

Abstract
RNA molecules transmit the information encoded in the genome and generally reflect its content. Adenosine-to-inosine (A-to-I) RNA editing by ADAR proteins converts a genomically encoded adenosine into inosine. It is known that most RNA editing in human takes place in the primate-specific Alu sequences, but the extent of this phenomenon and its effect on transcriptome diversity are not yet clear. Here, we analyzed large-scale RNA-seq data and detected ∼1.6 million editing sites. As detection sensitivity increases with sequencing coverage, we performed ultradeep sequencing of selected Alu sequences and showed that the scope of editing is much larger than anticipated. We found that virtually all adenosines within Alu repeats that form double-stranded RNA undergo A-to-I editing, although most sites exhibit editing at only low levels (<1%). Moreover, using high coverage sequencing, we observed editing of transcripts resulting from residual antisense expression, doubling the number of edited sites in the human genome. Based on bioinformatic analyses and deep targeted sequencing, we estimate that there are over 100 million human Alu RNA editing sites, located in the majority of human genes. These findings set the stage for exploring how this primate-specific massive diversification of the transcriptome is utilized.
Consistency of genomic information flow is a basic concept in biology. In general, it is believed that the processed content of a gene (RNA) has the exact same sequence as its original DNA template. However, adenosine deaminases acting on RNA (ADARs), an essential family of RNA-modifying enzymes, can edit nucleotides in the RNA (Savva et al. 2012). Specifically, these enzymes can modify a genetically encoded adenosine (A) into an inosine (I) in double-stranded RNA structures. ADAR editing results in inosine, which replaces the genomically encoded adenosine, and is read by the cellular machinery as a guanosine (G) (Bass 2002; Nishikura 2010). Thus, sequencing of inosine-containing RNAs results in G where the corresponding genomic DNA reads A. The progress in sequencing techniques in recent years has brought about many reports of A-to-I editing in the human genome (Li et al. 2009; Bahn et al. 2012; Park et al. 2012; Peng et al. 2012; Ramaswami et al. 2012, 2013). These studies have identified a growing number of A-to-G mismatches in mRNA-sequencing data aligned to the genome, and used various algorithmic techniques to identify those mismatches originating from A-to-I editing. Analyses of various data sets have resulted in identification of thousands, and up to hundreds of thousands of editing sites. However, the overlap between the many reported sets is quite low (see Supplemental Tables 1, 2; Ramaswami et al. 2012), suggesting that the reported sites do not reflect the full scope of the A-to-I editing phenomenon.
The primate specific Alu sequences are the dominant short interspersed nuclear element (SINEs) in the primate genomes (International Human Genome Sequencing Consortium 2001; Cordaux and Batzer 2009). Humans have about a million copies of Alu, roughly 300 bp long each, accounting for ∼10% of their genome. Since these repeats are so common, especially in gene-rich regions (Korenberg and Rykowski 1988), pairing of two oppositely oriented Alus located in the same pre-mRNA structure is likely. Such pairing produces a long and stable dsRNA structure, an ideal target for the ADARs. Indeed, recent studies have shown that Alu repeats account for >99% of editing events found so far in humans (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004; Ramaswami et al. 2012, 2013).
Edited Alu sequences typically include a number of clustered edited sites (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004). This feature may be utilized to distinguish bona fide editing events from sequencing errors or misalignments due to duplication or genomic variability. Here, we refined the detection approach, focusing only on Alu editing, which allowed us to exploit the clustering property of editing sites. Analysis of large-scale RNA-seq data supplemented by targeted sequencing of Alu elements revealed that the majority of Alu elements form editable dsRNA structures, and nearly all adenosines expressed in such Alu repeats undergo A-to-I editing.
Results
To identify Alu RNA editing sites, we used two large RNA sequencing (RNA-seq) data sets. One is the Illumina Human Body Map (HBM) data comprising 16 tissues, and the other is the previously published deeply sequenced Han Chinese (YH) data (Peng et al. 2012). We aligned these two RNA data sets to the human genome (see Methods) and recorded all mismatches between the reads and the genomic reference within Alu repeats. All reads were aligned to the reference strand, and thus editing sites could appear as A-to-G if transcription is from the reference strand, or T-to-C, if transcription is from the reverse strand (hereby referred to as AG and TC, respectively). A total of 919,035 Alu elements (78.2% of all Alu elements in the human genome) were covered (see Methods). We used an algorithmic approach (Fig. 1; see Methods for details) that takes into account clustering of editing sites and removes misaligned reads, low-quality base calling, and known polymorphisms, to identify 1,586,270 editing sites (AG or TC mismatches) in 305,337 Alu repeats, the largest set reported to date (Fig. 2). The false positive rate is estimated by the number of mismatches of types G-to-A and C-to-T (GA and CT, respectively) detected by the same set of parameters, and is estimated to be 2.0%. Part of the YH data set is strand specific and allows us to verify that most of the identified mismatches are indeed AG (in the expressed strand): 478,301 AG sites compared with only 17,838 TC sites.
Figure 1.

Detection of A-to-I editing in Alu repeats. (A) Multiple alignment of reads to the reference genome reveals sites of A-to-I editing (red), as well as genomic polymorphisms and sequencing errors (yellow). Detection sensitivity is improved upon examining clusters of mismatches rather than looking at each site independently. Yet, at low coverage, many bona fide editing sites either do not show any AG mismatch, or show a weak signal indistinguishable from sequencing errors. The sites detected include the few strongly edited sites and a random sample of the weaker sites. (B) Ultradeep coverage enables the full scope of editing to be revealed, showing all sites that support editing, typically at very low levels (<1%).
