

Abstract
Malaria elimination has recently been reinstated as a global health priority but current therapies seem to be insufficient for the task. Elimination efforts require new drug classes that alleviate symptoms, prevent transmission and provide a radical cure. To develop these next generation medicines, public-private partnerships are funding innovative approaches to identify compounds that target multiple parasite species at multiple stages of the parasite lifecycle. Here, we review the cell-, chemistry- and target-based approaches used to discover new drug candidates that are currently in clinical trials or undergoing preclinical testing.
Introduction
Malaria is a devastating infectious disease that is characterized by intermittent high fevers and, in the case of cerebral malaria, neurological complications such as brain injury and coma. It affects pregnant women and children disproportionately with 85% of deaths occurring in children under 5. It is caused by protozoan parasites of the genus Plasmodium, which are transmitted to humans by the bites of female Anopheles mosquitoes. There are four main Plasmodium species that cause disease in humans: P. falciparum, P. vivax, P. ovale and P. malariae. In addition, a simian parasite, P. knowlesi, occasionally infects humans1. P. falciparum causes the most deaths whereas P. vivax is the most widespread. There is no sterilizing immunity against malaria and the disease can be fatal, although symptoms in repeatedly infected individuals tend to decrease over time. Efforts to develop an effective vaccine have been unsuccessful but drugs are able to cure infections. Malaria imposes a heavy social burden that has delayed economic development in regions where it is endemic. It also causes hundreds of thousands of deaths worldwide each year, with estimates ranging between 660,000 and 1,238,000 in 20102, 3, with the highest mortality occurring in Africa.
Malaria was once found throughout many regions of the world including North America and Northern Europe. It was eliminated from North America, Europe and parts of Asia and South America during the 1950s and 1960s following a global campaign that relied heavily on the new synthetic insecticide, dichlorodiphenyltrichloroethane (DDT), and powerful new synthetic drugs, such as chloroquine and sulfadoxine-pyrimethamine. When the parasites became resistant to these drugs and DDT use was restricted because of environmental and health hazards, malaria returned to many areas and the number of deaths peaked at 1.8 million in 20043. Nevertheless, because of novel, more effective medicines (BOX 1), improved vector control, increased funding and public awareness, the mortality rate has recently declined by ~30%, suggesting that it is time to consider new malaria elimination or even eradication campaigns. However, malaria is a complex disease and might prove more difficult to eradicate than viral diseases, such as smallpox, which have been successfully eradicated, mainly because effective vaccines exist.
Box 1. Currently used drug regimens.
The first-line treatment for P. falciparum infections in regions where chloroquine resistant parasites are present is a combination therapy of artemisinin derivatives with partner drugs that ideally have longer half-lives than artemisinin-derivatives. Artemether-lumefantrine (Coartem) and amodiaquine-artesunate (Coarsucam) are the most widely used, whereas dihydroartemisinin-piperaquine (Euartesim) and artesunate-pyronaridine (Pyramax) are the most recently approved77. Several reformulations with doses specific for children and pregnant women are in clinical trials77. Artemisinin derivatives are hypothesized to interact with Fe-II species in the parasite’s food vacuole, and early ring stage parasites combat this by slowing down hemoglobin digestion55. Artemisinins are fast acting and very potent against blood-stage parasites and show activity against early sexual stages of the parasite, which is important for blocking transmission. Their major limitation is a short half-life, which is why they are partnered with longer lasting drugs.
Chloroquine, a 4-aminoquinoline, is still the recommended treatment for P. vivax infections because resistance has not fully developed in contrast to P. falciparum111. In areas where chloroquine resistant P. vivax is endemic, artemisinin combination therapies are recommended for first-line treatment (except artesunate and sulfadoxine-pyrimethamine, which is ineffective owing to resistance), in particular those with partner drugs with long half-lives (e.g. dihydroartemisinin and piperaquine and artesunate and amodiaquine). In all cases, a 15-day course of primaquine is required to prevent relapse and provide a radical cure.
For travelers visiting areas with endemic malaria transmission, the CDC recommends atovaquone-proguanil (Malarone), choloroquine, doxycycline or mefloquine, with specific recommendations depending on individual and regional factors.
All species of the parasite have a complicated lifecycle that involves molecular interactions with both the vertebrate and invertebrate host (BOX 2). Although the parasite transitions between several developmental forms in the human host, all disease symptoms are caused by the repeated lysis and invasion of erythrocytes by the asexual blood-stage parasites. Therefore nearly all past and current therapies target the blood-stage parasite.
Box 2. The Plasmodium life cycle- multiple opportunities for intervention.
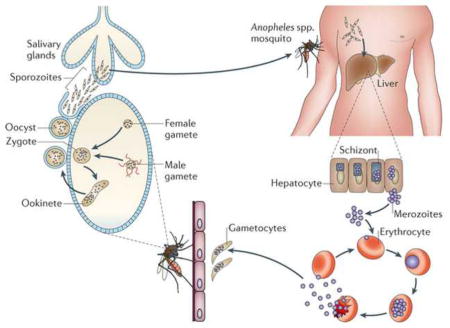
Plasmodium spp. have a complicated life cycle that is not well characterized at the molecular level. Human infection begins after transmission of sporozoites into the bloodstream, following the bite of an infected female Anopheles mosquito. Motile sporozoites migrate to the liver and take up residence in liver hepatocytes. For all Plasmodium spp., parasites incubate and multiply forming liver schizonts, which eventually burst, releasing thousands of merozoites into the bloodstream 2–16 days after initial infection112. P. vivax and P. ovale can remain dormant in the liver as hypnozoites, but can re-emerge and begin a blood-stage infection months to years after initial infection (relapsing malaria). Once they reach the blood stream merozoites attach and invade red blood cells and through DNA replication and schizogony produce 8–32 new merozoites per red blood cell. This 48-hour growth cycle is followed by red blood cell rupture and merozoite re-invasion, expanding the infection further and leading to symptoms of the disease. This cycle of asexual reproduction can persist indefinitely in the absence of chemotherapy.
In a process that is not completely understood, a small fraction of the haploid asexual parasites differentiate into male and females gametocytes within the red blood cell113. It is possible that secreted parasite factors induce this differentiation114, 115. These asymptomatic, non-replicating forms can persist for weeks and are responsible for malaria transmission. After ingestion of gametocytes by a mosquito during a blood meal, they differentiate into male and female gametes that fuse to form a zygote in the midgut of the mosquito. Meiosis occurs in the zygote, which then develops into a motile ookinete that migrates through the gut wall and eventually forms an oocyst. In the oocyst multiple rounds of DNA replication take place producing thousands of sporozoites116. These sporozoites migrate to the salivary glands and are transmitted to the next human host during a blood meal. Remarkably, few of the molecular regulators that control this intricate cycle are known—indeed, the factors that determine if a parasite becomes male or female are unknown.
Although effective antimalarials are currently available (BOX 1) that could be used in eradication campaigns, there are two major problems. The first is the potential emergence of resistance to artemisinin and its derivatives, which are the most effective drugs available today. Artemisinins (for example artemether and artesunate) comprise the only known drug class that works effectively against multidrug resistant parasites although reports of prolonged parasite clearance times in artemisinin treated patients (BOX 3) have raised concerns that the fragile gains of the last few years might be lost. The second problem is that only one drug, primaquine, can completely eliminate P. vivax and P. ovale and thus provide a radical cure. P. vivax and P. ovale infections are challenging to treat because they form dormant liver stages (hypnozoites) that are refractory to most drugs. Primaquine, an 8-aminoquinoline, requires repeated dosing (up to 15 days) and is toxic to individuals with glucose-6-phosphate dehydrogenase deficiency4, a common condition in malaria endemic regions. This limits the use of primaquine by the over 2.85 billion people at risk for P. vivax infection in Central and Southeast Asia, and in Central and South America5. Therefore new drugs with activity against all stages of the parasite life cycle and with new mechanisms of action are needed to help fulfill the ultimate goal of elimination6, 7.
Box 3. Artemisinin resistance.
The first signs of artemisinin resistance came from a study conducted in 2008, in which parasite clearance times after initial artesunate monotherapy were slower in patients from the Eastern Thai-Cambodian border (84 hours) than from the Thai-Myanmar border in the West (48 hours)117. Furthermore, the proportion of slow-clearing infections (defined as a parasite half-life of ≥6.2 h) on the Thai-Myanmar border increased from 0.6% in 2001, to 20% in 2010 approaching the rate of 42% observed in Cambodia between 2007 and 2010118. This suggests resistance has spread and is now present in western Thailand as well. At present, the genes which confer this delayed clearance are unknown, although studies have suggested that a region of chromosome 13 is involved119, 120. Further work indicated that four single-nucleotide substitutions on chromosomes 10, 13, and 14 are strongly correlated with delayed parasite clearance times121. These parasite loci seem to be under positive selection in these regions and could thus be responsible for resistance, although further studies will be needed to narrow down the exact genes involved. It has been proposed that parasite resistance is caused by an allele that down-regulates metabolism in the early stages of the erythrocytic life-cycle and increases it during the later stages, which would allow parasites to circumvent the drug and survive for long enough for the short-lived artemisinins to become degraded122. Although this is disturbing, there has been no clear increase in mortality in regions where reduced parasite clearance rates are observed. The spread of artemisinin resistance from the Thai-Cambodia border (as was seen with choloroquine and sulfadoxine-pyrimethamine) would be devastating for malaria treatment worldwide.
Concerns about artemisinin resistance have led universities and research institutes, funding agencies, governments, non-governmental organizations, the military and public-private partnerships to work together to find possible replacement medicines. Although several drugs are active against blood stage parasites, the ultimate goal is to develop a new compound that blocks all stages of the parasite life cycle, including transmission and the liver stages. In addition, because compliance is an issue, the ideal drug would be potent enough to work in a single, curative dose — described as a Single Exposure, Radical Cure and Prophylaxis (SERCaP) treatment8, 9 and inexpensive to manufacture ($0.15/dose), especially as many antimalarials treatments are paid for by nonprofits and governments. This target product profile has provided a framework for drug-discovery campaigns.
