

As part of a regulatory loop linking cell metabolism, growth, and proliferation, CIP2A promotes mTORC1-mediated cell growth and autophagy inhibition but is itself down-regulated by autophagy.
Abstract
mTORC1 (mammalian target of rapamycin complex 1) integrates information regarding availability of nutrients and energy to coordinate protein synthesis and autophagy. Using ribonucleic acid interference screens for autophagy-regulating phosphatases in human breast cancer cells, we identify CIP2A (cancerous inhibitor of PP2A [protein phosphatase 2A]) as a key modulator of mTORC1 and autophagy. CIP2A associates with mTORC1 and acts as an allosteric inhibitor of mTORC1-associated PP2A, thereby enhancing mTORC1-dependent growth signaling and inhibiting autophagy. This regulatory circuit is reversed by ubiquitination and p62/SQSTM1-dependent autophagic degradation of CIP2A and subsequent inhibition of mTORC1 activity. Consistent with CIP2A’s reported ability to protect c-Myc against proteasome-mediated degradation, autophagic degradation of CIP2A upon mTORC1 inhibition leads to destabilization of c-Myc. These data characterize CIP2A as a distinct regulator of mTORC1 and reveals mTORC1-dependent control of CIP2A degradation as a mechanism that links mTORC1 activity with c-Myc stability to coordinate cellular metabolism, growth, and proliferation.
Introduction
Macroautophagy (hereafter autophagy) is a tightly regulated catabolic process, in which damaged organelles and macromolecules are sequestered into autophagic vesicles that deliver them to lysosomes for degradation and recycling (Xie and Klionsky, 2007; Mehrpour et al., 2010; Mizushima and Komatsu, 2011). Autophagic recycling activity is low under optimal conditions but can be rapidly activated in response to starvation, cytotoxic drugs, or other forms of cellular stress. Under such conditions, autophagy promotes cell survival by sustaining metabolic homeostasis and preventing accumulation of damaged organelles and proteins. The essential role of autophagy in promoting survival of cancer cells exposed to metabolic and therapeutic stress may provide a window of opportunity for exploitation of autophagy as a therapeutic target in cancer (Amaravadi et al., 2011).
mTORC1 (mammalian target of rapamycin [TOR; mTOR] complex 1) kinase is the major negative regulator of autophagy (Corcelle et al., 2009; Jung et al., 2010; Efeyan et al., 2012). It serves as a signaling nexus that integrates information regarding cellular stress and availability of nutrients and growth factors to maintenance of the appropriate balance between anabolic (e.g., protein synthesis) and catabolic (e.g., autophagy) processes. The signaling pathways promoting mTORC1 activation are induced by numerous mitogenic factors and oncoproteins via the class I phosphoinositide-3 kinase (PI3K)–Akt pathway, whereas various cellular stresses inhibit the mTORC1 activity via activation of AMP-activated protein kinase (AMPK). The mTORC1 homodimer consists of mTOR kinase, regulatory-associated protein of TOR (raptor), mammalian lethal with Sec13 protein 8, disheveled, Egl-10, pleckstrin domain–containing mTOR-interacting protein, and proline-rich Akt substrate of 40 kD. The best-characterized mTORC1 substrates, S6K1 (S6 kinase 1) and 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1), regulate mRNA translation at multiple levels. The autophagy-associated targets of mTORC1 include Ulk1 (unc-51–like kinase 1) and Atg13, both of which are essential for the initiation of autophagosome formation (Jung et al., 2010).
Contrary to our rapidly emerging knowledge of autophagy-regulating kinases, current understanding of phosphatases involved in this process is very limited. Thus, we screened a human phosphatome siRNA library for regulators of autophagosome accumulation in human MCF7 breast carcinoma cells expressing EGFP-tagged microtubule-associated protein light chain 3 (EGFP-LC3) as an autophagosomal marker. We identified 61 genes whose targeting increased the accumulation of EGFP-LC3–positive autophagosomes in optimal growth conditions and 17 genes whose targeting decreased the number of autophagosomes induced by siramesine, a putative anticancer agent that inhibits autophagosome turnover (Ostenfeld et al., 2008). Bioinformatics analyses of the candidate genes resulted in the prioritization of four PP2A (protein phosphatase 2A)-related genes as leading hits for further investigation. Biochemical and cell biological analyses of the regulation of autophagy and mTORC1 signaling by these proteins identified PP2A regulatory subunit 3A (PPP2R3A or PR72/130) as an mTORC1-independent activator of autophagy and CIP2A (cancerous inhibitor of PP2A) as an mTORC1-associated allosteric inhibitor of mTORC1-associated PP2A activity and potent inhibitor of autophagy. Moreover, we observed that the inhibition of mTORC1 activity led to a rapid and selective autophagic degradation of CIP2A and disappearance of Myc, an oncoprotein whose PP2A-mediated dephosphorylation and degradation are inhibited by CIP2A (Junttila et al., 2007). Because CIP2A promotes tumorigenesis and associates with cancer progression (Côme et al., 2009), we tested whether CIP2A levels correlated with mTORC1 activity in primary human breast cancer. Tissue microarray (TMA) analysis of 210 cancer samples revealed a highly significant, positive correlation between CIP2A expression and phosphorylation of mTORC1 substrate S6K1. Collectively, these data uncovered enhanced mTORC1 activity as a novel mechanism by which CIP2A can promote tumor growth and the subsequent inhibition of autophagy as a positive feedback loop that stabilizes CIP2A and c-Myc, thereby further enhancing cell proliferation and tumor progression.
Results
Identification of autophagy-regulating phosphatases by siRNA screens
To identify phosphatases that either enhance or inhibit autophagy, we performed two parallel siRNA screens. In screen 1, we looked for phosphatases required for repression of constitutive autophagy and, in screen 2, for those required for autophagy induction. For this purpose, we used an siRNA library targeting the human phosphatome and a cell-based imaging assay in MCF7-EGFP-LC3 breast carcinoma cells. Applying a scoring system and criteria outlined in Table 1 and Fig. S1, we identified 61 candidate phosphatases whose depletion increased the number of autophagic vesicles (EGFP-LC3 puncta) in untreated cells and 17 phosphatases that when depleted decreased EGFP-LC3 puncta formation in siramesine-treated cells (Table 1). Carbohydrate, acid, and dual-specificity phosphatases were enriched among the candidates identified in screen 1, whereas serine/threonine protein phosphatases and their modulators as well as dual-specificity phosphatases were overrepresented in the candidate list from screen 2 (Fig. 1 A). Because very little is known about the phosphatase-mediated regulation of autophagy, it is not surprising that most of the candidate genes identified in our screens have not been previously implicated in autophagy. Nonetheless, the well-defined general autophagy-regulating phosphatases, PTEN (phosphatase and tensin homologue; Arico et al., 2001) and MTMR14 (myotubularin related protein 14)/Jumpy (Vergne et al., 2009), were among the candidates, thereby validating the approach. Our candidate list contained also four subunits of the PP2A complex, whose phosphatase activity has been reported to regulate autophagy by several mechanisms (Yorimitsu et al., 2009; Bánréti et al., 2012; Iovino et al., 2012), and one (CIP2A) that was recently shown to inhibit bortezomib-induced autophagy (Yu et al., 2013).
Table 1.