Figure 2.

Mismatch distributions along the detection pipeline. (A) Even a simple count of all mismatches in high-quality base pairs of sequencing reads data of Alu repeats shows a significant enrichment of editing-derived mismatch types (AG and TC). (B) Applying a strict statistical model to filter out probable sequencing errors further increases the fraction of AG/TC mismatches, but results in the loss of most of the estimated true editing signal as well. (C) In this study, we focused on the full Alu repeats rather than single genomic sites. This improves the statistical power, with only a minor reduction in the signal. As a result, we found that virtually all Alu repeats are dominated by AG/TC mismatches. (D–F) The same pipeline applied to mismatches located in the common L1 retroelement. Clearly, the strong propensity for A-to-I RNA editing is unique to the Alu repeat. However, some enrichment of AG/TC mismatches is nevertheless observed, attesting to some editing activity in the L1 repeats.
Control: Mismatches in L1 (LINE-1) repeats
Nearly half of the human genome is composed of mobile elements. However, editing takes place almost exclusively in the Alu repeat due to its exceptional ability to form dsRNA structures (Morse et al. 2002; Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004). To verify that our results, showing vast numbers of editing sites, are indeed unique to the Alu repeats, and to exclude the possibility of sequencing errors leading to AG and TC clusters, we applied the identical editing detection algorithm (see Methods) to another type of retroelements, the L1 repeats, which are also very common in the human genome, with nearly a million copies, accounting for 18% of the human genome.
Overall, 603,850 (63.44%) L1 elements were covered (most were partially covered; see Supplemental Fig. 1), yielding a total of 2,713,306 mismatch locations. Applying the same filtering and clustering approach, we found 74,926 L1 elements having a dominant mismatch type. While we did observe some enrichment of AG/TC mismatches (corresponding, probably, to low levels of A-to-I editing in L1 repeats; Athanasiadis et al. 2004), the number of such sites is two orders of magnitude lower than the corresponding number in Alu repeats (using the same expression data; note, however, that L1 repeats are less deeply covered, as they tend to reside in intergenic regions; see Supplemental Figs. 1, 2). Furthermore, no clear signal is seen when looking for repeats with a single dominant mismatch type (see Fig. 2; Supplemental Fig. 3). These results strongly support the idea that massive A-to-I editing is limited to the Alu repeats.
Editing site motif
According to previous studies (Lehmann and Bass 2000; Eggington et al. 2011), the editing site has a strong aversion to G at the −1 position (upstream of the editing site), while it has preference for G at the +1 position (downstream). Our editing site set is in accordance with this preference (Fig. 3), and editing is more pronounced at sites with a stronger editing signal. In addition we have looked at the location of the editing sites with respect to the Alu sequence consensus. The results are presented in Figure 4, for each of the eight most edited Alu families. Clearly, hotspots for editing are observed. As expected, Alu elements for which the expressed strand is the consensus strand undergo more editing due to the poly(A) regions which are targeted by ADARs (Carmi et al. 2011).
Figure 3.

Distribution of downstream (A) and upstream (B) nucleotides for editing sites detected in the HBM data sets. Edited sites are split into three groups according to their editing level: low level ≤10%, high level ≥40%, and medium level >10% and <40%. A clear signature of the ADAR sequence preference is observed (low G upstream of the site, and some enrichment downstream from the site). The preference is stronger at sites with high editing levels.
Figure 4.

Distribution of editing events along the consensus for the eight most edited Alu subfamilies (UCSC Genome Browser annotation). The number of edited Alu repeats of each family is given. Clearly, there are hotspots for editing in each of the families.
Noise is appreciably reduced upon using updated dbSNP data
SNPs that were deposited into dbSNP based on RNA data alone are called cDNA SNPs. Only a single GA site and no CT sites were found as known cDNA SNP sites, while the putative editing set contains 1146 AG and TC sites known as cDNA SNPs. These are likely to be RNA-editing sites mistakenly identified as genomic polymorphisms (Eisenberg et al. 2005; Gommans et al. 2008).
After we initiated this study, dbSNP was updated, and we used this to further validate our editing detection. Reassuringly, we found that only 0.69% (6,809) of the putative editing sites overlap the list of newly discovered gSNPs (dbSNP135 vs. dbSNP131), compared with 8.9% (1763) of GA and CT sites that passed our filter. This result is encouraging, suggesting much of our noise could be due to gSNPs yet to be annotated as rare SNPs.
Editing level by tissue
While known protein-modifying editing sites are enriched in synaptic genes (Rosenthal and Seeburg 2012) expressed mainly in the brain, Alu editing in the brain was not exceptionally high compared with other tissues (Fig. 5). The average editing level over all adenosines (fraction of Alu adenosines that are deaminated into inosines) is estimated by the fraction of read bases showing G (C), where the reference genome reads A (T) (before filtering). Notably, editing levels in the brain are high, but not exceptionally high compared with other tissues. The levels presented are underestimates due to two factors: First, hyperedited reads fail to align to the genome (Carmi et al. 2011). Second, as the data are not stranded, some of the reference genome reads could have been transcribed from the antisense strand.
Figure 5.
