

Abstract
Signal transducer and activator of transcription STAT5 and its upstream activating kinase JAK2 are essential mediators of cytokine signaling. Their activity is normally tightly regulated and transient. However, constitutive activation of STAT5 is found in numerous cancers and a driving force for malignant transformation. We describe here the identification of the synthetic chalcone α-Br-2′,3,4,4′-tetramethoxychalcone (α-Br-TMC) as a novel JAK/STAT inhibitor. Using the non-transformed IL-3-dependent B cell line Ba/F3 and its oncogenic derivative Ba/F3-1*6 expressing constitutively activated STAT5, we show that α-Br-TMC targets the JAK/STAT pathway at multiple levels, inhibiting both JAK2 and STAT5 phosphorylation. Moreover, α-Br-TMC alters the mobility of STAT5A/B proteins in SDS-PAGE, indicating a change in their post-translational modification state. These alterations correlate with a decreased association of STAT5 and RNA polymerase II with STAT5 target genes in chromatin immunoprecipitation assays. Interestingly, expression of STAT5 target genes such as Cis and c-Myc was differentially regulated by α-Br-TMC in normal and cancer cells. While both genes were inhibited in IL-3-stimulated Ba/F3 cells, expression of the oncogene c-Myc was down-regulated and that of the tumor suppressor gene Cis was up-regulated in transformed Ba/F3-1*6 cells. The synthetic chalcone α-Br-TMC might therefore represent a promising novel anticancer agent for therapeutic intervention in STAT5-associated malignancies.
Introduction
The signal transducer and activator of transcription STAT5 is a key regulator of immune responses, cell proliferation, differentiation, survival and oncogenesis [1], [2]. STAT5 proteins normally reside as latent transcription factors in the cytoplasm of unstimulated cells. Following cytokine, growth factor or hormone stimulation, the receptor-associated JAK2 tyrosine kinase gets auto-phosphorylated (trans-phosphorylation) before it phosphorylates the receptor intracellular domain, creating docking sites for STAT5, which is in turn phosphorylated by activated JAK2. STAT5 phosphorylation allows its dimerization and translocation into the nucleus where it binds to specific recognition sites, and ultimately activates transcription of specific target genes (e.g. c-Myc, Pim-1, Bcl-x, Cis) [1], [3]–[5]. STAT5 activity is regulated through multiple post-translational modifications [1], [6]–[8] and through interactions with cofactors, other transcription factors or STAT5 itself (tetramerization) [1], [9]–[15]. STAT5 activation under physiological conditions is rapid and transient. Attenuation of the STAT5 response is controlled mainly through dephosphorylation by specific phosphatases (SHP-1) and a negative feedback loop mediated by proteins of the SOCS family (CIS, SOCS-1/-3) [16], [17]. However, constitutively activated STAT5 (caSTAT5) has been observed in a wide variety of human cancers, from hematologic malignancies (leukemia, lymphoma, myeloma) to solid tumors such as breast cancer, squamous cell carcinoma of the head and neck (SCCHN) or melanoma [16]–[18]. Constitutive activation of STAT5 directly contributes to oncogenesis, mainly through the up-regulation of oncogenes driving cell proliferation and survival (Bcl-x, c-Myc, Pim-1) [4], [19]–[23], but also through the down-regulation of tumor suppressor genes and inhibitors of the pathway, such as SHP-1 and CIS/SOCS family members, via epigenetic silencing [17], [18], [24]–[28].
Therefore, the JAK/STAT pathway in general and STAT5 proteins in particular represent attractive targets for therapeutic intervention in human cancers [18], [29]–[31]. Numerous inhibitors of the Janus Kinase family members (JAK1, JAK2, JAK3, Tyk2, and the JAK2V617F active mutant responsible for myeloproliferative neoplasia) and of STAT5 have been described [30], [32]–[45]. Most of the JAK inhibitors are small molecules acting as tyrosine kinase inhibitors by targeting their ATP-binding catalytic domain. The JAK1/JAK2 inhibitor Ruxolitinib has been approved by the U.S. Food and Drug Administration for the treatment of myelofibrosis while several others are being currently evaluated in clinical trials for the treatment of various diseases [32], [33]. Reported STAT5 inhibitors target STAT5 activity at multiple levels, from its activation by phosphorylation to its DNA binding activity, independently of upstream activating kinases [30], [34], [35], [38], [39], [45]. We were the first to show that deacetylase inhibitors such as sodium butyrate, trichostatin A (TSA) or suberoylanilide hydroxamic acid (SAHA) inhibit IL-3-mediated STAT5 transcriptional activity in the mouse pro-B cell line Ba/F3 [46]. We demonstrated that deacetylase inhibitors interfere neither with STAT5 initial activation (phosphorylation or nuclear translocation) nor with STAT5 binding to DNA in vivo. Instead, these drugs prevent the proper assembly of the basal transcription machinery ultimately resulting in inhibition of transcription [46], [47]. The deacetylase inhibitor SAHA (Vorinostat) has been approved for the treatment of cutaneous T cell lymphoma while many other deacetylase inhibitors are currently evaluated in clinical trials for the treatment of various types of cancers [48]. Therefore our findings support the attractive possibility that this class of drugs might be efficient in targeting STAT5-associated cancers. Accordingly, tyrosine kinase inhibitor and deacetylase inhibitor combination therapies have recently proven to be more effective in the treatment of cancers with constitutive active STAT5 [49]–[53]. More recently, we turned to the identification of new types of STAT5 inhibitors, with a particular focus on natural and synthetic chalcones.
Chalcones, which are naturally abundant in fruits, vegetables and spices, are α,β-unsaturated carbonyl compounds well known for their multiple biological activities as antioxidant, antibacterial, antifungal, anti-inflammatory and anticancer agents [54]. Natural and synthetic chalcones have been shown to inhibit the function of a variety of tyrosine kinases (JAKs, ERKs, Src) and transcription factors (NF-κB, STAT3) implicated in the response to inflammation and the control of cell proliferation and survival [54]–[58]. We therefore hypothesized that the JAK2/STAT5 signaling pathway might be affected by chalcones. We previously described the synthesis of α-X-2′,3,4,4′-tetramethoxychalcones (α-X-TMCs) with different X substituents (X = H, F, Cl, Br, I, CN, Me, p-NO2-C6H4, Ph, p-OMe-C6H4, NO2, CF3, COOEt, COOH) at the α-position of the α,β-unsaturated carbonyl unit [59]. We formerly showed that these α-X-TMCs have distinct electrophilic reactivities [59]–[61] which correlate well with their anti-inflammatory activity [59]. In this study, the ability of four α-X-TMCs (with X = CN, Br, Cl, H) to regulate STAT5 function was investigated. Five natural chalcones, namely 2′-hydroxy-3,4,4′-trimethoxychalcone (HTMC), 2′,4′-dihydroxy-3,4-dimethoxychalcone (DHDMC), 2′,3,4′-trihydroxy-4-methoxychalcone (THMC), butein, and isoliquiritigenin (ISL), as well as curcumin, another natural product carrying two α,β-unsaturated carbonyl groups, were also investigated (Figure 1A).
Figure 1. α-Br-TMC inhibits IL-3-mediated induction of STAT5-dependent and -independent genes in Ba/F3 cells.

(A) Structure of the natural and synthetic chalcones used in this study. Second-order rate constants k2 values of compounds obtained using cysteamine in TRIS-HCl buffer pH 7.4: ethylene glycol 20:80 are taken from refs. [59], [60]. (B) Ba/F3 cells were pre-treated 30 minutes with 0.2 µM (TSA) or 20 µM (all other compounds) candidate inhibitors and stimulated 60 minutes with 5 ng/mL IL-3, as described in Materials and Methods. Following cell harvest, expression of the STAT5 target gene Cis and of the housekeeping gene 36b4 were measured by quantitative RT-PCR, as described in Materials and Methods. Together with TSA, α-Br-TMC was the only compound able to inhibit expression of the STAT5 target gene Cis. (C) Ba/F3 cells were pre-treated 30 minutes with the indicated concentrations of TSA and α-Br-TMC and further stimulated with 5 ng/mL IL-3 for 30 minutes. Expression of STAT5-dependent (Cis, Osm, c-Myc) and -independent (JunB, Ho-1, 36b4) genes was analyzed by quantitative RT-PCR. DMSO (vehicle) was adjusted to 0.02% final concentration in all conditions. Curcu., curcumin.
We describe here α-Br-2′,3,4,4′-tetramethoxychalcone (α-Br-TMC) as a novel inhibitor of the STAT5 signaling pathway. We show that α-Br-TMC inhibits both JAK2 and STAT5 phosphorylation, leading to the inhibition of expression of downstream STAT5-dependent and -independent genes in IL-3-stimulated Ba/F3 cells. Moreover, α-Br-TMC alters the mobility of STAT5A and STAT5B proteins in SDS-PAGE, both in cells expressing regulated (Ba/F3) and constitutively active (Ba/F3-1*6 and K562) STAT5, presumably reflecting a change in STAT5 post-translational modifications. Nevertheless, α-Br-TMC treatment affects differentially STAT5 transcriptional activity in normal and cancer cells. Altogether, this study identified a novel inhibitor of STAT5 signaling targeting both STAT5 and its upstream activating kinase JAK2, and affecting the regulation of downstream oncogenes and tumor suppressor genes.