Phosphatase genes identified as putative regulators of autophagy
| Symbol | Family | Gene full name | ID | Score |
| Screen 1 | ||||
| ACP6 | AP | Acid phosphatase 6, lysophosphatidic | 51205 | 3.47 |
| PTPRH | PTP | Protein tyrosine phosphatase, receptor type, H | 5794 | 3.47 |
| PPAPDC1A | Other LP | Phosphatidic acid phosphatase type 2 domain-containing 1A | 196051 | 3.33 |
| ENTPD3 | NP | Ectonucleoside triphosphate diphosphohydrolase 3 | 956 | 3.30 |
| G6PC3 | CP | Glucose 6 phosphatase, catalytic, 3 | 92579 | 3.30 |
| ACP5 | AP | Acid phosphatase 5, tartrate resistant | 54 | 3.13 |
| DUSP21 | DUSP | Dual-specificity phosphatase 21 | 63904 | 3.08 |
| KIAA1524 | PPP-M | CIP2A | 57650 | 3.05 |
| HINT2 | NP | Histidine triad nucleotide binding protein 2 | 84681 | 2.97 |
| PPP1R14A | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit 14A | 94274 | 2.97 |
| PTPN18 | PTP | Protein tyrosine phosphatase, nonreceptor type 18 (brain derived) | 26469 | 2.97 |
| PTPN3 | PTP | Protein tyrosine phosphatase, nonreceptor type 3 | 5774 | 2.97 |
| MTMR14 | PIP | Myotubularin-related protein 14, Jumpy, FLJ22405 | 64419 | 2.88 |
| LPPR2 | Other LP | Lipid phosphate phosphatase-related protein type 2 | 64748 | 2.88 |
| PIP3AP | PIP | Phosphatidylinositol-3-phosphate–associated protein; PIP3AP | 378766 | 2.88 |
| SSH3 | DUSP | Slingshot homologue 3 (Drosophila melanogaster) | 54961 | 2.88 |
| DUSP16 | DUSP | Dual-specificity phosphatase 16 | 80824 | 2.80 |
| NUDT10 | PP | Nudix (nucleoside diphosphate–linked moiety X)-type motif 10 | 170685 | 2.80 |
| NUDT11 | PP | Nudix (nucleoside diphosphate–linked moiety X)-type motif 11 | 55190 | 2.80 |
| PPP1CA | PPP | Protein phosphatase 1, catalytic subunit, α isoform | 5499 | 2.80 |
| PPP1R1B | PPP | Protein phosphatase 1, regulatory (inhibitor) subunit 1B; DARPP-32 | 84152 | 2.80 |
| PPP1R3F | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit 3F | 89801 | 2.80 |
| ANP32C | Other M | Acidic (leucine rich) nuclear phosphoprotein 32 family, member C | 23520 | 2.72 |
| CHP | Other M | Calcium binding protein P22 | 11261 | 2.72 |
| DUSP9 | DUSP | Dual-specificity phosphatase 9 | 1852 | 2.72 |
| PPM1J | PPP | Protein phosphatase, Mg2+/Mn2+ dependent, 1J | 333926 | 2.72 |
| PPP1R16B | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit | 26051 | 2.72 |
| PTPRN | PTP | Protein tyrosine phosphatase, receptor type, N | 5798 | 2.72 |
| CHTF18 | NP | Chromosome transmission fidelity factor 18 homologue | 63922 | 2.63 |
| DUSP4 | DUSP | Dual-specificity phosphatase 4 | 1846 | 2.63 |
| NT5C2 | NP | 5-Nucleotidase, cytosolic II | 22978 | 2.63 |
| PPP1R14B | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit 14B | 26472 | 2.63 |
| PPP1R1A | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit 1A | 5502 | 2.63 |
| PPP3R1 | PPP-M | Protein phosphatase 3, regulatory subunit B, α | 5534 | 2.63 |
| SGPP1 | Other P | Sphingosine-1-phosphate phosphatase 1 | 81537 | 2.63 |
| ACPP | AP | Acid phosphatase, prostate | 55 | 2.55 |
| ATP6V0E2 | Other P | ATPase, H+ transporting V0 subunit e2 | 155066 | 2.55 |
| PTPN20B | PTP | Protein tyrosine phosphatase, nonreceptor type 20B | 26095 | 2.55 |
| ILKAP | PPP | Integrin-linked kinase–associated serine/threonine phosphatase 2C | 67444 | 2.55 |
| PALD | Other P | Paladin; KIAA1274 | 27143 | 2.55 |
| PPP3CB | PPP | Protein phosphatase 3, catalytic subunit, β isozyme | 19056 | 2.55 |
| PPP3R2 | PPP-M | Protein phosphatase 3, regulatory subunit B, β | 5535 | 2.55 |
| PPP2R3B | PPP-M | Protein phosphatase 2, regulatory subunit B, β | 28227 | 2.55 |
| ALPP | AlkP | Alkaline phosphatase, placental | 250 | 2.47 |
| DUSP14 | DUSP | Dual-specificity phosphatase 14 | 11072 | 2.47 |
| DUSP2 | DUSP | Dual-specificity phosphatase 2 | 1844 | 2.47 |
| DUSP22 | DUSP | Dual-specificity phosphatase 22 | 56940 | 2.47 |
| G6PC | CP | Glucose-6-phosphatase, catalytic subunit | 2538 | 2.47 |
| PHACTR4 | PPP-M | Phosphatase and actin regulator 4 | 65979 | 2.47 |
| PPM1A | PPP | Protein phosphatase, Mg2+/Mn2+ dependent, 1A | 5494 | 2.47 |
| PPP1R14D | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit 14D | 54866 | 2.47 |
| TPTE2 | PIP | Transmembrane phosphoinositide 3-phosphatase and tensin homologue 2 | 93492 | 2.47 |
| ATP6V0E | Other P | ATPase, H+ transporting, lysosomal 9 kD, V0 subunit e1 | 8992 | 2.38 |
| CDC14A | DUSP | CDC14 cell division cycle 14 homologue A (Saccharomyces cerevisiae) | 8556 | 2.38 |
| NAP1L2 | Other | Nucleosome assembly protein 1–like 2 | 4674 | 2.38 |
| OCRL | Other | Oculocerebrorenal syndrome of Lowe | 4952 | 2.38 |
| PFKFB1 | CP | 6-Phosphofructo-2-kinase/fructose-2, 6-biphosphatase 1 | 5207 | 2.38 |
| PPP4R1 | PPP-M | Serine/threonine protein phosphatase 4 regulatory subunit 1 | 9989 | 2.30 |
| G6PC2 | CP | Glucose-6-phosphatase, catalytic, 2 | 57818 | 2.22 |
| NUDT14 | PP | Nudix (nucleoside diphosphate–linked moiety X)-type motif 14 | 256281 | 2.22 |
| PNKP | NP | Polynucleotide kinase 3-phosphatase | 11284 | 2.22 |
| Screen 2 | ||||
| PPP2R1A | PPP-M | Protein phosphatase 2, regulatory subunit A, α | 5518 | 2.50 |
| PPM1G | PPP | Protein phosphatase, Mg2+/Mn2+ dependent, 1G | 5496 | 2.50 |
| PPP1R12A | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit 12A | 4659 | 2.50 |
| PPP1R3A | PPP-M | Protein phosphatase 1, regulatory (inhibitor) subunit 3A | 5506 | 3.00 |
| PPP2R3A | PPP-M | Protein phosphatase 2, regulatory subunit B, α | 5523 | 3.00 |
| PTPRJ | PTP | Protein tyrosine phosphatase, receptor type, J | 5795 | 3.00 |
| MTM1 | PIP | Myotubularin 1 | 4534 | 3.00 |
| ANP32B | Other M | Acidic (leucine rich) nuclear phosphoprotein 32 family, member B | 10541 | 3.00 |
| DUSP5 | DUSP | Dual-specificity phosphatase 5 | 1847 | 3.25 |
| MTMR6 | PIP | Myotubularin-related protein 6 | 9107 | 3.25 |
| PPP1CC | PPP | Protein phosphatase 1, catalytic subunit, γ isoform | 5501 | 3.25 |
| PTPRE | PTP | Protein tyrosine phosphatase, receptor type, E | 5791 | 3.25 |
| PTEN | PIP | Phosphatase and tensin homologue | 5728 | 3.25 |
| G6PC2 | CP | Glucose-6-phosphatase, catalytic, 2 | 57818 | 3.33 |
| CTDSP1 | Other P | CTD (C-terminal domain, RNA polymerase II, polypeptide A) small phosphatase 1 | 58190 | 3.33 |
| PSPH | PPP | Phosphoserine phosphatase | 5723 | 3.33 |
| SGPP2 | Other LP | Sphingosine-1-phosphate phosphatase 2 | 130367 | 3.50 |
MCF7-EGFP-LC3 breast carcinoma cells were transfected with a human phosphatome siRNA library targeting 318 genes. After 56 h in normal culture conditions (screen 1) or after treatment for additional 3 h with siramesine (screen 2), the cells were scored for EGFP-LC3 puncta accumulation on the scale from 0 to 5, where control siRNA scored 1 and siramesine scored 5 (Fig. S1). The candidates were requested to reach a score ≥2 in screen 1 and a score ≤4 in screen 2 with a minimum of 2/3 siRNAs in three independent screens. The score values represent means of three independent screens performed with two independent siRNAs. P < 0.0001 in all cases when compared to control siRNA (screen 1) or siramesine (screen 2). AlkP, alkaline phosphatase; AP, acid phosphatase; CP, carbohydrate phosphatase; DUSP, dual-specificity phosphatase; LP, lipid phosphatase; M, modulator; NP, nucleotide phosphatase; P, phosphatase; PIP, phosphatidylinositol phosphatase; PPP, phosphoprotein phosphatase; PTP, phosphotyrosine phosphatase.
Figure 1.

Bioinformatics analysis of candidate autophagy-regulating genes and identification of PP2A as a complex regulator of autophagy. (A) All analyzed 318 siRNA target genes were categorized into phosphatase families and marked according to the ability of their corresponding siRNAs to regulate EGFP-LC3 accumulation. PIP, phosphatidylinositol phosphatase; PTP, phosphotyrosine phosphatase; DUSP, dual-specificity phosphatase; PPP, (Ser/Thr) phosphoprotein phosphatase. (B) Canonical pathways with significant enrichment of candidate genes were identified by hypergeometric method using the Ingenuity Pathway Analysis software. Blue bars indicate the −log of the p-values (Fisher’s exact test) of the enrichment (left vertical axis). The orange line indicates the percentage of the identified autophagy-regulating genes of all genes in the respective pathway (right vertical axis). The threshold line indicates the position of the P = 0.05. See Table S1 for genes associated with these pathways. AMPK, AMP-activated protein kinase; ERK, extracellular signal–regulated kinase; ROS, reactive oxygen species; ILK, integrin-linked kinase; iCOS, inducible T cell co-stimulator. (C) Schematic representation of the PP2A holoenzyme that is composed of a catalytic subunit (PP2Ac), a scaffold protein (PR65), and a variable regulatory B subunit that interacts with the complex via PR65-α/β HEAT domains (Huntington/elongation/A subunit/TOR). CIP2A interacts with PP2A holoenzyme and modulates its activity toward specific substrates. The gene names for the identified autophagy-regulating candidates and the corresponding proteins (bold) are indicated. (D) Quantification of EGFP-LC3 puncta in MCF7-EGFP-LC3 cells 56 h after transfection with the indicated siRNAs. When indicated, cells were exposed to 100 nM rapamycin for the last 3 h. RPTOR (raptor) and BECN1 (beclin1) siRNAs served as positive and negative controls, respectively. (E) Autophagic flux was measured as the ratio between luciferase activities in MCF7-RLuc-LC3WT and MCF7-RLuc-LC3G120A cells transfected with the indicated siRNAs 56 h earlier and left untreated or treated with 100 nM rapamycin for the last 3 h. Error bars are SDs for a representative (n = 5) triplicate experiment with a minimum of 4 × 10 randomly chosen areas/sample analyzed (D) or three independent experiments (F). *, P < 0.05; **, P < 0.01; ***, P < 0.001, as compared with similarly treated control siRNA-transfected samples.
