

Abstract
Inosine is an abundant RNA modification in the human transcriptome and is essential for many biological processes in modulating gene expression at the post-transcriptional level. Adenosine deaminases acting on RNA (ADARs) catalyze the hydrolytic deamination of adenosines to inosines (A-to-I editing) in double-stranded regions. We previously established a biochemical method called “inosine chemical erasing” (ICE) to directly identify inosines on RNA strands with high reliability. Here, we have applied the ICE method combined with deep sequencing (ICE-seq) to conduct an unbiased genome-wide screening of A-to-I editing sites in the transcriptome of human adult brain. Taken together with the sites identified by the conventional ICE method, we mapped 19,791 novel sites and newly found 1258 edited mRNAs, including 66 novel sites in coding regions, 41 of which cause altered amino acid assignment. ICE-seq detected novel editing sites in various repeat elements as well as in short hairpins. Gene ontology analysis revealed that these edited mRNAs are associated with transcription, energy metabolism, and neurological disorders, providing new insights into various aspects of human brain functions.
RNA molecules contain a wide variety of chemical modifications that are introduced enzymatically after transcription (Bjork 1995; Grosjean 2005; Suzuki 2005). Inosine (I) is an abundant type of RNA modification found in the double-stranded regions of RNAs (dsRNA) of metazoans and is formed through the hydrolytic deamination of adenosines to inosines (A-to-I editing) catalyzed by adenosine deaminase that acts on RNA (ADAR) (Bass 2002). Functional ADAR is required for normal development in vertebrates (Higuchi et al. 2000; Wang et al. 2000; Wang et al. 2004) and normal behavior in invertebrates (Jepson and Reenan 2008). A number of pathogenic mutations in ADAR (also known as ADAR1) gene are associated with dyschromatosis symmetrica hereditaria (DSH) (Tojo et al. 2006; Keller et al. 2008) and Aicardi-Goutieres syndrome (AGS) (Rice et al. 2012). In addition, a lack of A-to-I editing has been associated with several neurological disorders (Maas et al. 2006), including malignant gliomas (Maas et al. 2001) and amyotrophic lateral sclerosis (ALS) (Kawahara et al. 2008). The functional importance of A-to-I editing is also indicated by the fact that both ADAR and ADARB1 (also known as ADAR2) are essential enzymes in mouse (Higuchi et al. 2000; Wang et al. 2000). To date, a large number of A-to-I editing sites have been identified biochemically or predicted bioinformatically in coding regions, introns, and untranslated regions of mRNAs in the human transcriptome (Wulff et al. 2011). Most A-to-I editing sites reside in Alu repeat elements in the untranslated regions and introns, and A-to-I editing is frequent and prominent in the transcriptomes of humans and other primates (Eisenberg et al. 2005; Paz-Yaacov et al. 2010). A-to-I editing results in the modulation of gene expression, including amino acid alterations (Higuchi et al. 1993; Burns et al. 1997; Hoopengardner et al. 2003; Levanon et al. 2005), alternative splicing (Rueter et al. 1999), prevention of aberrant exonization (Sakurai et al. 2010), nuclear retention (Chen et al. 2008), nonsense-mediated mRNA decay (NMD) (Agranat et al. 2008), RNA interference (Bass 2006), variations in the 3′ UTR (Osenberg et al. 2009), altered translation (Hundley et al. 2008), and miRNA-mediated translational repression (Borchert et al. 2009). In addition, A-to-I editing also occurs in pre-miRNAs in dsRNA regions that modulate the processing and target specificity of the miRNA (Kawahara et al. 2007a,b). However, the exact function of most A-to-I editing sites remains elusive, and many researchers believe that there are still large numbers of novel editing sites that remain to be discovered in the human transcriptome.
The most conventional method used to identify A-to-I editing sites is comparison of cDNA sequences with the corresponding genomic sequence (Burns et al. 1997; Paz et al. 2007). As inosine (I) can base-pair with cytidine (C), inosines are converted to guanosines (G) in the cDNA by reverse-transcription and PCR (I-to-G replacements). Therefore, if adenosine (A) in the genomic sequence is partially or completely replaced with G at the corresponding site in the cDNA sequence (A-to-G replacement), that site is a candidate for A-to-I editing. This method can be applied to genome-wide screening of A-to-I editing sites using a deep sequencing method called RNA–DNA differences (RDDs) (Li et al. 2011; Bahn et al. 2012; Peng et al. 2012). In an analysis of human B cells from 27 individuals, more than 10,000 RDD sites, including non-A-to-G sites, were reported as putative editing sites that did not match the corresponding sites in the human genome (Li et al. 2011). However, up to 94% of these RDD sites were estimated to be false positives due to mapping errors of short sequence tags to the reference sequence (Kleinman and Majewski 2012; Lin et al. 2012; Pickrell et al. 2012). In fact, it is difficult to distinguish between G derived from genuine I and G resulting from mapping errors, sequencing errors, and noise or unfavorable amplification of pseudogenes. Mapping errors have been decreased to some extent by a pipeline that reduces false positives (Peng et al. 2012). However, the most recent report (Piskol et al. 2013) revealed that a considerable amount of mapping errors persist even in the improved RDD sequencing method. In particular, short RNA sequences are inevitably mapped erroneously to pseudogenes with high sequence similarity to the original gene.
To overcome these difficulties in identifying A-to-I editing sites, we previously established a biochemical method called “inosine chemical erasing” (ICE) to directly and definitively identify inosines in RNA strands (Sakurai et al. 2010). This method is based on detecting the erased G signals originating from inosines in the sequence chromatogram of cDNAs following the cyanoethylation of inosines (Fig. 1A). The ICE method does not require genomic DNA as a reference and can discriminate inosines from the G signals arising from SNPs, sequencing errors, and pseudogenes. However, this method can only be applied to specific sequences of interest, which produces considerable limitations and bias on the identification and discovery of novel sites.
Figure 1.

Biochemical identification of A-to-I editing sites by ICE-seq. (A) Chemistry of inosine cyanoethylation. Acrylonitrile adducts to the N1 position of inosine to form N1-cyanoethylinosine (ce1I). (B) Outline of ICE-seq. Schemes without (CE−) or with (CE+ or CE++) cyanoethylation of RNA are shown on the left and right, respectively. RNA and cDNA are indicated by gray and black arrows, respectively. (i) Cyanoethylation and fragmentation. The I in the RNA strand is specifically cyanoethylated to form ce1I (CE+). RNAs are partially digested by mild alkaline treatment. (ii) First strand cDNA synthesis. RNAs are reverse-transcribed with a random primer. RNA bearing an A at the editing site is converted to T in the cDNA in both conditions (CE− or CE+). In the CE− condition, RNA bearing I is transcribed to C in the cDNA. In the CE+ condition, first strand cDNA extension is arrested at the ce1I site (red arrow). (iii) Second strands are synthesized to obtain double-stranded cDNAs which are then subjected to the end-repair reaction and adaptor ligation. (iv) Gel purification of 300-bp cDNAs for PCR amplification. The amplified cDNAs with 300 bp are gel-purified again. In this step, cDNAs arrested at ce1I are discarded. (v) The cDNAs for CE−, CE+, and CE++ conditions are sequenced from both ends by a GA2 sequencer. Data processing of these reads identifies inosines by detecting erased reads upon cyanoethylation.
To obtain a global and agnostic view of A-to-I editing in the human transcriptome with unbiased detection, we have developed a new strategy using the ICE method combined with deep sequencing, which we call ICE-seq. Poly(A)+ RNAs, which were left untreated or were treated with acrylonitrile to cyanoethylate inosines, were converted to cDNAs and then analyzed by deep sequencing. After mapping sequence reads to the reference sequence, we were able to accurately detect erased reads derived from inosines by comparison to the control sample [untreated poly(A)+ RNA]. ICE-seq can rigorously exclude A-to-G mismatches generated by inevitable mapping errors, unidentified SNPs, and sequencing noise.
