

Abstract
The ARF/INK4A (Cdkn2a) locus includes the linked tumour suppressor genes p16INK4a and p14ARF (p19ARF in mice) that trigger the antiproliferative activities of both RB and p53. With beta cell self-replication being the primary source for new beta cell generation in adult animals, the network by which beta cell replication could be increased to enhance beta cell mass and function is one of the approaches in diabetes research. In this review, we show a general view of the regulation points at transcriptional and posttranslational levels of Cdkn2a locus. We describe the molecular pathways and functions of Cdkn2a in beta cell cycle regulation. Given that aging reveals increased p16Ink4a levels in the pancreas that inhibit the proliferation of beta cells and decrease their ability to respond to injury, we show the state of the art about the role of this locus in beta cell senescence and diabetes development. Additionally, we focus on two approaches in beta cell regeneration strategies that rely on Cdkn2a locus negative regulation: long noncoding RNAs and betatrophin.
1. Introduction
Expansion and proliferation of insulin-secreting beta cells in pancreatic islets is a key highly regulated mechanism for establishing, maintaining, and adapting islet function to meet organism physiological demands. Understanding how the pieces of this mechanism fit together could improve development of islet replacement approaches; given that both type 1 and type 2 diabetes result from reduced beta cell mass and impaired beta cell functions. Islet beta cells expand in neonatal humans, mice, and other species, but this proliferation decays thereafter, which may promote pandemic (type 2) forms of diabetes mellitus [1]. Beta cell mass can be expanded by increasing beta cell replication, enlarging beta cell size, decreasing beta cell death, and promoting beta cell neogenesis. Murine beta cells proliferate slowly after birth, but this process can be accelerated under various conditions, including obesity, pregnancy, and stimulation by different beta cell mitogens, such as glucose, amino acids, insulin, prolactin, placental lactogen, and glucagon-like peptide-1. In addition to differentiation from pre-existing adult beta cell progenitors or transdifferentiation from another cell lineage, beta cell self-replication is considered to be the primary source for new beta cell generation in adult animals [2]. One of the approaches in diabetes research is investigating the function of beta cell cycle activators and inhibitors to elucidate the network by which beta cell replication could be increased to enhance beta cell mass and function.
For the understanding of beta cell replication machinery it is important to focus on the cell cycle phases and checkpoints: the G1-S transition, S phase checkpoint, G2 to M transition and the Mitotic checkpoint (Figure 1). Checkpoint regulation mechanisms act through negative intracellular signals that arrest the cell cycle, rather than through the removal of positive signals that stimulate cell-cycle progression. Negative signals prevent cycle transition at the initiation of proliferation, replication and mitosis until the cellular conditions are the adequate for cell cycle progression. Sequential activation/inactivation of cyclin-dependent kinases (Cdks) is the primary means of cell cycle regulation. Cyclins confer substrate specificity and regulation to Cdk/cyclin complexes. According to that, in early G1 phase Cdk4 and/or Cdk6 are activated by D-type cyclins and initiate phosphorylation of the retinoblastoma protein (Rb) family. This triggers the release of E2F transcription factors and the subsequent activation and transcription of E2F responsive genes (including E- and A-type cyclins) required for cell-cycle progression. In the late G1 phase, Cdk2 binds to cyclin E and completes the phosphorylation of Rb, reinforcing the activation of E2F mediated transcription. These events lead to transition from the G1/S boundary point to S phase initiation.
Figure 1.