To identify canonical signaling pathways showing significant enrichment of the identified candidate genes, we examined these by Ingenuity Pathway Analysis software. Many of the pathways identified in this manner (Fig. 1 B), such as insulin receptor, Akt, AMPK, and mitogen-activated protein kinase pathways, are known potent regulators of autophagy (Behrends et al., 2010). The obverse analysis examining known autophagy-controlling pathways (illustrated in Fig. S2 for insulin receptor signaling) disclosed protein phosphatases PP1 and PP2A as key regulators of these pathways.
CIP2A and PPP2R3A have opposite effects on autophagy
Prompted by the multiple PP2A hits identified in our screens and PP2A’s prominent role in the control of major autophagy-regulating signaling cascades, including the Akt–mTORC1 signaling axis (Westermarck and Hahn, 2008; Bononi et al., 2011), we focused our subsequent experiments on this abundant Ser/Thr protein phosphatase. PP2A exists in two general forms, a heterodimer core enzyme and a heterotrimeric holoenzyme composed of catalytic (PP2A catalytic subunit [PP2Ac]), scaffold (PP2Aa/PR65), and variable regulatory B subunits that generate numerous distinct PP2A holoenzymes with spatially and temporally determined specific functions (Fig. 1 C). PP2A-related candidates whose depletion induced spontaneous accumulation of EGFP-LC3 puncta included regulatory subunit 3B (PPP2R3B or PR48) and PP2A inhibitor CIP2A (encoded by the KIAA1524 gene; hereafter referred to as CIP2A), whereas siRNAs targeting scaffolding subunit PR65-α (PPP2R1A) and regulatory subunit 3A (PPP2R3A or PR72/130) inhibited siramesine-induced EGFP-LC3 puncta accumulation (Table 1). Importantly, these siRNAs down-regulated their cognate targets without significantly compromising cell viability (Fig. S3, A and B), which could otherwise constitute a confounding factor for our analysis.
Consistent with their identification in screen 1, PPP2R3B and CIP2A RNAi increased the number of EGFP-LC3 puncta, which in the case of PPP2R3B could be further enhanced by inhibition of mTORC1 signaling by rapamycin (Fig. 1 D). Because augmented amounts of EGFP-LC3 puncta can be caused by either increased formation or reduced turnover of autophagosomes, we monitored the effect of the candidate siRNAs on autophagic flux using a luciferase reporter–based assay for LC3 turnover. Down-regulation of CIP2A expression strongly stimulated LC3 turnover, which could be only marginally enhanced by rapamycin (Fig. 1 E). In this respect, the behavior of CIP2A siRNA resembled that of inactivation of mTORC1 by depletion of raptor (Fig. 1, D and E). Conversely, PPP2R3B siRNA, which was a strong inducer of EGFP-LC3 puncta (Fig. 1 D), affected neither constitutive nor rapamycin-induced autophagic flux (Fig. 1 E).
RNAi of PPP2R1A and PPP2R3A, which inhibited siramesine-induced EGFP-LC3 puncta formation (Table 1), reduced also rapamycin-induced EGFP-LC3 puncta formation significantly and showed a tendency to inhibit rapamycin-induced LC3 turnover (Fig. 1, D and E). The autophagy inhibitory effect of PPP2R3A siRNA was indistinguishable from that of depletion of an essential autophagy protein beclin1, whereas the effect of PPP2R1A siRNA was clearly weaker and it increased EGFP-LC3 puncta in normal growth conditions (Fig. 1, D and E). These data identify CIP2A as a potent inhibitor and regulatory subunit 3A as a mediator of autophagic flux, whereas the autophagy-regulating roles of scaffolding subunit PR65-α and regulatory subunit 3B appear more complex.
CIP2A regulates autophagy in an mTORC1-dependent manner
To elucidate the molecular basis by which the identified PP2A-regulating proteins control autophagy, we monitored the phosphorylation status of T389-S6K1, a known PP2A target in the autophagy-regulating mTORC1 pathway (Bononi et al., 2011). CIP2A siRNA induced a significant reduction in the level of P-T389-S6K1, whose basal level and amino acid starvation-induced reduction were unaffected by siRNAs targeting PPP2R1A, PPP2R3A, or PPP2R3B siRNAs (Figs. 2 A and S3 C). CIP2A depletion had an even more pronounced inhibitory effect on phosphorylation of another mTORC1 substrate, 4E-BP1, and T421/S424 sites in S6K1 (Fig. 2, A and B). A similar requirement of CIP2A for mTORC1 signaling was observed in U-2-OS osteosarcoma cells (Fig. S3 C). Overall, these data indicate that CIP2A suppresses autophagy by enhancing mTORC1 signaling. This conclusion was compellingly endorsed by the ability of siRNA-resistant CIP2Ar cDNA to suppress dephosphorylation of mTORC1 targets and EGFP-LC3 puncta accumulation upon CIP2 depletion (Fig. 2, B and C). Contrary to CIP2A, the three autophagy-regulating PP2A subunits identified in our screen appear to regulate autophagy in an mTORC1-independent manner.
Figure 2.

CIP2A regulates the mTORC1 pathway in a c-Myc– and Akt-independent manner. (A) Representative immunoblots of indicated proteins from whole-cell lysates of MCF7 cells transfected with indicated siRNAs for 56 h and subjected to 0–40 min of amino acid starvation [−AA]). (B) Representative immunoblots of the indicated proteins from whole-cell lysates of MCF7-EGFP-LC3 cells stably infected with control or CIP2Ar (siRNA-resistant CIP2A)-encoding lentivirus, transfected with the indicated siRNAs for 53 h, and treated with 100 nM rapamycin (Rapa) for 0–20 min. (C) MCF7-EGFP-LC3 cells stably infected with control or CIP2Ar-encoding lentivirus were transfected with indicated siRNAs for 48 h and analyzed for EGFP-LC3 puncta formation. (D) MCF7-EGFP-LC3 cells transfected with control (−) and CIP2A siRNAs together with an empty vector (−) or MYC-S62A cDNA as indicated were analyzed 56 h later for EGFP-LC3 puncta (left) and expression of indicated proteins (right). (E) Representative immunoblots (left) of indicated proteins from whole-cell lysates of MCF7 cells treated with the control (C), RPTOR (RP), or CIP2A (CIP) siRNAs for 56 h and densitometric quantification of three independent experiments (right). Error bars show SDs for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared with Student’s t test as indicated (C and D) or with control siRNA-transfected samples (E). Black lines indicate that intervening lanes have been spliced out.
CIP2A regulates autophagy and mTORC1 activity independent of c-Myc and Akt
Prompted by the demonstrated robust regulation of autophagy and mTORC1 activity by CIP2A as well as CIP2A’s reported relevance in breast cancer (Côme et al., 2009), we turned our focus on this candidate. First, we tested whether the reported ability of CIP2A to stabilize c-Myc contributed to the altered mTORC1 activity and autophagy observed here (Junttila et al., 2007). For this purpose, we took advantage of a phosphosilencing c-Myc–S62A mutant that is unresponsive to CIP2A-mediated stabilization (Junttila et al., 2007). Transient expression of this mutant in MCF7-EGFP-LC3 cells affected neither autophagy nor mTORC1 inhibition induced by CIP2A siRNA (Fig. 2 D). Thus, CIP2A-mediated regulation of mTORC1 activity and autophagy are independent of its effects on cellular c-Myc.
CIP2A has also been reported to suppress Akt-associated PP2A in hepatocellular carcinoma cells (Chen et al., 2010). Thus, we addressed the effect of CIP2A on the Akt–mTORC1 signaling axis in MCF7 cells by comparing the activation of this pathway in cells transfected with control, RPTOR, and CIP2A siRNAs. Whereas both RPTOR and CIP2A siRNAs effectively inhibited the phosphorylation of mTORC1 targets (T389-S6K1 and T37/41-4E-BP1), neither siRNA had a significant effect on the phosphorylation status of Akt (S473 or P-308) or an Akt target (T1462-Tsc2) that regulates mTORC1 activity (Fig. 2 E; Tee et al., 2003). Thus, CIP2A is likely to inhibit PP2A at the level of mTORC1 or its substrates rather than at the level of Akt in MCF7 cells. Accordingly, amino acid starvation exhausted the mTORC1 pathway activity clearly faster in CIP2A-depleted cells than in control cells (Fig. 2 A). Similar Akt-independent regulation of mTORC1 was observed in U-2-OS cells (Fig. S3 C).
mTORC1 regulates autophagy via an inhibitory phosphorylation of unc-51–like kinase, which in turn phosphorylates S318 in an autophagy-regulating Atg13 protein (Nazio et al., 2013). In concordance with the ability of RPTOR and CIP2A siRNAs to inhibit mTORC1 activity and induce autophagy, both siRNAs significantly reduced the phosphorylation of S318-Atg13 (Fig. 2 E). Probably reflecting a difference in their mechanisms of action at the level of mTORC1, raptor depletion reduced the phosphorylation of mTOR at S2448 significantly, whereas CIP2A depletion had only a mild inhibitory effect on this phosphorylation (Fig. 2 E).
