

Abstract
A primary mode of regulating receptor tyrosine kinase (RTK) signaling is to control access of ligand to its receptor. Many RTK ligands are synthesized as transmembrane proteins. Frequently, the active ligand must be released from the membrane by proteolysis before signaling can occur. Here, we discuss RTK ligand shedding and describe the proteases that catalyze it in flies and mammals. We focus principally on the control of EGF receptor ligand shedding, but also refer to ligands of other RTKs. Two prominent themes emerge. First, control by regulated trafficking and cellular compartmentalization of the proteases and their ligand substrates plays a key role in shedding. Second, many external signals converge on the shedding proteases and their control machinery. Proteases therefore act as regulatory hubs that integrate information that the cell receives and translate it into precise outgoing signals. The activation of signaling by proteases is therefore an essential element of the cellular communication machinery.
Ligands for many receptor tyrosine kinases (e.g., EGFR) are synthesized as transmembrane proteins and released by proteases (e.g., ADAMs). Regulated access of ligand to receptor, over distance and time, helps control signaling.
Cells must talk to one another. This principle applies throughout the tree of life: from unicellular bacteria, to the trillions of cells that coordinate to make a mammal. Communication between cells requires dedicated machinery, capable of relaying information across membranes. Transmembrane proteins are therefore essential for signaling. Understanding how this is regulated is paramount. In mammals, receptor tyrosine kinases (RTKs) and their ligands are important examples of such machinery (Schlessinger 2000), controlling many biological processes including development, immunity, tissue repair, and metabolic homeostasis (Ullrich and Schlessinger 1990). They are transmembrane proteins with an extracellular ligand-binding motif and an intracellular kinase domain. As discussed in other chapters, a common mode of RTK activation involves receptor dimerization induced by ligand binding (Lemmon and Schlessinger 2010).
Regulated access of ligand to receptor, over distance and time, is key to controlling signaling. Ligands are frequently synthesized as transmembrane forms; when they remain membrane-tethered and cannot diffuse, the range over which they can operate is limited to adjacent cells (Massague and Pandiella 1993; Singh and Harris 2005). Other ligands are soluble secretory proteins. This enables paracrine and endocrine signaling—communication between nonadjacent cells. A more complex mode of signaling exploits the characteristics of both of the above. Ligand is synthesized as a transmembrane precursor, which is then shed from the cell surface by proteolysis. This adds an additional and stringent regulatory step to a signaling network (Massague and Pandiella 1993).
This chapter will focus on RTK ligand cleavage and its regulation. We shall highlight how shedding is often critical for signaling, and describe the protease families that catalyze ligand release in flies and mammals. An emergent theme is that regulated trafficking and compartmentalization of ligand and protease modulate signaling. Another theme will be the range of stimuli that impinge on shedding.
The epidermal growth factor receptor (EGFR) is an excellent model RTK to illustrate the regulation of ligand proteolysis because the requirement for ligand cleavage in signaling is well established, and the major physiological sheddases have been identified (Blobel 2005). Where warranted, physiological evidence for the role of ligand shedding in the regulation of other RTKs will also be discussed. Whereas we shall deal mostly with ADAM proteases (“a disintegrin and metalloprotease”), which represent the canonical mammalian RTK ligand shedding machinery, the rhomboid family of intramembrane proteases will also be discussed.
THE EGFR—A PARADIGM FOR RTK REGULATION BY LIGAND CLEAVAGE
EGFR signaling has many developmental and physiological roles in flies and mammals (Shilo 2003; Sibilia et al. 2007). EGFR ligands in both species have the same domain organization and topology (Fig. 1) (Schneider and Wolf 2009). They are usually type I transmembrane proteins with an amino-terminal extracellular domain (ectodomain); therein lies a conserved motif called the EGF domain that is responsible for receptor binding. The EGF domain occurs in all EGFR ligands, but also in other contexts (Davis 1990). For cleavage to occur, the transmembrane ligand precursor must be trafficked into the same membranous compartment as its shedding protease, allowing proteolytic activation into a secreted ligand. We will now compare and contrast ligand shedding in flies and mammals.
Figure 1.