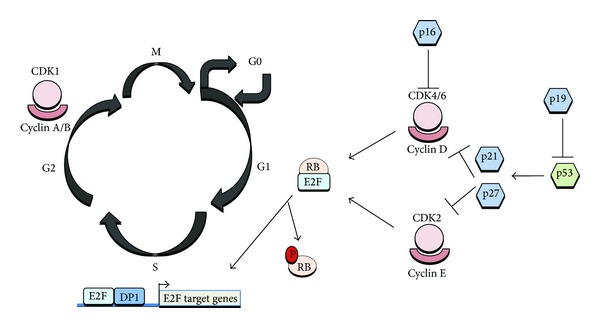
Mammalian cell cycle regulation. p16Ink4b sequesters Cdk4 or Cdk6 inhibiting interactions with type D cyclins and preventing phosphorylation of pRB. Inactivation of CDK4/6 promotes Rb/E2F1 association triggering G1/S transition. Phosphorylation of pRB is essential for passage through the restriction point in G1. The cyclin D1-Cdk4 complex specifically phosphorylates the pRB protein leading to sequential phosphorylation by cyclin E-Cdk2 and release of free E2F. The phosphorylation of pRB, and relief of transcriptional inhibition by pRB induces S-phase entry. p53-dependent regulation by p21 and p27 contributes to checkpoint maintenance at later timepoints. Cdc2-Cyclin A/B binding contribute to phosphorylation of proteins involved in G2/M transition.
Through S phase progression Cdk2 binds to cyclin A that contributes to DNA replication. During the G2/M transition, Cdk1/cyclin A complex is required for the initiation of prophase. Finally, Cdk1/cyclin B complexes are actively involved in the completion of mitosis. The negative regulation of Cdk/cyclin complexes relies on two families of Cdk inhibitors; the INK4 family (p16INK4a, p15INK4b, p18INK4c, p19INK4d) specifically bind to Cdk4 and Cdk6 and prevent D-type cyclin activity and the Cip/Kip family (p21Cip1/Waf1/Sdi1, p27Kip1, p57Kip2) inhibits Cdk2/cyclin E, Cdk2/cyclin A, Cdk1/cyclin A, as well as Cdk1/cyclin B activity (for review see [3]).
The p53 gene product is another key cell cycle check-point regulator at both the G1/S and G2/M and it has been shown to activate transcription of number of cell cycle genes and its essential role is to arrest cells in G1 after genotoxic damage allowing DNA repair prior to DNA replication and cell division. In response to massive DNA damage, p53 triggers the apoptotic cell death pathway [4].
Here we give an overview about the regulation points of ARF/INK4A locus transcription in beta cell, specifically in proliferation processes that give rise to cellular senescence and the development of diabetes. We state the more striking studies that shed a light in the approach of pancreatic beta cell proliferation and regeneration.
2. ARF/INK4A Locus Regulation in Beta Cells
Candidate gene and genome-wide association studies (GWAS) have identified several loci associated with type 2 diabetes and related traits. Specifically, genetic variants at CDKN2A/B locus have been associated with type 2 diabetes in many ethnic populations. A number of publications in the last five years confirm and validate that CDKN2A/B is a locus associated with genetic risk of type 2 diabetes development [5–8]. Among the strongest linked variants is rs10811661, located 125 kb upstream of the CDKN2A and CDKN2B genes (for review, [9]). This polymorphism appears to be associated to type 2 diabetes in almost all the ethnic cohorts studies [10–18] and people carrying the TT genotype of this variant showed impaired insulin release and impaired glucose tolerance [19].
The ARF/INK4A (Cdkn2a) locus spans around 35 kilobases on human chromosome 9p21.25 that includes the linked tumor suppressor genes p16INK4a and p14ARF (p19ARF in mice) that trigger the anti-proliferative activities of both RB and p53. While p14ARF is transcribed from exon 1b and exon 2, p16Ink4A is transcribed from exon 1a localized 20 kb downstream of 1b and exons 2 and 3. As described in more detail below and shown in Figure 2, p16Ink4a binds to CDK4/6 inhibiting its kinase activity thereby preventing Rb phosphorylation (pRb), while p14Arf inhibits the ubiquitin ligase activity of MDM2, thereby stabilizing p53. Rb remains associated with transcription factor E2F1 localizing it to the cytoplasm and thus preventing transcription of E2F1 target genes that, as mentioned above, are crucial for the G1/S transition [20].
Figure 2.