Materials and Methods
Chemicals
Chalcones were prepared by published procedures [59], [60]. Dimethyl sulfoxide (DMSO), trichostatin A (TSA) and curcumin were purchased from SIGMA (D2650, T8552 and B6938 respectively). Imatinib was from Cayman Chemical (No. 13139). Compounds were dissolved in DMSO at a final concentration of 1 mM (TSA) or 100 mM (other compounds).
Thiol assay
Kinetic thiol assay for TSA (Biotrend BN0742) was performed as described [60] using 500-fold excess cysteamine as a thiol substrate and an incubation time of 48 hours. The presence of the addition product was detected by liquid chromatography-mass spectrometry (not shown). Second-order rate constants k2 values for chalcones and curcumin are taken from the literature [59], [60].
Cell lines and drug treatments
All cell lines were grown in RPMI 1640 (PAN-Biotech P04-16500) supplemented with 10% heat-inactivated fetal calf serum (FCS; PAN-Biotech), penicillin/streptomycin (PAN-Biotech) (thereafter designated as RPMI-based medium) and cultivated at 37°C under 5% CO2 in a humidified incubator. K562 cells (a kind gift from Daniela Männel, University of Regensburg, Germany; [62]) were maintained in RPMI-based medium. The non-tumorigenic immortalized interleukin-3 (IL-3)-dependent mouse pro-B cell line Ba/F3 (a kind gift from Jacqueline Marvel, IFR 128 BioSciences Gerland-Lyon Sud, France; [63]) was grown in RPMI-based medium supplemented with 2 ng/mL rmIL-3 (ImmunoTools). The Ba/F3-1*6 cell line stably expressing the constitutively active mouse STAT5A-1*6 mutant [64] was generated according to German GenTSV (genetic engineering safety regulations; authorization AZ.55.1-8791.7.52) by electroporating Ba/F3 cells with a pcDNA3-based expression vector allowing expression of a mSTAT5A-1*6-FLAG fusion protein. Stably transfected cells were selected in IL-3-free medium in the presence of 500 µg/mL G418 (PAA). Individual IL-3-independent clones were isolated and characterized to verify mSTAT5A-1*6 transgene expression and constitutive activity, as originally described [64]. Clone Ba/F3-1*6 F7 was used for this study. Cells were maintained in RPMI-based medium supplemented with 500 µg/mL G418.
For cytokine stimulation of Ba/F3 cells, cells were washed twice in RPMI 1640 and rested in RPMI-based medium for 12 hours before addition of 5 ng/mL IL-3. Duration of IL-3 stimulation was contingent upon the downstream assay, as indicated in the figure legends. For chalcone and other inhibitor treatments, Ba/F3 cells were incubated with compounds or DMSO (vehicle) for 30 minutes prior to cytokine stimulation. Ba/F3-1*6 and K562 cells were treated for 60–90 minutes with the various compounds or DMSO (vehicle), as indicated. With the exception of cell viability assays (see below), all experiments were performed in the presence of equal amounts of DMSO.
Gene expression analysis by quantitative RT-PCR
Following inhibitor and cytokine treatments, cells were harvested, lysed in the iScript RT-qPCR sample preparation reagent (170–8899, Bio-Rad Laboratories) and subjected to cDNA synthesis using the iScript cDNA Synthesis kit (170–8891, Bio-Rad Laboratories), following the manufacturer's instructions. Quantitative PCR was performed on a RotorGene Q (Qiagen) with SYBR Green I and HotStarTaq (Qiagen). Data were normalized to either S9 ribosomal mRNA (Ba/F3 and Ba/F3-1*6 cell lines) or human Lamin A/C (LMNA) mRNA (K562 cell line), and expressed as relative mRNA levels, as previously described [3], [46], [47]. Real-time PCR primers specific for the following mouse (m) or human (h) cDNAs have been described: S9 (m), Cis (m), c-Myc (m/h), Pim-1 (m), Osm (m), c-Fos (m), Jun-B (m), 36b4 (m) and LMNA (h) [46], [65]. Forward (fwd) and reverse (rev) real-time PCR primers specific for human Cis cDNA were 5′- CTGCTGTGCATAGCCAAGACC-3′ (fwd) and 5′-GTGCCTTCTGGCATCTTCTGC-3′ (rev), and for mouse Ho-1 cDNA were 5′-GCTGGTGATGGCTTCCTTGT-3′ (fwd) and 5′-GGTTCTGCTTGTTGCGCTCT-3′ (rev). Data shown are representative of at least three independent experiments.
Cytotoxicity assays
WST-1 assays (11 644 807 001, Roche) were performed as described in the manufacturer's protocol. The tetrazolium salt WST-1 is cleaved into the formazan dye by mitochondrial dehydrogenase enzymes. Changes in metabolically active mitochondrial dehydrogenases as a result of TSA- or α-Br-TMC-induced cytotoxicity was evaluated by measuring formazan dye production upon incubation with the WST-1 reagent. Rested Ba/F3 and growing Ba/F3-1*6 and K562 cells were pre-treated for 30 minutes with the indicated TSA and α-Br-TMC concentrations or with DMSO (vehicle), as in our gene expression analysis experiments. WST-1 reagent was added to the cells either alone (Ba/F3-1*6, K562) or together with IL-3 (Ba/F3). Absorbance was measured in a microplate reader (Mithras LB 940, Berthold Technologies) at 450/620 nm after incubation at 37°C under 5% CO2 in a humidified incubator for 90 minutes. This duration of treatment was chosen as it is both optimal for the WST-1 assay (not shown) and close to the maximal duration of inhibitor treatment in our gene expression studies. A positive control for no mitochondrial enzyme activity (1% Triton X-100) was included in every experiment. Data are expressed as a percentage of cytotoxicity relative to DMSO (vehicle). Small molecule inhibitor treatment was carried out 2 to 3 times for each cell line, and the WST-1 assay was performed in duplicate. Results of one representative experiment are shown.
Cell viability assays
Equal cell numbers of growing Ba/F3, Ba/F3-1*6 and K562 cells were incubated for 24 and 48 hours in the presence of the indicated TSA and α-Br-TMC concentrations. Since very low DMSO concentrations were used (0.0005% to 0.01% final), no DMSO vehicle control was included. The total number of living and dead cells was evaluated by Trypan Blue exclusion. Viable cell number, reflecting cell proliferation and survival, was plotted as a function of time for each treatment. Data shown are representative of at least three independent experiments.
Quantitative chromatin immunoprecipitation (ChIP) assays
Chromatin immunoprecipitation was performed as previously described [3], [46], [47] with the following modifications: sonication was performed on a BRANSON SONIFIER 250 and cell samples were subjected to 6 pulses of 60 seconds each (output control: 5; duty cycle: 60%); Immunoprecipitated genomic DNA was purified on a Nucleospin clean-up column (740609, Macherey-Nagel) following their recommended protocol for SDS-containing samples. STAT5 and RNA polymerase II antibodies, as well as real-time PCR primers specific for the STAT5 binding sites of the mouse Cis gene and for the transcription start site (tss) of the mouse Cis and Osm genes, have been described [46]. Real-time PCR primers specific for the mouse Osm STAT5 binding sites (−184/−122 relative to tss) were 5′-CATCATCCTTGGGCGTGGGGC-3′ (fwd) and 5′-CGCTCCTCCTCCCGTTTTCTTCG-3′ (rev). Data shown are representative of at least three independent experiments.
Protein analysis
Whole-cell Brij protein lysis, nuclear and cytosolic protein extractions, and Western-blot analyses were performed as described [46], [65]. Antibodies used for detection of the respective proteins and their relevant dilutions were: pSTAT5 (#9351, Cell Signaling Technology; 1∶1000), STAT5A (L-20, sc-1081, Santa-Cruz Biotechnology; 1∶1000), STAT5B (G-2, sc-1656, Santa-Cruz Biotechnology; 1∶200), STAT5A+B (C-17, sc-835, Santa-Cruz Biotechnology; 1∶1000), pJAK2 (Cell Signaling Technology, #3771; 1∶200), JAK2 (#3230, Cell Signaling Technology; 1∶500), α-tubulin (DM1A, sc-32293, Santa-Cruz Biotechnology; 1∶200), Anti-Rabbit IgG-Peroxidase (SIGMA A0545; 1∶10,000), Anti-Mouse IgG-Peroxidase (SIGMA A8924; 1∶10,000). Apparent molecular weight of detected proteins was as predicted by the antibody manufacturers, i.e. 125 kDa for JAK2, 90 kDa for STAT5 (with STAT5A running slightly slower than STAT5B in SDS-PAGE, as confirmed by the STAT5A+B immunoblots) and 55 kDa for α-tubulin. Chemoluminescence detection was performed using Amersham ECL Prime (RPN2232, GE Healthcare Life Sciences) or SuperSignal West Femto (34095, Thermo Fisher Scientific) for weaker signals, and images were captured on an ImageQuant LAS 4000 mini imaging system (GE Healthcare Life Sciences). Immunoblots shown are representative of at least three independent experiments.