Topology of epidermal growth factor receptor (EGFR) ligands. EGFR ligands are type I transmembrane proteins with an extracellular (luminal) amino-terminus and a cytoplasmic carboxyl terminus. The domain structure of various EGFR ligands is indicated. (A) Drosophila Spitz, and the mammalian EGFR ligands TGF-α, Betacellulin, Epiregulin, and Epigen have a basic structure containing an amino-terminal prodomain and a bioactive EGF domain (indicated in blue). (B) Amphiregulin and HB-EGF contain a heparin binding motif amino terminal to the EGF domain (indicated in green); this facilitates binding to extracellular proteoglycans. Proteolytic cleavage occurs within the juxtamembrane domain between the EGF domain and the TMD; proteolytic removal of the amino-terminal prodomain also occurs (A,B). (C) Epidermal growth factor (EGF) contains additional EGF domains. The EGF domain closest to the membrane can activate the EGFR, whereas the remaining eight EGF domains cannot. The role of these is unclear, although they may play a role in regulating cell–cell adhesion. Cleavage liberates the bioactive EGF domain from the transmembrane precursor; depending on the tissue/context, the other EGF modules may either remain on the soluble molecule, or are cleaved off.
REGULATION OF EGFR SIGNALING IN DROSOPHILA BY LIGAND PROTEOLYSIS
Flies have a relatively simple EGFR pathway: they have a single receptor and only four ligands (Shilo, 2003). As in mammals, most of the ligands (Spitz, Gurken, and Keren) are synthesized as transmembrane precursors, whereas Vein, which resembles neuregulins, is soluble (Freeman 1998). Spitz, a TGF-α-like molecule, is the most important EGFR-activating ligand; the others play subsidiary or tissue-specific roles (for example, Brown et al. 2007). For all three membrane-tethered ligands, their activation requires cleavage by rhomboid proteases (Urban et al. 2002).
Rhomboids are integral membrane proteins that contain six or seven transmembrane domains (Fig. 2) (Freeman 2009). First identified in flies, genetic analysis showed that rhomboid-1 was involved in EGFR signaling but its precise role was obscure (Ruohola-Baker et al. 1993; Sturtevant et al. 1993; Freeman 1994). Genetic mosaic experiments indicated that it acted in the signal-emitting cell, rather than the EGFR-expressing cell, implying a role in signal generation (Wasserman et al. 2000). This enigma was resolved when rhomboids were shown to be serine proteases (Urban et al. 2001) controlled by a catalytic dyad (Lemberg et al. 2005). Rhomboids cleave their substrates within or close to the upper part of the transmembrane domain (Urban et al. 2003; Strisovsky et al. 2009), but this raises the question of water accessibility for a proteolytic reaction in a membrane environment. This was resolved by high-resolution crystal structures of bacterial rhomboids. The catalytic site lies in a hydrophilic depression, just inside the lipid bilayer (Wang et al. 2006; reviewed in Lieberman and Wolfe 2007). In addition to regulating EGFR signaling in flies, rhomboids exist in all kingdoms of life. They regulate processes as diverse as quorum sensing in bacteria, host invasion in Plasmodium, and mitochondrial homeostasis in eukaryotes (Urban and Dickey 2011), but their roles in mammals are less well understood.
Figure 2.
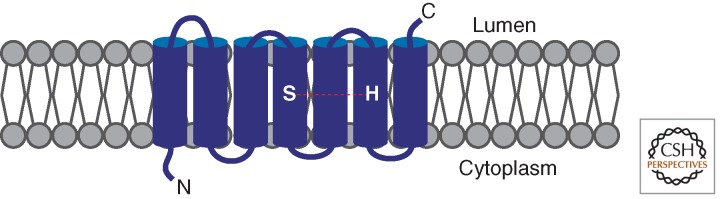
Domain structure of a rhomboid protease. Secretase rhomboids are polytopic transmembrane proteins with a cytoplasmic amino terminus and six or seven transmembrane domains. The catalytic serine and histidine residues are positioned within the upper third of transmembrane helices 4 and 6, respectively.
Regulation of Drosophila EGF Ligand Cleavage by Trafficking
Transcription is a primary regulator of rhomboid in flies (Bier et al. 1990). Superimposed on this, ligand cleavage in Drosophila is controlled by the compartmentalization of the growth factor and rhomboid (Fig. 3) (Lee et al. 2001). As for all proteins that enter the secretory pathway, Drosophila Spitz is synthesized in the endoplasmic reticulum (ER). Spitz is retained in the ER by a mechanism that depends on phospholipase C-γ (Schlesinger et al. 2004). Thus, Spitz is spatially separated from its shedding protease, rhomboid-1, which is Golgi-resident. This segregation is overcome by a trafficking partner called Star, whose role is to escort Spitz to the Golgi, where Spitz is cleaved by rhomboid-1 (Fig. 3) (Lee et al. 2001).