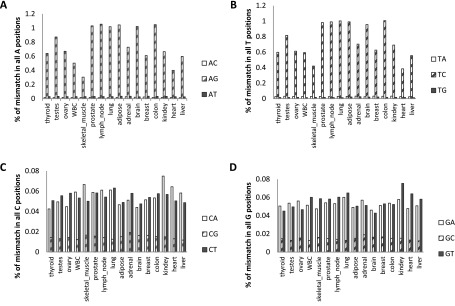
Average editing levels per tissue in HBM data. For each tissue, the total mismatches (before filtering) are grouped for each of the four bases and presented according to the mismatch type. Although in A (T) positions, only one type of mismatch is dominant (G or C, accordingly), at C and G the picture is very different, exhibiting a lower number of mismatches (note the different scale) with a more even distribution. (A) A reference positions with non-A reads, per tissue. (B) T reference positions with non-T reads, per tissue. (C) C reference positions with non-C reads, per tissue. (D) G reference positions with non-G reads, per tissue.
Detection efficacy increases with coverage
The more reads covering a given genomic site, the more accurate our estimate of the editing level in this site is. Specifically, a low level of editing requires more reads to guarantee its detection. It is thus expected that coverage limits the number of editing sites discovered. In order to estimate the scope of this effect, we looked at the fraction of sites detected as edited as a function of their coverage. Obviously, looking only at the raw number of mismatches, the number of all sites exhibiting mismatches of all types increases with coverage, due to the increased probability of finding a sequencing error in at least one of the covering reads. Interestingly, though, following our filtering and clustering procedure (see Methods), the number of most types of mismatches saturates (data shown for GA and CT only), but the number of sites showing AG and TC mismatches keeps growing even for very high coverage, which is attained in only a tiny fraction of the expressed transcriptome (Fig. 6A; Supplemental Fig. 4). This result suggests that, given sufficient coverage, the number of editing sites to be detected is a great deal larger than was reported previously.
Figure 6.

Editing detection is sensitive to sequencing coverage. (A) The average number of adenosines in an Alu repeat showing evidence for editing increases with the available coverage (number of reads supporting the examined nucleotide), with no sign of saturation (HBM data). A number of mismatch sites of types other than AG/TC saturate at a relatively low coverage (after applying the statistical model to filter sequencing errors). As the typical coverage in RNA-seq is much lower than 1000 reads, this suggests that previous counts of editing sites are grossly underestimated. (B) Fraction of Alu repeats showing evidence of editing (i.e., dominated by AG/TC mismatches). Again, strong dependence on coverage is observed, and atypically high coverage is required for detection in most of the Alu repeats. Our ultradeep MiSeq experiment reached saturation with all Alu repeats detected at a coverage of 1000 reads (coverage is defined as the median read coverage for the adenosines and thymines in the given Alu repeat). Based on these calculations, we estimate the total number of A-to-I editing sites in the human genome to exceed 100 million sites. (C) Number of different transcript variants per Alu, as a function of the reads' coverage. No saturation is observed even for ultrahigh coverage.
Ultradeep sequencing of selected Alu
The low overlap between the various reported data sets, including the present one (Supplemental Material), and the lack of saturation in the number of detected sites as a function of sequencing coverage (Fig. 6A; Supplemental Fig. 4) suggest that even the enlarged set of editing sites that we detected does not exhaust the full scope of the phenomenon. We thus set out to quantify the global potential of Alu editing. We started by using the editing sites found using the HBM data set to further understand the genomic architectural criteria required for Alu repeats to form a dsRNA and undergo editing. In agreement with previous studies (Athanasiadis et al. 2004; Blow et al. 2004; Kim et al. 2004; Levanon et al. 2004), we found (Supplemental Fig. 5) that the edited Alu repeats tend to have at least one neighboring inverted Alu (from any Alu subfamily) closer than 3500 bp, and are not highly divergent from the Alu consensus sequence (see Supplemental Material). We further hypothesized that all 761,244 Alu repeats that meet these criteria (henceforth termed “editable” Alus) are indeed edited if expressed, and may be detected if sufficiently covered by sequencing reads. We used several approaches to test this hypothesis. First, we identified 15,806 editable Alus that are located in RefSeq exons, amplified the sequences, and Sanger-sequenced 48 Alus randomly selected from this group in two tissues. Thirty of these 96 experiments resulted in amplicons that were successfully sequenced, 23 of which (77%) showed a clear editing signal. In addition, we used the deeply sequenced YH data set and found that, as coverage increases, the fraction of editable Alu repeats that is detected as edited approaches unity (Fig. 6B). Finally, we selected a subset of Alu elements for PCR amplification and carried out ultradeep sequencing.
To this end, we randomly chose 28 Alu elements from the editable Alu list that exhibited editing in the HBM data, and an additional 52 for which there was no evidence of editing. We amplified each of them by PCR using cDNA derived from the Human Brain Reference RNA as the template (see Methods). We sequenced these 80 Alus with ultrahigh coverage (75/80 were expressed and recovered in the tissue tested, with average and median coverage of 13,511 and 11,919 reads, respectively), and examined the detectable editing signal as a function of coverage. The unusually high coverage allowed for reliable and accurate quantification of low editing levels. Focusing on ultradeeply covered regions (>5000 reads), we observed a clear distinction between the AG/TC mismatches (likely to result from RNA editing) and all other mismatches (occurring due to sequencing errors, misalignments, somatic mutations, etc.) (Fig. 7). Surprisingly, we found that virtually all A's exhibit AG mismatches and virtually all T's exhibit TC mismatches in all of these regions, given sufficient coverage. For a large fraction of human genes, expression of both strands was reported (Yelin et al. 2003; Katayama et al. 2005). Consistently, we find that for each Alu studied, individual reads exhibit either AG or TC mismatches but rarely both (Supplemental Fig. 6). This suggests that both strands of the Alu repeats are transcribed and virtually all of the transcribed adenosine sites are edited.
Figure 7.