Results
The α-Br-chalcone α-Br-TMC inhibits expression of IL-3-induced STAT5-dependent and -independent genes in mouse Ba/F3 cells
In our initial screening for potential STAT5 inhibitors we chose a variety of natural and synthetic chalcones characterized towards their chemical reactivity in Michael additions with thiols. The respective second-order rate constants (k2) of α-X-TMCs (with X = CN, Br, Cl, H) using cysteamine as substrate [59]–[61] revealed differences of more than four orders of magnitude (Figure 1A), pointing to large variations in electrophilic reactivity. We also included the natural product curcumin, which is a weak electrophile (k2 = 0.0660 M−1s−1) [60], and the synthetic lysine deacetylase inhibitor trichostatin A (TSA), which serves as a reference compound for STAT5 inhibition [46], [47]. A kinetic thiol assay on TSA, which contains an α,β,γ,δ-unsaturated carbonyl unit, revealed very low amounts of the expected thiol adduct by mass spectrometry after 48 hours incubation with 500-fold excess of cysteamine (data not shown). This indicates that TSA main activity is not based on an electrophilic behavior, as anticipated from its electron-rich carbonyl functionality.
To identify novel inhibitors of the STAT5 signaling pathway, these ten natural and synthetic chalcones as well as curcumin were tested for their ability to suppress IL-3-mediated induction of the STAT5 target gene Cis. The gene Cis was selected for the initial screen as it is a well characterized STAT5-dependent gene [3], [46], [47], [66] and a potential therapeutic target in STAT5-associated cancers [67].
Ba/F3 cells withdrawn from IL-3 for 12 hours (STAT5 pathway turned off) were pre-treated for 30 minutes with 0.2 µM TSA or with 20 µM natural and synthetic chalcones or curcumin, before being stimulated with IL-3 for 1 hour (Figure 1B). Out of the 10 compounds tested, one (α-Br-2′,3,4,4′-tetramethoxychalcone thereafter called α-Br-TMC) exerted an 80% inhibition of IL-3-mediated induction of Cis expression, comparable to that exerted by TSA, while the housekeeping gene 36b4 remained unaffected (Figure 1B). In the following experiments, we therefore focused on characterizing α-Br-TMC activity, using TSA as a reference inhibitory compound.
First, we ensured the absence of α-Br-TMC-mediated cytotoxicity in the investigated time frame (up to 90 minutes treatment) in IL-3-stimulated Ba/F3 cells (Figure 2A). No cytotoxicity was detected up to 10 µM α-Br-TMC. However, strong cytotoxicity was evidenced at an α-Br-TMC concentration of 100 µM (Figure 2A). Thus α-Br-TMC was further used at concentrations not above 10 µM. We also investigated the long term effect of α-Br-TMC treatment on cell proliferation and survival of IL-3-growing Ba/F3 cells (Figure 2B). Similarly to TSA, α-Br-TMC impaired cell growth and viability in a dose-dependent manner. Ba/F3 cells cultured in 5 µM α-Br-TMC for 24 and 48 hours stopped dividing and died, while cells grown in 0.5 µM α-Br-TMC showed limited cell death and reduced cell proliferation (Figure 2B and data not shown).
Figure 2. Effect of α-Br-TMC on cytotoxicity and viability of normal (Ba/F3) and transformed (Ba/F3-1*6, K562) cells.

(A) Cells were pre-treated 30 minutes with 0.001, 0.01, 0.1 and 1 µM TSA or with 0.1, 1, 10 and 100 µM α-Br-TMC before starting the WST-1 assay. IL-3 (5 ng/mL) was added to rested Ba/F3 cells at the same time as the WST-1 reagent to mimic the IL-3 stimulation conditions used in other assays. DMSO (vehicle) concentration was adjusted to 0.1% final in all conditions. OD measurement was performed after 90 minutes incubation with the WST-1 reagent, and the percentage of cytotoxicity was normalized to the vehicle control. (B) Growing Ba/F3, Ba/F3-1*6 and K562 cells were incubated for 24 and 48 hours in the presence of the indicated concentrations of TSA and α-Br-TMC. Cell viability was measured by Trypan Blue exclusion assay.
We then analyzed expression of a series of STAT5 target genes (Cis, Osm, c-Myc) as well as STAT5-independent genes (JunB, Ho-1, 36b4) in dose-response experiments using 0.4 to 10 µM α-Br-TMC. Rested Ba/F3 cells were incubated 30 minutes with the respective compounds and stimulated with IL-3 for 30 minutes (Figure 1C). Induction of all three STAT5 target genes was inhibited by α-Br-TMC in a dose-dependent manner. In addition, the IL-3-inducible STAT5-independent gene JunB, while unaffected by TSA as expected [46], was also inhibited by α-Br-TMC in a dose-dependent manner (Figure 1C). It is thought that JunB is induced through the JAK2/MAPK pathway [68], [69], therefore suggesting that α-Br-TMC is targeting the upstream JAK/STAT pathway rather than STAT5 itself. α-Br-TMC was shown to up-regulate the Nrf2-dependent anti-inflammatory HO-1 protein levels in the mouse macrophage cell line RAW264.7 [59]. In Ba/F3 cells however, although Ho-1 gene expression was slightly up-regulated by IL-3 and TSA, no dose-dependent effect of α-Br-TMC was observed (Figure 1C), suggesting that the Nrf2-dependent anti-inflammatory response pathway is - if at all - only modestly activated and not influenced by α-Br-TMC. In contrast to IL-3-regulated genes, expression of the control gene 36b4 remained unaffected by α-Br-TMC treatment (Figure 1C), further supporting specificity of action.
Together, these gene expression data suggest that α-Br-TMC might target the JAK/STAT pathway for inhibition. We therefore monitored the effect of α-Br-TMC on STAT5 and JAK2 protein phosphorylation.
α-Br-TMC inhibits both JAK2 and STAT5 phosphorylation
STAT5 proteins are expressed in two forms, STAT5A and STAT5B, with both redundant and unique functions and encoded by two related genes. STAT5A and STAT5B exhibit 91% identity in their amino acid sequence, the C-terminal transactivation domain being most divergent [3]. STAT5 phosphorylation in IL-3-stimulated Ba/F3 cells pre-treated with either 0.2 µM TSA or 0.4 to 10 µM α-Br-TMC was investigated by Western-blot using a phospho-STAT5-specific antibody (pSTAT5) (Figure 3A). IL-3-induced STAT5 phosphorylation was not affected by TSA, in agreement with our previous report [46]. By contrast, IL-3-induced STAT5 phosphorylation was decreased by α-Br-TMC in a dose-dependent manner. Interestingly, both STAT5A and STAT5B proteins showed a dose-dependent downward mobility shift in SDS-PAGE, with STAT5B being more affected than STAT5A (Figure 3A). This mobility shift suggests that α-Br-TMC induces a protein modification targeting both STAT5 proteins. Analysis of cytosolic and nuclear fractions of unstimulated and stimulated α-Br-TMC-treated cells, revealed that this α-Br-TMC-mediated protein modification is taking place in the cytoplasm of unstimulated cells (Figure 3B) and hence is IL-3-independent. Analysis of cytosolic and nuclear fractions of α-Br-TMC-treated cells also confirmed that STAT5 phosphorylation and nuclear translocation are reduced (Figure 3B), thus supporting the idea that STAT5 activation is impaired upon α-Br-TMC treatment. Of note, the detection of shifted bands by the pSTAT5 antibody in the cytosolic and nuclear fractions of α-Br-TMC-treated IL-3-stimulated cells, suggests that the modified STAT5 protein can be to some extent phosphorylated at the expected tyrosine residue (Y694/699 in STAT5A/B respectively) and translocated into the nucleus.
Figure 3. α-Br-TMC inhibits both JAK2 and STAT5 phosphorylation.

Ba/F3 cells were pre-treated 30 minutes with the indicated concentrations of TSA and α-Br-TMC (A) or with 10 µM α-Br-TMC and DMSO vehicle (B, C) and further stimulated with 5 ng/mL IL-3 for 30 minutes (A), 15 minutes (B) or 5 minutes (C). Final DMSO concentration was 0.02% in (A) and 0.01% in (B, C). Protein whole cell extracts (panels A, C) or cytosolic (C) and nuclear (N) extracts (panel B) were prepared as described in Materials and Methods and analyzed by Western-blot using antibodies specific for phospho-STAT5 (pSTAT5), phospho-JAK2 (pJAK2), STAT5A, STAT5B, STAT5A and B, JAK2 and α-tubulin (loading control and cytosolic-specific marker). SDS-PAGE in (C) was shorter, explaining why the STAT5 mobility patterns (α-Br-TMC-induced shift and STAT5A and B doublet) are not as apparent as on immunoblots in (A, B).