In this Review, we discuss the cell-, chemistry- and target-based approaches that are currently leading the way to the discovery of the next generation of antimalarial drugs. We will describe compounds that are in the drug development pipeline (Table 1) and the methods available to discover novel chemically validated targets and novel chemical drug classes with broad activity against multiple stages of the parasite life cycle.
Table 1.
Novel antimalarial compounds in preclinical and clinical development
| Compound (product name) | Chemical class | Presumed Target or MOA | Therapeutic activity | |||
|---|---|---|---|---|---|---|
|
| ||||||
| CH | PR | TB | RC | |||
| Preclinical
| ||||||
| DSM26583 | triazolopyrimidine | DHOD | x | |||
| 21A092 | pyrazole | unknown | x | |||
| MMV39004834 | aminopyridine | unknown | x | |||
| NPC-1161B64 | 8-aminoquinoline | unknown | x | x | x | |
| ELQ-30068 | Quinolone-3-Diarylether | cytochrome bc1 | x | x | x | |
| BCX4945131 | Immucillin-G | PNP | x | |||
| RKA18261 | 1,2,4,5-tetraoxane | hemoglobin digestion | x | |||
| P21876 | Diaminopyridine | DHFR | x | x | ||
|
| ||||||
| Phase I
| ||||||
| DSM26583 | triazolopyrimidine | DHOD | x | |||
|
| ||||||
| Phase IIa
| ||||||
| GNF156 (KAF156)42 | imidazolopiperazine | cyclic amine resistance locus (Pfcarl) | x | x | x | |
| NITD609 (KAE609)24 | spiroindolone | sodium transporter (PfATP4) | x | x | ||
| OZ43958 | 1,2,4-trioxolane | hemoglobin digestion | x | x | x | |
| Ferroquine72 | 4-aminoquinoline | hemozoin formation | x | |||
|
| ||||||
| Phase IIb/III
| ||||||
| Tafenoquine132 | 8-aminoquinoline | unknown | x | x | x | |
| Nauclea pobeguinii 98, 133 | natural product extract | unknown | x | |||
| Argemone mexicana100 | natural product extract | unknown | x | |||
| OZ277 (RBx11160)54 | 1,2,4-trioxolane | hemoglobin digestion | x | |||
Abbreviations: CH, chemotherapeutic; PR, prophylactic; RC, radical cure; TB, transmission blocking; DHOD, dihydroorotate reductase; DHFR, dihydrofolate reductase; PNP, purine nucleoside phosphatase. Source: http://www.clinicaltrials.gov, http://www.mmv.org. MOA; mechanism of action
Finding new drug candidates
Diverse strategies exist for the development of novel antimalarial drugs. Although efforts have been made to improve existing molecules (for example by modifying a scaffold to work against parasites that have acquired resistance to the parent scaffold), most new classes of antimalarial compounds have come from high-throughput screens. In this approach, a large compound library is screened to identify compounds that are active against the parasite in what is called a “phenotypic” or “whole-cell” assay (see below; Figure 1). Alternatively, the library can be screened for activity against “targets,” which typically are proteins that are crucial for parasite survival. Such screens often use biochemical assays (for example to detect ATP hydrolysis in a kinase activity assay). After screening, the most promising scaffolds are identified through cheminformatic analysis. Further selection is based on potency, cost, ease of synthesis, toxicity and novelty (Figure 1). Once scaffolds are selected, additional derivatives are often synthesized and tested against the whole parasite or the specific protein target. These tests can reveal Structure Activity Relationships (SAR), which predict the effect of chemical modifications on the properties of the molecule (for example its bioavailabilty and plasma concentrations). Several rounds of iterative SAR analyses can lead to candidates for efficacy testing and eventually lead compound identification. The type of assay and the number of compounds that are screened is typically balanced with cost. Until recently, malaria drug discovery has focused on finding replacements for compounds that are active against blood-stage P. falciparum, such as artemisinin. Now, with the development of cellular screens that can identify compounds active against all stages of the parasite lifecycle10–15, as well as assays available to test the stage specificity of candidates (Figure 2), the focus of the field has shifted.
Figure 1. Drug development strategy for the identification of novel antimalarials.
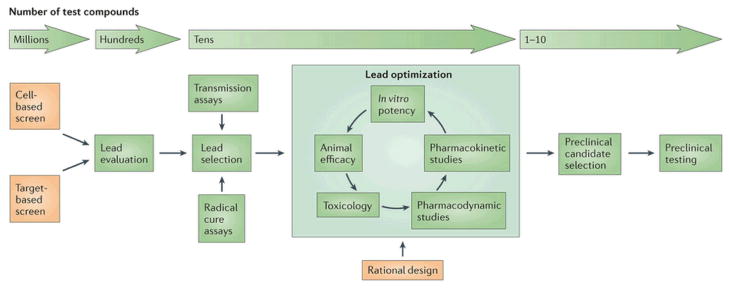
At the screening-stage millions of compounds can be screened (1,000 – 2,000,000 compounds per screening campaign). When hits are identified, (on average, a hit rate of 1.0% is observed) they are ranked based on a number of criteria (such as potency, ease of synthesis, known limitations to their use, novelty) to determine a possible lead compound. These compounds are tested in transmission and radical cure assays which are low throughput, time consuming and expensive and are therefore only applied to a handful of compounds. Following lead selection, multiple chemical derivatives of the lead are synthesized with the goal of maximizing potency and bioavailability while reducing cross-reactivity with possible human targets (lead optimization). The best candidate is selected for preclinical testing, an expensive and time-consuming process that involves assessing safety and finding the optimal doses that can be used in phase I human trials.
Figure 2. Assays available for determining the stage of activity and potency of potential antimalarial compounds.

To detect likely prophylactic activity, P. berghei or P. yoelli sporozoites are seeded onto human hepatoma cells and then the infection rate is imaged41 or detected enzymatically123. To detect prophylactic in vivo activity, rodent malaria sporozoites are injected into a mouse shortly before or after the mouse has been treated with the compound. The infection can be visualized with luciferase (with genetically modified parasites) or by measuring reduction in blood stage parasitemia and/or improved survival. The radical cure potential of a compound is tested using the hypnozoite-forming monkey model, P. cynolmolgi11. In the in vitro assay, P. cynolmolgi sporozoites are seeded onto primary monkey hepatocytes and imaged to determine the ratio between large, rapidly developing schizonts and dormant “small forms” (thought to be hypnozoites). Radical cure agents will eliminate all parasites including hypnozoites, whereas prophylactic compounds will act against growing schizonts only. In the in vivo model, monkeys are infected with P. cynolmolgi sporozoites followed by treatment with a compound that eliminates all blood stage parasites (e.g. chloroquine) and then by a potential radical cure compound124. The monkeys are then monitored over several months to measure reductions in the frequency of hypnozoite-caused relapses. The IC50 of P. falciparum blood stages is typically measured in a parasite proliferation assay20. P. vivax blood stage sensitivity is determined using a schizont maturation assay using parasites taken directly from infected patients125. The in vivo efficacy of blood-stage compounds is typically measured in mice infected with P. berghei or P. yoelli, although SCID mice can be infected with P. falciparum126. Transmission-blocking activity can be assayed in vitro by looking either at the viability127 or the development128 of purified P. falciparum gametocytes or ookinetes52. The ability of gametocytes to infect mosquitoes is measured using a standard membrane feeding assay (with P. falciparum)129,130 or by direct feeding from an infected mouse(with P. berghei or P. yoelli)53. After feeding the number of oocysts per mosquito midgut is counted to determine drug efficacy.
Notes: Pf, P. falciparum; Pv, P. vivax; Pb, P. berghei; Py, P. yoelli; Pc, P. cynomolgi.
Whole-cell based approaches
Cellular screening is the traditional approach for identifying compounds with antimicrobial activity16. This involves exposing the microorganism (in this case a culture of the parasite) to the test compound (a pure chemical compound or a natural product extract) and after a short incubation period, the culture is examined to determine if the compound is capable of killing the microorganism. Although it might seem straightforward, the challenge is to execute the assay reproducibly and cost-effectively for a very large number (typically millions) of compounds in a very small test volume. Technological advances in liquid handling, image analysis, assembly of pure chemical libraries and high-throughput automation now allow the screening of millions of individual compounds in 384 or 1536 well plates in a way that would have been unimaginable 20 years ago17. In the last 5 years, high-throughput, cellular-based screens have identified hundreds of previously unknown chemical compounds that have the potential to treat malaria.
Blood-stage screens
The first large-scale cellular screens were conducted by groups with the capacity for high-throughput screening: The Genomics Institute of the Novartis Research Foundation (GNF), GlaxoSmithKline (GSK) and St. Jude Children’s Research Hospital (SJCRH) beginning in 200818–20. These screens identified compounds that inhibit proliferation of P. falciparum blood-stages using a 384 or 1536-well format (Figure 2). Because parasites grow in anucleated human erythrocytes, increases in nucleic acid content in the well over a 72 hour period can be used as a read-out for parasite growth. Here, fluorescent DNA-binding dyes identify wells containing compounds that inhibit parasite growth19–21. Alternatively, parasite viability can be measured based on the activity of parasite lactate dehydrogenase18. Using these approaches, over 4 million chemicals have been screened in both academic and commercial settings and between 0.4% and 1.0% of the compounds showed activity against P. falciparum blood stages18–20.