Mismatch fraction distribution. Even before applying any statistical filters or analysis, a marked distinction is evident between AG/TC mismatches and other types of mismatches, provided there is sufficiently deep coverage. Presented are the distributions of the mismatch fractions (percent of reads that exhibit the mismatch among all reads supporting the site) for all (high quality, Q ≥ 30) mismatches seen in our MiSeq experiment at sites with high coverage (≥5000 reads, allowing for an accurate assessment of the mismatch fraction). Most mismatches are likely to result from sequencing errors and occur at fractions <0.1%, consistent with the sequencing quality. The AG/TC mismatches span a different range of mismatch fractions, where the bulk of the distribution lies in the range 0.1%–1%, but some sites are edited with stronger efficiencies, up to those showing close to 100% editing in a few sites. This separation of scales allows identification of editing sites, provided an accurate assessment of the mismatch fraction (requiring ultradeep coverage) is available. MM, mismatch. The y-axis shows the normalized probability density P(−log[MM fraction]).
Taking into account expression of both strands, this brings the total number of editable genomic sites in the set of editable Alus to 105.7 Mbp, representing 1.5% of the entire bases in the human genome. It should be noted that additional Alu elements not belonging to the “editable Alu” set are also heavily edited (Supplemental Material), suggesting that the actual number of editing sites is even higher. Their editing could be explained by too-strict cutoff values used for defining the editable Alus, or possibly, hybridization with Alu repeats in trans (Neeman et al. 2005) or polymorphic Alu insertions (Stewart et al. 2011; Witherspoon et al. 2013).
Editing levels follow a log-normal distribution
As virtually all adenosines in editable Alus undergo editing, the question arises regarding the editing levels at these sites. To address this issue, we looked at the fraction of AG/TC mismatches (unfiltered) for all A's and T's in the 52 randomly selected Alus that were covered by at least 5000 reads. This high coverage allowed us to accurately determine the editing level. We found (Fig. 7) the editing levels to approximately follow a wide log-normal distribution with a sizable fat tail at the high editing level range. Most sites are edited at low levels (<1% of transcripts), with the median editing level being 0.475%. Notably, the distribution for other types of mismatches exhibits a different pattern, which allows detection of almost all editing sites with high confidence, given sufficient read coverage.
As most sites are edited at low levels, typically only a fraction of the supporting reads exhibit the editing signature. Previous studies, limited by coverage, have generally identified the few highly edited sites along with a much larger number of weak sites, which is still a small sample of the full abundance of such sites. This explains the very low overlap between the data sets provided by previous studies.
Editing detection as a function of coverage
In order to estimate the dependence of editing detection on coverage, we selected 30 Alu repeats out of the 52 randomly selected Alus that were covered by at least 1000 reads. We then sampled fractions of increasing size from these reads and applied the detection algorithm to see how many sites are detected, and if the Alu repeat shows AG/TC as a dominant mismatch. Here, coverage was defined as the median number of reads supporting all A's and T's in the respective Alu repeat. The results are presented in Supplemental Figure 7 and show that with a median coverage of 1000 reads, virtually all Alu are detected as edited. Furthermore, the number of detected sites on a given Alu continues to rise with no sign of saturation.
Editing site motif in the deep-sequenced Alus
Editing sites were separated into three groups according to their editing level, and the identity of the base one position upstream and downstream was checked. The high and medium level sites show the expected editing motif. In particular, G is depleted in the upstream site and enriched in the downstream site. Note that in this case, the low editing level sites are actually almost all adenosines of the Alu repeat, and thus reflect the features of the Alu sequence rather than preferences of editing. Note that the TA dinucleotide is depleted in Alu repeats in general. Still, T appears quite frequently upstream of strong editing sites, suggesting a strong preference for it by the editing enzymes. Supplemental Figure 8 presents the upstream and downstream base distribution for all sites, weighted by their editing levels, and normalized by the underlying Alu neighbor distribution, in accordance with previous results (Kleinberger and Eisenberg 2010; Eggington et al. 2011).
Immense transcript diversity due to Alu editing
The large number of editing sites naturally leads to a staggering number of different transcripts per Alu-harboring gene (Barak 2009; Paz-Yaacov et al. 2010). Indeed, we found the number of different variants to grow with coverage, with no sign of saturation (Fig. 6C). Looking at the information included in each Alu repeat in terms of its editing pattern, Shannon's information entropy (Methods) shows that the effective number of bits per Alu ranges between 4.8 and 8.8. The number of inosines per transcribed Alu varies considerably among the randomly selected Alus, ranging between 0.24 and 11.5 inosines per transcript (Supplemental Material; Supplemental Fig. 9).
Out of the 23,357 human RefSeq genes, 67.4% include editable Alus, and 12.4% include such elements in their exons. Many genes include more than one editable Alu. Thus, the diversity at the transcript level is even much larger than the one observed for each single Alu repeat. In order to demonstrate the full scope of potential editing on a given gene, we selected three genes (RILPL1, PSEN1, and KIF1B), each harboring a large number of Alu repeats (76, 104, and 207, respectively), and deeply sequenced 14, 23, and 73 of these repeats, respectively. As expected, we found virtually all covered adenosines (372, 1128, and 3858, respectively) to exhibit editing, bringing the number of editing sites in these gene to thousands.