STAT5 activation by IL-3 relies on the initial activation of the cellular tyrosine kinase JAK2 [70]. Our gene expression data revealing an inhibitory effect of α-Br-TMC on expression of the JAK2/MAPK-dependent JunB gene (Figure 1C) suggest that α-Br-TMC might also target the upstream JAK2 tyrosine kinase. We therefore investigated whether IL-3-induced phosphorylation of JAK2 was inhibited by α-Br-TMC. Ba/F3 cells were treated as before with 10 µM α-Br-TMC and briefly stimulated with IL-3 to analyze JAK2 phosphorylation by Western-blot using a pJAK2-specific antibody (Figure 3C). Following α-Br-TMC treatment, JAK2 phosphorylation was strongly diminished. By contrast to STAT5A and STAT5B proteins, JAK2 protein mobility was not affected by α-Br-TMC (Figure 3C).
Altogether, these experiments demonstrated that the α-Br-chalcone α-Br-TMC has the ability to inhibit the JAK/STAT pathway at multiple levels, by targeting both JAK2 and STAT5 proteins in IL-3-stimulated Ba/F3 cells.
α-Br-TMC inhibits STAT5-mediated transcriptional activity
The effect of α-Br-TMC on STAT5 transcriptional activity was further investigated by chromatin immunoprecipitation (ChIP). We previously showed that the deacetylase inhibitor TSA inhibits STAT5-mediated transcription at a step subsequent to STAT5 binding to its target genes by preventing recruitment of the transcriptional machinery [46]. STAT5 and RNA polymerase II ChIP assays were performed on TSA- and α-Br-TMC-treated Ba/F3 cells and protein recruitment to the mouse Cis and Osm promoters was examined (Figure 4). In agreement with our former report [46], RNA polymerase II recruitment to the Cis and Osm promoters was abrogated upon TSA treatment, without notably affecting STAT5 DNA binding activity. Although the level of nuclear pSTAT5 was strongly reduced in α-Br-TMC-treated cells (Figure 3B), the proportion of STAT5 bound to the Cis and Osm promoters was only moderately affected (20% and 58% reduced respectively; Figure 4A and B). This indicates that the nuclear pool of STAT5 proteins in α-Br-TMC-treated Ba/F3 cells might still support efficient binding to target sites. On the other hand, in correlation with the reduced Cis and Osm mRNA levels (Figure 1), recruitment of the RNA polymerase II to the Cis and Osm transcription start sites was markedly impaired (55% and 94% respectively; Figure 4A and B), pointing to an α-Br-TMC-mediated transcriptional inhibition of the Cis and Osm STAT5 target genes in Ba/F3 cells.
Figure 4. RNA polymerase II recruitment to the Cis and Osm promoters is impeded in Ba/F3 cells treated with α-Br-TMC.

Ba/F3 cells were pre-treated 30 minutes with 0.2 µM TSA or 10 µM α-Br-TMC before being stimulated with 5 ng/mL IL-3 for 30 minutes. DMSO (vehicle) was adjusted to 0.02% in all conditions. Chromatin immunoprecipitation (ChIP) was performed as described in Materials and Methods using antibodies directed against STAT5 or RNA polymerase II (RNA Pol II) proteins. Co-precipitated genomic DNA was analyzed by quantitative PCR using primers specific for the STAT5 binding sites (STAT5 ChIP) or the transcription start site (RNA Pol II ChIP) of the mouse Cis (A) and Osm (B) genes.
α-Br-TMC differentially affects JAK/STAT signaling in cells transformed by constitutively active STAT5
We showed that α-Br-TMC can inhibit IL-3-mediated JAK2 and STAT5 activation in normal mouse Ba/F3 cells. We next addressed whether α-Br-TMC can also hinder JAK/STAT activity in cancer cells exhibiting constitutive activation of STAT5. For this purpose, we compared gene expression patterns of normal Ba/F3 cells to those of transformed Ba/F3-1*6 and K562 cells.
Ba/F3-1*6 cells stably express a mutant form of mouse STAT5A (so-called 1*6) with two amino acids substitutions within its N-terminal and C-terminal transactivation domains respectively. These point mutations result in constitutive STAT5 phosphorylation, nuclear localization and transactivation properties [64]. Expression of constitutively active STAT5A-1*6 is sufficient to confer IL-3-independent growth to Ba/F3 cells in vitro and tumorigenicity to bone marrow cells in vivo [64], [71]. Importantly, JAK2 is not activated in Ba/F3-1*6 cells as a consequence of IL-3-independent growth [4], [64]. The mechanism of constitutive activation of STAT5A-1*6 is unclear. Whether basal JAK2 activity or an unidentified tyrosine kinase is responsible for STAT5A-1*6 phosphorylation is unknown [4], [64]. Increased stability of the phosphorylated STAT5A-1*6 mutant via an uncharacterized mechanism also contributes to its constitutive activity [64]. The human K562 leukemia cell line expresses a constitutively active BCR-ABL tyrosine kinase. BCR-ABL oncogenic fusion protein constitutively phosphorylates STAT5A and STAT5B proteins, directly contributing to oncogenesis [72]–[74].
Ba/F3, Ba/F3-1*6 and K562 cells were treated with 0.2 µM TSA or 10 µM α-Br-TMC for 90 minutes, with Ba/F3 being stimulated 60 minutes with IL-3 following 30 minute inhibitor pre-treatment, as before. In addition, K562 cells were treated with 1 µM of the BCR-ABL specific inhibitor Imatinib, as a positive control for inhibition of STAT5 phosphorylation [75]. Expression of STAT5 target genes (Cis, Osm, c-Myc, Pim-1), of JAK2/MAPK-regulated STAT5-independent genes (JunB, c-Fos), and of a housekeeping control gene (36b4) was evaluated by quantitative RT-PCR (Figure 5). As predicted upon expression of constitutively active STAT5-1*6 [46], expression of the STAT5 target genes was up-regulated in growing Ba/F3-1*6 cells in comparison to unstimulated Ba/F3 cells (Figure 5A–B). As anticipated, expression of the STAT5 target genes in all three cell lines was inhibited by TSA (Figure 5A–C). Similarly, STAT5 target genes were down-regulated in Imatinib-treated K562 cells (Figure 5C). Expression of all STAT5-target genes investigated in IL-3-stimulated Ba/F3 cells, including Cis, Osm, c-Myc, Pim-1 (Figure 5A) as well as Spi2.1 and SOCS-1 (not shown) was inhibited by α-Br-TMC. While expression of STAT5 target genes was down-regulated in α-Br-TMC-treated Ba/F3 cells (Figure 5A), distinct effects of α-Br-TMC were noted in Ba/F3-1*6 and K562 cells (Figure 5B–C). For instance, expression of Cis and Osm was up-regulated in α-Br-TMC-treated Ba/F3-1*6 (Figure 5B) and K562 (Figure 5C) cells. c-Myc and Pim-1 mRNA levels were partially reduced in α-Br-TMC-treated Ba/F3-1*6 cells (Figure 5B), while c-Myc mRNA level was slightly increased in α-Br-TMC-treated K562 cells (Figure 5C). Therefore, the α-Br-2′,3,4,4′-tetramethoxychalcone exerts distinct effects on STAT5 target gene expression in normal Ba/F3 vs. its transformed counterpart Ba/F3-1*6, as well as in the oncogenic K562 cell line.
Figure 5. α-Br-TMC exerts distinct effects in normal and cancer cells.

Ba/F3 (A), its caSTAT5-transformed counterpart Ba/F3-1*6 (B) and human leukemic K562 (C) cells were treated 90 minutes with 0.2 µM TSA, 10 µM α-Br-TMC or 1 µM Imatinib. Ba/F3 cells (A) were stimulated with 5 ng/mL IL-3 after an initial 30 minute drug pre-treatment (hence subjected to a 60 minute IL-3 stimulation). DMSO (vehicle) final concentration was adjusted to 0.02% in all conditions. Expression of STAT5-dependent (Cis, Osm, c-Myc, Pim-1) and -independent (JunB, c-Fos, 36b4) genes was analyzed by quantitative RT-PCR. Gene expression data were normalized to mouse ribosomal S9 (A, B) or to human Lamin A/C (LMNA) (C) housekeeping gene-encoded mRNAs. (A, B) Normalized data are presented with adjusted Y-axis scale for a direct comparison of mRNA levels in the respective normal and transformed Ba/F3 and Ba/F3-1*6 cell lines.