Because many interested researchers may not have access to sophisticated screening equipment, such as ultra-accurate liquid dispensing and robotic compound plate storage, the results from many of the large-scale, blood-stage screens, including specific structures of the effective chemicals and their inhibition constants, have been made available online (https://www.ebi.ac.uk/chemblntd)22. This includes data from a collection of 13,000 compounds designated the Tres Cantos Antimalarial set (TCAMS)18. Based on these public screening results, a selection of 400 unique compounds with blood-stage antimalarial activity was created. These compounds have been resynthesized and this “malaria box” of potential chemical starting points can be obtained from Medicines for Malaria Venture (MMV)22. The malaria box concept allows biologists who can not resynthesize compounds to participate in the drug discovery process and to help discover how the compounds are working. MMV requests that results of tests using the malaria box are made public and encourages collaboration between groups.
Drug candidates from blood stage screens
One of the first novel drug classes identified using modern cellular screening methods were the spiroindolones. The lead for this class, a racemic spiroazepineindole23 (Figure 3), was identified in a screen of ~10,000 natural compounds for their activity against proliferating, blood-stage P. falciparum. Several hundred derivatives, including various enantiomers, were synthesized and further evaluated in blood-stage parasite proliferation tests, pharmacokinetic tests and in animal models of malaria. The most promising molecule, KAE609 (also called NITD609)24, is potent enough to cure P. berghei-infected mice with a single oral 100 mg/kg dose and prevents transmission25, as indicated by reduced oocyst numbers in a standard membrane-feeding assay (Figure 2). The spiroindolone class has a novel mechanism of action, which was identified by in vitro evolution and whole-genome scanning. It is believed to target the outer membrane transporter, PfATP424, which was initially annotated as a calcium ion pump but now is reported to be important for maintaining sodium homeostasis in the parasite26. Inhibition of this enzyme by KAE609 presumably increases intracellular sodium concentration leading the parasite to swell up and die. KAE609 is the first candidate in 20 years with a novel mechanism of action to enter into clinical trials. Through the combined efforts of a global consortium (the NGBS consortium: the Novartis Institute for Tropical Diseases; GNF, the Biomedical Primate Research Center and the Swiss Tropical and Public Health Institute), and with the help of industry and private-public partnerships (specifically the Wellcome Trust and MMV), KAE609 has progressed very quickly from the screening stage in 2007 to clinical trials27, where it is now in Phase II (http://www.mmv.org/research-development/rd-portfolio).
Figure 3. Chemical structures of new antimalarial candidates.

Lead compound chemical structures developed through optimization of chemical classes identified using A) cellular-based screens B) target-based screens or C) chemistry-based approaches. For the cellular screen, the compound classes are novel and all are derived from leads that have potent activity against parasite blood stages. DSM265 and P218 were derived from target-based screens against PfDHOD and PfDHFR respectively. OZ439 was rationally designed to have the parasite killing activities of artemisinins, but with a longer half life. References: TCMDC-13904629; NITD609 (KAE609)23, 24; GNF156 (KAF156)42. GSK105771431; MMV39004834; DSM26583; P21876; OZ43958.
GSK also identified chemical classes for further development by structural clustering of the lead compounds from the TCAMS. By filtering for properties such as high bioavailability and parasite potency28, 47 scaffolds were identified, five of which were chosen for SAR analysis and lead identification, and the rest were made publicly available with an invitation for collaboration28. One of these scaffolds (represented by TCMDC-139046 in Figure 3) contains an indoline core29 and is a known inhibitor of the human serotonin receptor (5-hydroxytryptamine subtype 2c), limiting its potential usefulness as a antimalarial drug because of likely adverse side-effects. However, through divergent SAR analysis the team was able to create additional compounds with selective and potent activity against P. falciparum and with reduced activity against the human receptor, although in vivo efficacy still needs to be improved. A second class, the cyclopropyl carboxamides30, have potent in vitro activity against P. falciparum and at least one (GSK1057714) has in vivo activity in the P. falciparum SCID mouse model (Figure 2)31. However, in vitro resistance against these compounds emerged rapidly, raising concerns about resistance development in the field. Therefore, if these are to enter into development, further optimization will be needed.
Other leads from blood-stage screens
Since the first high-throughput screens using blood-stage parasites, several other screens have been implemented that focus on specialized compound libraries or parasite lines. In a first example of such a focused approach, a screen of a commercially available kinase library32 using a parasite proliferation assay33 identified 3,5-Diaryl-2-aminopyridines as a new class of antimalarial compound. One member of this series, MMV390048 (previously named compound 1534), cures 100% of P. berghei-infected mice (Figure 2) after a single 30 mg/kg dose. Although the cellular target remains unpublished, MMV390048 (Figure 3) has progressed to preclinical trials because of its high antiparasitic potency (25 nM IC50), 7–8 hour half-life and good oral bioavailability.
In a second example, genetically modified parasites were used to identify compounds active against PfABCG2, one of the parasite’s ABC transporters35. A library of 2,816 approved drugs was screened against wild-type P. falciparum and against a recombinant strain lacking PfABCG2. This screen identified the antihistamine ketotifen as a potential antimalarial. The IC50 for the mutant strain was higher than the IC50 for the wild-type strain, suggesting that PfABCG2 is involved in the antimalarial activity of ketotifen. Previous studies with ketotifen showed that it has some anti-relapse activity in P. cynolmolgi-infected monkeys36, and causal prophylaxis against P. yoelli-infected mice37 (Figure 2). Furthermore it reduces oocyst formation in both P. falciparum and P. yoelli mouse models35. Recently, the ketotifen metabolite norketotifen (which has improved pharmacokinetic properties) was shown to be active against both blood- and early-liver stages38. Thus, further lead optimization of ketotifen might identify new clinical candidates.
Liver stage screens
Recent progress has been made in the development of high-throughput screens for the identification of compounds that are active against the sexual and liver stages of the parasite life cycle39. It can be argued that these are the most important life-cycle stages when it comes to eradication because of the population bottleneck that occurs during these stages15. Specifically, there may be billions of parasites in a human blood stage infection, but only a limited number (<50) of ookinetes, hypnozoites or early liver forms exist during transmission and reinfection. Resistance is thus less likely to emerge. Since the first report of a liver stage screen40, several groups have established liver-stage assays (Figure 2). For example, high-content imaging has been used to determine the efficacy of drugs with known blood-stage activity on P. yoelli-infected hepatocytes41. One candidate that was identified in this screen was GNF179, a member of the imidazolopiperazine class. This compound is believed to act through the cyclic amine resistance locus protein (Pfcarl; Pf3D7_0321900), which was identified after in vitro evolution and whole-genome scanning. Pfcarl contains several transmembrane domains but its function and whether or not it is the actual target of GNF179 remain unknown. Further lead-optimization of the imidazolopiperazine class led to KAF156 (also called GNF156, Figure 3) as a clinical candidate that is currently in Phase II clinical trials42, 43. KAF156 is slightly less potent than KAE609 but it provides prophylactic protection in animal models and has activity against gametocytes (http://www.mmv.org/research-development/rd-portfolio). Similar liver-stage screens with other libraries have been run44, 45 but have not yet yielded new preclinical candidates.
Radical cure assays
For P. vivax and P. ovale infections, a subset of parasites can remain undetected in the liver in a dormant state, called a hypnozoite, for months to years before re-activation, which results in a blood stage infection. Thus, this developmental stage is an important parasite reservoir, especially for maintaining transmission of P. vivax and P. ovale in endemic countries. In previous eradication campaigns, regions where P. falciparum had been eliminated P. vivax persisted at low levels46 presumably due to the hypnozoite reservoir seeding new infections. This highlights that in terms of a radical cure, it is crucial that the drug is capable of eliminating this reservoir.
The radical cure activity of primaquine, the only currently known drug capable of eliminating hypnozoites, was identified in the 1950s by dosing hundreds of P. cynolmolgi (a hypnozoite producing simian strain) infected monkeys with various aminoquinoline derivatives47 and determining which animals no longer relapsed. Primaquine is a prodrug that must be metabolized to become active. Because it is unclear which of the many metabolic products work against hypnozoites, its mechanism of action remains unknown48. There have been reports of primaquine resistance49, 50, which could help in identifying the primaquine target. However the amount of primaquine resistance remains controversial because it is difficult to rule out incomplete patient adherence to the 15-day dosing schedule or patient metabolic deficiencies as a cause for low drug potency. As there are no validated targets for radical cure compounds, target-based screens cannot be initiated.
The development of cellular screens that detect activity against hypnozoites are difficult and has been hindered by the fact that P. vivax cannot be maintained in cell culture. Recently, a low-throughput in vitro assay (Figure 2) was developed using the P. cynomolgi monkey model where P. cynomolgi sporozoites from mosquitoes fed on infected monkeys are used to infect primary monkey hepatocytes followed by image analysis to determine infection levels11. This assay can distinguish compounds that are active against “small-form” parasites (which are believed to be the dormant hypnozoites) from compounds that only prevent infection or parasite proliferation in the liver. However, validation of this model is difficult: P. cynomolgi sporozoites do not freeze well, and obtaining fresh ones is challenging given constraints on primate research. Furthermore, molecular markers that distinguish a hypnozoite from an early liver schizont do not yet exist and it has not yet been demonstrated that clearing small forms in vitro predicts anti-hypnozoite activity. Nevertheless, this is a promising first step to an assay that examines compounds targeting this important developmental stage
Transmission screens
Because infectious gametocytes (infectious to the mosquito) can persist in the patient long after symptoms have resolved, a good lead compound should also kill gametocytes in order to disrupt transmission. If the gametocytes remain viable and mosquitoes are present, then the person can infect his neighbors as well as re-infect himself. Several groups are developing high-throughput screens to identify compounds active against gametocytes (Figure 2). These assays are particularly difficult because the process of gametocytogenesis is not well understood. Because gametocytes do not divide during the 12 days it takes them to mature from the early to the late stage, of which the latter is transmissible, methods based on the detection of inhibited parasite proliferation cannot be used. Therefore, alternative high-throughput techniques to detect gametocyte death have been developed. Several of these assays use overall ATP hydrolysis to measure viability, with loss of this activity used as an indicator for gametocyte death10. Alternatively, it is possible to selectively count parasites expressing a gametocyte-stage specific GFP tag by flow cytometry12. The first gametocyte screen was conducted using alamar blue13, an oxidation-reduction indicator that both fluoresces and changes color in response to chemical reduction of growth medium that occurs when gametocytes are metabolically active, and other screens have been implemented that involve lactate dehydrogenase51. Because these assays only attempt to predict whether a compound might block transmission by killing gametocytes, effort has been devoted to finding more predictive assays that measure viability in later stages, such as the ookinete 52. The gold standard test is feeding mosquitoes on infected, treated blood (standard membrane feeding assay for P. falciparum), or on infected, treated mouse (direct feeding assay) (Figure 2). These assays (reviewed in 53) are difficult to automate. Interestingly, primaquine is the only drug known to block transmission in humans but given that it is a prodrug, it functions poorly in cellular assays and even in the standard membrane feeding assay.