Combined with the sites identified by the conventional ICE method, we report here the identification of 19,791 novel editing sites in the human transcriptome and 1258 mRNAs in which A-to-I editing had not previously been detected. This study demonstrates the importance of biochemical identification of A-to-I editing sites and the effectiveness of ICE-seq.
Results
Outline of ICE-seq
We developed a new strategy using the ICE method combined with deep sequencing, which we have named ICE-seq. Before starting ICE-seq analysis, a high-quality data set of A-to-I editing sites validated by the ICE method must be isolated for optimization of several ICE-seq parameters. As an extension of our previous analysis (Sakurai et al. 2010), we analyzed 1686 regions in human brain cDNA by the ICE method and identified 13,884 editing sites (Supplemental Table S1A), including 8847 novel sites (Supplemental Table S1B). Within the coding sequences (CDS), 10 editing sites were found (see Table 1). Most of these sites have a low (<50%) editing frequency (Supplemental Fig. S1). Combined with our previous data (Sakurai et al. 2010), 17,745 editing sites (Supplemental Table S1C) have now been confirmed by the ICE method, which provides a data set of sufficient size with which to optimize ICE-seq analysis.
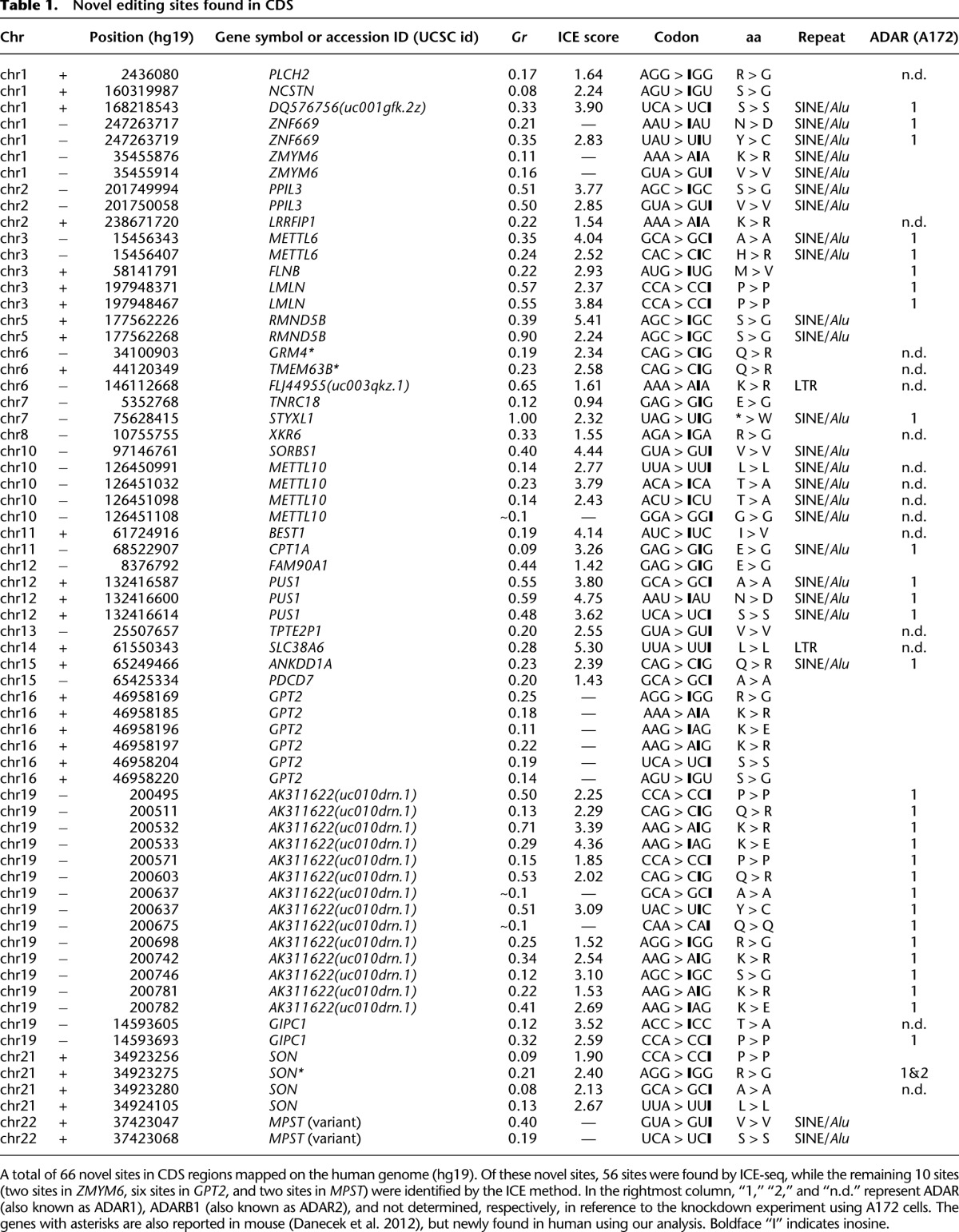
Table 1.
Novel editing sites found in CDS
The principles and procedures for ICE-seq analysis are outlined in Figure 1B. The first step of the procedure is the same as that of the ICE method. As reported previously (Sakurai et al. 2010), inosines (I) in the RNA strand are specifically cyanoethylated by acrylonitrile to form N1-cyanoethylinosine (ce1I) (Fig. 1A). Since this reaction is performed at 70°C, almost all I residues will be converted to ce1I, even in stable double-stranded regions. Poly(A)+ RNA was treated with acrylonitrile to cyanoethylate inosines under the following two conditions: CE+, mild treatment with acrylonitrile; or CE++, strong treatment with acrylonitrile. Untreated RNA (CE−) was used as a control. Subsequent steps in the analysis follow the standard protocols for deep sequencing of mRNAs. After the fragmentation of poly(A)+ RNA, first strand cDNA was synthesized. In this step, the N1-cyanoethyl group of ce1I blocks strand extension, thereby eliminating cDNAs derived from edited RNA segments. After second strand synthesis, both ends of the strands are repaired and capped with adaptors. The 300-bp cDNA fragments are then prepared for deep sequencing. After mapping the sequence reads to the reference sequences (human transcriptome of USCS genes and the human genome), the missing reads in CE+ or CE++ conditions can be identified by comparing them with the CE− reads.
ICE-seq analysis of the human brain transcriptome
Poly(A)+ RNA from adult human brain tissue was subjected to cyanoethylation under mild condition (CE+), strong condition (CE++), or control condition (CE−). The RNAs prepared in these three conditions were converted to cDNAs and subjected to deep sequencing. Because most editing sites are assumed to be located in repetitive regions, such as Alu sequences, each cDNA was sequenced for 75 nt from both ends. This sequence length effectively prevents misalignment of the reads to the reference sequences. Approximately 400–500 million tags were obtained for each treatment (Fig. 2A). More than 200 million tags were mapped to the human genome (hg18) or transcriptome (UCSC gene) for each sample. The distribution of gene expression estimated by the coverage of the reads is shown in Figure 2B,C. Approximately 63% of the detected genes had more than 20 times the coverage (∼1 RPKM) (Fig. 2C; Supplemental Table S2). The scatter plot of RPKM values in CE− versus CE+ or CE++ showed good correlation, and little difference was observed between these plots (Fig. 2D), indicating that there was no unfavorable bias due to the cyanoethylation of RNAs, and ensuring the reproducibility of cluster generation and deep sequencing in each run.
Figure 2.