Both JAK2-mediated MAPK-dependent genes JunB and c-Fos were induced by IL-3 in Ba/F3 cells (Figure 5A). Their expression was either unaffected or upregulated by TSA respectively, as previously reported [46]. Expression of JunB and c-Fos was inhibited by α-Br-TMC, probably as a consequence of JAK2 inhibition (Figure 5A). In contrast to Ba/F3 cells, JunB and c-Fos genes were expressed at background levels in Ba/F3-1*6 cells (Figure 5B), most likely as a result of the absence of JAK2/MAPK activation [4]. Treatment of Ba/F3-1*6 cells with α-Br-TMC did not affect JunB expression while c-Fos was slightly up-regulated (Figure 5B), although mRNA levels remained low in comparison to that detected in IL-3-stimulated Ba/F3 cells (Figure 5A). The observation that α-Br-TMC was able to up-regulate the expression of c-Fos - and of other genes - in Ba/F3 cells transformed with STAT5A-1*6 but not in Ba/F3 parental cells, suggests that α-Br-TMC might target other factor(s) involved in gene regulation in Ba/F3-1*6 cells.
To further evaluate the effect of α-Br-TMC on normal and transformed cells, cytotoxicity and cell viability assays were performed as before (Figure 2A–B). The effect of α-Br-TMC on proliferation and survival of Ba/F3-1*6 and K562 cells was comparable to that observed with normal Ba/F3 cells. All cells died in the presence of 5 µM α-Br-TMC, while reduced proliferation and minimal cell death were observed in the presence of 0.5 µM α-Br-TMC over 48 hours (Figure 2B and data not shown). On the other hand, α-Br-TMC-induced cytotoxicity (at 100 µM) was reduced by approximately 50% in the transformed Ba/F3-1*6 and K562 cell lines in comparison to normal Ba/F3 cells (Figure 2A; 75% cytotoxicity in Ba/F3 cells vs. 47% and 42% in Ba/F3-1*6 and K562 cells respectively). This indicates a distinct chemosensitivity of the cancer cell lines to α-Br-TMC. Together with the observation that α-Br-TMC differentially affects gene expression in the transformed cells, our results suggest that its activity is regulated in a distinct manner in the investigated normal vs. transformed cell lines.
In conclusion, our gene expression analysis demonstrated that α-Br-TMC differentially regulates expression of STAT5 target genes in normal and cancer cells. Since we showed that α-Br-TMC hinders STAT5 transcriptional activity in Ba/F3 cells by inhibiting both STAT5 and JAK2 phosphorylation and possibly by modifying STAT5 proteins, we examined the effect of α-Br-TMC on JAK2 and STAT5 proteins in the transformed Ba/F3-1*6 and K562 cells.
α-Br-TMC-induced STAT5 protein modification differentially modulates STAT5 activity in normal and cancer cells
STAT5 phosphorylation in Ba/F3-1*6 and K562 cells was assessed by Western-blot analysis as before, on cells treated with 0.4 to 10 µM α-Br-TMC or 0.2 µM TSA. Both cell lines were also treated with 1 µM Imatinib, as a positive control for pSTAT5 inhibition in K562 cells and a negative control for Ba/F3-1*6 cells (Figure 6A). As expected, TSA did not affect STAT5 phosphorylation in Ba/F3-1*6 and K562 cells. Similarly, the BCR-ABL inhibitor Imatinib did not affect STAT5 phosphorylation in Ba/F3-1*6 cells, while drastically inhibiting STAT5 phosphorylation in K562 cells, as previously reported [75]. As observed in Ba/F3 cells, treatment of Ba/F3-1*6 and K562 cells with α-Br-TMC led to a decrease in STAT5 phosphorylation and a downward mobility shift of both STAT5A and STAT5B proteins, in a dose-dependent manner (Figure 6A and 6B). Again, STAT5B appeared to be more affected than STAT5A.
Figure 6. α-Br-TMC inhibits the STAT5 signaling pathway in both a JAK2-dependent and -independent manner.

(A) Ba/F3-1*6 and K562 cells were treated for 60 minutes with the indicated compounds. Protein whole cell extracts were analyzed by Western-Blot using antibodies specific for pSTAT5, STAT5A, STAT5B, STAT5A and B and α-tubulin as a loading control. (B) Ba/F3 cells were pre-treated 30 minutes with 10 µM α-Br-TMC and stimulated with 5 ng/mL IL-3 for 5 minutes. Ba/F3-1*6 and K562 cells were treated with 10 µM α-Br-TMC for 90 minutes. DMSO (Veh.) was adjusted to 0.01% in all conditions. Protein whole cell extracts were analyzed by Western-Blot using antibodies specific for pSTAT5, pJAK2, STAT5A, STAT5B, STAT5A and B, JAK2 and α-tubulin (loading control).
We next assessed the effect of α-Br-TMC on JAK2 phosphorylation in Ba/F3-1*6 and K562 cells (Figure 6B). In agreement with previous reports [4], [64], pJAK2 was barely detectable in Ba/F3-1*6 cells. The absence of JAK2 phosphorylation in Ba/F3-1*6 cells is also in agreement with our gene expression data, revealing only basal expression levels of the JAK2/MAPK-dependent genes c-Fos and JunB (Figure 5B). The observation that STAT5 proteins undergo α-Br-TMC-mediated dephosphorylation and mobility shift in the absence of JAK2 supports the idea that these modifications occur in a JAK2-independent manner. Finally, like in Ba/F3 cells, JAK2 phosphorylation in K562 cells was inhibited upon α-Br-TMC treatment (Figure 6B), further indicating that α-Br-TMC targets both JAK2 and STAT5 proteins.
Discussion
In the search for novel STAT5 inhibitors, we examined the effect of a series of natural and synthetic chalcones as well as of curcumin, another natural α,β-unsaturated carbonyl compound, on JAK2/STAT5 activation and expression of downstream STAT5 target genes in normal (Ba/F3) and caSTAT5-transformed (Ba/F3-1*6, K562) cell lines.
We show here that the synthetic α-Br-2′,3,4,4′-tetramethoxychalcone (α-Br-TMC) is a potent inhibitor of both JAK2 tyrosine kinase and STAT5 transcription factor activities. None of the natural products investigated (curcumin and five natural chalcones) affected STAT5-mediated transactivation of the Cis gene in IL-3-stimulated Ba/F3 cells at a concentration of 20 µM. Amongst the four synthetic chalcones α-X-TMCs (with X = CN, Br, Cl, H) tested, α-Br-TMC was the only active STAT5 inhibitor. We showed before that α-Br-TMC, α-Cl-TMC and to a lesser extent α-H-TMC exhibit anti-inflammatory activities in RAW364.7 cells, in particular inhibiting both Nrf2- and NF-κB-dependent pathways [59]. We could not detect an effect of α-Br-TMC on the expression of the Nrf2-dependent Ho-1 gene in Ba/F3 cells, possibly due to the short duration of treatment. Nevertheless, taken together, our data indicate that α-Br-TMC is able to target multiple pathways controlling inflammation, proliferation and survival. The natural compound sophoraflavanone G was recently reported to exhibit a similar pleiotropic activity against both the JAK/STAT signaling pathway and pathways controlling inflammation and infection in murine and human cell lines [36]. Since interconversion between flavanones and 2′-OH-chalcones was recently evidenced in cell culture [76], it might explain why these two classes of molecules exhibit similar biological activities.
In accordance with the pleiotropic activity of α-Br-TMC, we found that it targets the JAK2/STAT5 pathway at multiple levels (Figure 7). First, α-Br-TMC inhibits JAK2 tyrosine kinase activation, thus indirectly inhibiting activation of pathways downstream of JAK2, in particular STAT5 and mitogen-activated protein kinase (MAPK). Second, α-Br-TMC inhibits constitutive STAT5 phosphorylation in cells showing basal JAK2 activation (Ba/F3-1*6). Although we cannot rule out that basal JAK2 activation is responsible for the constitutive STAT5 phosphorylation in Ba/F3-1*6 cells [4], our data suggest that α-Br-TMC also inhibits STAT5 phosphorylation - directly or indirectly - in a JAK2-independent manner. Third, α-Br-TMC might alter a post-translational modification on STAT5 proteins, as revealed by a downward mobility shift in SDS-PAGE. α-Br-TMC treatment changed the mobility of both STAT5A and STAT5B proteins in SDS-PAGE, although STAT5B appears more strongly affected than STAT5A. Moreover, this effect takes place within the cytoplasm and independently of IL-3 stimulation and STAT5 phosphorylation. In fact, the faster-migrating STAT5 forms could still be phosphorylated and translocated into the nucleus to some extent. Since this protein modification correlates with a reduced phosphorylation of STAT5A-1*6, possibly independent of JAK2, it is tempting to speculate that α-Br-TMC-induced STAT5 modification interferes - directly or indirectly - with STAT5 activation and hence STAT5 transcriptional activity.