Medicinal chemistry-based approaches
Directed, chemistry-based approaches base the design of new compounds on known chemical structures of antimalarials that have been successful in the clinic. The compounds are modified to optimize their therapeutic properties and to reduce limitations to their use (for example resistance, bioavailability) using SAR and whole-cell or biochemical assays. Although this approach has been successful, (one example is the synthetic ozonides, which are based on artemisinin (see below)), the major limitation is that no model compound exists that acts during all stages of the parasite life cycle and thus a template for a SERCaP molecule is lacking.
Synthetic ozonides
One of the best examples of the chemistry-based approach is the development of the synthetic ozonides. Ozonides (synthetic peroxides) retain the endoperoxide bridge that gives artemisinin its potent blood-stage activity but also contain a bulky amantadine ring54, which increases their stability in plasma. It is hypothesized that their activity results from the peroxide bond being reduced by ferrous iron and heme, which are liberated through the digestion of hemoglobin by the parasite 55. This reduction produces carbon-centered radicals that alkylate heme and parasite proteins, ultimately leading to parasite death. The first-generation synthetic ozonide, OZ27754, is as potent as artesunate in vitro and has increased activity in the P. berghei mouse model, with the ability to completely cure mice with three 10 mg/kg oral doses. OZ277 (a 1,2,4-trioxolane) was the first synthetic ozonide evaluated in the clinic but after the phase II results showed only 70% efficacy after 7 days of treatment, its development was deprioritized56. Nevertheless, RBx11160, as it is now known, has been approved in India since April 2012 as Syrniam (a combination with piperaquine)57.
A newer generation synthetic ozonide, OZ439 (Figure 3), has now been developed; OZ439 is potent and fast-acting, as well as being pharmacologically active for longer and having improved bioavailability compared to the current artemisinin derivatives and OZ27758. Importantly, OZ439 is able to cure and prevent P. berghei blood-stage mouse infections with a single 30 mg/kg dose and blocks mosquito infection (transmission) in the in vitro membrane feeding assay59. Encouraging results from Phase I clinical trials confirm this single-dose potency, and combined with its longer half-life, this synthetic compound class is very attractive60. However, as with artemisinins, ozonides are only active against blood stages and as their activity is based on the endoperoxide bridge found in artemisinins, they might be less effective against artemisinin-resistant parasites, although whether parasites are truly resistant is a matter of debate (see Box 3).
Another synthetic ozonide chemical class, the 1,2,4,5-tetraoxanes, which also contain the endoperoxide bridge, are also being developed61. The lead compound, RKA182, reduces P. berghei parasitemia in mice to undetectable levels 24 hours after treatment. This compound is currently in preclinical trials and although it has greater stability than OZ27761, its antimalarial activity is inferior to OZ43962.
Other improvements on known scaffolds
There are also efforts to design new versions of primaquine (the only 8-aminoquinoline in clinical use) that lack some of its undesirable toxic properties, such as inducing hemolytic anemia in patients with glucose-6-phosphate deficiency4. Tafenoquine is a 3-phenoxy-substituted 8-aminoquinoline (previously named WR238605) that is 4–100 times more potent than primaquine and has a longer half-life63. It was identified in 1993 and it is in a dose range phase II trial as a single dose anti-relapse agent. Similarly, NPC1161C64 and its enantiomer NPC1161B, both 8-aminoquinolines, seem to be promising candidates but it is unclear whether these closely related molecules will have similar levels of side effects as primaquine. Furthermore, assays to measure primaquine resistance are lacking65 and the genes that confer resistance are unknown65, which might pose problems in the future for drugs that are designed using primaquine as a scaffold. Another disadvantage of this class is that some patients do not respond to primaquine therapy, possibly owing to differences in primaquine metabolism66.
Substantial effort has also been devoted to the development of novel antimalarials based on the pyridone scaffold of endochins, whose antimalarial activity was first recognized in the 1940s67. Endochins have been optimized extensively to improve their oral pharmacokinetic properties and have now yielded the new preclinical candidate ELQ-30068. Based on their structure (quinolone-3-diarylethers), it is likely that these target mitochondrially-encoded cytochrome bc1, similar to atovaquone.
Hybrid molecules
Another rational chemistry-based approach is to design hybrid molecules that combine several chemical groups that provide stability, solubility, potency or other attractive features69. Two molecules, each with their own antimalarial activity, can be covalently linked, producing a single hybrid molecule with dual activity. The need for parasites to digest human hemoglobin in asexual blood stages has always represented a vulnerability for the parasite that has been targeted by drug designers. Furthermore, the parasite must also convert the resulting reactive heme molecule to nonreactive hemozoin. Dual-function acridones contain a heme-targeting acridone group that provides potent blood stage activity and a chemosensitizing component that counteracts resistance to existing aminoquinoline antimalarial drugs70. The lead compound, T3.5, shows synergy with chloroquine, piperaquine, quinine and amodiaquine in vitro, when used to treat aminoquinoline-resistant parasites. In combination with primaquine, it also reduces parasitemia in P. yoelli-infected mice70. Similarly, ferroquine is an organometallic compound composed of a lipid-targeting, ferrocenyl group covalently linked to a 4-aminoquinoline and a basic alkylamine71. Ferroquine is designed to simultaneously disrupt membranes and to prevent hemozoin formation72. Although interesting from a chemical biology and chemical synthesis point of view, the same antimalarial effect of hybrid molecules might be achieved with a combination therapy. Furthermore, some hybrids have the same liabilities as their component molecules (e.g. the 4-aminoquinolines’ hERG channel activation liability 73, which for some drugs, has been implicated in cardiac arrhythmias74).
Target-based approaches
Target-based drug discovery is another popular approach for lead identification, although its popularity seems to be waning owing to disappointing results of this approach in finding next generation antibacterial drugs75. Targets are typically proteins with essential cellular functions, the inhibition of which results in cell death. Target molecules can be identified using chemical inhibition studies (a chemically-validated target). Alternatively, a target can be chosen because it is expected to be essential for parasite viability, which is usually determined by genetic knockdown experiments (a genetically-validated target) or by the fact that it is highly conserved and is therefore likely to have an essential function. Once a target is selected, the recombinant protein is produced and a biochemical assay is developed that can be used to identify compounds that inhibit the target’s function. The process can be further enhanced if crystal structures of the protein are available to guide the design of potential inhibitors and to increase selectivity and specificity against the parasite protein, relative to the host protein.
Key targets
Examples of chemically-validated targets in P. falciparum include dihydrofolate reductase (DHFR) and cytochrome bc1, which are inhibited by the antifolates (pyrimethamine and proguanil) and atovaquone, respectively. These drugs work well clinically and pyrimethamine is currently used in combination therapy with artesunate and sulfadoxine2. Inhibition of DHFR prevents folate synthesis, which is essential for DNA replication in the parasite. DHFR inhibitors show specificity for parasite over human DHFR. Because DHFR targeting has been successful in the past, an in silico method was developed to design new drugs that are capable of binding to the DHFR enzyme bearing mutations that confer resistance to pyrimethamine and proguanil76. Co-crystal structures of DHFR complexed with its substrate, known inhibitors and lead candidates were used to guide the development of a lead that binds to both wild-type DHFR and the mutated form. This lead, P218 (Figure 3) is specific for Plasmodium DHFR and has an IC50 in the nanomolar range against P. falciparum expressing wild-type or mutant DHFR. This diaminopyridine was designed so that it binds DHFR in the same place as the natural substrate (dihydrofolic acid), in the hope that resistance mutations that emerge would not be tolerated. P218 has now advanced to pre-clinical trials77.
The mitochondrial enzyme dihydroorotate dehydrogenase (DHOD) (Figure 3) has long been recognized as a potential antimalarial target because it catalyzes the fourth step in the essential de novo pyrimidine biosynthesis pathway78,79. The first high-throughput screen to directly measure activity against recombinant DHOD was conducted in 200580 and although this screen identified several potential DHOD inhibitors, they lacked adequate activity in parasite proliferation assays. A new chemical class from the original screen, the triazolopyrimidines, was identified with potent activity in whole-cell assays (P. falciparum IC50 = 79nM) and >5000 fold specificity for parasite over human DHOD, yet this class was inactive in the P. berghei in vivo model81. After a series of chemical modifications81, 82 and analysis of drug-enzyme co-crystal structures to further optimize binding, a potent lead was discovered that has an IC50 of 40–50 nM against drug-sensitive and drug-resistant P. falciparum, including those resistant to chloroquine, atovaquone, and the antifolate, pyrimethamine83. The molecule, DSM265 (Figure 3), shows a similar potency as chloroquine in the humanized SCID mouse model. DMS265 is the first DHOD inhibitor to enter pre-clinical trials83.