Figure 3.

Regulated Spitz trafficking controls EGFR activation in Drosophila. Spitz is synthesized in the endoplasmic reticulum (ER) as a transmembrane precursor. Exit of Spitz from the ER to the Golgi requires the chaperone protein, Star (illustrated in red). On entry to the Golgi, Spitz encounters rhomboid (illustrated in blue) and undergoes proteolysis within the transmembrane domain. Spitz can now be secreted, thereby facilitating EGFR activation on a nearby cell.
Recent evidence suggests the potential for even more intricate trafficking regulation. Another rhomboid, rhomboid-3, can cleave Spitz in both the ER and the later secretory pathway (Yogev et al. 2008). Spitz cleavage in different compartments exerts a radically different outcome on signaling. In the late secretory pathway, rhomboid-3 cleavage of Spitz is analogous to rhomboid-1: it is Star-dependent and leads to EGFR activation (Yogev et al. 2008). In contrast, ER cleavage of Spitz is an inactivating step because it leads to ER retention. In addition, it has been reported that proteolysis of Star by rhomboid-3 inhibits ER exit of Spitz, thereby attenuating EGFR activation (Tsruya et al. 2007).
In summary, the basis for maintaining control over EGFR activation in the fly involves keeping growth factor and enzyme apart, until signaling is required.
REGULATION OF EGFR SIGNALING IN MAMMALS
Mammalian EGFR Ligands
Although the mechanisms of regulation are different in mammals, the logic of regulated trafficking and compartmentalization is conserved. Regulation of shedding is more complex in mammals and includes posttranslational regulation of the shedding protease (Blobel 2005; Murphy 2008). Mammals have four members of the EGF receptor family: ErbB1–B4. Of these, only ErbB1 and ErbB4 are truly analogous to fly EGFR in that they bind ligand and are active RTKs; ErbB2 cannot bind ligand, whereas ErbB3 lacks kinase activity (Yarden and Sliwkowski 2001). Although ErbB2 and ErbB3 cannot therefore signal autonomously, they can form productive heterodimers with ErbB1 and ErbB4, thereby diversifying signaling properties (Citri et al. 2003).
For convenience, mammalian EGFR ligands can be separated into two classes, based on receptor-binding preferences (Harris et al. 2003). The first class, comprising ligands that bind to ErbB1, are amphiregulin (AREG), betacellulin (BTC), epidermal growth factor (EGF), epigen, epiregulin (EPR), heparin-binding EGF (HB-EGF), and transforming growth factor α (TGF-α) (Massague and Pandiella 1993; Schneider and Wolf 2009). Members of the neuregulin family (Nrg1-4) form the second group of ligands. These can bind to ErbB3/ErbB4 and include the multiple and complex splice variants of Nrg1 (Falls 2003). In addition, some members of the first class also show activity on ErbB4: BTC, HB-EGF, and EPR (Harris et al. 2003).
As shown in Figure 1, with the exception of EGF, most ligands have a domain structure resembling Drosophila Spitz. They are type I transmembrane proteins (their amino termini are on the luminal side of the membrane) and contain an amino-terminal prodomain, followed by a single EGF domain, a transmembrane domain, and cytoplasmic tail. Amphiregulin and HB-EGF have a heparin-binding motif located amino terminal to the EGF domain (Fig. 1) (Cook et al. 1991; Higashiyama et al. 1991). This allows binding to proteoglycans in the extracellular matrix and provides an extra mechanism to control ligand diffusion after cleavage (Piepkorn et al. 1998). EGF itself is somewhat unusual, because it contains eight EGF repeats amino terminal to the actual bioactive EGF domain (the module closest to the TMD) (Fig. 1).
ADAM PROTEASES
ADAM Protease Domain Structure
Mammalian EGFR ligand cleavage requires ADAM proteases instead of rhomboids. These are single-pass transmembrane proteins and their active sites are located on the extracellular side of the membrane, not within it (Fig. 4). Substrate cleavage typically occurs on the cell surface or within the late secretory pathway (Murphy 2008). Mammalian genomes contain multiple ADAM genes, a total of 21 in humans, of which 13 are predicted to be catalytically active (Blobel 2005). Rodent genomes have additional ADAMs, many of which are testes-specific (Puente and Lopez-Otin 2004).