Sequence statistics of ICE-seq. (A) Numbers of mapped tags used for ICE-seq in CE−, CE+, and CE++ conditions. The reference genome and cDNA sequences used here are hg18 and the UCSC gene, respectively. (B) Distribution histogram of gene expression estimated by the read coverage. Gene expression represented by the Log2(RPKM) value for the reference transcriptome sequence is compiled at 0.1 Log2(RPKM) intervals under conditions of CE− (blue), CE+ (yellow), and CE++ (magenta). (C) Pie chart of gene expression estimated by the read coverage. A total of 63% of human genes are detected with more than 20 times coverage (∼1 RPKM). (D) Scatter plots of logarithmic RPKM values in CE− versus CE+ or CE++. Their coefficients of determination (R2) are 0.998 and 0.995, respectively.
After the mapping process, mapping quality was checked by a global scan of the ICE-seq reads (Fig. 3). Many reads were mapped onto exons. When two known editing sites (Q/R and Q/Q) in exon 11 of the GRIA2 mRNA (Fig. 3A; Sommer et al. 1991) were examined, a specific decrease in G-base counts was clearly detected. In addition, we also observed a specific reduction in the amount of reads near the editing sites in exon 11 in the CE+ and CE++ samples (downward peak in Fig. 3A). At the I/M site in GABRA3 mRNA (Fig. 3B; Ohlson et al. 2007), a decrease in the G-base count was evident and a reduced amount of reads were observed surrounding the editing sites in the CE+ and CE++ samples. In the case of the multiple editing sites in the 3′ UTR of the BPNT1 mRNA (Fig. 3C; Levanon et al. 2004), the G-base counts at each position decreased and the amount of reads in the CE+ and CE++ samples was extensively reduced at 26 adenosine positions, including 16 sites reported by Levanon et al. (2004) or validated by the ICE method in this study. These observations demonstrate that ICE-seq can detect editing sites reliably.
Figure 3.

Genome-wide views of the regions with editing sites piled with the mapped reads of ICE-seq. (A) Q/R and Q/Q sites in exon 11 of GRIA2 mRNA. (B) I/M site in GABRA3 mRNA. (C) Editing cluster in 3′ UTR of BPNT1 mRNA. Top panel shows histograms of the mapped reads under CE− conditions at genomic positions. Genome number, positions, and scale of length are indicated. Middle panel shows close-up views of the regions with editing sites piled with the mapped reads in conditions of CE− (blue), CE+ (orange), and CE++ (pink). Bottom panel shows the decreased read ratio upon cyanoethylation (CE++) (gray downward peaks) and editing sites with the decreased ratio of G-base counts upon cyanoethylation (CE++) (red bars).
Agnostic genome-wide identification of novel A-to-I editing sites
To process the massive amount of data produced by mapped reads, we developed a MapReduce pipeline for A-to-I editing, termed FastPass (Supplemental Fig. S2). Briefly, the data from mapped reads were compressed by extracting their start and end positions based on genomic reference (hg18) and stored in binary files (bpos file). The base counts at each mismatched site were extracted from the mapped reads with mismatches against the reference sequence and stored in another binary file (bdiff file). Using this data analysis process, the size of the data set was successfully reduced to approximately one-tenth of the original data set. Further analyses to narrow down the candidates for the editing sites were performed using these size-reduced and indexed binary files.
Using the CE− data set, we were able to detect all mismatches mapped to the transcriptome and genome references if more than eight aligned reads at each site had different bases from those of the reference sequence. When mapped to the reference transcriptome of UCSC genes, we obtained 462,583 mismatch sites, 53,912 of which were A-to-G mismatch sites and initial candidates for A-to-I editing sites without any filtering processes (Supplemental Fig. S3), while the rest of the sites (408,671) were non-A-to-G sites. When compared with the genome reference, we obtained 744,182 mismatch sites, 94,713 of which were A-to-G mismatch sites and 97,441 of which were T-to-C mismatch sites. Since transcriptional direction cannot be determined from the deep sequencing data, both A-to-G and T-to-C mismatch sites were considered prospective candidates for A-to-I editing when mapped to the reference genome. The remaining 552,028 sites were non-A-to-G and non-T-to-C mismatch sites.
Next, the change in G-base counts (Ng) upon cyanoethylation was assessed by calculating ΔNg, the logarithmic value of the decrease in Ng in CE+ (or CE++) compared with CE−. As shown in Figure 4A, the ΔNg for CE+ versus ΔNg for CE++ was plotted. In this scatter plot, 45 known CDS editing sites (light blue points, listed in Supplemental Table S3) were identified in the lower-left region (quadrant III). In this region, we also detected 1963 editing sites in non-CDS (pink points, listed in Supplemental Table S4) that were validated by the ICE method. In contrast, SNPs (yellow and orange points) stayed near the origin, and their ΔNg was not markedly changed upon cyanoethylation. These data demonstrated that editing sites can be clearly discriminated from SNPs, as well as from G contamination caused by any other reasons. In fact, as candidates for a novel editing site, a number of nonannotated A-to-G sites (dark blue points, listed in Supplemental Table S5A) can be seen in the lower-left region of Figure 4A. For having a negative control, the same analysis was performed at non-A-to-G mismatch sites that were randomly chosen from the mapped genome (Fig. 4B). Most points (ΔNn) remained near the origin because they are allelic SNPs, and very few of these points were observed in quadrant III, suggesting that this analysis can robustly identify novel editing sites. To identify candidate editing sites, the distance of each point from the origin was measured and designated as the “ICE score.” As shown in the histogram of ICE scores (Fig. 4C), most A-to-G SNPs reside within an ICE score lower than 1.75. In fact, among 25,285 A-to-G gSNPs, there are only 241 sites having an ICE score higher than 1.75. The P-value for an ICE score of 1.75 was calculated to be 0.00953. Namely, 99% of gSNPs reside within an ICE score lower than 1.75. Most of the known editing sites exhibited an ICE score higher than 1.75 (Fig. 4C). The ICE scores of novel editing candidate sites fell within the same range as the known editing sites. As a negative control, the ICE scores of the non-A-to-G sites without annotations fell in the same range as those from SNPs and other mismatch sites (Fig. 4D).
Figure 4.

Separation of A-to-I editing sites from false-positive sites and SNPs. (A) Scatter plot of ΔNg(CE+) versus ΔNg(CE++). The 40 known editing sites in CDS (Supplemental Table S3) and 1963 editing sites validated by the ICE method in non-CDS (Supplemental Table S4) are indicated by light blue and magenta points, respectively. cSNPs and gSNPs are indicated by yellow and orange points, respectively. Dark blue points are nonannotated A-to-G sites. (B) Scatter plot of ΔNn(CE+) versus ΔNn(CE++). cSNPs and gSNPs are shown as yellow and orange points, respectively. Dark blue points are nonannotated mismatched sites. (C) Histogram of the ICE scores for A-to-G sites from SNPs (yellow), known editing sites (magenta), and nonannotated sites (blue). (D) Histogram of the ICE scores for non-A-to-G sites from SNPs (yellow) and nonannotated sites (blue).
To filter out false-positive candidates, 1963 known editing sites in non-CDS (validated by the ICE method) were used as positive controls to optimize various parameters, such as base-call quality, mismatch tolerance, mapping quality, and neighboring mutations (see Supplemental Methods). The threshold of each parameter was set to include known editing sites and exclude SNP sites (gSNPs) as negative controls. Through these filtering algorithms, the number of A-to-G sites was narrowed to 5680 sites, which accounted for 10.5% of the initial population in the reference transcriptome of UCSC genes (Supplemental Fig. S3). When mapped to the genome reference, 13,993 (14.8%) A-to-G sites and 14,481 (14.9%) T-to-C sites were chosen from the initial population (Supplemental Fig. S3). Finally, from both reference sequences, 16,575 sites that had more than 20 total read counts, more than 10 Ng, a Gr (ratio of Ng in total read counts) higher than 0.1, and an ICE score higher than 1.75 were included. By removing the sites whose genomic positions overlapped between the two references and sites included in dbSNP137, 14,393 A-to-G sites (Supplemental Table S5A), consisting of 14,225 non-CDS sites, 39 known CDS sites, and 129 candidates in CDS (Supplemental Table S5B), were finally identified by ICE-seq analysis.