Volume of Alu editing activity compared with recoding sites
Most editing sites in Alu repeats are very weak, but due to their immense number, the accumulated editing events in these sites vastly outnumber those in known functional recoding sites. Using our data sets, we quantified the volume of editing activity in these two groups of editing sites (Methods). Based on current knowledge, the most important function of ADARB1 (ADAR2) is editing of the critical Q/R site in the glutamate receptor, as point mutation at this site can rescue lethality in mice (Higuchi et al. 2000). However, in the brain tissue only 77 edited reads are found in this critical site and 426 editing events were detected in all 22 recoding sites examined (all well-characterized recoding sites; Table S3 of Li et al. 2009), compared with 586,581 events in Alu repeats. That is, the critical glutamate receptor editing activity accounts for only 0.013% of A-to-I deamination reactions in the primate brain cell, and all recoding sites examined, combined, explain no more than 0.073% of all A-to-I editing activity in this tissue. Looking at other tissues we found editing in all 22 recoding sites to range between <0.001% and 0.4% (Supplemental Table S11).
Alu editing within lincRNA
It was recently demonstrated that Alu repeats within lincRNAs can undergo editing (Kapusta et al. 2013) and are involved in gene expression regulation via the Staufen-mediated mRNA decay (SMD) mechanism (Gong and Maquat 2011). We selected seven such Alu repeats and verified that they, too, undergo extensive RNA editing (Supplemental Material), possibly affecting the SMD process.
Discussion
It was suggested that inosine-containing transcripts are subject to specific regulation, affecting the fate of the RNA molecule (Rueter et al. 1999; Scadden and Smith 2001; Zhang and Carmichael 2001; Prasanth et al. 2005; Liang and Landweber 2007; Scadden 2007). However, given the wide scope of Alu editing in the human transcriptome, a large fraction of human transcripts contains inosines, suggesting that additional factors must be involved in these RNA regulations. It is possible that Alu editing is occasionally recruited for creating novel transcriptomics (Lev-Maor et al. 2007). In addition, editing by ADAR can unwind the multitude of dsRNA structures (Bass and Weintraub 1988) that exist in the primate genome.
Our results render superfluous catalogs of Alu adenosines exhibiting evidence of editing, as all adenosines in editable Alu repeats would be included given sufficient coverage. Rather, the characteristics of editable Alus should be used (and refined), and more careful measurements of editing rates per site and per Alu should be derived.
With these results, A-to-I editing becomes the most comprehensively characterized post-transcriptional modification. The massive number of Alu-editing events dwarfs the few known functional and conserved editing events within the coding regions, and accounts for >99% of all ADAR deamination reactions. The fact that such a heavy burden was not selected against raises the possibility that a fraction of the plethora of Alu-editing sites may have been utilized in ways that remain unknown (Mattick 2009; Paz-Yaacov et al. 2010) in the course of primate evolution.
Methods
RNA-seq data
RNA-seq data from the Illumina Human BodyMap 2.0 Project (GEO accession number GSE30611, HBM) was analyzed to find RNA-editing sites within Alu repeats. The data, generated on HiSeq 2000 instruments, consist of RNA-seq of 16 human tissue types: adrenal, adipose, brain, breast, colon, heart, kidney, liver, lung, lymph, ovary, prostate, skeletal muscle, testes, thyroid, and white blood cells. Two different read lengths were used for each tissue (2 × 50 bp paired-end and 1 × 75 bp single-read data). In addition, three libraries were generated from a mixture of the total RNA from the 16 human tissues [poly(A)-selected mRNA, poly(A)-selected mRNA with normalization, and total RNA without poly(A) selection] and sequences with 100 bp single reads. Overall, the Human Body Map data set includes 5,015,542,166 reads.
Furthermore, we analyzed a second RNA-seq data set (Sequence Read Archive accession SRA043767.1, YH) (Peng et al. 2012). The RNA used in this set was derived from a lymphoblastoid cell line of a male Han Chinese individual (YH) and sequenced on Illumina GA IIx and HiSeq 2000 machines. Three different read lengths were used: 2 × 75 bp and 2 × 100 bp paired-end reads for poly(A)+, and 2 × 90 bp paired-end reads for poly(A)−. In total, this set includes 1,167,280,060 reads.
Identification of edited Alus in RNA-seq data
Alignment of short reads is prone to exhibiting artifacts that might look as if they represent RNA modifications (Schrider et al. 2011; Kleinman and Majewski 2012; Lin et al. 2012; Pickrell et al. 2012). This is even more of an issue when dealing with the highly repetitive Alu elements. Thus, we took a highly conservative approach, taking into account only reads that were unambiguously aligned. We aligned both data sets (HBM and YH) to the human genome (hg19), using Bowtie aligner (Langmead et al. 2009) with liberal parameters that allow mismatch detection (−n 3, −l 20, −k 20, −e 140 −−best). These parameters allow having up to three mismatches in 20-base-long “seeds” and overall a score of 140 to all mismatches in an ungapped read. Up to 20 possible alignments per read are reported in a best-to-worst order. With these parameters, we considered for all downstream analysis only reads for which a single alignment was found. Using these parameters, 2.3 G of the HBM reads and 670 M of the YH reads were uniquely aligned. Next, we continued with reads that overlapped Alu repeat regions. A total of 919,035 Alu elements (78.19% of all Alu elements in the human genome) were covered (846,269 [72%] were covered by the HBM alone; 646,670 [55%] by YH data alone). Most Alu elements are only partially covered (Supplemental Fig. 2). Detailed information on the alignments per sequencing lane is presented in Supplemental Table S3.
Following alignment, we collected all mismatches between the above reads to the reference genome residing in Alu elements. Mismatches in read positions with quality phred score <30 were discarded, as were genomic locations which appear as genomic SNP in dbSNP (SNP build 131). Mismatches from all runs were merged, leading to a total of 25,103,998 mismatches, out of which 9,831,333 (39.2%) were AG (A in the genome with G in the corresponding position in some of the RNA-seq reads) and 9,416,730 were TC (37.5%). All together, we found 6,784,377 genomic locations exhibiting mismatches (HBM 4,126,430; YH 3,198,951), most of which were AG and TC: 1,938,752 AGs (28.6%) and 1,878,218 TCs (27.7%).