Figure 4.

Domain structure and activation of ADAM metalloproteases. ADAMs are type I transmembrane proteins containing extracellular (luminal) amino termini and cytoplasmic carboxyl termini. (A) ADAMs are synthesized as a zymogen proform that lacks proteolytic activity, because the prodomain binds within the active site cleft. Removal of the prodomain by autocatalysis or by the proportein convertase, furin, is required before the enzyme can be active. (B) After processing, the prodomain may remain bound to the active site, and may require displacement before the ADAM can be active. When activated by signals, ADAMs cleave their substrates with a region just outside the membrane (within the juxtamembrane region).
ADAMs contain a complicated domain structure (Fig. 4). Beyond the amino-terminal signal peptide, they contain a bifunctional prodomain. During biogenesis, it acts as a chaperone (Leonard et al. 2005) and subsequently maintains the enzyme in a zymogen (inactive) form during transit through the secretory pathway (Gonzales et al. 2008). Before ADAMs reach the cell surface, the inhibitory prodomain must be removed. Some ADAMs remove their prodomain autocatalytically (Murphy 2009), whereas for others (including ADAMs 10 and 17), it is removed in the late Golgi by the proprotein convertase furin (Peiretti et al. 2003a).
Adjacent to the prodomain is the catalytic heart of the enzyme, the metalloprotease domain, which contains the zinc-coordinating HEXHH catalytic motif (Blobel 2005). Next to this lies a disintegrin domain that derives its name from a similarity to an integrin-binding protein secreted in snake venom (Blobel and White 1992). In some ADAMs, extracellular matrix interactions with this domain may influence cell–cell adhesion (White 2003). The cysteine-rich region, which may influence substrate recognition (Smith et al. 2002), is located further toward the carboxyl terminus, followed by an EGF domain and transmembrane domain. On the other side of the membrane lies the cytoplasmic tail, which may be important for sensing cytoplasmic signals that regulate trafficking and control enzyme activity. Regulation of ADAM trafficking and activity is discussed later.
Physiological Importance of ADAMs in EGFR Ligand Shedding
It has been proposed that the membrane-tethered versions of some ligands can initiate juxtamembrane activation of the EGFR (Singh and Harris 2005). However, as in Drosophila, the importance of ligand shedding is very clear. First, as described below, ADAM mutant mice essentially phenocopy the EGFR ligand knockouts; second, pharmacological inhibition of metalloproteases blocks EGFR activation (Dong et al. 1999).
Although several ADAMs can cleave EGF ligands, physiological evidence suggests that TACE (tumor necrosis factor α-converting enzyme) also known as ADAM17, and ADAM10 are the major mammalian EGFR ligand sheddases. ADAM10 and TACE orthologs also exist in Drosophila, although they do not appear to be involved in EGFR ligand processing (Delwig and Rand 2008). In mammals, robust genetic and biochemical data suggest that TACE is most important: TACE knockout mice phenocopy many aspects of individual EGFR ligand knockouts, particularly TGF-α and HB-EGF knockouts. This includes defects in epithelial maturation, premature eyelid opening, hair follicle defects, lung branching morphogenesis defects, and heart valve malformations (Luetteke et al. 1993; Mann et al. 1993; Peschon et al. 1998). The importance of shedding is further highlighted by experiments in mice expressing a noncleavable version of HB-EGF. These animals show severe heart failure and enlarged heart valves: a phenocopy of the HB-EGF knockouts (Iwamoto et al. 2003; Yamazaki et al. 2003). Experiments using primary and immortalized cells (embryonic fibroblasts and keratinocytes) from TACE-null mice confirm that it sheds TGF-α, HB-EGF, amphiregulin, and epiregulin (Merlos-Suarez et al. 2001; Sunnarborg et al. 2002; Sahin et al. 2004).
ADAM10 knockout mice die in utero, because of Notch-related defects (on ligand binding, the receptor Notch is cleaved sequentially by ADAM10, then γ secretase; this triggers transcription of Notch target genes) (Hartmann et al. 2002). Because of this lethality, ADAM10’s contribution to ligand shedding in individual tissues has not been examined. However, experiments using immortalized embryonic fibroblasts show that it is required for EGF and BTC cleavage (Sahin et al. 2004).