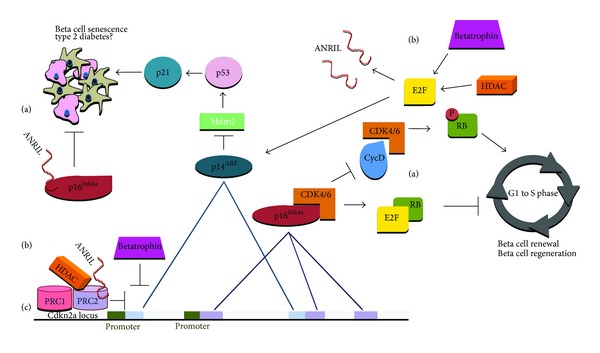
(a) Cell cycle regulation by p16Ink4b/p14ARF. p16Ink4b inhibits CDK4/6 activity, preventing RB phosphorylation, thus preventing G1/S transition. Inactivation of CDK4/6 promotes Rb/E2F1 association down-regulating transcription of E2F1 target genes, which would trigger G1/S transition. p14ARF inhibits MDM2 activity thereby stabilizing p53 and promoting beta cell senescence. (b) Regulation of beta cell Cdkn2a locus. Binding of PRC1 and PRC2 complex proteins to the p16/p14ARF promoter results in formation of heterochromatin leading to suppression of transcription. HDACs also repress Cdkn2a locus but they conform a regulatory feedback loop: release of E2F1 which up-regulates p14ARF leading to an inhibition of Mdm2 and inducing senescence through p21-p53 pathway. Betatrophin promotes beta cell proliferation by inhibiting Cdkn2a and increasing expression levels of cyclins and E2F1 (through a still unknown mechanism). lncRNA ANRIL, transcribed from the Cdkn2a locus, directly binds to the p16Ink4b transcript and also recruits the PRC complexes to repress the transcription of genes at this locus. ANRIL is induced by E2F1 after DNA damage. (c) Cdkn2a locus showing exons of p16Ink4b/p14ARF genes involved in alternate splicing. Promoter regions are shown in green.
Beta cells express most of the known cell cycle inhibitors, including p16INK4a, p18INK4c, p21CIP1, p27Kip1, p53, and Rb. In contrast, and according to the negative regulation model explained in the introduction, there is much less redundancy of cell cycle activators in the beta cell. This model converges in mouse and human models. For example, rodent beta cells express only CDK4 and not CDK6, whereas most other cell types express both of these proteins. Mouse beta cells express all three D cyclins, D1, D2, D3, but the mRNA expression of D2 is significantly higher than both D1 and D3 with only D2 detectable by immunohistochemistry [2]. In order to establish another regulation point of this pathway in beta cells, Fiaschi-Taesch and colleagues [21, 22] have delineated the repertoire of G1/S regulatory proteins present in the adult human islet and have used this information to develop what they call the “human islet G1/S proteome”. These studies state that although the G1/S molecules are mainly considered to be nuclear proteins, they are present principally in the cytoplasm, where possibly they would not be able to regulate cell cycle progression. Furthermore, the only nuclear G1/S molecules are the cell cycle inhibitors, pRb, p57, and p21. p16Ink4a remains in the nucleus in only 8.4% of beta cells under basal conditions. Which in turns becomes nuclear in 16.1% under induction of proliferation. Cell cycle activators as cyclins or Cdks, necessary to drive β-cell proliferation are present in the cytoplasm, not in the nuclear compartment.
Regarding the role of E2F in beta cells, it is well known that the increased expression of E2F contributes to the uncontrolled proliferation of cancer cells, but there is increasing evidence for a Cdk4–E2F1–pRB-specific role in metabolism. To that extent, there is the finding that some specific polymorphisms in the Cdk4 gene could contribute to type 2 diabetes-associated obesity [23]. In that sense, studies E2F1−/− mice show impaired postnatal pancreatic growth that triggers a reduction in pancreatic size with the subsequent impaired glucose homeostasis [24]. Moreover, the CDK4-pRB-E2F1 pathway is activated by glucose through the insulin pathway in beta cells, leading to increased Kir6.2 expression that induces insulin secretion [25–27].