We then filtered the reads using a probabilistic model. For each genomic site, we calculated the probability that the observed mismatches in the reads in this genomic base pair could result from sequencing errors, assuming an a priori sequencing error rate of 0.001 (associated with the phred score cutoff of 30). Controlling for the multiple testing over all Alu nucleotides, we applied the Benjamini Hochberg correction to produce a set of putatively modified nucleotides, setting the desired false detection rate at 0.05. The resulting set consisted of 2,384,699 mismatch locations. This set was enriched with AG and TC mismatches, as 77.9% of these locations exhibited mismatches of these types (947,392 AGs [39.7%] and 911,094 TCs [38.2%]).
As Alu editing often affects numerous neighboring sites, we used our results to look for Alu elements containing clusters of editing sites. For this purpose, we looked only for Alu elements that were dominated by a single type of mismatch, i.e., Alu elements in which the number of mismatches of the most common mismatch type is higher than the number of mismatches of all other types combined. The results, for all types of mismatches, are presented in Figure 2C. We detected 305,337 Alu elements (HBM: 235,212; YH: 194,106) harboring clusters of AG or TC mismatches, all together containing 1,586,270 (HBM: 993,052; YH: 818,078) mismatch sites. In comparison, only 24,858 Alu elements harbor clusters of GA and CT mismatches, containing only 32,536 such mismatches, suggesting that this set of mismatches is dominated by editing, with a false positive rate of 8.1% at the Alu level and 2.0% at the site level (Fig. 2).
Strand selectivity of mismatches supports the editing model
Of the 694,523 AG/TC putative editing sites that reside in RefSeq genes, 90.58% of AG sites were found at locations where “A” (and not “T”) is encoded in the genome on the same DNA strand from which the RefSeq is expressed. (Thus, the RNA is likely to have been transcribed from the same strand, and had an A, not T, at this location.) Similarly, 90.33% of TC sites were found in reads aligned to the strand opposing the one to which the RefSeq transcript is aligned (thus, the RNA is likely to have been transcribed from the strand opposite to the reference genome, and also had an A at this location). This strong strand asymmetry further supports the notion of these sites being editing sites rather than genomic SNPs, sequencing errors, or other artifacts. The remaining 11% could be explained as the outcome of antisense transcription coming from the DNA strand not reported in RefSeq, a common event in the human genome (Yelin et al. 2003). In comparison, other mismatch types were evenly distributed between the two strands.
The YH data include strand-specific sequencing results. Analyzing these data shows a very high correlation of the AG/TC putative editing sites with the sequenced strand (Supplemental Fig. 10).
Mismatches attributed to editing have higher read-quality scores
In order to reduce the levels of sequencing errors, we discarded all read-base-pairs with quality scores lower than 30. The average score for the remaining sites was slightly above 38. Here we look at the average quality score for sites with mismatches that passed our filtering and clustering scheme. Reassuringly, we find that AG/TC mismatches are in fact supported by reads that are, on average, of higher quality than sites matching the reference genome. In comparison, mismatch sites of other types are, on average, of lower quality than sites matching the reference genome (Supplemental Fig. 11). This increases our confidence in that the AG/TC sites are not a result of some sequencing bias.
Editing sites, unlike noise, are evenly distributed along the reads
In order to avoid mismatches that result from misalignments at splicing junction regions (Lin et al. 2012) we trimmed the 5 bp at each end of the read if they included any mismatch (Lin et al. 2012). We next checked that the remaining mismatches are not biased toward the ends of the reads, which might suggest alignment artifacts due to exon borders. Indeed, the AG/TC sites are rather evenly distributed along the reads, and are even slightly depleted toward the read ends, as the alignments are more sensitive to mismatches in this region. However, this depletion only decreases the size of the observed editing effect. In comparison, other types of mismatches (GA/CT) show a pronounced increase toward the read ends, suggesting many of these mismatches, despite the above trimming, could be attributed to alignment artifacts (Fig. 8; Supplemental Fig. 12).
Figure 8.

Mismatch distribution along the reads. (A) AG/TC sites are evenly distributed along the reads and are even slightly depleted toward the read ends, as the alignments are more sensitive to mismatches in this region. (B) Other types of mismatches (GA/CT) show a pronounced increase toward the read ends, suggesting many of these mismatches, albeit trimming, could be attributed to alignment artifacts. Reads are 75 bp long.
Validation by ultrahigh coverage of selected targets
Based on the results from the HY and HBM data, we hypothesized that all adenosines in editable Alu repeats are edited to some extent. In order to test this hypothesis, we aimed at achieving ultrahigh coverage to examine the full potential of the editing phenomenon. We thus decided to target a small sample of Alu elements and subjected it to deep sequencing using MiSeq technology.
We randomly chose 52 editable Alu repeats that were not shown to be edited in the HBM data, and 28 editable repeats that were shown to be edited in the HBM data and sequenced them to attain ultrahigh coverage. A target library was prepared. Primers were designed such that they will be outside the Alu repeat, but as close as possible to the Alu boundary (primers used for each Alu repeat are given in the Supplemental Material), and two tissues were chosen (total brain reference and cerebellum). The library was sequenced using 150 × 2 paired-end reads (allowing for coverage of most of each Alu by the two paired-end reads).