A number of other ADAMs can also cleave EGFR ligands (Edwards et al. 2008). For example, ADAMs 8, 9, 12, and 15 can process HB-EGF; EGF can be shed by ADAMs 8, 9, 12, and 19 and betacellulin by ADAMs 8, 12 and 19 (Izumi et al. 1998; Asakura et al. 2002; Schafer et al. 2004). Although these ADAMs are not major physiological EGFR ligand sheddases, there may be pathological contexts in which their deregulated expression or activity contributes to cleavage (Sahin et al. 2004; Horiuchi et al. 2007a). Related to this, ADAM10 can cleave TACE substrates when the activity of the latter is inhibited (Le Gall et al. 2009).
REGULATION OF ADAMS
EGFR ligand cleavage is not the only biological role of ADAM proteases. ADAM10 and TACE also regulate processes as diverse as inflammation, Notch signaling, cell adhesion, amyloid precursor protein processing, neuronal migration, and angiogenesis (Blobel 2005; Edwards et al. 2008). TACE itself was identified as the physiological sheddase for TNF (tumor necrosis factor) (Black et al. 1997); when secreted, this cytokine is the primary regulator of inflammatory responses (Palladino et al. 2003). For TACE alone, more than 50 substrates have been identified (Edwards et al. 2008). This raises an important question: How can a seemingly promiscuous sheddase control a given signaling pathway with the necessary precision?
Stimuli that Trigger ADAM Shedding
ADAM10 and TACE are controlled by distinct signaling circuits. TACE can be activated rapidly and powerfully by multiple signals including pharmacological stimuli like phorbol esters, which operate via protein kinase C (Fig. 5) (Rovida et al. 2001; Doedens et al. 2003). TACE is also activated by a more physiological but complex process known as transactivation (Fig. 5) (Gschwind et al. 2001; discussed by Ullrich and colleagues). Transactivation occurs when G-protein coupled receptor (GPCR) stimulation indirectly triggers activation of the EGFR. In outline, GPCR agonists (e.g., bombesin, angiotensin II, serotonin, hormones) trigger a rather poorly defined signaling cascade that activates TACE, resulting in ligand processing (principally HB-EGF) and consequent EGFR activation (Fig. 5). It is now clear that EGFR transactivation plays critical physiological roles in angiogenesis, heart development, and neurogenesis (Gschwind et al. 2001). It is also implicated extensively in tumor growth, invasion and metastasis (Lappano and Maggiolini 2011). TACE activity can also be induced by many other pathways including toll receptors, multiple kinases, and even cleaved TACE substrates (Diaz-Rodriguez et al. 2002; Soond et al. 2005). In summary, TACE lies at the heart of a complex regulatory network, responsible for multiple potent signaling events.
Figure 5.

Transctivation of the EGFR by G protein-coupled receptors (GPCRs). Activation of GPCRs by agonists triggers a signaling cascade involving second messengers including Ca2+ and protein kinase C (PKC). This induces TACE cleavage of EGFR ligands (including HB-EGF), culminating in EGFR activation. How these signals trigger TACE activation remains unclear (see text). Phorbol esters such as PMA can also trigger TACE via a mechanism that involves PKC.
ADAM10 is also regulated by a different complement of signals. It is not affected by phorbol esters, but can be activated by calcium ionophores, calmodulin inhibition, and the metalloprotease-activating drug p-aminophenylmercuric acetate (Sanderson et al. 2005; Horiuchi et al. 2007a). More physiologically, activation of purinergic receptors can also trigger ADAM10 activity (Le Gall et al. 2009), as can GPCRs (Lemjabbar and Basbaum 2002). For both TACE and ADAM10, the cytoplasmic tail may be required for receiving input stimuli, although the mechanism remains unclear (Horiuchi et al. 2007a).
How do these diverse stimuli activate TACE? Despite extensive study, this is still disappointingly unclear. Proposed mechanistic effects on TACE can be divided into three classes: regulation of enzyme activity on the plasma membrane, regulated access of enzyme to substrate, and control of TACE trafficking. Given the diversity of signals that can control TACE, it is quite possible that different mechanisms are used in different contexts.
Impact of Stimuli on TACE Activity on the Plasma Membrane
Signals may impinge directly on the activity of mature TACE on the plasma membrane. A recent study from Blobel and colleagues has suggested that phorbol esters can trigger a conformational change in the TACE active site, thereby enhancing proteolytic activity (Le Gall et al. 2010). This suggests a model whereby prodomain removal from TACE is not sufficient to render it fully active—an additional conformational rearrangement of the active site architecture is also required. It will be interesting to establish how signals trigger these potential active site conformational changes, whether this impacts directly on TACE or on a cofactor, and how this class of mechanism integrates with other forms of control.