CIP2A regulates cell size, proliferation, and morphology
Having established CIP2A as a positive regulator of mTORC1 activity and inhibitor of autophagy, we investigated the effect of CIP2A on other mTORC1-controlled cellular processes. For this purpose, we created MCF7-EGFP-LC3 cells stably depleted for CIP2A by lentiviral expression of CIP2A shRNA. Consistent with increased autophagic flux, stably depleted cells had lower levels of p62/SQSTM1 (p62/sequestosome-1), a well-established autophagy substrate, and an over sixfold increase in EGFP-LC3 puncta as compared with control shRNA-transduced cells (Fig. 3 A). Supporting the hypothesis that CIP2A acts at the level of mTORC1, CIP2A-depleted cells were significantly smaller in size than control cells (Fig. 3 B). Moreover, CIP2A depletion resulted in a strong reduction in cell proliferation in untreated cells but not in cells in which mTORC1 was inhibited by rapamycin (Fig. 3 C). In line with its autophagy-promoting activity, CIP2A depletion sensitized MCF7 cells to inhibitors of lysosomal (concanamycin A [ConA]) and proteasomal (MG132) function but protected them against lysosomotropic siramesine and DNA-damaging etoposide treatments (Fig. 3 D).
Figure 3.

CIP2A regulates cell size, proliferation, and survival. (A, top left) Quantification of EGFP-LC3 puncta in MCF7-EGFP-LC3 cells infected with control or CIP2A lentiviral shRNAs. Representative immunofluorescence images (right) and immunoblots (bottom) are shown. (B) Mean cell diameters of MCF7-EGFP-LC3 cells infected as in A were measured with a cytometer (NucleoCounter NC-3000; Chemometec AS) equipped with Via1-Cassettes (Chemometec AS). (C) MCF7 cells infected with shRNAs as in A were plated in equal density and left untreated or treated with 100 nM rapamycin (Rapa) for 72 h. The viable cells were either visualized after fixation and crystal violet staining (left; representative figure, n = 4) or trypsinized and counted with NucleoCounter NC-3000 equipped with Via1-Cassettes (right). (D) MCF7 cells infected with shRNAs as in A were left untreated or treated with 2 nM ConA, 2 µM MG132, 5 µM siramesine (Sira), or 100 µM etoposide (Etop) for 48 h, and the metabolic activity (cell density) was measured by the MTT assay (left), and cell death was measured by the LDH assay (right). (E) Light microscopic pictures of MCF10A cells at passage 8 after infection with control or CIP2A-encoding lentivirus. (F) Representative immunoblots of the indicated proteins from whole-cell lysates of MCF10A (left) and HEK-293 (right) cells infected as in E and treated with 100 nM rapamycin for the indicated times. Black lines indicate that intervening lanes have been spliced out. Error bars are SDs for three independent experiments (B) or a representative (n ≥ 3) triplicate experiment (C and D). *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared by Student’s t test with similarly treated control shRNA-transfected cells or as indicated. Bars: (A and E) 20 µm; (C) 1 cm.
To mimic the cancer-associated increase in CIP2A expression, we transduced nonmalignant MCF10A mammary epithelial cells, which have negligible levels of endogenous CIP2A protein, with a CIP2A-encoding lentiviral vector. Ectopic CIP2A expression changed the morphology of MCF10A cells from a flat and evenly distributed monolayer to cells with numerous protrusions, an appearance characteristic of transformed cells (Fig. 3 E). Supporting its role in mTORC1 complex, CIP2A increased S6K1 phosphorylation and spontaneously increased S6K1 phosphorylation in MCF10A and HEK-293 kidney cells (Fig. 3 F). Thus, ectopic expression of CIP2A is sufficient to increase mTORC1 activity in nonmalignant cells.
CIP2A expression correlates with mTORC1 activity in primary breast cancer
To determine whether CIP2A-mediated regulation of mTORC1 could be germane to CIP2A’s role as an oncoprotein, we examined a tandem set of TMA slides comprising 33 normal breast and 210 breast cancer tissue samples in duplicates for CIP2A expression and P-T389-S6K1 immunoreactivity by immunohistochemistry. CIP2A protein expression showed a highly significant (P < 0.0001) positive correlation with P-T389-S6K1 (Fig. 4). To substantiate the validity of this correlation, we included in the analysis three additional breast cancer–related proteins with no expected association with CIP2A, i.e., ErbB2 receptor tyrosine kinase (Hynes and Lane, 2005), cathepsin B (Rafn et al., 2012), and γ-glutamylcyclotransferase (Gromov et al., 2010). No significant correlations were found between the expression of these proteins and CIP2A (Fig. 4).
Figure 4.

CIP2A expression correlates with mTORC1 activity in primary human breast tumors. TMAs including 33 normal breast tissues and 210 primary breast carcinoma samples were analyzed by semiquantitative immunohistochemistry for the expression of the indicated proteins and mean scores (arbitrary units [a.u.]) for duplicate samples were averaged and plotted according to mean CIP2A expression (left). The red lines are the regression lines, and the stippled red lines define the 95% confidence intervals. Statistical analysis of data was performed using NCSS 2007 and Prism version 5. Examples of the staining of breast cancer (a) and normal (b) cores indicated in the P-T389-S6K1/CIP2A plot are shown on the right. rsp, Spearman’s correlation coefficient.
CIP2A associates with the mTORC1 complex
To characterize the molecular mechanism underlying CIP2A-dependent regulation of mTORC1 activity, we investigated whether this regulatory effect was caused by direct physical association between CIP2A and mTORC1. First, we tested the ability of in vitro translated full-length CIP2A to bind mTORC1 complexes immunoprecipitated with anti-HA antibodies from MCF7 cells expressing ectopic HA-raptor or HA-S6K1. CIP2A associated relatively strongly with the HA-raptor complex and weakly with the HA-S6K1 complex (Fig. 5, A and B). As a result of the presence of PP2A in reticulocyte lysates used for in vitro translation of CIP2A, we were not able to definitely conclude whether these associations required PP2A.
Figure 5.

Dynamic interaction of the CIP2A, PP2A, and mTORC1 complex regulates mTORC1 activity. (A and B) In vitro translated full-length CIP2A was incubated with purified human PP2A complex and HA-Raptor (A) or HA-S6K1 (B) purified from transiently transfected MCF7 cells. Protein complexes were immunoprecipitated with anti-HA antibody and analyzed by immunoblotting as indicated. Nonspecific IgG served as a control. Note that human reticulocyte (RC) lysate used for in vitro translation of CIP2A contained detectable amounts of PP2Ac. (C) Endogenous CIP2A protein complexes immunoprecipitated from MCF7 cells were analyzed by immunoblotting and PP2A activity assay. Immunoblots of corresponding cell lysates are shown on the right. (D) Endogenous raptor protein complexes immunoprecipitated from MCF7 cells stably infected with control (Ctr) or CIP2A shRNA lentiviruses (Fig. 3 A) and starved for amino acids for 20 min when indicated were analyzed by immunoblotting and PP2A activity assay. Immunoblots of corresponding cell lysates are shown on the right. (E) Raptor protein complexes immunoprecipitated from MCF7 cells treated with control (C) or PPP2R1A (R1A) siRNAs for 56 h were analyzed by immunoblotting with the indicated antibodies. Immunoblots of corresponding cell lysates are shown on the left. (F) HA-S6K1 protein complexes immunoprecipitated from MCF7 cells stably infected with control or CIP2A shRNA lentiviruses (Fig. 3 A), transiently transfected with HA-RPS6K1 cDNA and starved for amino acids for 20 min when indicated, were analyzed by immunoblotting. Immunoblots of corresponding cell lysates are shown on the right. (G) Enzymatic activity of a purified PP2A holoenzyme incubated with human reticulocyte lysate with or without in vitro translated full-length CIP2A. Error bars are SDs for four (D) independent experiments or a representative triplicate experiment (C and G). (H) Representative confocal images of untreated and amino acid–starved (4 h) MCF7 cells stained for endogenous CIP2A and raptor. Black lines indicate that intervening lanes have been spliced out. −AA, amino acid starvation; IP, immunoprecipitation; a.u., arbitrary unit. Bar, 10 µm.
The significance of the observed in vitro association of CIP2A and mTORC1 was strongly supported by the ability of CIP2A and raptor antibodies to bring down endogenous raptor and CIP2A, respectively, from MCF7 cell lysates (Fig. 5, C and D). Depletion of PPP2R1A resulted in the dissociation of PP2A holoenzyme and CIP2A from the raptor complex, strongly suggesting PP2A-dependent association of CIP2A and mTORC1 (Fig. 5 E). Contrary to endogenous raptor, relatively large amounts of ectopic HA-S6K1 coprecipitated only minute amounts of CIP2A, supporting stronger association of CIP2A with mTORC1 than its substrates (Fig. 5 F).
CIP2A, raptor, and HA-S6K1 coimmunoprecipitated also endogenous PP2Ac and PP2A activity (Fig. 5, C and D; and not depicted). Notably, 20-min amino acid starvation, which leads to almost complete dephosphorylation of mTORC1 substrates (Fig. 2 A), was associated with only a modest increase in mTORC1-bound PP2A activity without a marked effect on the association of CIP2A and mTORC1 (Fig. 5, C and D). The existence of PP2A activity in CIP2A immunoprecipitates and the insensitivity of mTORC1-associated PP2A activity to CIP2A depletion (Fig. 5 C), suggested an allosteric regulatory role rather than direct PP2A inhibitory action for CIP2A. This hypothesis was further supported by the inability of recombinant CIP2A to influence phosphatase activity of purified PP2A holoenzyme in vitro (Fig. 5 G). Finally, we investigated whether CIP2A and raptor exist in the same cellular compartment and can thereby interact in living cells. Both proteins were abundant in the cytoplasm and formed small punctuate structures with partial colocalization (Fig. 5 H).
These data demonstrate for the first time that the CIP2A–PP2A complex associates with mTORC1. The strong CIP2A-mediated regulation of the phosphorylation status of mTORC1 targets (Figs. 2, A, B, and G; and 3 E) combined with its inability to inhibit PP2A activity toward a phosphopeptide with PP2A recognition sequence suggests that CIP2A functions as a context-specific allosteric inhibitor of PP2A activity in mTORC1 complex.