Validation of editing sites: technical details
Multiplex sequencing was used to validate editing sites in Alu regions in the brain. A Human Brain Reference Total RNA (HBRR) sample pooled was obtained from brain samples of 23 individuals (Ambion, 6050), and a cerebellum sample from Biochain Institute. cDNA was synthesized in 20 µL reactions, each containing ∼5 μg of RNA, 4 µL of iScript advanced reaction master mix, and 1 µL of iScript advanced reverse transcriptase (Bio-Rad). Regions of interest were amplified and attached to oligos for multiplexing using 10 µL PCR reactions containing 5 µL of iQ SYBR Green Supermix (Bio-Rad), 5 ng of cDNA template, and 100 nM each of the forward and reverse primers. The following PCR program was used: 95°C for 5 min, 35 cycles of 95°C for 30 sec, 60°C for 30 sec, and 72°C for 60 sec, and finally 72°C for 5 min. The cerebellum and HBRR PCR reactions were then separately pooled, and the amplicons purified using QIAquick Gel Extraction Kit (Qiagen). Barcodes and sequencing adapters were attached using 50 µL PCRs each containing 5 µL of a 1000× dilution of the pooled amplicons, 25 µL KAPA2G Fast Multiplex Mix (2×) (Kapa Biosystems), and 200 nM of primers at 95°C for 5 min, three cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 1 min, 10 cycles of 95°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min, and finally 72°C for 5 min. The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen), and sequenced using Illumina MiSeq.
Ultradeep sequencing results
The resulting reads were mapped to the Alu target database, using BWA aligner (Li and Durbin 2009) with parameters that allow finding of mismatches (−n 10 −o 5 −e 2 −i 2 −l 10 −k 2). Alignment to the full genome yielded similar results. We then filtered out alignments with insertions or deletions, kept only pairs of reads that both mapped to the same Alu, and took only the parts of the reads that were mapped to the Alu repeats. A total of 75 Alu elements (out of 80, 50/52, and 25/28 from the editable Alu repeats that were not shown to be edited in the HBM data and the editable repeats that were shown to be edited in the HBM, respectively) were covered; most Alus were covered by 5000 reads, and more than half were covered by more than 10,000 reads (Supplemental Fig. 13).
We applied here the same filtering procedure used in the large RNA-seq data: All covered reads at each position were identified for mismatch positions comparing to the reference genome. Bases with quality phred score <30 and genomic SNPs (SNP build 131) were filtered out. Mismatched positions from all runs were merged (filtering out positions with only one non-reference read, unless it was the only read). All mismatch sites were rated for significance (using an a priori sequencing error rate of 0.002).
We then turned to clustering and looked for a single dominant type of mismatch. However, as many Alus were deeply covered (>5k reads), we found that most Alus contained both AG and TC editing sites, i.e., both strands, sense and antisense, were expressed at a level detectable by the large number of reads available. Accordingly, we modified our definition of an Alu with a dominant type of mismatch to one in which a single type of mismatch on both strands (e.g., AG and TC, or GA and CT) dominates, meaning that the number of mismatches of this type is more than twice the number of all other mismatches combined.
We found that 73 of the 75 covered Alus exhibited AG/TC as the dominant mismatch type, compared with none for GA/CT. These Alu elements include 4154 putative editing sites (2084 AG and 2070 TC). Note that most of the reads are still aligned with no editing sites at all (Supplemental Fig. 14). In order to verify that the appearance of both AG and TC mismatches in the same Alu repeat is indeed due to antisense expression, we looked at all reads exhibiting exactly three mismatches, and plotted, for each Alu repeat, the number of such reads with only AG, only TC, or mixed mismatch types (Supplemental Fig. 15). In addition, we note that although 51,212 of the 1,013,366 reads (5.05%) exhibit three or more AG mismatches, and 38,466 reads (3.79%) exhibit three or more TC mismatches, only 20 reads (0.002%) show both types together (P-value < 1 × 10−99), suggesting a strong anti-correlation between the two events. That is, if a read was transcribed from the reference strand of the genome and edited to contain three AG mismatches, it is very unlikely to also contain three TC mismatches, as its T base pairs cannot be edited. In addition, Supplemental Figure 6 presents the distribution of the number of TC events per read, stratified by the number of AG events in the same read. As observed for the AG editing signal intensity, the residual TC events seen in these reads are distributed exponentially, as expected for independent sequencing errors.
Shannon's information entropy
Shannon's Entropy (Shannon 1948) is a standard measure of the information content. It quantifies the extent to which the data encoded in a message (in our case, edited signal) is unpredictable. For example, if all messages are fully edited, than there is only one possible outcome and the entropy is zero. If all messages are either fully edited or fully unedited, then there are two possibilities and the entropy is a single bit. If there are many possible outcomes, with partial correlations among them, one needs to resort to the full formula
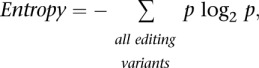 |
where p is the probability (relative occurrence) of each variant. We introduce Shannon's entropy to quantify in a compact manner how correlated the various editing events are within a single Alu repeat.
Editing activity
Editing activity in Alu was calculated by counting the total number of AG and TC mismatches for all genomic A and T sites within Alu elements expressed in one sample (brain). As usual, the number of reads aligned to a given site are assumed to be proportional to the expression level. That is, if 10 reads were aligned to a given site, and four of them were edited, we counted four deamination reactions. We apply the process for all Alu sites and sum the results. After removing the expected noise levels (calculated in the same way for the CT/GA mismatches) we have an unnormalized measure for the global number of deamination reactions at Alu sites in this specific brain tissue. We compare these data with the results of the same procedure applied to the well-characterized recoding sites (Li et al. 2009; Supplemental Table S3).