The Timps (tissue inhibitors of metalloproteases) are another class of ADAM regulators that impact directly on enzymatic activity. These soluble proteins inhibit matrix metalloproteases and ADAMs by inserting their amino-terminal wedge-shaped cleft into the active site of the enzyme (Wisniewska et al. 2008). There are four Timps in mammals; of these, ADAM10 can be inhibited by Timp1 and Timp3, whereas TACE is inhibited only by Timp3 (Murphy 2011). The impact of Timp on TACE shedding of EGF ligands is unclear; although some evidence suggests that loss of Timp3 affects EGFR ligand shedding in vivo (Murthy et al. 2010), a recent study suggests that loss of Timp3 in mouse embryonic fibroblasts has little impact on phorbol ester-triggered TACE shedding (Le Gall et al. 2010). Interestingly, the biggest physiological impact of loss of Timp3 is its effect on TNF shedding. As described above, as well as regulating EGFR signaling, TACE controls inflammation and apoptosis via cleavage of TNF. Reflecting this, deletion of Timp3 in mice causes uncontrolled TNF shedding by TACE, resulting in excessive inflammation caused by TNF perturbation (Mohammed et al. 2004; Guinea-Viniegra et al. 2009). Perhaps control by Timps of TACE activity is a means of regulating specificity in the face of multiple stimuli.
Tetraspanins: Spatial Control of ADAM Shedding
Another way of controlling TACE activity on the plasma membrane is by modulating access to substrates or regulators. This can be achieved by tetraspanins, which form a large family of proteins with four transmembrane domains in mammals (Yanez-Mo et al. 2011). The function of tetraspanins is diverse and not well understood in many cases but a common theme is the organization of membrane proteins into defined microdomains (Hemler 2005). They have been found in association not only with ADAMs (particularly ADAM10), but also EGF ligands and the EGFR itself, suggesting that they may have complex influences on signaling (Imhof et al. 2008; Murayama et al. 2008).
Tetraspanins can inhibit or promote ligand proteolysis, depending on the context. For instance, binding of tetraspanins, including CD9, CD81, and CD82, to ADAM10 promotes ligand shedding (Arduise et al. 2008). How this promotes cleavage is unclear, although an obvious possibility is regulating access of enzyme to substrate. In the case of tetraspanin-12, other possibilities include enhanced prodomain removal, or stabilizing the active enzyme (Xu et al. 2009).
In contrast, the tetraspanin CD9, which can bind to TACE and also to EGFR ligands, reduces shedding (Higashiyama et al. 1995; Imhof et al. 2008). By inhibiting ligand cleavage, tetraspanins may regulate switching from shedding to juxtamembrane signaling. In addition to corralling membrane proteins on the cell surface, CD9 may also influence ligand biogenesis/trafficking through the secretory pathway (Berditchevski and Odintsova 2007). Overall, it appears that rather than behaving as specific regulators of shedding, tetraspanins may represent a spatial anchoring network for many membrane proteins. They may control access of enzyme to substrate within membrane microdomains, or by coordinating access of signaling proteins that regulate ADAM activity.
Impact of Stimuli on TACE Trafficking
Clearly, before TACE can reach the cell surface as an active protease, it must first transit through the secretory pathway. Significant fractions of endogenous TACE are found within intracellular compartments (Schlondorff et al. 2000; Soond et al. 2005). This suggests that TACE biogenesis or trafficking is rate-limiting and may require chaperones or cofactors. Indeed, whereas many glycoproteins transit through the secretory pathway quite rapidly (within minutes to hours) (Ward and Kopito 1994; Jansens et al. 2002), TACE trafficking is considerably slower (Schlondorff et al. 2000). Consistent with this, enhanced trafficking and furin cleavage has been suggested as a mechanism whereby signals activate TACE (Soond et al. 2005).