CIP2A protein stability is regulated by mTORC1, p62/SQSTM1, ubiquitination, and autophagy
Prolonged amino acid starvation and rapamycin treatment reduced protein levels of CIP2A as effectively as those of p62/SQSTM1, a well-established receptor for autophagic cargo and substrate for autophagic degradation (Fig. 6 A). Thus, we investigated the effect of mTORC1 activity and autophagy on CIP2A stability in further detail. Neither amino acid starvation nor rapamycin reduced CIP2A or p62 mRNA levels in a significant manner. (Fig. 6 B). Instead, inhibition of autophagy by depletion of an essential autophagy kinase Ulk1 effectively reverted CIP2A loss upon both treatments (Fig. 6 C). Consistent with autophagy-dependent degradation of CIP2A upon mTORC1 inhibition, inhibition of lysosomal degradation by ConA effectively inhibited rapamycin-induced loss of CIP2A, even when de novo protein synthesis was inhibited by cycloheximide (Fig. 6 D). Furthermore, immunocytochemistry of rapamycin-treated MCF7 cells revealed reduced CIP2A levels specifically in cells with numerous LC3 and p62/SQSTM1 puncta indicative of activated autophagy, and inhibition of autophagic degradation by ConA resulted in a partial colocalization of CIP2A with these puncta (Figs. 6 E and S4).
Figure 6.

CIP2A and c-Myc are degraded in an autophagy-dependent manner. (A) Representative immunoblots of the indicated proteins from whole-cell lysates of MCF7 cells subjected to 100 nM rapamycin or amino acid starvation for 0–24 h. (B) qPCR analysis of CIP2A and MYC mRNA levels in MCF7 cells treated as in A. AA-starv, amino acid starvation; a.u., arbitrary unit. Error bars show SDs for three independent experiments. (C) Representative immunoblots of the indicated proteins from whole-cell lysates of MCF7 cells treated with ULK1 or SQSTM1 siRNAs for 54 h before the exposure to fresh medium (0) or amino acid starvation (−AA) for 12 h. Ctr, control. (D) Representative immunoblots of the indicated proteins from whole-cell lysates of MCF7 cells treated for 0–5 h with the indicated combinations of 100 µM cycloheximide (CHX), 100 nM rapamycin, and 2 nM ConA. (E) Representative confocal images of MCF7 (left and middle) and MCF7-LC3-EGFP (right) cells left untreated or treated with 100 nM rapamycin for 4 h or 100 nM rapamycin and 2 nM ConA for 8 h, fixed, and stained with antibodies against CIP2A. Nuclei in MCF-LC3-EGFP cells were visualized with Hoechst. Green arrowheads indicate cells with many LC3-positive puncta and low CIP2A levels, and yellow arrowheads show cells in which LC3 and CIP2A colocalize. Bars, 10 µm. (F) Representative immunoblots of an endogenous CIP2A protein complex immunoprecipitated from lysates of MCF7 cells left untreated or treated for 4 h with 100 nM rapamycin (Rapa) and 2 nM ConA as indicated. Mouse IgG served as a negative control. (G) MCF7 cells were treated with 100 nM rapamycin, 2 nM ConA, and 2 µM MG132 for 24 h as indicated. Endogenous CIP2A was immunoprecipitated (IP) in stringent conditions (no coimmunoprecipitation of p62) and analyzed by immunoblotting using antibodies against CIP2A, monoubiquitin, and polyubiquitin. Short (<100 kD), medium (>150 kD), and long (100–130 kD) exposures are shown. (H) Proteins from MCF7 cell lysates were immunoprecipitated with the indicated antibodies in stringent conditions (see G) and analyzed by immunoblotting for CIP2A and monoubiquitin. (I) Lysates from MCF7 cells transfected with the indicated ubiquitin-HA constructs were subjected to anti-CIP2A immunopurification followed by immunoblotting with antibodies against CIP2A and HA. (J) Lysates from MCF7 cells transfected with the CIP2A-FLAG construct (Junttila et al., 2007) and the indicated ubiquitin-HA constructs were subjected to anti-FLAG immunopurification in denaturing conditions with 0.1% SDS followed by immunoblotting with the indicated antibodies. Black lines indicate that intervening lanes have been spliced out. Ub, ubiquitin; WT, wild type.
Having established autophagy-dependent degradation of CIP2A, we next examined whether p62/SQSTM1, which recruits ubiquitinated proteins and protein aggregates to autophagosomal degradation (Bjørkøy et al., 2005), was responsible for targeting CIP2A to autophagosomes. Supporting this hypothesis, p62/SQSTM1 depletion inhibited starvation- and rapamycin-induced loss of CIP2A as effectively as autophagy inhibition by ULK1 siRNA (Fig. 6 C), and endogenous p62/SQSTM1 associated with immunopurified endogenous CIP2A (Fig. 6 F). Immunopurification of CIP2A in stringent conditions, at which p62 was not coprecipitated, revealed abundant mono- and polyubiquitination, which was increased by the blockade of lysosomal degradation by ConA (Fig. 6, G and H). Also, a detectable amount of CIP2A was precipitated with a monoubiquitin antibody (Fig. 6 H). These data strongly suggest that CIP2A was modified by ubiquitin moieties in so far unknown lysine residues. Blockade of proteasomal degradation by MG132 resulted in a smaller but detectable increase in CIP2A ubiquitination (Fig. 6 G), which might reflect either MG132-induced autophagy or the ability of proteasome to degrade ubiquitinated CIP2A.
To investigate CIP2A ubiquitination in further detail, we expressed HA-tagged wild-type ubiquitin and its seven mutants (K6, K11, K27, K29, K33, K48, and K63), in which all but the indicated lysine are mutated to arginine, in MCF7 cells (Lim et al., 2005). Immunoblot analysis of endogenous CIP2A immunopurified from these cells revealed that CIP2A was modified by different types of ubiquitins, lysine 6–, 11–, and 48–linked modifications being most abundant in repeated experiments (Fig. 6 I). This conclusion was further supported by identification of wild-type and K6 ubiquitin modifications and their rapamycin-induced increase in ectopically expressed CIP2A after immunoprecipitation in denaturating conditions (Fig. 6 J). These data suggest that CIP2A is a target of mTORC1-regulated ubiquitin modifications whose precise nature remains to be defined and that ubiquitination may regulate the p62/SQSTM1-dependent autophagic degradation of CIP2A.
CIP2A coordinates mTORC1 activity and c-Myc stability to promote cell proliferation
c-Myc oncoprotein stimulates cell division by regulating numerous genes that control cellular metabolism, and its activity is essential for translating the effects of mTORC1 pathway on growth of the cell mass to cell proliferation (DeBerardinis et al., 2008). However, little is known about the molecular mechanisms that coordinate the activities of these two growth-promoting proteins with supplemental functions. Because CIP2A enhances the activity of mTORC1 and stabilizes c-Myc (Fig. 3 F), we speculated that the control of CIP2A stability by mTORC1 could provide such a link. Accordingly, the loss of CIP2A upon mTORC1 inhibition by rapamycin or amino acid starvation was accompanied by an apparent reduction in c-Myc protein levels (Fig. 6 A), which at least in the case of rapamycin treatment could not be explained by changes in MYC mRNA levels (Fig. 6 B). Instead, inhibition of autophagy by ULK1 siRNA effectively increased c-Myc expression and inhibited its starvation- and rapamycin-induced loss (Fig. 6 C and not depicted). The loss of c-Myc protein upon mTORC1 inhibition may thus be mediated indirectly via autophagic degradation of CIP2A and subsequent dephosphorylation-induced proteasomal degradation of c-Myc.
Discussion
Phosphatase-mediated regulation of autophagy
Here, we report a global view of phosphatase-mediated regulation of autophagy. Using an imaging-based siRNA screen based on the formation of EGFP-LC3 puncta (autophagic vesicles) in MCF7 cells, we identified 61 and 17 phosphatase-related genes as putative inhibitors or inducers of autophagy, respectively. This high number that corresponds to almost 20% of all targeted genes could be partially explained by the fact that the human genome contains a considerably smaller number of phosphatases than kinases. Because appropriate regulation of cellular signaling networks depends on a delicate balance between kinase and phosphatase activities, dysregulation of individual phosphatases is likely to have much more frequent effects on cellular functions, especially on autophagy, which is swiftly induced even by minor disturbances in cellular homeostasis. In support of our screen approach, the few well-established general autophagy-regulating phosphatases PTEN (Arico et al., 2001), MTMR14 (Vergne et al., 2009), and PP2A (Yorimitsu et al., 2009) as well as phosphatases recently identified as autophagy regulators in specific contexts, e.g., sphingosine-1-phosphate phosphatase 1 (Lépine et al., 2011) and calcineurin (Dwivedi et al., 2009), were identified as autophagy regulators here. Moreover, the increased accumulation of autophagosomes upon depletion of glucose-6-phosphatase catalytic subunits G6PC, G6PC2, and G6PC3 and vacuolar H+-ATPase subunits ATP6V0E and ATP6V0E2 (all identified in screen 1) could have been predicted based on their essential functions in glucose homeostasis and acidification of lysosomes, respectively. And finally, the bioinformatics analysis revealed a significant enrichment of the candidate genes in major signaling pathways known to regulate autophagy. Even though the hits were chosen with stringent criteria, we cannot exclude the inclusion of false positives caused by off-target effects of the siRNAs. The list of putative autophagy-regulating phosphatases presented here can, however, serve as a valuable resource for researchers working on autophagy signaling, provided that the hits are considered as real autophagy modulators only after proper validation.