A similar discovery effort identified the carboxamide chemical class as DHOD inhibitors. Recombinant DHOD from P. falciparum, P. berghei and P. vivax were screened against the 208,000 Genzyme compound library and hits were validated in cell proliferation assays against P. falciparum84. Further optimization resulted in a lead (Genz667348) that is efficacious in the P. berghei blood-stage model and is undergoing further development for preclinical trial selection85. Although the triazolopyrimidines and the carboxamides both target DHOD, structural analysis of the drug binding pocket suggests these two classes bind to overlapping yet distinct sites on DHOD83.
While these former approaches used biochemical assays, an in silico molecular docking approach was used to identify potential inhibitors that disrupt the interaction between the carboxy terminal tail of myosin A and the myosin tail interacting protein (MTIP) of the parasite. This interaction is required for erythrocyte invasion86 and is thus essential. Potential inhibitors were identified using an algorithm that computationally docked 300,000 possible inhibitors to the crystal structure of MTIP. After ranking, 15 promising molecules with the appropriate druglike properties were physically obtained and subsequently tested for their ability to block parasite proliferation. Further optimization of a urea-pyrazole scaffold has yielded a molecule (21A092) that has advanced to preclinical studies87. However, given the high rate of positives random compounds show in cellular proliferation screens it is possible that the parasiticidal activity of this class is a result of binding to another cellular target.
New Target discovery
It is likely that a number of new chemically-validated targets will be available for target-based screening campaigns in the future. These are the proteins that were identified as targets of the promising compounds identified in the aforementioned cellular screens. Although discovering how a compound acts in the cell has traditionally been very challenging in malaria parasites, targets can now be identified by creating parasites that are resistant to a compound, and then comparing the genomes of resistant progeny clones to the genomes of their sensitive parent clones41, 88. This method has led to the identification of a handful of novel24, 89, 90 and previously known targets91, 92. Interestingly, the protein biosynthetic pathway appears to be a rich source of novel, chemically-validated targets with three tRNA synthetases having been identified as targets of antimalarial compounds89, 90, 93, some (for example the natural products cladosporin and halofuginone) with demonstrated activity across multiple stages89, 94. It is interesting to note that a number of the targets that have been discovered by this approach (e.g. PfATP4, PfCARL, lysyl tRNA synthetase) were not on lists of the most desirable targets predicted based on structure-based drug-ability and essentiality95, and that few proteins, which were hypothesized to be good targets, have yielded compounds that have progressed into development with the exception of the well known antimalarial targets, DHOD, DHFR and cytochrome bc1 (see above). Although many hypothetical targets can look attractive from a structural biology point of view, it is possible that some of these proteins are simply too abundant in the cell and thus the inhibition of all cellular copies is difficult to achieve at physiologically-relevant inhibitor concentrations.
Natural products, new formulations and combination therapies
Two of the most effective antimalarials, artemisinin and quinine, are natural products derived from traditional herbal remedies that are used to treat fevers. A third, atovaquone, is a synthetic version of the natural product lapichol96. There are other fever-reducing folk remedies that could yield a next-generation antimalarial. For example, the bark of the plant Nauclea pobeguinii, from the Democratic Republic of Congo, can substantially reduce parasitemia in mice infected with rodent malaria97 and has shown efficacy in man in phase IIb clinical trials98. It is interesting to note that the chemical structure of a possible active ingredient, strictosamide, is similar to that of the spiroindolones99. A second natural product from the Argemone mexicana plant, has also shown efficacy in a Phase II clinical trial100.
Drug formulation, for example in a pill or as a liquid suspension, can affect the drug’s usefulness. Thus, efforts have been made to reformulate traditional antimalarial compounds, including antibiotics. For example, an artemether/lumefantrine tablet works well with adults, but may be rejected by children because of the bitter taste. Thus, sweet-tasting liquid formulations needed to be designed for children101. In addition, antimalarials cannot be given ethically as monotherapies because of the risk of resistance emergence, so the choice of partner drugs must be carefully considered and is often determined by how long and when a compound is active and whether parasite resistance is likely to develop. More effective medicines can thus be created by the combination of new antimalarial compounds or the novel combination of traditional approved drugs. Multiple novel combination therapies, including pyronaridine-artesunate102, azithromycin-chloroquine103, dihydroartemisinin-piperaquine104 and sulfadoxine-pyrimethamine + amodiaquine (SP+AQ)105 are in clinical trials. It is possible that a SERCaP medicine could be created by formulating a medicine that contains three different molecules that are independently active against blood, liver and transmission stages.
Future perspectives
Malaria elimination will ultimately require an integrated strategy, including new and old drugs, vaccines, vector control and public health measures. Although the task seems daunting, new scientific discoveries could rapidly change the outlook. For example, traditionally, vector control strategies have depended on pesticides (indoor residual spraying and insecticide treated nets), and although these interventions decrease the number of malaria infections, they are insufficient for elimination in endemic regions106. New vector control strategies such as genetically modified mosquitoes and more recently, the colonization of mosquitoes with Wolbachia (which renders them refractory to Plasmodium spp. infection107) could help with elimination. An effective vaccine would also be very helpful, however, there is currently no vaccine available, and the most developed vaccine (RTS,S) showed disappointing preliminary results in Phase III trials, which reported a 16.8% vaccine efficacy that declined to 0% over 4 years108, 109. Although these trials provided a proof-of-principle for blood-stage vaccines, second generation vaccines with greater efficacy are urgently needed. Pre-erythrocytic vaccines, attenuated parasites, and multisubunit vaccines are all being explored110.
Even though elimination could potentially be accomplished with the existing arsenal of drugs, it might be less costly if more effective drugs that interrupt transmission, were available. Although target-based drug discovery could be a useful approach for finding a SERCaP drug, it should be noted that currently known targets fall short of the SERCaP requirements. Furthermore, cellular screens that could lead to the identification of small molecules with SERCaP activity are difficult to implement and are not available to the average researcher, nor can they be used routinely during lead optimization steps. One source of problems is that often the organisms used for testing (such as P. vivax sporozoites) or specialized assays are located thousands of miles away from the chemical libraries and hi-tech screening equipment. Nevertheless, the recent progress suggests that a SERCaP could realistically be developed, especially now that funding agencies have made this a priority. Although special challenges might be associated with eliminating metabolically quiescent hypnozoites and very late stage gametocytes, targets central to all stages of the life cycle could nevertheless exist.
Considering the high mortality and morbidity caused by malaria, there is no question that new drugs are needed. It is an exciting time for malaria drug discovery; the combination of new and innovative screens to identify compounds with broad-range activity is hoped to yield new insight into chemicals with broad-range activity. With the support of various funding agencies the time is ripe to take advantage of these opportunities and to discover drugs that will lead the way to the global eradication of malaria.
Key points.
Although malaria continues to endanger 40% of the world’s population, and is estimated to be responsible for up to a million deaths per year, the number of cases reported by the World Health Organization has declined.
Some fear that these fragile gains will be reversed if parasites become resistant to artemisinins, currently the only class of antimalarial that works effectively against all drug-resistant parasite strains. Ever slowing response times to artemisinin monotherapies and the risk that these compounds loose effectiveness has spurred new search for replacement therapies.
The World Health Organization and several non-profit, non-governmental organizations have made the elimination of malaria a long term public health goal. This has generated interest in developing novel antimalarial compounds that can not only eliminate the symptoms of malaria, but remove all parasites from the body and prevent the spread of malaria.
In recent years sophisticated and powerful cellular and phenotypic screening methods have identified drug candidates active against different stages of the parasite’s lifecycle and at least two of these novel classes of antimalarials are being tested for efficacy in humans.
For known, validated antimalarial “targets”, structure guided drug design has yielded drug candidates with better potency and activity against drug resistant malaria parasites.
Insightful chemical design has also resulted in new drug candidates that have improved potency, or remain for a longer period of time in the patient’s bloodstream.
Acknowledgments
E. Winzeler and E. Flannery acknowledge support from NIH R01 AI090141 and F32AI102567 respectively. All authors also gratefully acknowledge support from the Bill and Melinda Gates Foundation (OPP1054480) and the Medicines for Malaria Venture.