TACE Trafficking Regulators
A controversy concerns the role of the TACE cytoplasmic tail. In response to signals, the TACE cytoplasmic tail is phosphorylated by kinases, including extracellular-signal-regulated kinase (ERK), and this has been suggested to enhance TACE trafficking (Fan et al. 2003; Soond et al. 2005; Xu and Derynck 2010). Some studies, however, have reported that the TACE tail is dispensable for signaling (Horiuchi et al. 2007a). In other approaches, it has been shown that the tails of many ADAMs possess SH3-binding sites; this has encouraged the search for binding partners (Seals and Courtneidge 2003). Yeast two-hybrid screens have identified TACE tail-interacting proteins, several of which contain SH3 domains and/or PDZ domains (Zheng et al. 2002; Peiretti et al. 2003b; Tanaka et al. 2004). Disappointingly, however, the physiological importance of these interactions remains to be shown. Overall, there is probably insufficient evidence to make a definitive judgment about the significance of the cytoplasmic tail of TACE; perhaps its role is different in different cellular contexts.
MAMMALIAN RHOMBOIDS AND RTKS
iRhoms: Pseudoprotease Regulation of RTK Ligand Shedding
As mentioned above, although TACE activity and trafficking is highly controlled, physiologically important regulators are lacking. However, we and others have recently shown that a protein called iRhom is essential for TACE trafficking (Adrain et al. 2012; McIlwain et al. 2012; Siggs et al. 2012). iRhoms are a metazoan subfamily of rhomboid-like proteins that lack key catalytic residues, rendering them proteolytically inactive (Fig. 6) (Adrain and Freeman 2012).
Figure 6.

Regulation of RTK signaling by iRhoms. (A) Comparison of an active rhomboid and an iRhom. In comparison with an active rhomboid (left), iRhoms (right) contain an extended cytoplasmic amino terminus and a globular cysteine-rich domain called the iRhom homology domain (iRHD) within the lumen of the ER. All iRhoms have a conserved proline residue immediately amino terminal to the serine in TMD 4; this renders iRhoms proteolytically inactive. (B) Drosophila iRhom regulates ER-associated degradation of EGFR ligands. Spitz is normally trafficked out of the ER by Star and encounters rhomboid in the Golgi (Fig. 3). However in the presence of iRhom, Spitz is retained in the ER and instead, shunted into the ER-associated degradation (ERAD). This results in its dislocation from the ER membrane and degradation by the proteasome. As a result, no Spitz enters the Golgi for cleavage and EGFR signaling is attenuated. (C) Regulation of TACE trafficking by mammalian iRhom2. TACE is synthesized in the ER as an inactive zymogen containing the prodomain (the TACE prodomain is indicated in orange). iRhom is required for trafficking of TACE into the Golgi, where it undergoes prodomain cleavage by furin. Active TACE can then cleave its substrates in the late Golgi or on the cell surface.
There are two mammalian iRhoms (also called RHBDF1 and RHBDF2). iRhom1/RHBDF1 has been broadly implicated in growth control of cancer cells and EGFR signaling, although the mechanistic basis for this is unclear (Nakagawa et al. 2005; Yan et al. 2008). Recently, however, the cellular function of mammalian iRhoms has been revealed: iRhom2 is essential for the export of TACE from the ER, and thereby for the shedding of TACE substrates (Adrain et al. 2012; McIlwain et al. 2012). In macrophages the primary role of TACE is TNF release, and accordingly iRhom2 mutant mice do not secrete TNF in response to immune challenge. Processing of the TACE substrate HB-EGF, is also defective in iRhom2-null cells, suggesting that mammalian iRhom can act as a positive regulator of the EGFR. Therefore iRhom is an important trafficking regulator required for the ER exit of TACE. How signals impact on the ability of iRhom to control TACE activation remains to be established.
In Drosophila, iRhom is also implicated in trafficking regulation, but in a different manner. It controls EGFR signaling, but unlike its effects on TACE, it is an inhibitor rather than an activator of trafficking. Flies null for iRhom show a severe activity phenotype (Zettl et al. 2011), which phenocopies the effect of rhomboid overexpression in the central nervous system (Foltenyi et al. 2007). At the cellular level, iRhom binds to EGFR ligands in the ER, and directs them into ERAD (ER-associated degradation); this blocks their onward trafficking and prevents them from activating EGFR signaling (Zettl et al. 2011). ERAD is a fundamental cellular quality control process, whereby misfolded proteins in the ER are retrotranslocated into the cytoplasm and degraded by the proteasome (Smith et al. 2011). In Drosophila, iRhoms exploit this mechanism to regulate signaling.