CIP2A regulates mTORC1 pathway and autophagy at the level of mTORC1
We focused our further analysis on CIP2A because it has recently emerged as a promising target for cancer therapy. Until now, the oncogenic potential of CIP2A has been attributed to its ability to inhibit PP2A-mediated dephosphorylation of c-Myc oncoprotein and Akt kinase (Junttila et al., 2007; Chen et al., 2010). Our data provide several lines of evidence that point to an additional oncogenic function of CIP2A via direct regulation of the mTORC1 pathway and autophagy. First, CIP2A depletion in MCF7 and U-2-OS cells induced dephosphorylation of well-defined mTORC1 target sites in mTORC1 substrates, without significant changes in major phosphorylation sites in its upstream effectors including Akt. Second, CIP2A depletion in MCF7 cells triggered a massive autophagy response that could not be significantly enhanced by pharmacological inhibition of mTORC1. Third, the effect of CIP2A siRNA on mTORC1 substrate phosphorylation and autophagy could be effectively reverted by ectopic expression of CIP2A. Fourth, ectopic expression of CIP2A in MCF10A and HEK-293 cells enhanced mTORC1 substrate phosphorylation. And finally, the relatively strong physical associations of endogenous and recombinant CIP2A proteins with endogenous mTORC1 provided strong molecular evidence that CIP2A functions in the mTORC1 complex.
CIP2A has been identified as a selective inhibitor of PP2A-mediated dephosphorylation of two specific substrates, but its mechanism of action as a PP2A inhibitor has remained somewhat elusive, and it has never been challenged by biochemical assays using recombinant full-length CIP2A protein. Here, we show for the first time that recombinant CIP2A has no direct inhibitory effect on purified PP2A holoenzyme. Instead, our data support a model in which CIP2A functions as an allosteric PP2A inhibitor. This model is further supported by the relatively high PP2A activity coimmunoprecipitating with endogenous CIP2A and data showing that in spite of CIP2A’s potent ability to stabilize the phosphorylation of specific PP2A substrates in living cells, endogenous CIP2A completely failed to inhibit mTORC1-associated PP2A activity toward a PP2A-specific phosphopeptide in vitro. Thus, CIP2A is more likely to regulate the substrate availability than the activity of the phosphatase.
CIP2A induces a positive feedback loop that enhances tumorigenesis
In addition to demonstrating that CIP2A effectively inhibits autophagy, we show here that CIP2A is itself degraded by autophagy. This conclusion is based on the rapid Ulk1-dependent and ConA-sensitive disappearance of CIP2A upon autophagy activation. In line with this finding, CIP2A is rapidly degraded upon proteasome inhibition (Chen et al., 2010), a known activator of autophagy (Lamark and Johansen, 2010). The ubiquitin–proteasome system and autophagy are two major cellular pathways for protein degradation. Until recently, the selective degradation of target proteins was thought to occur exclusively via the proteasome. Emerging evidence suggests, however, that autophagy can also serve as an alternative route to eliminate specific proteins (Johansen and Lamark, 2011). The discovery of p62/SQSTM1 as an autophagic cargo receptor that guides ubiquitinated substrates for autophagic degradation has provided mechanistic insight into this process. Thus, it is interesting to note that the degradation kinetics of CIP2A upon autophagy induction closely followed those of p62/SQSTM1 and that depletion of p62/SQSTM1 inhibited the autophagic degradation of CIP2A as effectively as that of Ulk1. These data together with our demonstration of CIP2A ubiquitination and physical association between p62/SQSTM1 and CIP2A strongly suggest that p62/SQSTM1 guides CIP2A to autophagic degradation, thereby suppressing tumorigenesis. Notably, this model is highly homologous to recently reported p62/SQSTM1-dependent autophagic degradation of Dishevelled, which leads to the attenuation of oncogenic Wnt signaling (Gao et al., 2010). Furthermore, mTORC1- and autophagy-regulated stability of CIP2A provides a molecular explanation for the coordinated activation of mTORC1 and c-Myc, which together alter cellular metabolism to allow excessive cell growth and proliferation (Fig. 7).
Figure 7.
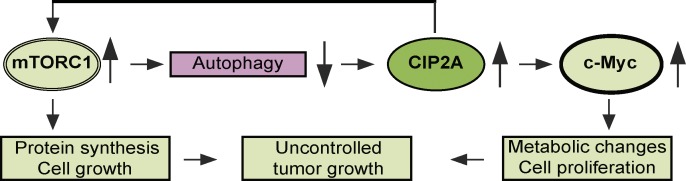
Proposed model for how CIP2A controls cell metabolism and proliferation and vice versa. The molecules and processes regulated positively and negatively by CIP2A are marked pink and light green, respectively.
Collectively, the data presented here reveal a signaling module that is likely to play an important role in the regulation of cell growth, metabolism, and tumorigenesis (Fig. 7). Because CIP2A is a potent activator of mTORC1 and inhibitor of autophagy, its autophagic degradation creates a feedback loop in which autophagy reduces cellular CIP2A levels, which in turn leads to reduced mTORC1 signaling and prolonged autophagy. Vice versa, high levels of CIP2A inhibit mTORC1-associated PP2A, thereby enhancing mTORC1 signaling and inhibiting autophagy, which in turn leads to the stabilization of CIP2A and prolonged activation of the mTORC1 pathway. Importantly, this signaling module is closely connected to c-Myc stability. CIP2A-mediated reduction in autophagic activity leads to the accumulation of c-Myc, thereby further contributing to the well-established tumor suppressor function of autophagy. Together with the TMA data showing significant correlation between CIP2A expression and mTORC1 activity in human breast cancer, these results reveal CIP2A as a relevant oncoprotein involved in the regulation of cell growth, metabolism, and tumorigenesis.
Materials and methods
Cell culture and reagents
MCF7S1 (referred to as MCF7), a highly TNF-sensitive subclone of MCF7 human breast carcinoma cells (Jäättelä et al., 1995), transfected MCF7 subclones, and U-2-OS human osteosarcoma cells were cultured in RPMI 1640 with GlutaMAX (Life Technologies) supplemented with 6% heat-inactivated FCS (Biological Industries). HEK-293 was cultured in RPMI 1640 supplemented with 10% FCS and 1% nonessential amino acids. MCF10A human breast epithelial cells purchased from ATCC were grown in DMEM-F12 (Life Technologies) with 10% heat-inactivated horse serum, 10 µg/ml insulin, 20 ng/ml EGF, 100 ng/ml cholera toxin, 500 ng/ml hydrocortisone, 2 ml/500 ml bovine pituitary extract (Sigma-Aldrich), and antibiotics. Ubiquitin-HA and c-Myc–S62A constructs were purchased from Addgene.
Amino acid starvation was performed in Hanks’ balanced salt solution containing 1 g/liter glucose (Gibco). Other reagents were purchased from Sigma-Aldrich if not otherwise stated.
Autophagy and cell viability
EGFP-LC3 puncta formation was assessed in MCF7-EGFP-LC3 cells (Høyer-Hansen et al., 2007) fixed in 3.7% formaldehyde by counting the number of puncta per 10 cells in a minimum of four randomly chosen areas of the well using an inverted confocal laser-scanning microscope (Axiovert 100M; Carl Zeiss) with 20× magnification. Renilla luciferase (RLuc) reporter assay for autophagic flux was performed in live cells essentially as described previously (Farkas et al., 2009). In brief, RLuc-LC3WT– and RLuc-LC3G120A–expressing cells were plated 24 h before the analysis at a density of 3.6 × 104 cells/cm2 in the even and uneven numbered columns in 96-well plates, respectively. Enduren (Promega) was added at a final concentration of 50 nM for the last 5 h, and the cells were treated as indicated for the last 3 h before the luminescence was measured in a flash plate reader (Varioskan; Thermo Electron Corporation). The autophagic flux was expressed as the ratio in luminescence between neighboring wells containing RLuc-LC3WT– and RLuc-LC3G120A–expressing cells, and the ratio in the untreated control cells was set to 100%.
The cell density was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay in 96-well plates. At the end of the experiment, cells were incubated with the MTT solution (final concentration of 10%; Sigma-Aldrich) for 2 h after which the medium was replaced with 100 µl MTT solvent (0.1 N HCL in isopropanol), and the formation of MTT formazan was measured at a wavelength of 570 nm.
Cell death was quantitated by lactate dehydrogenase (LDH) release assay (Roche) essentially as described previously (Foghsgaard et al., 2001). In brief, cells were seeded in 96-well plates in 100 µl of growth medium and treated as described. At the end of the treatment, 50 µl of medium was transferred to a new plate for the measurement of the released LDH, remaining medium was removed, cells were lysed in 100 µl of 1% Triton X-100 for 30 min at 37°C, and 50 µl of the lysate was transferred to a new plate for the measurement of total LDH. Then 50-µl LDH reaction mixture (Roche) was added to the plates with growth medium and cell lysates and incubated for 20 min at 25°C before determining the absorbance (490 nm) by microplate reader (VersaMax; Molecular Devices). Cytotoxicity (percentage) was measured as Abs[sample] × 100/(Abs[sample] + Abs[lysate]).
RNAi and lentiviral expression
The siRNA library used in the screen was the Silencer Select siRNA Library V3 (Ambion; Invitrogen) supplemented with nontargeting control siRNA (Rohde et al., 2005) and CIP2A siRNAs (Junttila et al., 2007) described previously. In all screens, cells were transfected with 8 nM siRNA in 96-well plates using Oligofectamine (Invitrogen) according to manufacturer’s protocols. For all subsequent experiments, the two most effective siRNAs for each target were pooled and added at a total concentration of 20 nM. Validated shRNA lentiviral vectors (MISSION; Sigma-Aldrich) and a lentiviral vector encoding for siRNA-resistant CIP2Ar (Junttila et al., 2007) were amplified and infected according to standard protocols. CIP2A-FLAG and lentiviral CIP2A plasmids were provided by J. Westermarck (University of Turku and Åbo Akademi University, Turku, Finland).