However, six of the 45 known editing sites in CDS detected by the ICE-seq analysis (Supplemental Table S3) were excluded by this filtering procedure. To identify as many novel editing sites in CDS as possible, we decreased the threshold of parameters (more than eight Ng and an ICE score higher than 1.4) at the cost of an increased false-positive rate, and repeated the filtering procedure with these parameters, resulting in the inclusion of all of the 45 known editing sites in CDS (see Supplemental Methods). A total of 99 additional candidates in CDS (Supplemental Table S6) were chosen using this lower threshold screening procedure.
ICE method and ICE-seq analyses identified 19,791 novel editing sites
ICE-seq analysis produced 14,393 A-to-G sites (Supplemental Table S5A) consisting of 11,046 novel editing candidates, 2652 known editing sites (2422 sites registered in DARNED [Kiran and Baranov 2010], 15 known but not registered in DARNED, and 215 sites previously reported by us [Sakurai et al. 2010]), and 695 sites identified by the ICE method in this study. A total of 11,046 novel editing candidates consist of 10,917 sites in non-CDS and 129 sites in CDS (Supplemental Table S5B). Together with 99 additional candidates (Supplemental Table S6), 228 CDS candidates in total were obtained in this study.
To validate the editing candidates obtained by ICE-seq, the ICE method was used to validate both non-CDS and CDS sites. First, 931 sites (Supplemental Table S7A) out of 14,225 A-to-G sites in non-CDS were randomly chosen and analyzed by the ICE method. For accurate validation, each editing site was designated as such only if its editing frequency was >10%. A total of 902 of the 931 sites were confirmed to be edited according to these criteria (Supplemental Table S7A). Hence, the accuracy of identifying editing candidates in non-CDS was estimated to be 96.9%. In this analysis, 29 sites in the non-CDS were either editing sites with lower editing frequency (<10%) or false-positive sites. Thus, 10,917 sites found in non-CDS were shown to be novel editing sites with 96.9% accuracy (Supplemental Table S7A). In other words, the false-positive rate of non-CDS sites is 3.1%. All 228 CDS candidate sites were analyzed by the ICE method. A total of 53 novel editing sites (Table 1) in CDS were identified, while 89 sites were false positives, and the remaining 86 sites were not validated due to failed amplification of cDNAs (Supplemental Table S8). In validating the CDS candidates by the ICE method, we also found three additional editing sites in ZNF669 (chr 1 − 247163717), METTL10 (chr 10 − 126451108), and SON (chr 21 + 34923280). Taken together with the 10 editing sites found by the ICE method (Supplemental Table S1B), 66 novel editing sites in CDS were identified (Table 1). In total, ICE-seq identified 11,639 novel editing sites (10,917 − 29 + 53 + 3 + 695) and the ICE method identified 8847 novel editing sites, including 695 sites that overlapped with the ICE-seq data. Thus, a total of 19,791 (11,639 + 8847 − 695) novel editing sites were identified in this study (Fig. 5A; Supplemental Table S9), distributed across 2114 genes, including 1258 newly identified genes that produce edited transcripts (Supplemental Table S10). These genomic sites with their editing frequency can be viewed on the UCSC Genome Browser (http://editing.cbrc.jp/).
Figure 5.

Statistical features of A-to-I editing sites detected by ICE-seq. (A) Venn diagrams show the number of novel editing sites detected by ICE-seq and the ICE method in this study (left panel), both from known and novel editing sites detected in this study (middle panel), and the number of editing sites detected by us and known/predicted sites deposited in DARNED (right panel). (B) Plot of the Gr value versus the G ratio at each editing site. The coefficient of determination (R2) is 0.77. (C) Editing frequency distribution of the sites detected by ICE-seq. The editing frequency of each site was calculated from the Gr value. The numbers of editing sites at each template ratio are compiled in the histogram. Gray and white boxes represent novel and known/predicted sites, respectively. (D) Base preference around the editing site presented by WebLogo using the full ICE-seq data set (left) and sites with >50% editing frequency (right). (E) Triplet preference of editing. Statistics of triplet sequences centered on the edited A were analyzed using the full ICE-seq data set (left) and sites with >50% editing frequency (right).
Statistical features of A-to-I editing sites in the human transcriptome
Since the Gr value in ICE-seq corresponds to the editing frequency at each site, we compared the Gr values of 1963 validated editing sites (Supplemental Table S4) with the G peak ratio in their Sanger sequencing chromatogram (Fig. 5B). The coefficient of determination (R2) of the plot was 0.77, indicating a strong correlation between the ICE-seq Gr value and the ICE method G peak ratio. The editing frequency distribution of the sites detected by ICE-seq is shown in Figure 5C. The new editing sites detected by ICE-seq tend to have higher editing frequencies compared with the editing frequencies of the sites identified by the ICE method (Supplemental Fig. S1). This result indicates that there still remains a number of novel editing sites with high editing frequency to be discovered in the entire transcriptome. Since the ICE method is based on RT-PCR, which has a much higher detection ability of minor transcripts than RNA-seq, it can detect inosines in transcripts with a very low expression level, if they can be amplified. If a much larger data set is provided for ICE-seq, most of the sites detected by the ICE method will be identified by ICE-seq.
Next, the base preferences adjacent to each of the sites identified in this study were examined (Fig. 5D). At the −1 position of the editing sites, the order of the base preferences was C ≈ U ≈ A ≫ G. At the +1 position of the editing sites, the order of the base preference was G > C ≈ A > U. In contrast, when the base preferences of sites with >50% editing frequency were analyzed, the base preference at the −1 position was U > C > A ≫ G, while the base preference at the +1 position was G ≫ C > A > U. At these sites, a U or C at the −1 position and a G at the +1 position were the most common. Similar base preferences were reported previously (Kim et al. 2004; Levanon et al. 2004; Sakurai et al. 2010). Further, triplet sequences centered on the edited A (Fig. 5E) showed the following preferences: CAG (18%) > UAG (13%) > AAG (10%). In contrast, GAC (1%), GAU (1%), and GAA (1%) were rarely edited. In editing sites with >50% editing frequency, the order of triplet preference was UAG (22%) > CAG (18%) > AAG (11%). The triplet preference of CDS editing sites was CAG (25%) > AAG (17%) > UAG (12%) (Supplemental Fig. S4A,B). These preferences correlate well with the base preferences adjacent to the editing site (Fig. 5D). The tendency toward reduced editing of the GAN triplet is remarkable. The UAG triplet is reported to be frequently edited in both mature and precursor forms of miRNAs (Kawahara et al. 2008). The data in this study revealed that the CAG and AAG triplets also serve as good substrates for editing, which may help to identify new editing sites in mRNAs as well as in noncoding RNAs, including miRNAs. In addition, we observed a weak base preference at the −2 position (Fig. 5D). Thus, we performed quadruplet logo analyses (Supplemental Fig. S4C,D). The top five quadruplets with the highest editing frequency were GUAG, CAAG, CUAG, UCAG, and CCAG.
