n=2). Scale bar, 5 μm. d, Microscopy of Giemsa-stained parasites treated with ~5× IC50 drug (125 nM KAI407, 15 nM KAI715, 150 nM KDU691 or 400 nM BQR695) or DMSO vehicle. Representative images from a single experimental replicate are shown (n=3). e, Measurement of merozoite viability via the merozoite release assay. The ability of merozoites to re-invade fresh RBCs after mechanical rupture of drug-arrested schizonts treated with 125 nM KAI407, 15 nM KAI715, 150 nM KDU691 or 400 nM BQR695 was compared to parasite re-invasion of untreated (DMSO) parasites. E-64 (grey), a known inhibitor of merozoite egress, was used as a control at 1 μM (means±s.d.; n=4).
Extended Data Figure 3 | Domain organization of P. falciparum phosphatidylinositol 4-kinase, amino acid alignment, and expression of GFP-PfPI4K. a, Schematic of the domain organization of PfPI4K: ARM, Armadillo-type fold; LKU, lipid kinase unique domain; BSM, Beta-signature motif; CAT, PI3/4-kinase catalytic domain. Amino acid boundaries are given below the schematic, and the locations of the resistance SNVs starred. The beta-signature motif is exclusive to PI4-kinases. b, Amino acid alignment of PI4KIIIβ from P. falciparum (Pf), P. vivax (Pv), P. berghei (Pb), P. yoelii (Py) and the human ortholog. Amino acids shaded in black are identical between species and those shaded grey are highly conserved. Resistance SNVs are highlighted in red. c, Fluorescence microscopy shows apical enrichment of GFP-PI4K in late schizonts. Representative images from a single experimental replicate are shown (n=3). Scale bar, 5 μm. d, Quantification of phosphatidylinositol phosphate (PIP) species in synchronized asexual blood-stage parasites treated with 500 nM KAI407 (+) or DMSO (−). PIP species were normalized to phosphatidylinositol (PI) and representative data (n=2) are shown as means±s.d.
Extended Data Figure 4 | Resistance to imidazopyrazine and quinoxaline compounds is mediated by genes encoding PfPI4K or PfRab11A. DNA microarray analysis of the parasite genomic DNA extracted from clonal lines evolved-to-resistance against KAI407, KAI715 or BQR695. Copy number variation (CNV) analysis is shown for all 14 chromosomes (Chr) and organelle-specific plasmids in the mitochondria (mito) and apicoplast (api) for a, KAI407-R lines c, KAI715-R lines, and e, BQR695-R lines. The inset box is a zoomed view of chromosome 5 centered on pfpi4k (indicated by black arrow). Key single nucleotide variations (SNVs) detected in the remaining b, KAI407-R d, KAI715-R, and f, BQR695-R parasite lines lacking significant CNV events. Results of direct DNA sequencing are given next to each detected mutation and the gene model is provided beneath the SNV analysis output for reference.
Extended Data Figure 5 | IC50 values for ZFN-edited lines and genome editing strategy to introduce a stop codon within the PfPI4K catalytic domain. a, Table summarizing IC50 values for PI4K mutants generated via ZFN editing. *IC50 values are represented as means±s.d. and were calculated from three independent experiments performed in quadruplicate. †Previously referred to as GNF179 (Meister et al.29) ‡Previously referred to as NITD246 (Rottmann et al.12). b, The ZFN plasmid contains an expression cassette for the ZFNs, which target a 34 base pair (bp) site on pfpi4k (thunderbolt), and a 1.7 kb homologous donor sequence with a single SNV introducing a stop codon at amino acid residue 1356 in the protein coding sequence. Although the edited mutation was detected in the bulk culture, analysis of cloned lines continued to show a mixed T/A peak at position 4068 (see chromatogram), suggesting a potential gene duplication. c, Whole-genome sequencing confirmed the direct sequencing results, yielding approximately equivalent numbers of reads encoding the wild type and stop codons. These result from a partial amplification (from 413,512 to 416,372 bp; green line in the normalized coverage) of the 3′ region of pfpi4k, with the edited stop codon within the downstream truncated copy.
Extended Data Figure 6 | Amino acid alignment and localization of PfRab11A. a, Comparison of amino acid identity (shaded in black) and similarity (shaded in grey) in ClustalW2 between the human (HsRab11a), P. falciparum (PfRab11A), and S. cerevisiae (ScYpt31) Rab11A homologs. The phosphate-binding P-loop and the switch regions that respond conformationally to the nucleotide-bound state of the protein are labeled above the amino acid alignment and consensus residues (100% identity) are indicated with an asterisk. Aspartate residue 139 (D139), which confers imidazopyrazine and quinoxaline resistance when mutated to tyrosine (Y), is labeled with an arrow. A negatively charged residue, either aspartate (D) or glutamate (E), occupies this position in all three species. b, Mapping of the resistance-conferring mutation (D139Y; yellow) within the X-ray crystallographic structure of PfRab11A (slate blue cartoon representation; PDB ID: 3BFK) in complex with GDP (green stick model). The structure was visualized in Pymol. Visualization of c, GFP-PfRab11A-WT and d, GFP-PfRab11A-D139Y by fluorescence microscopy in early (top panel) or segmented (bottom panel) schizonts (n=3). Hoechst 33342 was used to stain the nuclei of the daughter merozoites (blue) that develop during schizogony. A differential interference contrast (DIC) image of the parasitized red blood cell is shown. For each, representative images from a single experimental replicate are shown. Scale bar, 5 μm.