Figure 7. Model of inhibition of the JAK2/STAT5 pathway by α-Br-TMC.
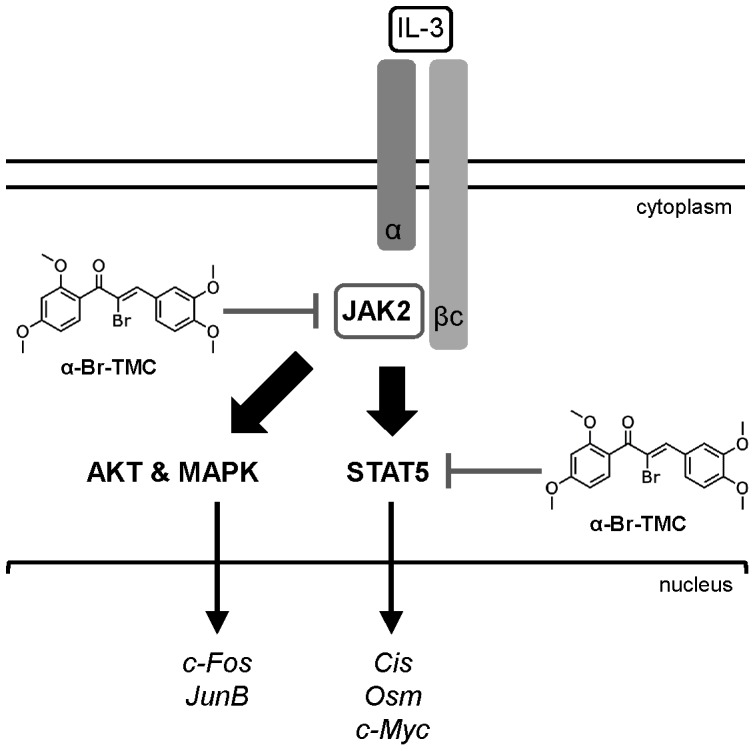
IL-3 binding to the α/βc chains of the IL-3 receptor leads to activation of the receptor-associated JAK2 tyrosine kinase by trans-phosphorylation. In turn, JAK2-mediated activation of the STAT5, MAPK and AKT pathways via phosphorylation (broad arrows) results in induced transcription of downstream target genes (thin arrows). We showed that α-Br-TMC inhibits JAK2 phosphorylation, hence impairing JAK2-regulated signaling pathways. In addition and independently of JAK2, α-Br-TMC inhibits STAT5 activity. Concomitantly, STAT5A and STAT5B protein mobility in SDS-PAGE is altered, indicating a change in their post-translational modification state induced by α-Br-TMC, which might be associated to STAT5 altered transcriptional activity.
As a result of reduced JAK2 and STAT5 activation, transcription of STAT5 target genes such as Cis and Osm was impaired, in agreement with a diminished recruitment of RNA polymerase II to their respective transcription start site. Somewhat surprisingly, STAT5 binding to DNA was marginally diminished upon α-Br-TMC treatment. It suggests that the modified nuclear STAT5 proteins still have the ability to bind to DNA and hence might compete with transcriptionally competent unmodified STAT5 proteins.
We found that although STAT5 proteins were similarly modified by α-Br-TMC in normal and transformed cells based on their mobility profile in SDS-PAGE, the consequence on expression of downstream target genes was different. Indeed, while expression of Cis and Osm was down-regulated in Ba/F3 cells, it was up-regulated in Ba/F3-1*6 and K562 cells. Meanwhile, expression of the c-Myc oncogene was reduced in both Ba/F3 and Ba/F3-1*6 cells but slightly increased in K562 cells. According to the differential effect of α-Br-TMC on gene expression, the three cell lines exhibited a distinct chemosensitivity to α-Br-TMC. These observations suggest that α-Br-TMC might target additional signaling and/or transcription factors in cells transformed by constitutively active STAT5 (mouse Ba/F3-1*6 and human K562), resulting in distinct gene expression patterns. The observation that basal expression of c-Fos in Ba/F3-1*6 cells in the absence of Jak2 activation was slightly increased upon α-Br-TMC treatment would support this hypothesis. On the other hand, we cannot exclude that α-Br-TMC-mediated modification of STAT5 protein differentially affects STAT5 transcriptional activity in normal and cancer cells. Given that STAT5 DNA binding activity was only weakly impaired in α-Br-TMC-treated cells, one could indeed envisage that DNA-bound STAT5 protein complexes exhibit distinct transcriptional activities depending on the cellular context. For instance, STAT5 is known to regulate transcription through interactions with positive and negative cofactors and with other transcription factors bound on adjacent sites, as well as through tetramerization [1], [9]–[11], which could be differentially regulated in transformed cells. Clearly, additional experiments are needed to determine whether STAT5 is also bound to the Cis promoter in Ba/F3-1*6 and K562 cells and further unravel the underlying molecular mechanism.
The question remains as to how α-Br-TMC inhibits JAK2 activation. We cannot rule out at this point that α-Br-TMC acts as a tyrosine kinase inhibitor. Indeed, its structure is reminiscent of that of AG490, a reversible substrate competitive tyrosine kinase inhibitor [77], [78]. In addition, the nature of the protein modification(s) targeting STAT5 upon α-Br-TMC treatment remains to be identified. Several lines of evidences support the idea that the divergent C-terminal transactivation domain of STAT5 is targeted for modification, including the observations that (i) α-Br-TMC inhibited STAT5 phosphorylation at Y694/699, (ii) α-Br-TMC-induced mobility shift of STAT5A was not as pronounced as that of STAT5B, (iii) α-Br-TMC differentially modulated STAT5 transcriptional activity in normal and transformed cells. On the other hand, the observation that STAT5 DNA binding activity was only slightly impaired, raises the possibility that the STAT5 DNA binding domain might be modified as well.
Chalcones in general, and α-Br-TMC in particular, present a highly electrophilic α,β-unsaturated carbonyl moiety that can potentially react with free sulfhydryl groups of cysteine residues in proteins and directly alter their function. On the other hand, electrophiles can inhibit the activation of transcription factors such as NF-κB and STAT3, possibly through S-glutathionylation of cysteine thiol groups, which is itself triggered by a rapid drop of intracellular glutathione [55], [79]. Three naturally occuring terpenes, which also contain an electrophilic α,β-unsaturated carbonyl group, induce S-glutathionylation of STAT3 thereby inhibiting its activity [80], [81]. Similarly to our α-Br-TMC compound, these molecules also inhibit the activation of JAK2 tyrosine kinase [80]. In agreement with the identification of several essential cysteine residues within JAK2 catalytic domain [82], activity of JAK2 - but also of STAT5 - is regulated by the synthetic glutathione disulfide mimetic NOV-002 [83], suggesting that both JAK2 and STAT5 can be S-glutathionylated. Finally, a review of the literature revealed that S-glutathionylation and redox regulation can either stimulate or inhibit protein activity [82], [83], possibly depending on the position of the modified cysteine within the functional domains of the targeted protein. Altogether, these observations raise the possibility that α-Br-TMC, as a potent Michael acceptor, might alter the function of STAT5 and JAK2 proteins, through either direct alkylation or indirect S-glutathionylation of accessible thiol groups, thereby inhibiting or stimulating their activity depending on the cellular context. The presence of cysteine residues in STAT5 proteins, both common and unique to STAT5A and STAT5B within their DNA binding and transactivation domains respectively, is consistent with that model.
On the other hand, the detection of a downward mobility shift of STAT5A and STAT5B upon α-Br-TMC treatment is not in full agreement with the covalent addition of one or several S-glutathione groups. Such downward shift is rather reminiscent of protein dephosphorylation and one could envision an alternative mode of action for α-Br-TMC. Beside tyrosine phosphorylation, STAT5 proteins can be phosphorylated on serine residues [84]. Interestingly, STAT5A and STAT5B are differentially phosphorylated on C-terminal serines (S725/730 in mSTAT5A/B; S779 in mSTAT5A) [84], resulting in either positive or negative transcriptional regulation depending on promoter and/or cellular context [85], [86]. It remains therefore possible that α-Br-TMC either inhibits the still-unknown kinase responsible for serine phosphorylation of STAT5 or activates a specific serine phosphatase, leading to the STAT5 mobility shift detected in SDS-PAGE and possibly to the alteration of its activity.
Whether α-Br-TMC acts as a tyrosine kinase inhibitor and whether the α-Br-TMC-induced STAT5 mobility shift involves S-glutathionylation and/or dephosphorylation of STAT5 and/or of other regulatory components of the STAT5 pathway is currently under investigation.
In conclusion, we identified the synthetic chalcone α-Br-TMC as a novel regulator of JAK2/STAT5 activity, targeting both STAT5 and its upstream activating kinase JAK2 (Figure 7). Its ability to down-regulate expression of the c-Myc oncogene while up-regulating expression of the tumor suppressor gene Cis in cancer cells, together with its previously described anti-inflammatory properties [59], potentially makes α-Br-TMC a promising novel therapeutic agent with pleiotropic anti-inflammatory and anticancer activities.