3. ARF/INK4A and Beta Cell Senescence
Emerging evidence indicates that proliferation of pancreatic beta cells is an important mechanism not only to maintain homeostasis in the endocrine pancreas but also for adapting islet function to changes in metabolic demands [9, 28, 29]. The inability of the beta cells to expand and compensate for the changing insulin demand can contribute to the pathogenesis of diabetes. Several studies suggest that beta cell proliferation declines with age [30, 31] and this age-dependent decline in the beta cell proliferation could curtail the ability of the endocrine pancreas to respond to metabolic changes. Furthermore, the cell-intrinsic genetic and epigenetic mechanisms regulating the age-dependent decline of beta cell proliferation [32].
On their review, Gunasekaran and cols compile contradictory studies on the age-related effects on the beta cell. While some of them found that insulin sensitivity decreases with age, others shown that plasma glucose clearance was found to be dependent on the waist-to-hip ratio and not age, with the exception of older people with pre-existing impaired glucose tolerance or type 2 diabetes. Even if these studies come into contradiction, there is evidence that with age, beta cells show decreased expression of cell cycle activators with simultaneous increases in expression of cell cycle inhibitors. Controversial findings and opinions regarding the balance of cell cycle inhibitors and activators have been found. Gunasekaran's compendium describes several studies showing that loss of a single cell cycle inhibitor does not accelerate beta cell cycle progression, whereas loss of multiple inhibitors enhances beta cell proliferation [2]. In the opposite sense, other groups describe that p16Ink4a and p19Arf expression (mRNA) was increased significantly with aging in pancreatic islets, but not other Cdk inhibitors examined, including p15, p18, p21, p27. This up regulation has been linked to reduction in the proliferative capacity of aged beta cells [32, 33].
Aging is associated with replicative senescence and p16Ink4a levels increase with aging in most mammalian tissues. The levels of p16Ink4a in an individual can be predicted by stochastic model that takes into consideration the subjects' age. According to this model, p16Ink4a levels exponentially increase with age in a p16Ink4a-dependent manner and reach a plateau.
Increased p16Ink4a levels in the pancreas during aging (independent of telomere shortening) inhibit the proliferation of beta cells and decrease their ability to respond to injury. While the beta cells of the p16Ink4a knockout mice were able to proliferate in response to injury, beta cells with ectopic expression of p16Ink4a showed reduced proliferative response confirming the association between p16Ink4a and beta cell senescence [20]. In addition, overexpression of p16Ink4a in transgenic mice caused a reduction of islet proliferation in younger more than in older animals.
Expression of p16Ink4a and p14ARF are regulated by promoter hypermethylation through proteins of the PRC1 and PRC2 complexes of the Polycomb group (PcG) of transcriptional repressor proteins. MLL1 and PcG directly control the Ink4a/Arf locus through chromatin epigenetic modifications and the loss of these repressive epigenetic marks leads to a shift of the replication timing of the locus, both in senescent and Polycomb mutant cells [34].
Bmi-1 is a transcription factor, member of the PcG repression complex 1 (PRC1) that inhibits senescence by inhibiting transcription of p16Ink4a. Thus Bmi-1 behaves as an oncogene and is a marker of tumor stem cells. Ezh2 (Enhancer of zeste homolog 2) belongs to the PRC2 complex, it is a histone methyltransferase which represses Ink4a/Arf in islet beta cells, with activity specific for histone H3 K27 [35, 36]. The ability of Bmi-1 to decrease transcription from the Ink4a locus depends on the presence of Ezh2 and other components of the PRC2 complex.