Glossary
- 3D7 parasites
A chloroquine-sensitive P. falciparum clone commonly used in the laboratory and whose genome was completely sequenced
- Causal prophylaxis
The prevention of a blood-stage infection by a therapeutic that prevents sporozoites from invading or developing within the liver
- Cheminformatic analysis
Computational analyses using systematic or common chemical names, used to group scaffold families and discover known activities, toxicities and sometimes targets
- Enantiomers
Molecules that are structurally equivalent, but are mirror images of one-another and therefore not super-imposable. It is common for one enantiomer of a drug to have more activity than the other
- Gametocyte
This is the sexual stage of parasite that develops from asexual parasites and differentiates into gametes in the mosquito. They are thus the parasite forms responsible for transmission
- High-content imaging
Automated microscopy, which collects images of cellular monolayers stained with antibodies. Computer algorithms are then used to automatically identify features such as number of cells in mitosis, number of cells, size of cells, or aberrant shape
- Eradication
Eradication is the complete removal of malaria parasites so that there is no transmission worldwide
- Elimination
Elimination means no local transmission of malaria
- Lead compound
This is a chemical compound often discovered in a screen that has pharmacological or biological activity. Its chemical structure is used as a starting point for chemical modifications that improve potency, selectivity, or pharmacokinetic parameters
- Hypnozoite
The dormant liver stage form of the parasite that forms when some P. vivax and P. ovale sporozoites invade hepatocytes. The hypnozoite does not replicate but can become activated weeks, months or years later, resulting in a blood-stage infection
- Merozoite
This is the infectious parasite that is released when a blood-stage schizont ruptures. The merozoite can bind to and invade an erythrocyte in a matter of seconds
- Pharmacophore
The core of a molecule that has unique atoms and has specific pharmacological properties
- Ookinete
This is the motile parasite form that develops from the zygote. Ookinetes are tetraploid as a result of meiosis in the zygote and develops into an oocyst on the midgut wall
- Radical cure
Treatment that eliminates the hypnozoite form of the parasite and thus prevents relapse with P. vivax or P. ovale infections
- Scaffold
This is the fixed part of a lead molecule on which chemical functional groups are substituted or exchanged
- Schizonts
Malaria parasites that have completed the process of DNA replication and syncytial nuclear division but which are still contained within a single red cell or hepatocyte
- Single Exposure, Radical Cure and Prophylaxis (SERCaP)
Treatment that would only need to be administered in one dose and which would eliminate blood-stage parasites (alleviating symptoms of malaria), and kill hypnozoites, preventing new infection from developing
- Sporozoite
The motile infectious form that is transmitted from the mosquito to the human, where they migrate from the dermis to a blood vessel and eventually invade a liver cell
- Standard membrane-feeding assay
An assay used to determine if a blood culture contains gametocytes that are infectious to mosquitoes, in which mosquitoes feed on human blood infected with P. falciparum parasites that is covered with parafilm is used
- Structure-activity relationship (SAR)
Relationship between the chemical structure of a molecule and its biological activity. The analysis of SAR allows scientists to identify the chemical groups responsible for a compound’s biological activity
- Target product profile
This is a set of guidelines that describes the ideal product. For antimalarial drugs, it might include pharmacokinetic and pharmocodynamics parameters, oral bioavailability, cost, potency, and activity against different lifecycle stages
Biographies
Erika Flannery Ph.D., M.P.H. is a Postdoctoral Fellow in the laboratory of Dr. Elizabeth Winzeler at the University of California San Diego School of Medicine. She received her Bachelor of Science in Cellular and Molecular Biology in 2004 from the University of Michigan. She earned her M.P.H. in Hospital and Molecular Epidemiology and her Ph.D. in Epidemiology from the University of Michigan School of Public Health where she studied horizontal gene transfer in the laboratory of Dr. Harry L.T. Mobley and host colonization rates with multi-drug resistant organisms with Dr. Betsy Foxman and Dr. Lona Mody. She is currently a Ruth L. Kirschstein NRSA award recipient. Her research interests include combining genomics and epidemiology to effectively identify pathogen factors contributing to antimalarial drug resistance in Plasmodium species.
Arnab K. Chatterjee Ph.D. completed a Bachelor of Arts in Chemistry and minor in Business from Northwestern University in 1997 and joined the California Institute of Technology in October 1998 to conduct his doctoral thesis research in the laboratory of Professor Robert H. Grubbs in the Division of Chemistry and Chemical Engineering. Upon completion of his doctoral research in September 2002, Dr. Chatterjee joined the Genomics Institute of the Novartis Research Foundation (GNF) as a Principal Investigator in the chemistry department. Since May 2012 he has been responsible for setting up and leading the chemistry group at the new California Institute of Biomedical Research in La Jolla, CA working across a wide-variety of disease areas. His research interests include application of novel synthetic methods to expedite the structural diversification in medicinal chemistry and cell-based lead optimization.
Elizabeth A. Winzeler Ph.D. received her Bachelor of Arts degree in Natural Sciences and Art from Lewis and Clark College in 1984. She obtained a Ph.D. in 1996 from the Department of Developmental Biology at the Stanford University School of Medicine. This was followed by a postdoctoral fellowship in the Department of Biochemistry. In 1999 she moved to San Diego to take a joint appointment at the Scripps Research Institute and the Genomics Institute of the Novartis Research Foundation (GNF). At GNF she developed a malaria drug discovery program and screening methods that have resulted in the identification of several novel antimalarial chemotypes. She recently moved to the University of California, San Diego, School of Medicine where she is a professor in the Department of Pediatrics.
References
- 1.Singh B, Daneshvar C. Human infections and detection of Plasmodium knowlesi. Clin Microbiol Rev. 2013;26:165–84. doi: 10.1128/CMR.00079-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. World Malaria Report 2012. World Health Organization; Geneva: 2012. pp. 1–276. [Google Scholar]
- 3.Murray CJ, et al. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–31. doi: 10.1016/S0140-6736(12)60034-8. This analysis is noteworthy in that it suggests that the number of malaria fatalities are underreported by the World Health Organization. [DOI] [PubMed] [Google Scholar]
- 4.Beutler E. The hemolytic effect of primaquine and related compounds: a review. Blood. 1959;14:103–39. [PubMed] [Google Scholar]
- 5.Guerra CA, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4:e774. doi: 10.1371/journal.pntd.0000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanner M, Savigny Dd. Malaria eradication back on the table. Bulletin of the World Health Organization. 2008;86:82–82. doi: 10.2471/BLT.07.050633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kappe SH, Vaughan AM, Boddey JA, Cowman AF. That was then but this is now: malaria research in the time of an eradication agenda. Science. 2010;328:862–6. doi: 10.1126/science.1184785. [DOI] [PubMed] [Google Scholar]
- 8.Alonso P, et al. A research agenda for malaria eradication: drugs. PLoS Med. 2011;8:e1000402. doi: 10.1371/journal.pmed.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burrows JN, Hooft van Huijsduijnen R, Mohrle JJ, Oeuvray C, Wells TN. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peatey CL, Spicer TP, Hodder PS, Trenholme KR, Gardiner DL. A high-throughput assay for the identification of drugs against late-stage Plasmodium falciparum gametocytes. Mol Biochem Parasitol. 2011;180:127–31. doi: 10.1016/j.molbiopara.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Dembele L, et al. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PLoS One. 2011;6:e18162. doi: 10.1371/journal.pone.0018162. This describes the development of a medium throughput assay that can be used to identify compounds with possible radical cure activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchholz K, et al. A high-throughput screen targeting malaria transmission stages opens new avenues for drug development. J Infect Dis. 2011;203:1445–53. doi: 10.1093/infdis/jir037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka TQ, Williamson KC. A malaria gametocytocidal assay using oxidoreduction indicator, alamarBlue. Mol Biochem Parasitol. 2011;177:160–3. doi: 10.1016/j.molbiopara.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucantoni L, Avery V. Whole-cell in vitro screening for gametocytocidal compounds. Future Med Chem. 2012;4:2337–60. doi: 10.4155/fmc.12.188. [DOI] [PubMed] [Google Scholar]
- 15.Derbyshire ER, Mota MM, Clardy J. The next opportunity in anti-malaria drug discovery: the liver stage. PLoS Pathog. 2011;7:e1002178. doi: 10.1371/journal.ppat.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–19. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 17.Macarron R, et al. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov. 2011;10:188–95. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 18.Gamo FJ, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–10. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 19.Guiguemde WA, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–5. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plouffe D, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A. 2008;105:9059–64. doi: 10.1073/pnas.0802982105. A description of the first ultrahighthroughput cellular screen for antimalarials and the source of methods for one of the publicly-available antimalarial datasets ( http://www.ebi.ac.uk/chemblntd) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–6. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guiguemde WA, et al. Global phenotypic screening for antimalarials. Chem Biol. 2012;19:116–29. doi: 10.1016/j.chembiol.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeung BK, et al. Spirotetrahydro beta-carbolines (spiroindolones): a new class of potent and orally efficacious compounds for the treatment of malaria. J Med Chem. 2010;53:5155–64. doi: 10.1021/jm100410f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rottmann M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–80. doi: 10.1126/science.1193225. Description and target discovery for the first new class of antimalarials to enter clinical trials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Pelt-Koops JC, et al. The spiroindolone drug candidate NITD609 potently inhibits gametocytogenesis and blocks Plasmodium falciparum transmission to anopheles mosquito vector. Antimicrob Agents Chemother. 2012;56:3544–8. doi: 10.1128/AAC.06377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spillman NJ, et al. Na(+) Regulation in the Malaria Parasite Plasmodiumfalciparum Involves the Cation ATPase PfATP4 and Is a Target of the Spiroindolone Antimalarials. Cell Host Microbe. 2013;13:227–37. doi: 10.1016/j.chom.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tse MT. Antimalarial drugs: Speeding to a new lead. Nat Rev Drug Discov. 2010;9:842. doi: 10.1038/nrd3301. [DOI] [PubMed] [Google Scholar]