Novel editing sites in CDS regions
We identified 66 novel editing sites in the CDS of 32 genes (Table 1). Regarding the distribution of these editing sites, 24 sites resided in SINE/Alu sequences, two sites in LTR regions, and the other 40 sites were found in non-repeat regions. Additionally, 41 sites resulted in altered amino acid assignment (Table 1). The N-to-D alteration found in PUS1 (variant) and ZNF699 is a novel type of amino acid alteration that has never before been documented in the reported editing sites. As shown in Supplemental Figure S5A–N, most of the novel editing sites identified in CDS were found in dsRNA regions. In fact, among 66 novel sites in CDS, 60 sites were found in apparent dsRNA structures which were predicted informatically. We also identified a Q-to-R site in CDK13 (Supplemental Fig. S5O) and an S-to-R site in NOVA1 (Supplemental Fig. S5P), both of which were reported (Maas et al. 2011; Irimia et al. 2012) during the review process for this manuscript. The related descriptions of these sites are shown in the Supplemental Results.
To determine which ADAR is responsible for editing these sites, ADAR or ADARB1 was down-regulated by siRNA in A172 glioblastoma cells. Among the genes expressed in A172 cells, 30 editing sites in CDS were detected by RT-PCR and direct sequencing (Table 1). All of the sites were edited by ADAR, while two sites in SON (chr 21 + 34923275, new; chr 21 + 34923319, known) could be edited by ADARB1 as well. The high editing frequency (92%) of the Q-to-R site in CDK13 (Maas et al. 2011) was also observed in A172 cells. This site was shown to be edited by ADAR. Unlike ADARB1, which has a high level of substrate specificity, ADAR exhibits broad substrate specificity with low editing frequency for dsRNAs (Dabiri et al. 1996; Melcher et al. 1996; Bahn et al. 2012). Thus, the Q-to-R site in CDK13 is one of the ADAR-targeted sites having a high editing frequency.
Novel editing sites in non-CDS regions
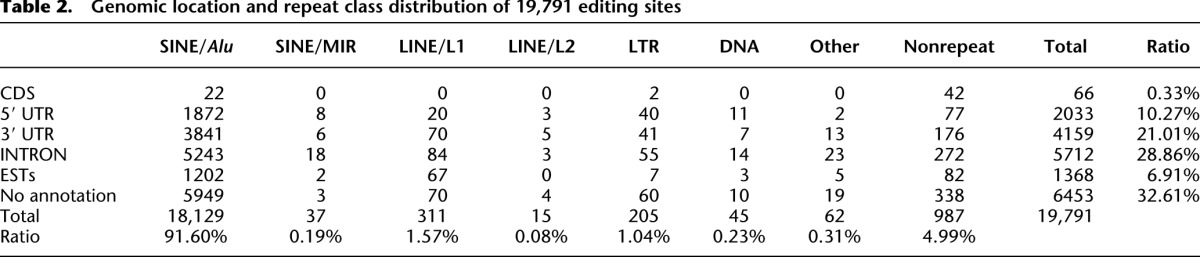
The genomic location and repeat class distribution of 19,791 editing sites are shown in Table 2. The majority of the editing sites are located in SINE/Alu sequences in 3′ UTRs, intronic regions, and intergenic regions, as reported previously (Kim et al. 2004; Levanon et al. 2004; Sakurai et al. 2010). Multiple inosines found in the 3′ UTR of the ATM and IFNAR2 mRNAs are illustrated as typical editing sites in SINE/Alu elements (Supplemental Fig. S6A–D). In most cases, multiple sites (>20) are edited in long dsRNA structures ∼300 bp in length that are formed by the plus and minus strands of a canonical SINE/Alu. A notable finding from our ICE-seq analysis is the identification of unique editing sites in various repeat elements other than SINE/Alu sequences in shorter dsRNA regions, in which a prediction of the editing sites by bioinformatic approaches is difficult. The novel editing sites detected by ICE-seq are distributed in various repeat regions, including SINE/MIR (Supplemental Fig. S7), LINE/L1 (Supplemental Fig. S8A–D), LINE/L2 (Supplemental Fig. S9), LTR (Supplemental Fig. S10A–D), DNA transposon (Supplemental Fig. S11A–D), 7SL RNA-like elements (Supplemental Fig. S12A,B), and 7SK RNA-like elements (Supplemental Fig. S13). In addition, regions without any annotation were also detected (Table 2). The length of the duplex structure (35–250 bp) formed by these repeat elements is shorter than that formed by SINE/Alu elements (∼300 bp). In most cases, those dsRNA structures are formed by plus and minus strands of the same repeat element (Supplemental Figs. S7–S10), similar to SINE/Alu elements (Supplemental Fig. S6).
Table 2.
Genomic location and repeat class distribution of 19,791 editing sites
In other cases, however, inosines are found in the dsRNA region formed by a repeat element and its complementary sequence next to the element. dsRNA can be formed by different classes of repeat elements. Three consecutive editing sites were found in a dsRNA region formed by an LTR and a SINE/MIR in the 3′ UTR of the LNP mRNA (Supplemental Fig. S10D). A single editing site was found in the 7SL-like element of the KIF1B mRNA (Supplemental Fig. S12B). Because 7SL RNA is an ancestor of SINE/Alu elements, the 7SL-like element is able to pair with the minus strand of the neighboring SINE/Alu due to their sequence similarity. Furthermore, novel editing sites are often found in single repeat elements that form a hairpin-like secondary structure. In fact, we found editing sites in the short hairpin structure formed by a DNA transposon in the 3′ UTR of the SLC7A5P1 mRNA (Supplemental Fig. S11D). Another intriguing editing site was found in a 7SK-like RNA element (Supplemental Fig. S13). An abundant noncoding RNA in the nucleus, 7SK RNA, is a negative regulator of transcription elongation (Diribarne and Bensaude 2009). A dispersed pseudogene family related to 7SK RNA has also been identified (Huang da et al. 2009). In the intronic region of MAN2A1, five editing sites were found to be clustered in the antisense strand of a 7SK pseudogene. Since the possible secondary structures around this region could not be predicted, the 7SK RNA or a 7SK-related transcript may form a duplex structure with this region in trans, which then serves as a substrate for editing.
Discussion
Using a biochemical method to identify A-to-I editing sites by inosine-specific cyanoethylation combined with Sanger sequencing (ICE method) and deep sequencing (ICE-seq), we identified here 19,791 novel editing sites in the human brain transcriptome. Combined with the sites identified previously by the ICE method (Sakurai et al. 2010), 29,843 sites were identified in total, including 6406 known or predicted editing sites (Fig. 5A) deposited in the DARNED database (Kiran and Baranov 2010). Over 42,000 A-to-I editing sites have been predicted or reported thus far, but, surprisingly, only 21% of the sites identified here overlapped with sites already in the database (Fig. 5A). This observation indicates that the exploration of A-to-I editing sites in the human transcriptome is not yet saturated and strongly suggests the importance of identifying editing sites biochemically, not merely predicting them bioinformatically. The conventional method used to identify inosines is based on the extraction of A-to-G RDDs obtained by comparing cDNA sequences with their matched genomic sequences. In this study, ∼54,000 A-to-G sites were detected when mapped to the reference transcriptome (CE− samples). This number was ∼12 times larger than the number of validated editing sites. Similarly, the numbers of A-to-G and T-to-C sites (95,000 sites), when mapped to the genome reference, were found to be 13 and 20 times larger, respectively, than the number of validated editing sites. Thus, ∼92%–95% of A-to-G (T-to-C) mismatch sites detected by deep sequencing without any filtering processes were not inosine sites. Most of these sites are thought to originate from unidentified SNPs and false-positive signals arising from inevitable mapping errors and/or sequencing noise. It is difficult to discriminate A-to-I editing sites accurately from such large numbers of RDDs by simple bioinformatic approaches or conventional deep sequencing alone (Piskol et al. 2013). We examined the degree to which the 29,843 total sites we identified using ICE-seq (including 19,791 novel sites) overlapped with RDD sites identified in other studies (Li et al. 2011; Bahn et al. 2012; Peng et al. 2012): only 30 sites (19 novel sites) of 10,210 RDDs by Cheung's group (Li et al. 2011), 1647 sites (or 939 sites) of 11,467 RDDs by Wang's group (Peng et al. 2012), and 1527 sites (933 novel sites) of 13,280 RDDs by Xiao's group (Bahn et al. 2012). Although the overlap rates were improved in the latter two groups, the limited overlap between our sites and the reported RDDs suggests that even improved RDD methods are not sufficient to detect A-to-I RNA editing with high accuracy. Alternatively, differences in the tissue type and sample sources used for these analyses might be another reason for the limited amount of overlap. Therefore, we performed a direct comparison of the results obtained by RDD and ICE methods using the same cell line, and illustrated the advantage of ICE-seq over the RDD method (see Supplemental Results; Supplemental Table S11; Supplemental Figs. S14–S16).