Data access
Sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRR1011286. Genome coordinates for editing sites (Dataset1) and Genome coordinates for edited Alu sites (Dataset2) are available in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank Khen Khermesh, Nurit Gal-Mark, Nurit Paz-Yaakov, and Karen Cesarkas for help with experimental procedures. This work was supported by the European Research Council (311257), the Legacy Heritage Biomedical Science Partnership, Israel Science Foundation (grant nos. 1466/10 [E.Y.L.] and 379/12 [E.E.]), Israel–US Binational Science Foundation (2011226), NSFGRFP (P.D.), and by the I-CORE Program of the Planning and Budgeting Committee and the Israel Science Foundation (grant nos. 41/11).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.164749.113.
Freely available online through the Genome Research Open Access option.
References
- Athanasiadis A, Rich A, Maas S 2004. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2: e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahn JH, Lee J-H, Li G, Greer C, Peng G, Xiao X 2012. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 22: 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak M 2009. Evidence for large diversity in the human transcriptome created by Alu RNA editing. Nucleic Acids Res 37: 6905–6915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55: 1089–1098 [DOI] [PubMed] [Google Scholar]
- Blow M, Futreal PA, Wooster R, Stratton MR 2004. A survey of RNA editing in human brain. Genome Res 14: 2379–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi S, Borukhov I, Levanon EY 2011. Identification of widespread ultra-edited human RNAs. PLoS Genet 7: e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Adamsky K, Cohen L, Amariglio N, Hirshberg A, Rechavi G, Levanon EY 2005. Identification of RNA editing sites in the SNP database. Nucleic Acids Res 33: 4612–4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommans WM, Tatalias NE, Sie CP, Dupuis D, Vendetti N, Smith L, Kaushal R, Maas S 2008. Screening of human SNP database identifies recoding sites of A-to-I RNA editing. RNA 14: 2074–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Maquat LE 2011. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470: 284–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner JC, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81 [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C 2013. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet 9: e1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et al. 2005. Antisense transcription in the mammalian transcriptome. Science 309: 1564–1566 [DOI] [PubMed] [Google Scholar]
- Kim DDY, Kim TTY, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A 2004. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res 14: 1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger Y, Eisenberg E 2010. Large-scale analysis of structural, sequence and thermodynamic characteristics of A-to-I RNA editing sites in human Alu repeats. BMC Genomics 11: 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman CL, Majewski J 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302. [DOI] [PubMed] [Google Scholar]
- Korenberg JR, Rykowski MC 1988. Human genome organization: Alu, lines, and the molecular structure of metaphase chromosome bands. Cell 53: 391–400 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann KA, Bass BL 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39: 12875–12884 [DOI] [PubMed] [Google Scholar]
- Lev-Maor G, Sorek R, Levanon EY, Paz N, Eisenberg E, Ast G 2007. RNA-editing-mediated exon evolution. Genome Biol 8: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon J-K, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM 2009. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324: 1210–1213 [DOI] [PubMed] [Google Scholar]
- Liang H, Landweber LF 2007. Hypothesis: RNA editing of microRNA target sites in humans? RNA 13: 463–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Piskol R, Tan MH, Li JB 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302. [DOI] [PubMed] [Google Scholar]
- Mattick JS 2009. Deconstructing the dogma: A new view of the evolution and genetic programming of complex organisms. Ann NY Acad Sci 1178: 29–46 [DOI] [PubMed] [Google Scholar]
- Morse DP, Aruscavage PJ, Bass BL 2002. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc Natl Acad Sci 99: 7906–7911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeman Y, Dahary D, Levanon EY, Sorek R, Eisenberg E 2005. Is there any sense in antisense editing? Trends Genet 21: 544–547 [DOI] [PubMed] [Google Scholar]
- Nishikura K 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Williams B, Wold BJ, Mortazavi A 2012. RNA editing in the human ENCODE RNA-seq data. Genome Res 22: 1626–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, Amariglio N, Eisenberg E, Rechavi G 2010. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc Natl Acad Sci 107: 12174–12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Cheng Y, Tan BC-M, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, et al. 2012. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 30: 253–260 [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Gilad Y, Pritchard JK 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL 2005. Regulating gene expression through RNA nuclear retention. Cell 123: 249–263 [DOI] [PubMed] [Google Scholar]
- Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, Li JB 2012. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods 9: 579–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal JJC, Seeburg PH 2012. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron 74: 432–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80 [DOI] [PubMed] [Google Scholar]
- Savva YA, Rieder LE, Reenan RA 2012. The ADAR protein family. Genome Biol 13: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD 2007. Inosine-containing dsRNA binds a stress-granule-like complex and downregulates gene expression in trans. Mol Cell 28: 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD, Smith CW 2001. Specific cleavage of hyper-edited dsRNAs. EMBO J 20: 4243–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Gout J-F, Hahn MW 2011. Very few RNA and DNA sequence differences in the human transcriptome. PLoS ONE 6: e25842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE 1948. A mathematical theory of communication. Bell Syst Tech J 27: 379–423 [Google Scholar]
- Stewart C, Kural D, Strömberg MP, Walker JA, Konkel MK, Stütz AM, Urban AE, Grubert F, Lam HYK, Lee W-P, et al. 2011. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet 7: e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherspoon DJ, Zhang Y, Xing J, Watkins WS, Ha H, Batzer MA, Jorde LB 2013. Mobile element scanning (ME-Scan) identifies thousands of novel Alu insertions in diverse human populations. Genome Res 23: 1170–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, et al. 2003. Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol 21: 379–386 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Carmichael GG 2001. The fate of dsRNA in the nucleus: A p54nrb-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 106: 465–475 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.