- 28.Calderon F, et al. An Invitation to Open Innovation in Malaria Drug Discovery: 47 Quality Starting Points from the TCAMS. ACS Medicinal Chemistry Letters. 2011;2:741–746. doi: 10.1021/ml200135p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calderon F, et al. A Divergent SAR Study Allows Optimization of a Potent 5-HT2c Inhibitor to a Promising Antimalarial Scaffold. ACS Medicinal Chemistry Letters. 2012;3:373–377. doi: 10.1021/ml300008j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rueda L, et al. Cyclopropyl Carboxamides: A New Oral Antimalarial Series Derived from the Tres Cantos Anti-Malarial Set (TCAMS) ACS Medicinal Chemistry Letters. 2011;2:840–844. doi: 10.1021/ml2001517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanz LM, et al. Cyclopropyl Carboxamides, a Chemically Novel Class of Antimalarial Agents Identified in a Phenotypic Screen. Antimicrobial agents and chemotherapy. 2011;55:5740–5745. doi: 10.1128/AAC.05188-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris CJ, Hill RD, Sheppard DW, Slater MJ, Stouten PF. The design and application of target-focused compound libraries. Comb Chem High Throughput Screen. 2011;14:521–31. doi: 10.2174/138620711795767802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duffy S, Avery VM. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am J Trop Med Hyg. 2012;86:84–92. doi: 10.4269/ajtmh.2012.11-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Younis Y, et al. 3,5-Diaryl-2-aminopyridines as a novel class of orally active antimalarials demonstrating single dose cure in mice and clinical candidate potential. J Med Chem. 2012;55:3479–87. doi: 10.1021/jm3001373. A novel class of compounds with potent cellular activity and a long half-life are described. [DOI] [PubMed] [Google Scholar]
- 35.Eastman RT, et al. A class of tricyclic compounds blocking malaria parasite oocyst development and transmission. Antimicrob Agents Chemother. 2013;57:425–35. doi: 10.1128/AAC.00920-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang W, Luo M, Zhou M, Pan X. Study on the treatment of Plasmodium cynomolgi infections of the macaque with ketotifen] Yao xue xue bao= Acta pharmaceutica Sinica. 1987;22:409. [PubMed] [Google Scholar]
- 37.Singh N, Puri S. Inhibition of the development of the hepatic stages of Plasmodium yoelii nigeriensis by antihistaminic agents. Annals of tropical medicine and parasitology. 1999;93:419. doi: 10.1080/00034989958438. [DOI] [PubMed] [Google Scholar]
- 38.Milner E, et al. Ketotifen is an antimalarial prodrug of norketotifen with blood schizonticidal and liver-stage efficacy. Eur J Drug Metab Pharmacokinet. 2012;37:17–22. doi: 10.1007/s13318-012-0080-2. [DOI] [PubMed] [Google Scholar]
- 39.Mazier D, Renia L, Snounou G. A pre-emptive strike against malaria’s stealthy hepatic forms. Nat Rev Drug Discov. 2009;8:854–64. doi: 10.1038/nrd2960. [DOI] [PubMed] [Google Scholar]
- 40.Carraz Ml, et al. A plant-derived morphinan as a novel lead compound active against malaria liver stages. PLoS medicine. 2006;3:e513. doi: 10.1371/journal.pmed.0030513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meister S, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science. 2011;334:1372–7. doi: 10.1126/science.1211936. A medium throughput screen is described that further delineates blood stage antimalarials into those with likely causal prophylactic activity and those without. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagle A, et al. Imidazolopiperazines: lead optimization of the second-generation antimalarial agents. J Med Chem. 2012;55:4244–73. doi: 10.1021/jm300041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu T, et al. Imidazolopiperazines: hit to lead optimization of new antimalarial agents. J Med Chem. 2011;54:5116–30. doi: 10.1021/jm2003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Derbyshire ER, Prudencio M, Mota MM, Clardy J. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc Natl Acad Sci U S A. 2012;109:8511–6. doi: 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.da Cruz FP, et al. Drug Screen Targeted at Plasmodium Liver Stages Identifies a Potent Multistage Antimalarial Drug. Journal of Infectious Diseases. 2012;205:1278–1286. doi: 10.1093/infdis/jis184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yekutiel P, Yekutiel P. In: III The Global Malaria Eradication Campaign. MAK, editor. S. Karger AG; Arnold-Böcklin-Strasse 25, CH-4011, Basel, Switzerland., Basel, Switzerland: 1980. 1980. [Google Scholar]
- 47.Schmidt LH, Fradkin R, Vaughan D, Rasco J. Radical cure of infections with Plasmodium cynomolgi: a function of total 8-aminoquinoline dose. Am J Trop Med Hyg. 1977;26:1116–28. doi: 10.4269/ajtmh.1977.26.1116. [DOI] [PubMed] [Google Scholar]
- 48.Vale N, Moreira R, Gomes P. Primaquine revisited six decades after its discovery. Eur J Med Chem. 2009;44:937–53. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 49.Chiang TY, Lin WC, Kuo MC, Ji DD, Fang CT. Relapse of imported vivax malaria despite standard-dose primaquine therapy: an investigation with molecular genotyping analyses. Clinical Microbiology and Infection. 2012;18:E232–E234. doi: 10.1111/j.1469-0691.2012.03820.x. [DOI] [PubMed] [Google Scholar]
- 50.Bright AT, et al. Genetic Analysis of Primaquine Tolerance in a Patient with Relapsing Vivax Malaria. Emerging infectious diseases. 2013;19:802. doi: 10.3201/eid1905.121852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Alessandro S, et al. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J Antimicrob Chemother. 2013 doi: 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- 52.Delves MJ, et al. A high-throughput assay for the identification of malarial transmission-blocking drugs and vaccines. Int J Parasitol. 2012;42:999–1006. doi: 10.1016/j.ijpara.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 53.Blagborough AM, et al. Assessing transmission blockade in Plasmodium spp. Methods Mol Biol. 2013;923:577–600. doi: 10.1007/978-1-62703-026-7_40. [DOI] [PubMed] [Google Scholar]
- 54.Vennerstrom JL, et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–4. doi: 10.1038/nature02779. A classic medicinal chemistry story describing the design and synthesis of the synthetic ozonide class that would later give rise to OZ439 and related molecules. [DOI] [PubMed] [Google Scholar]
- 55.Klonis N, et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A. 2011;108:11405–10. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olliaro P, Wells TN. The global portfolio of new antimalarial medicines under development. Clin Pharmacol Ther. 2009;85:584–95. doi: 10.1038/clpt.2009.51. [DOI] [PubMed] [Google Scholar]
- 57.Enserink M. If artemisinin drugs fail, what’s plan B? Science. 2010;328:846. doi: 10.1126/science.328.5980.846. [DOI] [PubMed] [Google Scholar]
- 58.Charman SA, et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A. 2011;108:4400–5. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delves M, et al. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS medicine. 2012;9:e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moehrle JJ, et al. First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol. 2013;75:524–37. doi: 10.1111/j.1365-2125.2012.04368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Neill PM, et al. Identification of a 1,2,4,5-tetraoxane antimalarial drug-development candidate (RKA 182) with superior properties to the semisynthetic artemisinins. Angew Chem Int Ed Engl. 2010;49:5693–7. doi: 10.1002/anie.201001026. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, et al. Comparative antimalarial activities and ADME profiles of ozonides (1,2,4-trioxolanes) OZ277, OZ439, and their 1,2-dioxolane, 1,2,4-trioxane, and 1,2,4,5-tetraoxane isosteres. J Med Chem. 2013;56:2547–55. doi: 10.1021/jm400004u. [DOI] [PubMed] [Google Scholar]
- 63.Peters W, Robinson BL, Milhous WK. The chemotherapy of rodent malaria. LI. Studies on a new 8-aminoquinoline, WR 238,605. Ann Trop Med Parasitol. 1993;87:547–52. doi: 10.1080/00034983.1993.11812809. [DOI] [PubMed] [Google Scholar]
- 64.Nanayakkara NP, et al. Antiparasitic activities and toxicities of individual enantiomers of the 8-aminoquinoline 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-4-methyl-5-[3,4-dichlorophenoxy]quinol ine succinate. Antimicrob Agents Chemother. 2008;52:2130–7. doi: 10.1128/AAC.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baird JK. Resistance to therapies for infection by Plasmodium vivax. Clin Microbiol Rev. 2009;22:508–34. doi: 10.1128/CMR.00008-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pybus BS, et al. CYP450 phenotyping and accurate mass identification of metabolites of the 8-aminoquinoline, anti-malarial drug primaquine. Malar J. 2012;11:259. doi: 10.1186/1475-2875-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winter R, et al. Optimization of endochin-like quinolones for antimalarial activity. Exp Parasitol. 2011;127:545–51. doi: 10.1016/j.exppara.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nilsen A, et al. Quinolone-3-Diarylethers: A New Class of Antimalarial Drug. Science Translational Medicine. 2013;5:177ra37. doi: 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muregi FW, Ishih A. Next-Generation Antimalarial Drugs: Hybrid Molecules as a New Strategy in Drug Design. Drug Dev Res. 2010;71:20–32. doi: 10.1002/ddr.20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kelly JX, et al. Discovery of dual function acridones as a new antimalarial chemotype. Nature. 2009;459:270–3. doi: 10.1038/nature07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dubar F, et al. The antimalarial ferroquine: role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem Biol. 2011;6:275–87. doi: 10.1021/cb100322v. [DOI] [PubMed] [Google Scholar]
- 72.Dubar F, Khalife J, Brocard J, Dive D, Biot C. Ferroquine, an ingenious antimalarial drug: thoughts on the mechanism of action. Molecules. 2008;13:2900–2907. doi: 10.3390/molecules13112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White NJ. Cardiotoxicity of antimalarial drugs. Lancet Infect Dis. 2007;7:549–558. doi: 10.1016/S1473-3099(07)70187-1. [DOI] [PubMed] [Google Scholar]
- 74.Fermini B, Fossa AA. The impact of drug-induced QT interval prolongation on drug discovery and development. Nature Reviews Drug Discovery. 2003;2:439–447. doi: 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]
- 75.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. Classic study on the perils of target-based drug discovery. Must read. [DOI] [PubMed] [Google Scholar]
- 76.Yuthavong Y, et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc Natl Acad Sci U S A. 2012;109:16823–8. doi: 10.1073/pnas.1204556109. Beautiful examples of how target-based drug discovery can work. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anthony MP, Burrows JN, Duparc S, Moehrle JJ, Wells TN. The global pipeline of new medicines for the control and elimination of malaria. Malar J. 2012;11:316. doi: 10.1186/1475-2875-11-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ittarat I, Asawamahasakda W, Meshnick SR. The effects of antimalarials on the Plasmodium falciparum dihydroorotate dehydrogenase. Exp Parasitol. 1994;79:50–6. doi: 10.1006/expr.1994.1058. [DOI] [PubMed] [Google Scholar]