In this study, ICE-seq was performed with a single biological replicate, because the quantity of poly(A)+ RNA from one individual was limited. Instead of replicates, we prepared three conditions, CE−, CE+, and CE++, from the same RNA at the same time. Each sample was independently prepared by cyanoethylation and cDNA synthesis. Thus, by utilizing the ΔNg values for CE+ and CE++, the false-positive rate can be reduced as much as possible. In addition, each sample was analyzed over several runs to confirm the reproducibility of cluster generation and deep sequencing by checking RPKM values (Fig. 2D). No unfavorable bias was detected in any run. In fact, the accuracy of candidate sites in non-CDS was estimated to be 96.9% by validation with the ICE method; thus the false-positive rate is calculated to be 3.1%, indicating the reliability of ICE-seq. In addition, all novel sites in CDS were confirmed by the ICE method. ICE-seq with a single biological replicate is a practical demonstration of the applicability to valuable clinical specimens with limited quantity. Also, no requirement for genomic DNA is another advantage of ICE-seq. Further analyses using various specimens will confirm the accuracy and performance of ICE-seq. In addition, ICE-seq will be improved to increase the sensitivity of detection by combining it with strand-specific RNA-seq and cutting-edge NGS technology.
Functional analyses of A-to-I editing in CDS will help to elucidate the physiological roles of ADAR, because alterations in amino acid assignment resulting from A-to-I editing in CDS may potentially modulate protein function. In addition to the ∼100 known editing sites in CDS (Supplemental Table S3), we identified 66 novel editing sites in CDS (Table 1), 41 of which cause a change in amino acid sequence. To identify functionally important editing sites, high editing frequency and evolutionary conservation at each site should be considered. SON is an essential DNA-binding protein localized to nuclear speckles and is involved in pre-mRNA splicing (Saitoh et al. 2004; Sharma et al. 2010). R-to-G editing (Supplemental Fig. S5A) may modulate the function of SON. BEST1 is another example of editing in CDS, with an I-to-V editing site contained in a short hairpin structure (Supplemental Fig. S5B). BEST1 is a bestrophin family protein that forms a Ca2+-dependent anion channel expressed in epithelial cells (Kunzelmann et al. 2011). Pathogenic mutation of this protein is associated with Best vitelliform macular dystrophy. Filamin B (FLNB), a cross-linker for actin filaments, regulates the cytoskeletal network and intracellular signaling pathways responsible for skeletal development (Stossel et al. 2001). Mutations in FLNB result in human skeletal disorders, such as boomerang dysplasia, which is characterized by disrupted vertebral segmentation, joint formation, and endochondral ossification. We identified an M-to-V editing site in exon 39 with a hairpin structure formed with neighboring introns in FLNB mRNA (Supplemental Fig. S5C).
In addition to the sites in CDS, 19,725 novel sites were found in non-CDS regions, including intergenic regions (no annotation), intronic regions, and the 5′ UTR and 3′ UTR. Although 32.61% of the novel sites mapped to intergenic regions (Table 2), some of these sites might reside on the elongated untranslated regions of the neighboring mRNAs. Alternatively, these sites might be included in genomic loci for poly(A)+ long noncoding RNAs (lncRNAs) or natural antisense transcripts. Recent studies reveal the functional aspects of lncRNAs (Sasaki and Hirose 2009; Wilusz et al. 2009; Gong and Maquat 2011), including transcriptional silencing, architectural functions in nuclear bodies, and mRNA decay. A-to-I editing may play a modulatory role in lncRNA function. A total of 28.86% of the novel sites were found in intronic regions (Table 2), the editing of which contributes to the modulation of alternative splicing (Lai et al. 1997; Lev-Maor et al. 2007; Agranat et al. 2010; Sakurai et al. 2010). We reported previously that intronic editing in pre-mRNA by ADAR plays a role in preventing aberrant exonization of antisense Alu sequences in the mature mRNA (Sakurai et al. 2010). By examining the novel sites found in this study, other instances of intronic editing preventing aberrant exonization may be found. Although little is known about the function of A-to-I editing in the 5′ UTR, ∼2000 novel editing sites were identified in 5′ UTRs (Table 2), which may reveal the functional roles of editing in this region.
Twenty-one percent of the identified novel editing sites were found in 3′ UTRs (Table 2), which are the functional domains of mRNAs responsible for translational repression and subcellular localization. There are multiple motifs and target sites in the 3′ UTR that are recognized by trans-factors including RNA-binding proteins and miRNAs. In fact, A-to-I editing in the 3′ UTR of DFFA mRNA creates a target site recognized by miR-513 (Borchert et al. 2009). When the miRNA response elements (MREs) in the 3′ UTR are edited, especially in the seed region, the changes modulate miRNA–MRE interactions. If an A–C mismatch in the seed region is edited, the I–C pairing stabilizes the interaction and enhances the efficacy of the miRNA (gain type). In contrast, if an A–U pairing in the seed region is edited, the I–U wobble pairing destabilizes the interaction and relieves translational repression (loss type). We hypothesize that there are a number of instances of both loss-type and gain-type editing changes in the human transcriptome. We examined the potential effects of the novel editing sites found in 3′ UTRs on miRNA-mediated translational repression. A total of 6467 miRNA–MRE pairs in 847 genes with only one editing site in the seed sequence were identified (Supplemental Table S12). Of these, 2963 pairs were stabilized or generated de novo by editing (gain type), while 3504 pairs were destabilized by the resultant I–U wobbling (loss type). As shown in a minimum free energy (MFE) plot of each miRNA–MRE pair (Supplemental Fig. S17), loss-type changes decrease and gain-type changes increase the MFE. Practically, there are a number of editing clusters in 3′ UTRs. If multiple editing changes occur in MREs, the modulatory effects of A-to-I editing on the thermodynamic stability of the miRNA–MRE pair become more complex, although these simulations can only be applied for a specific miRNA and its target mRNA in the same cell. The role of A-to-I editing in modulating translational repression mediated by miRNAs may be more important than previously thought.
In this study, we identified 2114 genes whose transcripts are edited with inosines. To characterize these genes, we performed gene ontology (GO) analysis (Supplemental Table S13). The predominant categories identified were nerve impulse, synapse, membrane, and ion binding, indicating that A-to-I editing plays important roles in human brain functions. Calcium binding motifs, glutamate receptors, and 5-HT2 type receptors were also enriched. DNA-binding motifs such as Kruppel-associated box and zinc finger motifs also appear to be concentrated, indicating that RNA transcription and gene expression are major pathways targeted by A-to-I editing. In particular, ZNF699, a zinc finger protein of unknown function, possesses two CDS editing sites resulting in Y-to-C and N-to-D alterations (Table 1; Supplemental Fig. S5M). Furthermore, a number of mitochondrial proteins and components of the respiratory chain and oxidative phosphorylation are also subject to A-to-I editing. Energy metabolism related to mitochondrial function may also be modulated by A-to-I editing, and disease-related genes, including those related to Alzheimer's, Huntington's diseases, and amyotrophic lateral sclerosis, were also found to have high GO scores. Interestingly, oxidative stress response genes and apoptosis pathways components were also enriched. Thus, the GO analysis provides insight into the phenotypic features of ADAR-null mice, and DSH or AGS patients.