Acknowledgments
We thank Jacqueline Marvel and Daniela Männel for providing the Ba/F3 and K562 cells respectively. We thank Belinda Jobst for her technical contribution to this project while completing an internship in our group. We are grateful to Joachim Griesenbeck for critically reading the manuscript and Fiona Kalinowski for kindly proof-reading the text.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft (Grant No. RA 2010/2-1 to AR), the Deutsche Krebshilfe (Grant No. 109750 to AR), institutional research funds (Foerderlinie C to AR), the Fonds der Chemischen Industrie (Liebig scholarship to SA) and the DAAD (Doctoral scholarship to NA). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Grimley PM, Dong F, Rui H (1999) Stat5a and Stat5b: fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev 10: 131–157. [DOI] [PubMed] [Google Scholar]
- 2. Wakao H, Gouilleux F, Groner B (1994) Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J 13: 2182–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Basham B, Sathe M, Grein J, McClanahan T, D'Andrea A, et al. (2008) In vivo identification of novel STAT5 target genes. Nucleic Acids Res 36: 3802–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nosaka T, Kawashima T, Misawa K, Ikuta K, Mui AL, et al. (1999) STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J 18: 4754–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mui AL, Wakao H, Kinoshita T, Kitamura T, Miyajima A (1996) Suppression of interleukin-3-induced gene expression by a C-terminal truncated Stat5: role of Stat5 in proliferation. EMBO J 15: 2425–2433. [PMC free article] [PubMed] [Google Scholar]
- 6. Van Nguyen T, Angkasekwinai P, Dou H, Lin F-M, Lu L-S, et al. (2012) SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol Cell 45: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wieczorek M, Ginter T, Brand P, Heinzel T, Krämer OH (2012) Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev 23: 293–305. [DOI] [PubMed] [Google Scholar]
- 8. Ma L, Gao J, Guan Y, Shi X, Zhang H, et al. (2010) Acetylation modulates prolactin receptor dimerization. Proc Natl Acad Sci U S A 107: 19314–19319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin JX, Leonard WJ (2000) The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 19: 2566–2576. [DOI] [PubMed] [Google Scholar]
- 10. Shuai K (2000) Modulation of STAT signaling by STAT-interacting proteins. Oncogene 19: 2638–2644. [DOI] [PubMed] [Google Scholar]
- 11. Lin J-X, Li P, Liu D, Jin HT, He J, et al. (2012) Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 36: 586–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, et al. (2011) Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol 12: 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Litterst CM, Kliem S, Marilley D, Pfitzner E (2003) NCoA-1/SRC-1 is an essential coactivator of STAT5 that binds to the FDL motif in the alpha-helical region of the STAT5 transactivation domain. J Biol Chem 278: 45340–45351. [DOI] [PubMed] [Google Scholar]
- 14. Pfitzner E, Jähne R, Wissler M, Stoecklin E, Groner B (1998) p300/CREB-binding protein enhances the prolactin-mediated transcriptional induction through direct interaction with the transactivation domain of Stat5, but does not participate in the Stat5-mediated suppression of the glucocorticoid response. Mol Endocrinol Baltim Md 12: 1582–1593. [DOI] [PubMed] [Google Scholar]
- 15. John S, Vinkemeier U, Soldaini E, Darnell JE Jr, Leonard WJ (1999) The significance of tetramerization in promoter recruitment by Stat5. Mol Cell Biol 19: 1910–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ward AC, Touw I, Yoshimura A (2000) The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 95: 19–29. [PubMed] [Google Scholar]
- 17. Valentino L, Pierre J (2006) JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 71: 713–721. [DOI] [PubMed] [Google Scholar]
- 18. Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19: 2474–2488. [DOI] [PubMed] [Google Scholar]
- 19. Liu CB, Itoh T, Arai K, Watanabe S (1999) Constitutive activation of JAK2 confers murine interleukin-3-independent survival and proliferation of BA/F3 cells. J Biol Chem 274: 6342–6349. [DOI] [PubMed] [Google Scholar]
- 20. Lord JD, McIntosh BC, Greenberg PD, Nelson BH (2000) The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J Immunol Baltim Md 1950 164: 2533–2541. [DOI] [PubMed] [Google Scholar]
- 21. Nosaka T, Kitamura T (2002) Pim-1 expression is sufficient to induce cytokine independence in murine hematopoietic cells, but is dispensable for BCR-ABL-mediated transformation. Exp Hematol 30: 697–702. [DOI] [PubMed] [Google Scholar]
- 22. Garçon L, Rivat C, James C, Lacout C, Camara-Clayette V, et al. (2006) Constitutive activation of STAT5 and Bcl-xL overexpression can induce endogenous erythroid colony formation in human primary cells. Blood 108: 1551–1554. [DOI] [PubMed] [Google Scholar]
- 23. Gesbert F, Griffin JD (2000) Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood 96: 2269–2276. [PubMed] [Google Scholar]
- 24. Chim C-S, Fung T-K, Cheung W-C, Liang R, Kwong Y-L (2004) SOCS1 and SHP1 hypermethylation in multiple myeloma: implications for epigenetic activation of the Jak/STAT pathway. Blood 103: 4630–4635. [DOI] [PubMed] [Google Scholar]
- 25. He B, You L, Uematsu K, Zang K, Xu Z, et al. (2003) SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A 100: 14133–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, et al. (2005) Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 24: 6406–6417. [DOI] [PubMed] [Google Scholar]
- 27. Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, et al. (2005) SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene 24: 6699–6708. [DOI] [PubMed] [Google Scholar]
- 28. Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, et al. (2005) STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A 102: 6948–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelson EA, Sharma SV, Settleman J, Frank DA (2011) A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget 2: 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Müller J, Sperl B, Reindl W, Kiessling A, Berg T (2008) Discovery of chromone-based inhibitors of the transcription factor STAT5. Chembiochem Eur J Chem Biol 9: 723–727. [DOI] [PubMed] [Google Scholar]
- 31. Luo C, Laaja P (2004) Inhibitors of JAKs/STATs and the kinases: a possible new cluster of drugs. Drug Discov Today 9: 268–275. [DOI] [PubMed] [Google Scholar]
- 32. Quintás-Cardama A, Verstovsek S (2013) Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res Off J Am Assoc Cancer Res 19: 1933–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warsch W, Walz C, Sexl V (2013) JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia. Blood 122: 2167–2175. [DOI] [PubMed] [Google Scholar]
- 34. Hayakawa F, Sugimoto K, Harada Y, Hashimoto N, Ohi N, et al. (2013) A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J 3: e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weber A, Borghouts C, Brendel C, Moriggl R, Delis N, et al. (2013) The inhibition of stat5 by a Peptide aptamer ligand specific for the DNA binding domain prevents target gene transactivation and the growth of breast and prostate tumor cells. Pharm Basel Switz 6: 960–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim B-H, Won C, Lee Y-H, Choi JS, Noh KH, et al. (2013) Sophoraflavanone G induces apoptosis of human cancer cells by targeting upstream signals of STATs. Biochem Pharmacol 86: 950–959. [DOI] [PubMed] [Google Scholar]
- 37. Ma L, Clayton JR, Walgren RA, Zhao B, Evans RJ, et al. (2013) Discovery and characterization of LY2784544, a small-molecule tyrosine kinase inhibitor of JAK2V617F. Blood Cancer J 3: e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, et al. (2011) The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 117: 3421–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, et al. (2012) The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations. Genes Cancer 3: 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanan EJ, van Abbema A, Barrett K, Blair WS, Blaney J, et al. (2012) Discovery of potent and selective pyrazolopyrimidine janus kinase 2 inhibitors. J Med Chem 55: 10090–10107. [DOI] [PubMed] [Google Scholar]
- 41. Kraus M, Wang Y, Aleksandrowicz D, Bachman E, Szewczak AA, et al. (2012) Efficacious intermittent dosing of a novel JAK2 inhibitor in mouse models of polycythemia vera. PloS One 7: e37207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang J, Ikezoe T, Nishioka C, Furihata M, Yokoyama A (2010) AZ960, a novel Jak2 inhibitor, induces growth arrest and apoptosis in adult T-cell leukemia cells. Mol Cancer Ther 9: 3386–3395. [DOI] [PubMed] [Google Scholar]
- 43. Ioannidis S, Lamb ML, Wang T, Almeida L, Block MH, et al. (2011) Discovery of 5-chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a novel inhibitor of the Jak/Stat pathway. J Med Chem 54: 262–276. [DOI] [PubMed] [Google Scholar]