Kotake et al. [37] showed that the removal of pRb from cells resulted in the loss of histone H3 K27 trimethylation leading to the loss of Bmi-1 recruitment to the Ink4a/Arf locus. Moreover, pRb is also shown to be necessary for Bmi-1 function in the transcription repression of Ink4a/Arf. There is a feedback loop between p16Ink4a and Rb: phosphorylation which results in increased p16Ink4a expression and inhibition of CDK4/6 [20]. Other regulatory feedback loop of the locus relays on histone deacetylases (HDAC), p53 is required for both HDAC and PcG to repress p14Arf expression [38], at the same time HDAC1 is involved in the release of E2F1 which in turn could up-regulate p14Arf, leading to an inhibition of Mdm2 activity, the subsequent activation of p53 and induction of senescence through p21 [20, 39]. Previously described by Wang and colleagues [40], HBP1 induces premature senescence through upregulating p16Ink4A expression in primary cells by targeting the p16Ink4A promoter, by interacting and recruiting p300/CBP, whereas HDAC4 represses HBP1-induced p16Ink4a expression, thus represses HBP1-induced premature senescence.
4. New Insights into INK4a/ARF Regulation of Beta Cell Proliferation
The factors explained below are well known to have a specific role in regulation of beta cell proliferation under physiological and pathological conditions. However, the molecular pathways in which they are involved are not fully studied, but they could be promising targets for expanding functional pancreatic islets in diabetes.
5. lncRNAs
Long non-coding RNAs (lncRNAs) are a new class of regulatory RNAs that are defined as transcribed RNA molecules ranging in length from 200 to 100,000 nucleotides and lacking protein-coding capacity. Islet lncRNAs show a marked cell-type specific expression pattern, Morán and colleagues [41] have integrated transcriptional and chromatin maps to systematically annotate lncRNA genes in human pancreatic islet cells and they state that some lncRNAs are dysregulated in type 2 diabetes or map to susceptibility loci. Furthermore, orthologous transcripts in mice are dynamically regulated in a similar manner as human islet lncRNAs. In the case of Cdkn2a, a long non-coding RNA, ANRIL (antisense non-coding RNA), also transcribed from the locus, is involved in the epigenetic regulation of the Cdkn2a locus by direct binding to the p16Ink4b transcript and recruiting the PRC complexes to repress the transcription of genes at this locus. ANRIL is targeted by PRC2 to the Ink4a/Arf/Ink4b locus [42]. Another regulation point regarding ANRIL and cell cycle inhibition is that ANRIL is induced by E2F1 transcription factor after DNA damage, and thus the elevated ANRIL levels suppress the expression of p14Arf and p16Ink4a at the late-stage of DNA damage response (DDR), forming a negative feedback loop to the DDR [43]. Recent studies showed that single nucleotide polymorphisms mapped in the ANRIL as well as in Cdkn2a locus sequences are linked to several pathologic conditions, including type 2 diabetes [39].
6. Betatrophin
Yi et al. [44] have described a peptide whose overexpression in mouse liver produces a secreted protein that significantly and specifically promotes pancreatic beta cell proliferation and beta cell mass expansion and, consequently, improves glucose tolerance. Hence, this peptide has been called betatrophin. Expression levels of cyclins (cyclins A1, A2, B1, B2, D1, D2, and F), CDKs (CDK1 and CDK2), and E2Fs (E2F1 and E2F2) increase, whereas cell cycle inhibitors (Cdkn1a and Cdkn2a) decrease in islets of betatrophin-injected mice compared to control-injected mice. The mechanism of action for betatrophin still remains unknown, if it acts directly or not on the beta cells for controlling their proliferation. Betatrophin is not a novel protein, it is also known as ANGPTL8, TD26, RIFL and Lipasin. Previously to the study of Yi and colleagues, it has been shown that betatrophin is expressed at the highest levels in liver and adipose tissue, and is up-regulated by feeding and suppressed by fasting [45, 46]. To date the betatrophin receptor has not been identified, and maybe other cofactors are acting in the specificity of the betatrophin effect on beta cell mass. However, promising opportunities are open with regard to betatrophin and beta cell mass regeneration.