Considering the close relationship between the emergence of large numbers of editing sites due to the explosive increase in Alu elements and primate evolution (Eisenberg et al. 2005; Paz-Yaacov et al. 2010), A-to-I editing is likely to actively affect higher-order biological processes in human and primates by modulating gene expression post-transcriptionally. Abnormal patterns or loss of editing have been frequently detected in neurological disorders (Maas et al. 2001, 2006; Kawahara et al. 2008). Therefore, A-to-I editing mediated by ADAR and ADARB1 appears to be required for sophisticated mental activity in the complex neural network of the brain. Although SNPs are generally used as markers to characterize individuals, differences in A-to-I editing sites may provide more sophisticated markers by which to assess individual characters and constitutions. Elaborate profiling of A-to-I editing in various tissues and cells may reveal the functional aspects of editing at each site in a complex network of gene expression. Based on the data set obtained in this study, genome-wide profiling of A-to-I editing (the editome) for various diseases including neurological disorders may provide effective measures for diagnostic purposes and medical applications. ICE-seq is a unique and practical method that is applicable to the genome-wide identification of A-to-I editing sites in tissues and clinical specimens without genomic DNAs. Further analyses using various specimens will confirm the accuracy and performance of ICE-seq.
Methods
Cyanoethylation of RNA
Cyanoethylation of RNA was performed essentially as described previously (Sakurai et al. 2010; Sakurai and Suzuki 2011). Total RNA (10 μg) isolated from human adult brain tissue or poly(A)+ RNA (0.5 μg) was incubated in 38 µL CE solution (50% ethanol, 1.1 M triethylammonium acetate [pH 8.6]) with 1.6 M acrylonitrile at 70°C for 15 min (CE+) or 30 min (CE++). As a reference, the same reaction was performed in the absence of acrylonitrile (CE−). The treated RNA was purified using an RNeasy MinElute kit (Qiagen) followed by ethanol precipitation.
ICE method
Primers for the ICE method were designed as described previously (Sakurai et al. 2010; Sakurai and Suzuki 2011). Two sets of primers (for two rounds of PCR) were designed to amplify a region 300–500 bp in length, including target inosine sites. The primer sets for the nested second PCR were designed inside the region amplified by the first round of PCR. The primers designed for 1686 regions with editing sites predicted by bioinformatics methods are listed in Supplemental Table S1D. The primers for validating 931 sites identified by ICE-seq are listed in Supplemental Table S7B. The PCR primers used for the second round of PCR were also used for sequencing. The ICE method was performed as described previously (Sakurai et al. 2010; Sakurai and Suzuki 2011).
ICE-seq
The cDNA for paired-end RNA-seq analysis using a Genome Analyzer II (Illumina) was performed basically as described in the manufacturer's protocol (Illumina RNA-seq PE Sample Prep protocol, v3.6). First, 100 ng of cyanoethylated poly(A)+ RNA was fragmented in 20 μL of 1× fragmentation buffer at 95°C for 5 min, and RNA fragments were recovered by ethanol precipitation. First strand cDNA synthesis was performed with random primer at 25°C for 10 min then 50°C for 50 min. Subsequently, second strand cDNA synthesis was performed with RNase H and DNA polymerase I. Purified cDNA was end-repaired and ligated with adaptors. After that, the 300-bp cDNA was purified by 2% agarose gel electrophoresis and then amplified by PCR with 20 cycles. The amplified cDNA was subjected to 2% agarose gel electrophoresis, and the 300-bp cDNA band was cut and extracted from the gel. The size and quality of the cDNA was confirmed using a DNA 1000 kit and Bioanalyzer (Agilent). The cDNAs for CE−, CE+, and CE++ conditions were then subjected to a Genome Analyzer II (Illumina) according to the manufacturer's protocol.
Data analysis
The detailed workflow for data processing and screening of A-to-I editing site is described in Supplemental Methods. For each ICE condition (CE−, CE+, and CE++), the sequence reads were mapped to the human NCBI Build 36 reference sequence (hg18) as well as to the transcriptome reference (UCSC Genes on hg18 version, a total of 66,803 genes) using BWA aligner (v5.1) (Li and Durbin 2009), allowing four and five mismatches to the genomic and transcriptome references, respectively. We did not use splice-aware mapping software such as TopHat (Silverberg et al. 2005) so as to avoid a mapping error caused by the exon-first approach, especially on pseudogene regions (Garber et al. 2011). To reduce mapping errors, local realignments were done by a Smith Waterman algorithm (Smith and Waterman 1981), and the realigned reads with <95% identities to the reference were discarded. Only the unique mapped reads or properly mapped read pairs were selected for further analyses. Base pileup with a double binomial test and RPKM calculation are done by the FastPass framework as described in Supplemental Methods. Potential sequence error, mapping error, and ambiguous reads were excluded, with thresholds determined by comparing the data set of dbSNP and known A-to-I editing sites which are determined by the ICE method. Finally, we chose the candidate sites bearing at least a 20-fold read depth at each ICE condition, and G-base counts decreased upon cyanoethylation with P-value <0.01 (ICE score 1.75). Parallelization of the process and data compression is done on a cluster computer environment. The compressed and reduced data were further analyzed with MySQL database to filter out the false-positive candidates.
Other procedures for preparation of poly(A)+ RNA, ICE-seq with details, read mapping and data processing, calculation of RPKM value, data screening of A-to-G sites, RNA interference, and GO analysis are described in the Supplemental Information.
Data access
The sequence data from this study have been submitted to the DDBJ Sequence Read Archive (DRA; http://trace.ddbj.nig.ac.jp/dra) under accession number DRA000478.
Supplementary Material
Acknowledgments
We thank members of the Suzuki laboratory for experimental assistance and fruitful discussions of this study. This work was supported by Grants-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports, and Culture of Japan and by a grant from the New Energy and Industrial Technology Development Organization (NEDO) (to T.S.).
Author contributions: M.S. and T.S. designed the research. M.S., T.Y., S.O., H.T., and H.K. performed cellular and biochemical experiments. H.U. developed all computational methods and performed in silico analyses assisted by T.M. A.T. and A.F. assisted with deep-seq analysis. M.S., H.U., and T.S. wrote the manuscript. T.S. supervised the study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.162537.113.