- 44. Jatiani SS, Cosenza SC, Reddy MVR, Ha JH, Baker SJ, et al. (2010) A Non-ATP-Competitive Dual Inhibitor of JAK2 and BCR-ABL Kinases: Elucidation of a Novel Therapeutic Spectrum Based on Substrate Competitive Inhibition. Genes Cancer 1: 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Müller J, Schust J, Berg T (2008) A high-throughput assay for signal transducer and activator of transcription 5b based on fluorescence polarization. Anal Biochem 375: 249–254. [DOI] [PubMed] [Google Scholar]
- 46. Rascle A, Johnston JA, Amati B (2003) Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol Cell Biol 23: 4162–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rascle A, Lees E (2003) Chromatin acetylation and remodeling at the Cis promoter during STAT5-induced transcription. Nucleic Acids Res 31: 6882–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gryder BE, Sodji QH, Oyelere AK (2012) Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem 4: 505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Evrot E, Ebel N, Romanet V, Roelli C, Andraos R, et al. (2013) JAK1/2 and Pan-deacetylase inhibitor combination therapy yields improved efficacy in preclinical mouse models of JAK2V617F-driven disease. Clin Cancer Res Off J Am Assoc Cancer Res 19: 6230–6241. [DOI] [PubMed] [Google Scholar]
- 50. Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, et al. (2009) Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 114: 5024–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guerini V, Barbui V, Spinelli O, Salvi A, Dellacasa C, et al. (2008) The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia 22: 740–747. [DOI] [PubMed] [Google Scholar]
- 52. Pietschmann K, Bolck HA, Buchwald M, Spielberg S, Polzer H, et al. (2012) Breakdown of the FLT3-ITD/STAT5 axis and synergistic apoptosis induction by the histone deacetylase inhibitor panobinostat and FLT3-specific inhibitors. Mol Cancer Ther 11: 2373–2383. [DOI] [PubMed] [Google Scholar]
- 53. Nguyen T, Dai Y, Attkisson E, Kramer L, Jordan N, et al. (2011) HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin Cancer Res Off J Am Assoc Cancer Res 17: 3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Batovska DI, Todorova IT (2010) Trends in utilization of the pharmacological potential of chalcones. Curr Clin Pharmacol 5: 1–29. [DOI] [PubMed] [Google Scholar]
- 55. Liu Y-C, Hsieh C-W, Wu C-C, Wung B-S (2007) Chalcone inhibits the activation of NF-kappaB and STAT3 in endothelial cells via endogenous electrophile. Life Sci 80: 1420–1430. [DOI] [PubMed] [Google Scholar]
- 56. Pandey MK, Sung B, Ahn KS, Aggarwal BB (2009) Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol Pharmacol 75: 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Funakoshi-Tago M, Tago K, Nishizawa C, Takahashi K, Mashino T, et al. (2008) Licochalcone A is a potent inhibitor of TEL-Jak2-mediated transformation through the specific inhibition of Stat3 activation. Biochem Pharmacol 76: 1681–1693. [DOI] [PubMed] [Google Scholar]
- 58. Liu Y, Gao X, Deeb D, Arbab AS, Dulchavsky SA, et al. (2012) Anticancer agent xanthohumol inhibits IL-2 induced signaling pathways involved in T cell proliferation. J Exp Ther Oncol 10: 1–8. [PMC free article] [PubMed] [Google Scholar]
- 59. Al-Rifai N, Rücker H, Amslinger S (2013) Opening or Closing the Lock? When Reactivity Is the Key to Biological Activity. Chem Eur J 19: 15384–15395. [DOI] [PubMed] [Google Scholar]
- 60. Amslinger S, Al-Rifai N, Winter K, Wörmann K, Scholz R, et al. (2013) Reactivity assessment of chalcones by a kinetic thiol assay. Org Biomol Chem 11: 549–554. [DOI] [PubMed] [Google Scholar]
- 61. Amslinger S (2010) The tunable functionality of alpha,beta-unsaturated carbonyl compounds enables their differential application in biological systems. ChemMedChem 5: 351–356. [DOI] [PubMed] [Google Scholar]
- 62. Lozzio CB, Lozzio BB (1975) Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45: 321–334. [PubMed] [Google Scholar]
- 63. Palacios R, Steinmetz M (1985) Il-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell 41: 727–734. [DOI] [PubMed] [Google Scholar]
- 64. Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, et al. (1998) Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol 18: 3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rascle A, Neumann T, Raschta A-S, Neumann A, Heining E, et al. (2009) The LIM-homeodomain transcription factor LMX1B regulates expression of NF-kappa B target genes. Exp Cell Res 315: 76–96. [DOI] [PubMed] [Google Scholar]
- 66. Matsumoto A, Masuhara M, Mitsui K, Yokouchi M, Ohtsubo M, et al. (1997) CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood 89: 3148–3154. [PubMed] [Google Scholar]
- 67. Elliott J, Hookham MB, Johnston JA (2008) The suppressors of cytokine signalling E3 ligases behave as tumour suppressors. Biochem Soc Trans 36: 464–468. [DOI] [PubMed] [Google Scholar]
- 68. Fujihara M, Ikebuchi K, Maekawa TL, Wakamoto S, Ogiso C, et al. (1998) Lipopolysaccharide-induced desensitization of junB gene expression in a mouse macrophage-like cell line, P388D1. J Immunol Baltim Md 1950 161: 3659–3665. [PubMed] [Google Scholar]
- 69. Santos FPS, Verstovsek S (2011) JAK2 inhibitors: what's the true therapeutic potential? Blood Rev 25: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Paukku K, Silvennoinen O (2004) STATs as critical mediators of signal transduction and transcription: lessons learned from STAT5. Cytokine Growth Factor Rev 15: 435–455. [DOI] [PubMed] [Google Scholar]
- 71. Schwaller J, Parganas E, Wang D, Cain D, Aster JC, et al. (2000) Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol Cell 6: 693–704. [DOI] [PubMed] [Google Scholar]
- 72. De Groot RP, Raaijmakers JA, Lammers JW, Jove R, Koenderman L (1999) STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood 94: 1108–1112. [PubMed] [Google Scholar]
- 73. Nieborowska-Skorska M, Wasik MA, Slupianek A, Salomoni P, Kitamura T, et al. (1999) Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J Exp Med 189: 1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ilaria RL Jr, Van Etten RA (1996) P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem 271: 31704–31710. [DOI] [PubMed] [Google Scholar]
- 75. Roskoski R Jr (2003) STI-571: an anticancer protein-tyrosine kinase inhibitor. Biochem Biophys Res Commun 309: 709–717. [DOI] [PubMed] [Google Scholar]
- 76. Simmler C, Hajirahimkhan A, Lankin DC, Bolton JL, Jones T, et al. (2013) Dynamic residual complexity of the isoliquiritigenin-liquiritigenin interconversion during bioassay. J Agric Food Chem 61: 2146–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Levitzki A, Mishani E (2006) Tyrphostins and Other Tyrosine Kinase Inhibitors. Annu Rev Biochem 75: 93–109. [DOI] [PubMed] [Google Scholar]
- 78. Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, et al. (1996) Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 379: 645–648. [DOI] [PubMed] [Google Scholar]
- 79. Xie Y, Kole S, Precht P, Pazin MJ, Bernier M (2009) S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 150: 1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Butturini E, Cavalieri E, de Prati AC, Darra E, Rigo A, et al. (2011) Two naturally occurring terpenes, dehydrocostuslactone and costunolide, decrease intracellular GSH content and inhibit STAT3 activation. PloS One 6: e20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Butturini E, de Prati AC, Chiavegato G, Rigo A, Cavalieri E, et al. (2013) Mild oxidative stress induces S-glutathionylation of STAT3 and enhances chemosensitivity of tumoral cells to chemotherapeutic drugs. Free Radic Biol Med. [DOI] [PubMed]
- 82. Mamoon NM, Smith JK, Chatti K, Lee S, Kundrapu K, et al. (2007) Multiple cysteine residues are implicated in Janus kinase 2-mediated catalysis. Biochemistry (Mosc) 46: 14810–14818. [DOI] [PubMed] [Google Scholar]
- 83. Townsend DM, He L, Hutchens S, Garrett TE, Pazoles CJ, et al. (2008) NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer Res 68: 2870–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Decker T, Kovarik P (2000) Serine phosphorylation of STATs. Oncogene 19: 2628–2637. [DOI] [PubMed] [Google Scholar]
- 85. Yamashita H, Xu J, Erwin RA, Farrar WL, Kirken RA, et al. (1998) Differential control of the phosphorylation state of proline-juxtaposed serine residues Ser725 of Stat5a and Ser730 of Stat5b in prolactin-sensitive cells. J Biol Chem 273: 30218–30224. [DOI] [PubMed] [Google Scholar]
- 86. Beuvink I, Hess D, Flotow H, Hofsteenge J, Groner B, et al. (2000) Stat5a serine phosphorylation. Serine 779 is constitutively phosphorylated in the mammary gland, and serine 725 phosphorylation influences prolactin-stimulated in vitro DNA binding activity. J Biol Chem 275: 10247–10255. [DOI] [PubMed] [Google Scholar]