In vitro translation and interactions
CIP2A was translated using TnT-coupled reticulocyte lysate system (Promega) according to the manufacturer’s protocol. CIP2A-containing reticulocyte lysate (20 µl) was incubated for 20 min at 20°C with 20 µl HA-S6K1 or HA-raptor bound to protein A beads, 5 µl BSA (100 µg/ml), and 10 µl PP2A holoenzyme dilution (10 U/ml; EMD Millipore) in the interaction assay buffer (Hepes, pH 7.4, 0.3% CHAPS, 5 mM EDTA, 2 mM EGTA, and 100 µM CaCl2). Beads were washed 2 × 5 min with interaction assay buffer, and bound proteins were analyzed by immunoblotting.
PP2A activity assay
PP2A activities were measured using the PP2A phosphatase assay kit (EMD Millipore). In brief, immunoprecipitated complexes or purified PP2A holoenzyme (EMD Millipore) were incubated for 15 min in 37°C in 100 µl reaction buffer (50 mM Tris-HCl, pH 7.0, 100 µM CaCl2, and 100 µM K-R-pT-I-R-R peptide) with or without okadaic acid. The reaction was developed with 100 µl of malachite green solution and incubated for 15 min at 37°C for absorbance (A620 nm) measurement. Relative activities were obtained by subtracting blank values or values for samples in which PP2A activity was inhibited by okadaic acid.
Immunodetection
The primary antibodies used included rabbit antibodies against P-Thr308-Akt (4056), P-Ser473-Akt (4060), P-Thr1462-TSC2 (3611), P-Ser2448-mTOR (2971), pan-S6K1 (2708), P-Thr389-S6K1 (9234), P-Thr421/Ser424-S6K1 (9204), P-Thr37/46-4E-BP1 (236B4), 4E-BP1 (9644), S6 (4857), and P-Ser235/Ser236-S6 (4856) obtained from Cell Signaling Technology, P-S318-ATG13 (600–401-c49) obtained from Rockland Immunochemicals, Inc., p62/SQSTM1 (ab31545) obtained from Abcam, polyubiquitin (U5379) purchased from Sigma-Aldrich, and PR65 (SC-15355) purchased from Santa Cruz Biotechnology, Inc. Mouse monoclonal antibodies were against CIP2A, PPPR1A, and c-Myc (9E10) obtained from Santa Cruz Biotechnology, Inc., P-T58/S62-c-Myc obtained from Cell Signaling Technology, LC3 (5F10) obtained from Nanotools, and monoubiquitin (F-11), HA, FLAG, GAPDH, and α-actin obtained from Sigma-Aldrich. Appropriate peroxidase (Dako)-, Alexa Fluor 488/594– and Alexa Fluor 647 (Invitrogen)–conjugated mouse and rabbit secondary antibodies were used for immunoblotting and immunocytochemistry.
For immunocytochemistry, cells were fixed in 4% paraformaldehyde in PBS at room temperature for 15 min and washed in PBS. After fixation, the cells were permeabilized with 0.2% Triton X-100 in PBS for 15 min at room temperature, washed with PBS, blocked with 3% BSA in PBS, and incubated with the primary antibodies in PBS containing 3% BSA and 0.5% Tween 20 for 18 h overnight at 4°C. Cells were washed 3 × 5 min in PBS and incubated with Alexa Fluor secondary antibodies diluted 1:2,000 in PBS containing 0.5% Tween 20 for 2 h and then washed 3 × 5 min in PBS. After the secondary antibody treatment, slides were mounted in ProLong gold antifade reagent (Molecular Probes). A fluorescence laser-scanning confocal microscope (LSM 510 META; Carl Zeiss) with objectives Plan Apochromat 63×/1.4 NA oil differential interference contrast and Plan Apochromat 40×/1.4 NA oil differential interference contrast was used. Image acquisition was made with ZEN acquisition software (2009; Carl Zeiss). The antibodies used are listed in the previous paragraph.
For interaction immunoprecipitations, cells were rinsed with ice cold PBS and lysed in lysis buffer (40 mM Hepes, pH 7.4, 2 mM EDTA, 0.3% CHAPS, and protease inhibitor cocktail obtained from Roche). Lysates were centrifuged at 2,000 g for 10 min before primary antibody (listed in this section) addition and incubated with rotation for 60 min at 4°C before addition of 1 vol of 50% protein G slurry and additional incubation for 30 min. Immunoprecipitates were washed 3 × 5 min with lysis buffer and 5 min with lysis buffer containing 175 mM NaCl and denatured by adding 1 vol of 2× Laemmli SDS sample buffer with β-mercaptoethanol and incubating at 96°C for 10 min. For ubiquitin immunoprecipitations, a published protocol (Lim et al., 2005) was followed. When specifically indicated, SDS-containing denaturating buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% NP-40, 5 mM EDTA, and protease inhibitor cocktail) was used. Immunoprecipitates were resolved in 4–12% NuPAGE Novex SDS-PAGE (Invitrogen) and analyzed by immunoblotting using antibodies listed in this section. ECL substrate (EMD Millipore) and an image reader (LAS-4000 Plus; Fujifilm) were used for luminescence detection of Western blots, and files were quantified with MultiGauge V3.2 (Fujifilm). Molecular weights were estimated using Novex Sharp Pre-stained Protein Standard (Invitrogen).
TMAs
Normal tissue (array catalog number MNO661) and breast cancer tissue (array catalog numbers BRC1501, BRC1502, and BRC1503) microarray slides were obtained from Pantomics, Inc., and their description as well as the histopathology of tissue cores present in the arrays can be found at Pantomics, Inc., tissue arrays. Antibody cross-reactivity was tested based on the FDA recommendation in the MNO661 array containing 33 types of normal tissues in duplicates. The BRC1501, BRC1502, and BRC1503 arrays contain 150 cores each, comprising 75 cases of normal, reactive, premalignant, and malignant (various grades and stages) tissues of the breast in duplicates. Collectively, this set of breast cancer TMAs totaled 210 nonoverlapping breast tumors and 15 normal breast tissue samples from surgical resections. They were fixed in 10% neutral buffered formalin for 24 h and processed using identical standard operating procedures. Sections were picked onto SuperFrost Plus slides (Menzel-Gläser). Each slide had >95% tissue core retention. The slides were stained as previously described (Moreira et al., 2010) using an appropriate primary antibody. Each set of slides was batch processed simultaneously for any given antibody using an automated stainer to minimize assay variation (Autostainer Plus; Dako).
An image analysis integrated system (Oncotopix; Visiopharm) was used to quantitate the stained TMA slides. After image acquisition with the Arrayimager software module (Visiopharm), the digital images from scanned TMA sections were submitted to segmentation and analysis by the Visiomorph analysis software module (Visiopharm), which is part of the Oncotopix system. The Visiomorph analysis software module was used to derive a combined score of staining intensity and percentage of immunopositivity for each core in a given TMA. A staining score was generated in this manner for each core. Statistical analyses of data were performed using NCSS 2007 (NCSS LLC) and Prism version 5 (GraphPad Software).
Quantitative PCR (qPCR)
qPCR analyses were performed according to standard protocols using High Capacity RNA-to-cDNA Master Mix (Applied Biosystems) and the following primer sets (QuantiTect; QIAGEN): ACTB, Hs_ACTB_1_SG; CIP2A (KIAA1524), Hs_KIAA_1_SG and Hs_p62/SQSTM1_SG; MYC, Hs_Myc_1_SG; PPP2R1A, Hs_PPP2R1A_1_SG; PPP2R3A, Hs_PPP2R3A_1_SG; and PPP2R3B, Hs_PPP2R3B_1_SG.
Bioinformatics and statistical analyses
The functional gene ontology, pathway annotations, and networks of the screening hits were analyzed using Pathway Analysis Software (Ingenuity Systems, Inc.). If not otherwise indicated, p-values were calculated with the two-tailed Student’s t test.
Online supplemental material
Fig. S1 provides the description of the scoring system used for the screens in Table 1 and Fig. 1. Fig. S2 shows a cartoon highlighting the positions of the identified candidate autophagy-regulating genes in insulin receptor signaling network. Fig. S3 shows the ability of PP2A-related candidate siRNAs presented in Fig. 1 (E and F) to deplete their target genes in MCF7 cells as analyzed by qPCR and their lack of toxicity in MCF7 cells as analyzed by the LDH assay as well as immunoblots analyses of their effect on the Akt–mTORC1 signaling pathway in MCF7 and U-2-OS cells. Fig. S4 shows confocal images of MCF7-LC3-EGFP cells immunostained for p62 and CIP2A that demonstrate the colocalization of LC3, p62, and CIP2A in cells treated with rapamycin and ConA for 8 h, supporting the autophagic degradation of CIP2A presented in Fig. 6. Table S1 provides a bioinformatics-based list of top candidate genes with the highest association with canonical pathways presented in Fig. 1 B. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201304012/DC1.
Supplementary Material
Acknowledgments
We thank J. Westermarck for valuable reagents, K. Grøn Henriksen for excellent technical assistance, and colleagues at the Danish Cancer Society Research Center for helpful discussions.