References
- Agranat L, Raitskin O, Sperling J, Sperling R 2008. The editing enzyme ADAR1 and the mRNA surveillance protein hUpf1 interact in the cell nucleus. Proc Natl Acad Sci 105: 5028–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agranat L, Sperling J, Sperling R 2010. A novel tissue-specific alternatively spliced form of the A-to-I RNA editing enzyme ADAR2. RNA Biol 7: 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahn JH, Lee JH, Li G, Greer C, Peng G, Xiao X 2012. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 22: 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL 2006. How does RNA editing affect dsRNA-mediated gene silencing? Cold Spring Harb Symp Quant Biol 71: 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork G. 1995. Biosynthesis and function of modified nucleosides. In tRNA: Structure, biosynthesis, and function (ed. Soll D, et al.), pp. 165–205. American Society for Microbiology, Washington, D.C. [Google Scholar]
- Borchert GM, Gilmore BL, Spengler RM, Xing Y, Lanier W, Bhattacharya D, Davidson BL 2009. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet 18: 4801–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB 1997. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387: 303–308 [DOI] [PubMed] [Google Scholar]
- Chen LL, DeCerbo JN, Carmichael GG 2008. Alu element-mediated gene silencing. EMBO J 27: 1694–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabiri GA, Lai F, Drakas RA, Nishikura K 1996. Editing of the GLuR-B ion channel RNA in vitro by recombinant double-stranded RNA adenosine deaminase. EMBO J 15: 34–45 [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Nellåker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ 2012. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol 13: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diribarne G, Bensaude O 2009. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol 6: 122–128 [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Nemzer S, Kinar Y, Sorek R, Rechavi G, Levanon EY 2005. Is abundant A-to-I RNA editing primate-specific? Trends Genet 21: 77–81 [DOI] [PubMed] [Google Scholar]
- Garber M, Grabherr MG, Guttman M, Trapnell C 2011. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods 8: 469–477 [DOI] [PubMed] [Google Scholar]
- Gong C, Maquat LE 2011. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470: 284–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H. 2005. Modification and editing of RNA: Historical overview and important facts to remember. In Topics in current genetics, Vol. 12, pp. 1–22. Springer-Verlag, New York. [Google Scholar]
- Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH 1993. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell 75: 1361–1370 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81 [DOI] [PubMed] [Google Scholar]
- Hoopengardner B, Bhalla T, Staber C, Reenan R 2003. Nervous system targets of RNA editing identified by comparative genomics. Science 301: 832–836 [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Hundley HA, Krauchuk AA, Bass BL 2008. C. elegans and H. sapiens mRNAs with edited 3′ UTRs are present on polysomes. RNA 14: 2050–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Denuc A, Ferran JL, Pernaute B, Puelles L, Roy SW, Garcia-Fernandez J, Marfany G 2012. Evolutionarily conserved A-to-I editing increases protein stability of the alternative splicing factor Nova1. RNA Biol 9: 12–21 [DOI] [PubMed] [Google Scholar]
- Jepson JE, Reenan RA 2008. RNA editing in regulating gene expression in the brain. Biochim Biophys Acta 1779: 459–470 [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K 2007a. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep 8: 763–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K 2007b. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K 2008. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res 36: 5270–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Backes C, Al-Awadhi M, Gerasch A, Kuntzer J, Kohlbacher O, Kaufmann M, Lenhof HP 2008. GeneTrailExpress: A web-based pipeline for the statistical evaluation of microarray experiments. BMC Bioinformatics 9: 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A 2004. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res 14: 1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiran A, Baranov PV 2010. DARNED: A DAtabase of RNa EDiting in humans. Bioinformatics 26: 1772–1776 [DOI] [PubMed] [Google Scholar]
- Kleinman CL, Majewski J 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Kongsuphol P, Chootip K, Toledo C, Martins JR, Almaca J, Tian Y, Witzgall R, Ousingsawat J, Schreiber R 2011. Role of the Ca2+-activated Cl- channels bestrophin and anoctamin in epithelial cells. Biol Chem 392: 125–134 [DOI] [PubMed] [Google Scholar]
- Lai F, Chen CX, Carter KC, Nishikura K 1997. Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol Cell Biol 17: 2413–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Maor G, Sorek R, Levanon EY, Paz N, Eisenberg E, Ast G 2007. RNA-editing-mediated exon evolution. Genome Biol 8: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, Rechavi G, Jantsch MF, Eisenberg E 2005. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res 33: 1162–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, Cheung VG 2011. Widespread RNA and DNA sequence differences in the human transcriptome. Science 333: 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Piskol R, Tan MH, Li JB 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302. [DOI] [PubMed] [Google Scholar]
- Maas S, Patt S, Schrey M, Rich A 2001. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci 98: 14687–14692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Kawahara Y, Tamburro KM, Nishikura K 2006. A-to-I RNA editing and human disease. RNA Biol 3: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Godfried Sie CP, Stoev I, Dupuis DE, Latona J, Porman AM, Evans B, Rekawek P, Kluempers V, Mutter M, et al. 2011. Genome-wide evaluation and discovery of vertebrate A-to-I RNA editing sites. Biochem Biophys Res Commun 412: 407–412 [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M 1996. A mammalian RNA editing enzyme. Nature 379: 460–464 [DOI] [PubMed] [Google Scholar]
- Ohlson J, Pedersen JS, Haussler D, Ohman M 2007. Editing modifies the GABAA receptor subunit α3. RNA 13: 698–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenberg S, Dominissini D, Rechavi G, Eisenberg E 2009. Widespread cleavage of A-to-I hyperediting substrates. RNA 15: 1632–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S, Barbash ZS, Adamsky K, Safran M, Hirschberg A, et al. 2007. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res 17: 1586–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, Amariglio N, Eisenberg E, Rechavi G 2010. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc Natl Acad Sci 107: 12174–12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, et al. 2012. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 30: 253–260 [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Gilad Y, Pritchard JK 2012. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 335: 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskol R, Peng Z, Wang J, Li JB 2013. Lack of evidence for existence of noncanonical RNA editing. Nat Biotechnol 31: 19–20 [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80 [DOI] [PubMed] [Google Scholar]
- Saitoh N, Spahr CS, Patterson SD, Bubulya P, Neuwald AF, Spector DL 2004. Proteomic analysis of interchromatin granule clusters. Mol Biol Cell 15: 3876–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Yano T, Kawabata H, Ueda H, Suzuki T 2010. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol 6: 733–740 [DOI] [PubMed] [Google Scholar]
- Sakurai M, Suzuki T 2011. Biochemical identification of A-to-I RNA editing sites by the inosine chemical erasing (ICE) method. Methods Mol Biol 718: 89–99 [DOI] [PubMed] [Google Scholar]
- Sasaki YT, Hirose T 2009. How to build a paraspeckle. Genome Biol 10: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Takata H, Shibahara K, Bubulya A, Bubulya PA 2010. Son is essential for nuclear speckle organization and cell cycle progression. Mol Biol Cell 21: 650–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverberg RF, Cheng ES, Aguirre JE, Bezaire JJ, Crawford TM, Meyer SS, Bier A, Campano B, Chen TC, Cottingham DA, et al. 2005. The TopHat experiment: A balloon-borne instrument for mapping millimeter and submillimeter emission. Astrophys J Suppl Ser 160: 59–75 [Google Scholar]
- Smith TF, Waterman MS 1981. Identification of common molecular subsequences. J Mol Biol 147: 195–197 [DOI] [PubMed] [Google Scholar]
- Sommer B, Kohler M, Sprengel R, Seeburg PH 1991. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67: 11–19 [DOI] [PubMed] [Google Scholar]
- Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS 2001. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2: 138–145 [DOI] [PubMed] [Google Scholar]
- Suzuki T. 2005. Biosynthesis and function of tRNA wobble modifications. In Topics in current genetics, Vol. 12, pp. 24–69. Springer-Verlag, New York. [Google Scholar]
- Tojo K, Sekijima Y, Suzuki T, Suzuki N, Tomita Y, Yoshida K, Hashimoto T, Ikeda S 2006. Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation. Mov Disord 21: 1510–1513 [DOI] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, Nishikura K 2000. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290: 1765–1768 [DOI] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K 2004. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279: 4952–4961 [DOI] [PubMed] [Google Scholar]
- Wilusz JE, Sunwoo H, Spector DL 2009. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev 23: 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff BE, Sakurai M, Nishikura K 2011. Elucidating the inosinome: Global approaches to adenosine-to-inosine RNA editing. Nat Rev Genet 12: 81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.