- 79.Reyes P, et al. Enzymes of purine and pyrimidine metabolism from the human malaria parasitePlasmodium falciparum. Mol Biochem Parasitol. 1982;5:275–290. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- 80.Baldwin J, et al. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280:21847–53. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 81.Gujjar R, et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J Med Chem. 2009;52:1864–72. doi: 10.1021/jm801343r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gujjar R, et al. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem. 2011;54:3935–49. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Coteron JM, et al. Structure-Guided Lead Optimization of Triazolopyrimidine-Ring Substituents Identifies Potent Plasmodium falciparum Dihydroorotate Dehydrogenase Inhibitors with Clinical Candidate Potential. Journal of Medicinal Chemistry. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patel V, et al. Identification and characterization of small molecule inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2008;283:35078–85. doi: 10.1074/jbc.M804990200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Booker ML, et al. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J Biol Chem. 2010;285:33054–64. doi: 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kortagere S, et al. Structure-based design of novel small-molecule inhibitors of Plasmodium falciparum. J Chem Inf Model. 2010;50:840–9. doi: 10.1021/ci100039k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Medicines for Malaria Venture. ( http://www.mmv.org)
- 88.Dharia NV, et al. Use of high-density tiling microarrays to identify mutations globally and elucidate mechanisms of drug resistance in Plasmodium falciparum. Genome Biol. 2009;10:R21. doi: 10.1186/gb-2009-10-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoepfner D, et al. Selective and specific inhibition of the plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–63. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Istvan ES, et al. Validation of isoleucine utilization targets in Plasmodium falciparum. Proc Natl Acad Sci U S A. 2011;108:1627–32. doi: 10.1073/pnas.1011560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong CK, et al. Identification and validation of tetracyclic benzothiazepines as Plasmodium falciparum cytochrome bc1 inhibitors. Chem Biol. 2011;18:1602–10. doi: 10.1016/j.chembiol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nam TG, et al. A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem Biol. 2011;6:1214–22. doi: 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhou H, Sun L, Yang X-L, Schimmel P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature. 2012 doi: 10.1038/nature11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Derbyshire ER, Mazitschek R, Clardy J. Characterization of Plasmodium liver stage inhibition by halofuginone. ChemMedChem. 2012;7:844–849. doi: 10.1002/cmdc.201200045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crowther GJ, et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol Biochem Parasitol. 2011;175:21–9. doi: 10.1016/j.molbiopara.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wells TN. Natural products as starting points for future anti-malarial therapies: going back to our roots. Malar J. 2011;10:S3. doi: 10.1186/1475-2875-10-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mesia K, et al. Antimalarial activity and toxicity evaluation of a quantified Nauclea pobeguinii extract. Journal of ethnopharmacology. 2010;131:10–16. doi: 10.1016/j.jep.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 98.Mesia K, et al. Antimalarial efficacy of a quantified extract of Nauclea pobeguinii stem bark in human adult volunteers with diagnosed uncomplicated falciparum malaria. part 1: A clinical phase IIA trial. Planta medica. 2011;78:211–218. doi: 10.1055/s-0031-1280359. [DOI] [PubMed] [Google Scholar]
- 99.Xu YJ, et al. Chromatographic profiling and identification of two new iridoid-indole alkaloids by UPLC-MS and HPLC-SPE-NMR analysis of an antimalarial extract from Nauclea pobeguinii. Phytochemistry Letters. 2012;5:316–319. [Google Scholar]
- 100.Graz B, et al. Argemone mexicana decoction versus artesunate-amodiaquine for the management of malaria in Mali: policy and public-health implications. Trans R Soc Trop Med Hyg. 2010;104:33–41. doi: 10.1016/j.trstmh.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 101.Abdulla S, Sagara I. Dispersible formulation of artemether/lumefantrine: specifically developed for infants and young children. Malar J. 2009;8 (Suppl 1):S7. doi: 10.1186/1475-2875-8-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tshefu AK, et al. Efficacy and safety of a fixed-dose oral combination of pyronaridine-artesunate compared with artemether-lumefantrine in children and adults with uncomplicated Plasmodium falciparum malaria: a randomised non-inferiority trial. The Lancet. 2010;375:1457–1467. doi: 10.1016/S0140-6736(10)60322-4. [DOI] [PubMed] [Google Scholar]
- 103.Chandra RS, et al. Creative solutions to extraordinary challenges in clinical trials: methodology of a phase III trial of azithromycin and chloroquine fixed-dose combination in pregnant women in Africa. Malaria journal. 2013;12:1–8. doi: 10.1186/1475-2875-12-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Keating GM. Dihydroartemisinin/Piperaquine: a review of its use in the treatment of uncomplicated Plasmodium falciparum malaria. Drugs. 2012;72:937–61. doi: 10.2165/11203910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 105.Schellenberg D, et al. The safety and efficacy of sulfadoxine-pyrimethamine, amodiaquine, and their combination in the treatment of uncomplicated Plasmodium falciparum malaria. The American journal of tropical medicine and hygiene. 2002;67:17–23. doi: 10.4269/ajtmh.2002.67.17. [DOI] [PubMed] [Google Scholar]
- 106.McGraw EA, O’Neill SL. Beyond insecticides: new thinking on an ancient problem. Nat Rev Microbiol. 2013;11:181–93. doi: 10.1038/nrmicro2968. [DOI] [PubMed] [Google Scholar]
- 107.Bian G, et al. Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science. 2013;340:748–51. doi: 10.1126/science.1236192. The authors present a novel, alternative approach to using drugs to control malaria. [DOI] [PubMed] [Google Scholar]
- 108.Agnandji ST, et al. First results of phase 3 trial of RTS, S/AS01 malaria vaccine in African children. N Engl J Med. 2011;365:1863. doi: 10.1056/NEJMoa1102287. [DOI] [PubMed] [Google Scholar]
- 109.D’Alessandro U. A phase 3 trial of RTS, S/AS01 malaria vaccine in African infants. Name: New England Journal of Medicine. 2012;367:2284–2295. doi: 10.1056/NEJMoa1208394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Riley EM, Stewart VA. Immune mechanisms in malaria: new insights in vaccine development. Nat Med. 2013;19:168–78. doi: 10.1038/nm.3083. [DOI] [PubMed] [Google Scholar]
- 111.WHO. Geneva: 2010. [Google Scholar]
- 112.Prudencio M, Rodriguez A, Mota MM. The silent path to thousands of merozoites: the Plasmodium liver stage. Nat Rev Microbiol. 2006;4:849–56. doi: 10.1038/nrmicro1529. [DOI] [PubMed] [Google Scholar]
- 113.Dixon MW, Thompson J, Gardiner DL, Trenholme KR. Sex in Plasmodium: a sign of commitment. Trends Parasitol. 2008;24:168–75. doi: 10.1016/j.pt.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 114.Mantel PY, et al. Malaria-Infected Erythrocyte-Derived Microvesicles Mediate Cellular Communication within the Parasite Population and with the Host Immune System. Cell Host Microbe. 2013;13:521–34. doi: 10.1016/j.chom.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Regev-Rudzki N, et al. Cell-Cell Communication between Malaria-Infected Red Blood Cells via Exosome-like Vesicles. Cell. 2013;153:1120–33. doi: 10.1016/j.cell.2013.04.029. [DOI] [PubMed] [Google Scholar]
- 116.Aly AS, Vaughan AM, Kappe SH. Malaria parasite development in the mosquito and infection of the mammalian host. Annu Rev Microbiol. 2009;63:195–221. doi: 10.1146/annurev.micro.091208.073403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dondorp AM, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Phyo AP, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. The Lancet. 2012;379:1960–1966. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cheeseman IH, et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012;336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miotto O, et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat Genet. 2013 doi: 10.1038/ng.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Takala-Harrison S, et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci U S A. 2013;110:240–5. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mok S, et al. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics. 2011;12:391. doi: 10.1186/1471-2164-12-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mwakingwe A, et al. Noninvasive real-time monitoring of liver-stage development of bioluminescent Plasmodium parasites. Journal of Infectious Diseases. 2009;200:1470–1478. doi: 10.1086/606115. [DOI] [PubMed] [Google Scholar]
- 124.Schmidt LH. Relationships between chemical structures of 8-aminoquinolines and their capacities for radical cure of infections with Plasmodium cynomolgi in rhesus monkeys. Antimicrob Agents Chemother. 1983;24:615–52. doi: 10.1128/aac.24.5.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Russell BM, et al. Simple in vitro assay for determining the sensitivity of Plasmodium vivax isolates from fresh human blood to antimalarials in areas where P. vivax is endemic. Antimicrobial agents and chemotherapy. 2003;47:170–173. doi: 10.1128/AAC.47.1.170-173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jimenez-Diaz MB, et al. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob Agents Chemother. 2009;53:4533–6. doi: 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lelievre J, et al. Activity of clinically relevant antimalarial drugs on Plasmodium falciparum mature gametocytes in an ATP bioluminescence "transmission blocking" assay. PLoS One. 2012;7:e35019. doi: 10.1371/journal.pone.0035019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Adjalley SH, et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc Natl Acad Sci U S A. 2011;108:E1214–23. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Miura K, et al. Qualification of standard membrane-feeding assay with Plasmodium falciparum malaria and potential improvements for future assays. PLoS One. 2013;8:e57909. doi: 10.1371/journal.pone.0057909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Churcher TS, et al. Measuring the blockade of malaria transmission--an analysis of the Standard Membrane Feeding Assay. Int J Parasitol. 2012;42:1037–44. doi: 10.1016/j.ijpara.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 131.Cassera MB, et al. Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model. PLoS One. 2011;6:e26916. doi: 10.1371/journal.pone.0026916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Crockett M, Kain KC. Tafenoquine: a promising new antimalarial agent. Expert Opinion on Investigational Drugs. 2007;16:705–715. doi: 10.1517/13543784.16.5.705. [DOI] [PubMed] [Google Scholar]
- 133.Mesia K, et al. Antimalarial efficacy of a quantified extract of Nauclea pobeguinii stem bark in human adult volunteers with diagnosed uncomplicated falciparum malaria. Part 2: a clinical phase IIB trial. Planta Med. 2012;78:853–60. doi: 10.1055/s-0031-1298488. [DOI] [PubMed] [Google Scholar]