7. Conclusions
Type 1 and 2 diabetes can be reversed by replacement of beta cell mass, as demonstrated by pancreas and islet transplantation [47]. However, it has limited applicability, given the shortage of organ donors and the need for chronic immunosuppression. Regeneration of beta cell mass is one promising approach to replace the deficit in beta cell mass in diabetic patients [32]. The results stated in this review support that modulation of the Ink4a/Arf locus plays a critical role in regulating pancreatic beta cell proliferation during aging and regeneration. A strategy of beta cell proliferation improvement based in Ink4a/Arf genes inhibition could help to develop new regeneration approaches. One of the main concerns about inhibiting the action of these genes is that they are anti-oncogenes, and could trigger tumour development [48]. To date, few studies show specific inhibitors of p16 and/or p19 genes. Recently, the direct derepression of p21 and p16Ink4a caused by loss of AP4 gene (a c-Myc transcription factor) in fibroblasts has been shown to be sufficient to mediate cellular senescence [49]. In beta cells, a recent and promising study shows that a bioavailable HNF4α (a nuclear receptor transcription factor) antagonist induced β-cell replication in rabbits and mice. Moreover, this compound promotes alpha, beta and delta cell replication in beta cell ablated mice, and repressed the expression of multiple cyclin-dependent kinase inhibitors, including p16Ink4a [50]. Understanding the regulation of Ink4a/Arf locus could reveal the molecular basis of reduced beta cell proliferation with aging and also be extremely useful in devising strategies for beta cell regeneration.
Acknowledgments
The authors apologize to all their colleagues whose work could not be cited due to space limitations. We thank members of the Annicotte's lab and Froguel's lab for discussions. The author's research is supported by the French Agence Nationale de la Recherche (ANR-10-LABX-46), Association pour la Recherche sur le Diabète, Lille2 University, Conseil Régional Nord-Pas de Calais and Lille Métropole Communauté Urbaine.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nature Clinical Practice. Endocrinology & Metabolism. 2007;3(11):758–768. doi: 10.1038/ncpendmet0647. [DOI] [PubMed] [Google Scholar]
- 2.Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging. 2011;3(6):565–575. doi: 10.18632/aging.100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28(33):2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- 4.Golias CH, Charalabopoulos A, Charalabopoulos K. Cell proliferation and cell cycle control: a mini review. International Journal of Clinical Practice. 2004;58(12):1134–1141. doi: 10.1111/j.1742-1241.2004.00284.x. [DOI] [PubMed] [Google Scholar]
- 5.Kuo JZ, Sheu WH, Assimes TL, et al. Trans-ethnic fine mapping identifies a novel independent locus at the 3′ end of CDKAL1 and novel variants of several susceptibility loci for type 2 diabetes in a Han Chinese population. Diabetologia. 2013;56(12):2619–2628. doi: 10.1007/s00125-013-3047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Daghri NM, Alkharfy KM, Alokail MS, et al. Assessing the contribution of 38 genetic loci to the risk of type 2 diabetes in the Saudi Arabian population. Clinical Endocrinology. 2013 doi: 10.1111/cen.12187. [DOI] [PubMed] [Google Scholar]
- 7.Cauchi S, Ezzidi I, El Achhab Y, et al. European genetic variants associated with type 2 diabetes in North African arabs. Diabetes & Metabolism. 2012;38(4):316–323. doi: 10.1016/j.diabet.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi F, Serizawa M, Yamamoto K, et al. Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes. 2009;58(7):1690–1699. doi: 10.2337/db08-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doria A, Patti M-E, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metabolism. 2008;8(3):186–200. doi: 10.1016/j.cmet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng F, Hu D, Gu C, et al. The relationship between five widely-evaluated variants in CDKN2A/B and CDKAL1 genes and the risk of type 2 diabetes: a meta-analysis. Gene. 2013;531(2):435–443. doi: 10.1016/j.gene.2013.08.075. [DOI] [PubMed] [Google Scholar]