This work was supported by Vilhelm Pedersen and Emil Aaltonen Foundations for P. Puustinen, the Swedish Research Council for A. Rytter, the Danish Medical Research Council for M. Mortensen and M. Jäättelä, and the Danish Cancer Society, European Commission FP7 (APO-SYS [Apoptosis Systems Biology Applied to Cancer and AIDS]), Lundbeck Foundation, and John and Birthe Meyer Foundation for M. Jäättelä.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- AMPK
- AMP-activated protein kinase
- ConA
- concanamycin A
- LDH
- lactate dehydrogenase
- mTOR
- mammalian target of rapamycin
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI3K
- phosphoinositide-3 kinase
- PP2Ac
- PP2A catalytic subunit
- qPCR
- quantitative PCR
- raptor
- regulatory-associated protein of TOR
- RLuc
- Renilla luciferase
- TMA
- tissue microarray
- TOR
- target of rapamycin
References
- Amaravadi R.K., Lippincott-Schwartz J., Yin X.M., Weiss W.A., Takebe N., Timmer W., DiPaola R.S., Lotze M.T., White E. 2011. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 17:654–666 10.1158/1078-0432.CCR-10-2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arico S., Petiot A., Bauvy C., Dubbelhuis P.F., Meijer A.J., Codogno P., Ogier-Denis E. 2001. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 276:35243–35246 10.1074/jbc.C100319200 [DOI] [PubMed] [Google Scholar]
- Bánréti A., Lukácsovich T., Csikós G., Erdélyi M., Sass M. 2012. PP2A regulates autophagy in two alternative ways in Drosophila. Autophagy. 8:623–636 10.4161/auto.19081 [DOI] [PubMed] [Google Scholar]
- Behrends C., Sowa M.E., Gygi S.P., Harper J.W. 2010. Network organization of the human autophagy system. Nature. 466:68–76 10.1038/nature09204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171:603–614 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bononi A., Agnoletto C., De Marchi E., Marchi S., Patergnani S., Bonora M., Giorgi C., Missiroli S., Poletti F., Rimessi A., Pinton P. 2011. Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011:1–26 10.4061/2011/329098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.F., Liu C.Y., Lin Y.C., Yu H.C., Liu T.H., Hou D.R., Chen P.J., Cheng A.L. 2010. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene. 29:6257–6266 10.1038/onc.2010.357 [DOI] [PubMed] [Google Scholar]
- Côme C., Laine A., Chanrion M., Edgren H., Mattila E., Liu X., Jonkers J., Ivaska J., Isola J., Darbon J.M., et al. 2009. CIP2A is associated with human breast cancer aggressivity. Clin. Cancer Res. 15:5092–5100 10.1158/1078-0432.CCR-08-3283 [DOI] [PubMed] [Google Scholar]
- Corcelle E.A., Puustinen P., Jäättelä M. 2009. Apoptosis and autophagy: Targeting autophagy signalling in cancer cells -‘trick or treats’? FEBS J. 276:6084–6096 10.1111/j.1742-4658.2009.07332.x [DOI] [PubMed] [Google Scholar]
- DeBerardinis R.J., Lum J.J., Hatzivassiliou G., Thompson C.B. 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7:11–20 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- Dwivedi M., Song H.O., Ahnn J. 2009. Autophagy genes mediate the effect of calcineurin on life span in C. elegans. Autophagy. 5:604–607 10.4161/auto.5.5.8157 [DOI] [PubMed] [Google Scholar]
- Efeyan A., Zoncu R., Sabatini D.M. 2012. Amino acids and mTORC1: from lysosomes to disease. Trends Mol. Med. 18:524–533 10.1016/j.molmed.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas T., Høyer-Hansen M., Jäättelä M. 2009. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy. 5:1018–1025 10.4161/auto.5.7.9443 [DOI] [PubMed] [Google Scholar]
- Foghsgaard L., Wissing D., Mauch D., Lademann U., Bastholm L., Boes M., Elling F., Leist M., Jäättelä M. 2001. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 153:999–1010 10.1083/jcb.153.5.999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C., Cao W., Bao L., Zuo W., Xie G., Cai T., Fu W., Zhang J., Wu W., Zhang X., Chen Y.G. 2010. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 12:781–790 10.1038/ncb2082 [DOI] [PubMed] [Google Scholar]
- Gromov P., Gromova I., Friis E., Timmermans-Wielenga V., Rank F., Simon R., Sauter G., Moreira J.M. 2010. Proteomic profiling of mammary carcinomas identifies C7orf24, a gamma-glutamyl cyclotransferase, as a potential cancer biomarker. J. Proteome Res. 9:3941–3953 10.1021/pr100160u [DOI] [PubMed] [Google Scholar]
- Høyer-Hansen M., Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., Bianchi K., Fehrenbacher N., Elling F., Rizzuto R., et al. 2007. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol. Cell. 25:193–205 10.1016/j.molcel.2006.12.009 [DOI] [PubMed] [Google Scholar]
- Hynes N.E., Lane H.A. 2005. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer. 5:341–354 10.1038/nrc1609 [DOI] [PubMed] [Google Scholar]
- Iovino S., Oriente F., Botta G., Cabaro S., Iovane V., Paciello O., Viggiano D., Perruolo G., Formisano P., Beguinot F. 2012. PED/PEA-15 induces autophagy and mediates TGF-β1 effect on muscle cell differentiation. Cell Death Differ. 19:1127–1138 10.1038/cdd.2011.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäättelä M., Benedict M., Tewari M., Shayman J.A., Dixit V.M. 1995. Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene. 10:2297–2305 [PubMed] [Google Scholar]
- Johansen T., Lamark T. 2011. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 7:279–296 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C.H., Ro S.H., Cao J., Otto N.M., Kim D.H. 2010. mTOR regulation of autophagy. FEBS Lett. 584:1287–1295 10.1016/j.febslet.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila M.R., Puustinen P., Niemelä M., Ahola R., Arnold H., Böttzauw T., Ala-aho R., Nielsen C., Ivaska J., Taya Y., et al. 2007. CIP2A inhibits PP2A in human malignancies. Cell. 130:51–62 10.1016/j.cell.2007.04.044 [DOI] [PubMed] [Google Scholar]
- Lamark T., Johansen T. 2010. Autophagy: links with the proteasome. Curr. Opin. Cell Biol. 22:192–198 10.1016/j.ceb.2009.11.002 [DOI] [PubMed] [Google Scholar]
- Lépine S., Allegood J.C., Park M., Dent P., Milstien S., Spiegel S. 2011. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 18:350–361 10.1038/cdd.2010.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K.L., Chew K.C., Tan J.M., Wang C., Chung K.K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C.A., et al. 2005. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25:2002–2009 10.1523/JNEUROSCI.4474-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrpour M., Esclatine A., Beau I., Codogno P. 2010. Overview of macroautophagy regulation in mammalian cells. Cell Res. 20:748–762 10.1038/cr.2010.82 [DOI] [PubMed] [Google Scholar]
- Mizushima N., Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell. 147:728–741 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- Moreira J.M., Ohlsson G., Gromov P., Simon R., Sauter G., Celis J.E., Gromova I. 2010. Bladder cancer-associated protein, a potential prognostic biomarker in human bladder cancer. Mol. Cell. Proteomics. 9:161–177 10.1074/mcp.M900294-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F., Strappazzon F., Antonioli M., Bielli P., Cianfanelli V., Bordi M., Gretzmeier C., Dengjel J., Piacentini M., Fimia G.M., Cecconi F. 2013. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 15:406–416 10.1038/ncb2708 [DOI] [PubMed] [Google Scholar]
- Ostenfeld M.S., Høyer-Hansen M., Bastholm L., Fehrenbacher N., Olsen O.D., Groth-Pedersen L., Puustinen P., Kirkegaard-Sørensen T., Nylandsted J., Farkas T., Jäättelä M. 2008. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy. 4:487–499 [DOI] [PubMed] [Google Scholar]
- Rafn B., Nielsen C.F., Andersen S.H., Szyniarowski P., Corcelle-Termeau E., Valo E., Fehrenbacher N., Olsen C.J., Daugaard M., Egebjerg C., et al. 2012. ErbB2-driven breast cancer cell invasion depends on a complex signaling network activating myeloid zinc finger-1-dependent cathepsin B expression. Mol. Cell. 45:764–776 10.1016/j.molcel.2012.01.029 [DOI] [PubMed] [Google Scholar]
- Rohde M., Daugaard M., Jensen M.H., Helin K., Nylandsted J., Jäättelä M. 2005. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 19:570–582 10.1101/gad.305405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee A.R., Anjum R., Blenis J. 2003. Inactivation of the tuberous sclerosis complex-1 and -2 gene products occurs by phosphoinositide 3-kinase/Akt-dependent and -independent phosphorylation of tuberin. J. Biol. Chem. 278:37288–37296 10.1074/jbc.M303257200 [DOI] [PubMed] [Google Scholar]
- Vergne I., Roberts E., Elmaoued R.A., Tosch V., Delgado M.A., Proikas-Cezanne T., Laporte J., Deretic V. 2009. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 28:2244–2258 10.1038/emboj.2009.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J., Hahn W.C. 2008. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 14:152–160 10.1016/j.molmed.2008.02.001 [DOI] [PubMed] [Google Scholar]
- Xie Z., Klionsky D.J. 2007. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9:1102–1109 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- Yorimitsu T., He C., Wang K., Klionsky D.J. 2009. Tap42-associated protein phosphatase type 2A negatively regulates induction of autophagy. Autophagy. 5:616–624 10.4161/auto.5.5.8091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.C., Hou D.R., Liu C.Y., Lin C.S., Shiau C.W., Cheng A.L., Chen K.F. 2013. Cancerous inhibitor of protein phosphatase 2A mediates bortezomib-induced autophagy in hepatocellular carcinoma independent of proteasome. PLoS ONE. 8:e55705 10.1371/journal.pone.0055705 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.