- 11.Chen G, Xu Y, Lin Y, et al. Association study of genetic variants of 17 diabetes-related genes/loci and cardiovascular risk and diabetic nephropathy in the Chinese She population. Journal of Diabetes. 2013;5(2):136–145. doi: 10.1111/1753-0407.12025. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Tang X, Liu Q, Wang Y. Association between type 2 diabetes and rs10811661 polymorphism upstream of CDKN2A/B: a meta-analysis. Acta Diabetologica. 2013;50(5):657–662. doi: 10.1007/s00592-012-0400-7. [DOI] [PubMed] [Google Scholar]
- 13.Landman GW, van Vliet-Ostaptchouk JV, Kleefstra N, et al. Association between 9p21 genetic variants and mortality risk in a prospective cohort of patients with type 2 diabetes (ZODIAC-15) Cardiovascular Diabetology. 2012;11, article 138 doi: 10.1186/1475-2840-11-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gamboa-Melendez MA, Huerta-Chagoya A, Moreno-Macias H, et al. Contribution of common genetic variation to the risk of type 2 diabetes in the Mexican Mestizo population. Diabetes. 2012;61(12):3314–3321. doi: 10.2337/db11-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nemr R, Almawi AW, Echtay A, Sater MS, Daher HS, Almawi WY. Replication study of common variants in CDKAL1 and CDKN2A/2B genes associated with type 2 diabetes in Lebanese Arab population. Diabetes Research and Clinical Practice. 2012;95(2):e37–e40. doi: 10.1016/j.diabres.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Gupta V, Vinay DG, Rafiq S, et al. Association analysis of 31 common polymorphisms with type 2 diabetes and its related traits in Indian sib pairs. Diabetologia. 2012;55(2):349–357. doi: 10.1007/s00125-011-2355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cauchi S, Meyre D, Durand E, et al. Post genome-wide association studies of novel genes associated with type 2 diabetes show gene-gene interaction and high predictive value. PLoS ONE. 2008;3(5) doi: 10.1371/journal.pone.0002031.e2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duesing K, Fatemifar G, Charpentier G, et al. Strong association of common variants in the CDKN2A/CDKN2B region with type 2 diabetes in French europids. Diabetologia. 2008;51(5):821–826. doi: 10.1007/s00125-008-0973-4. [DOI] [PubMed] [Google Scholar]
- 19.Hribal ML, Presta I, Procopio T, et al. Glucose tolerance, insulin sensitivity and insulin release in European non-diabetic carriers of a polymorphism upstream of CDKN2A and CDKN2B . Diabetologia. 2011;54(4):795–802. doi: 10.1007/s00125-010-2038-8. [DOI] [PubMed] [Google Scholar]
- 20.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. International Journal of Cancer. 2012;130(8):1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, et al. Human pancreatic beta-cell G1/S molecule cell cycle atlas. Diabetes. 2013;62(7):2450–2459. doi: 10.2337/db12-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, et al. Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human beta-cell replication: a revised model of human beta-cell G1/S control. Diabetes. 2013;62(7):2460–2470. doi: 10.2337/db12-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martín J, Hunt SL, Dubus P, et al. Genetic rescue of Cdk4 null mice restores pancreatic β-cell proliferation but not homeostatic cell number. Oncogene. 2003;22(34):5261–5269. doi: 10.1038/sj.onc.1206506. [DOI] [PubMed] [Google Scholar]
- 24.Fajas L, Annicotte J-S, Miard S, Sarruf D, Watanabe M, Auwerx J. Impaired pancreatic growth, β cell mass, and β cell function in E2F1−/− mice. Journal of Clinical Investigation. 2004;113(9):1288–1295. doi: 10.1172/JCI18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Annicotte J-S, Blanchet E, Chavey C, et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nature Cell Biology. 2009;11(8):1017–1023. doi: 10.1038/ncb1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fajas L, Blanchet E, Annicotte J-S. The CDK4-pRB-E2F1 pathway: a new modulator of insulin secretion. Islets. 2010;2(1):51–53. doi: 10.4161/isl.2.1.10338. [DOI] [PubMed] [Google Scholar]
- 27.Fajas L. Re-thinking cell cycle regulators: the cross-talk with metabolism. Frontiers in Oncology. 2013;3, article 4 doi: 10.3389/fonc.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D. Role of the forkhead protein FoxO1 in β cell compensation to insulin resistance. Journal of Clinical Investigation. 2006;116(3):775–782. doi: 10.1172/JCI24967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulkarni RN. The islet beta-cell. The International Journal of Biochemistry & Cell Biology. 2004;36(3):365–371. doi: 10.1016/j.biocel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of β-cells in aged adult mice. Diabetes. 2005;54(9):2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 31.Meier JJ, Butler AE, Saisho Y, et al. β-cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008;57(6):1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tschen S-I, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes. 2009;58(6):1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 34.Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, Djabali M. Polycomb mediated epigenetic silencing and replication timing at the INK4a/ARF locus during senescence. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005622.e5622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Gu X, Su I-H, et al. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes and Development. 2009;23(8):975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Different Ezh2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Molecular Cell. 2004;14(2):183–193. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- 37.Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes and Development. 2007;21(1):49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng Y, Kotake Y, Pei X-H, Smith MD, Xiong Y. p53 binds to and is required for the repression of Arf tumor suppressor by HDAC and polycomb. Cancer Research. 2011;71(7):2781–2792. doi: 10.1158/0008-5472.CAN-10-3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popov N, Gil J. Epigenetic regulation of the INK4B-ARF-INK4a locus: in sickness and in health. Epigenetics. 2010;5(8):685–690. doi: 10.4161/epi.5.8.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Pan K, Chen Y, Huang C, Zhang X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4a expression. Nucleic Acids Research. 2012;40(3):981–995. doi: 10.1093/nar/gkr818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moran I, Akerman I, van de Bunt M, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metabolism. 2012;16(4):435–448. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotake Y, Nakagawa T, Kitagawa K, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene. 2011;30(16):1956–1962. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wan G, Mathur R, Hu X, et al. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal. 2013;25(5):1086–1095. doi: 10.1016/j.cellsig.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell. 2013;153(4):747–758. doi: 10.1016/j.cell.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Ren G, Kim JY, Smas CM. Identification of RIFL, a novel adipocyte-enriched insulin target gene with a role in lipid metabolism. The American Journal of Physiology—Endocrinology and Metabolism. 2012;303(3):E334–E351. doi: 10.1152/ajpendo.00084.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang R. Lipasin, a novel nutritionally-regulated liver-enriched factor that regulates serum triglyceride levels. Biochemical and Biophysical Research Communications. 2012;424(4):786–792. doi: 10.1016/j.bbrc.2012.07.038. [DOI] [PubMed] [Google Scholar]
- 47.Dean PG, Kudva YC, Stegall MD. Long-term benefits of pancreas transplantation. Current Opinion in Organ Transplantation. 2008;13(1):85–90. doi: 10.1097/MOT.0b013e3282f2fd7f. [DOI] [PubMed] [Google Scholar]
- 48.Lashinger LM, Harrison LM, Rasmussen AJ, et al. Dietary energy balance modulation of Kras- and Ink4a/Arf+/--driven pancreatic cancer: the role of insulin-like growth factor-I. Cancer Prevention Research. 2013;6(10):1046–1055. doi: 10.1158/1940-6207.CAPR-13-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackstadt R, Jung P, Hermeking H. AP4 directly downregulates p16 and p21 to suppress senescence and mediate transformation. Cell Death & Disease. 2013;4 doi: 10.1038/cddis.2013.282.e775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SH, Piran R, Keinan E, Pinkerton A, Levine F. Induction of beta-cell replication by a synthetic HNF4alpha antagonist. Stem Cells. 2013;31(11):2396–2407. doi: 10.1002/stem.1496. [DOI] [PubMed] [Google Scholar]