Rather confusingly, therefore, the Drosophila and mammalian results imply that in different contexts, iRhoms can either inhibit or promote signaling. The mechanism underlying this dual function remains to be determined, but it is clear that iRhoms are ER-localized trafficking regulators that impact on, among other pathways, EGFR signaling (Fig. 6). More generally, they add another example of compartmentalization as a way of controlling of RTK signaling.
Active Mammalian Rhomboids
We have highlighted that ADAMs are the principal mammalian EGFR ligand sheddases. But whether they are the only sheddases remains to be proven. There are suggestions of other ligand-shedding activities, including serine proteases (Pandiella et al. 1992; Le Gall et al. 2004). Tissue-specific heterogeneity in the molecular weight forms of cleaved ligands also suggests that different sheddases exist (Dempsey et al. 1997). Furthermore, given that many signaling components are conserved between flies and mammals, a role for rhomboids in mammalian EGFR ligand cleavage is also possible.
Four mammalian rhomboid proteases localize to the secretory pathway, although physiological substrates have yet to be identified for most (Lohi et al. 2004). Of these, RHBDL2 is the best characterized. RHBDL2 shows a restricted expression pattern in the mouse including intestine, stomach, prostate, bladder, and skin. These are tissues where ErbB1 and one of its ligands, EGF, are expressed. Consistent with this, EGF is an efficient substrate of RHBDL2 (but not other rhomboids) in cell culture assays (Adrain et al. 2011). Although the physiological significance of RHBDL2 cleavage of EGF is unknown, RHBDL2 contributes to the shedding of EGF in some tumor cells (Adrain et al. 2011). In these cases, ADAM inhibition alone cannot block shedding, suggesting that there may be some therapeutic contexts where blocking ADAMs may not necessarily inhibit EGF cleavage. Despite the possible relationship between RHBDL2 and EGF, most mammalian EGF family ligands appear not to be cleaved by rhomboids, implying other, as yet unknown functions for these intramembrane proteases. Notably, at least two other substrates of RHBDL2 have been reported, although, again, their biological relevance remains obscure (Lohi et al. 2004; Pascall and Brown 2004).
OTHER RTKS REGULATED BY LIGAND PROTEOLYSIS
Cleavage of Ephrins
Although the importance for ligand cleavage is most studied in the EGFR, shedding also regulates other RTKs. The regulation of Eph signaling is particularly interesting because it is atypical. Rather than being an activating step, ADAM cleavage is required to switch off signaling. Regulation of axonal guidance requires cell–cell repulsion, driven by the interaction between Eph receptors on one cell and membrane-tethered ephrin ligand on an adjacent cell (Pitulescu and Adams 2010). As the Eph:ephrin interaction is multivalent and high affinity, receptor:ligand complexes favor cell–cell adhesion. But successful repulsion requires signal termination, which necessitates breaking Eph:ephrin complexes. One way in which this is achieved is via proteolysis.
ADAM10 is the ephrin protease (Hattori et al. 2000; Janes et al. 2005). To terminate signaling, ADAM10 recognizes assembled Eph:ephrin complexes and severs them by cleaving the ligand in trans on the adjacent cell; unbound ligands are ignored (Davis et al. 1994). Failure to cleave ephrins delays axon withdrawal (Hattori et al. 2000). The role of ADAM proteolysis in Eph signaling may be conserved in flies, as mutants in the ADAM10 homolog, Kuzbanian, show an axonal extension defect (Fambrough et al. 1996). Although the physiological context is yet unclear, the mammalian rhomboid protease RHBDL2, can also cleave ephrinB3, leading to the possibility of analogous rhomboid regulation of Eph signaling (Pascall and Brown 2004).
In some contexts, Eph:ephrin signaling is bidirectional in nature. As well as triggering RTK activity in the receptor-bearing cells, a signal is triggered in the ligand-expressing cell (Georgakopoulos et al. 2006). This is important for the role of ephrinB2 in cardiac valve maturation and axon path finding (Cowan et al. 2004). On ADAM10 cleavage of receptor-bound ephrin, the transmembrane stub is further cleaved by γ-secretase and the carboxy-terminal fragment thus generated drives an Src-associated signaling cascade (Georgakopoulos et al. 2006).
Shedding of Ligands of the PDGF Family
Some members of the PDGF (platelet-derived growth factor) receptor subfamily are regulated by ligand shedding, including cKit/SCFR (Huang et al. 1992), FLT3/Flk2 (Horiuchi et al. 2009), and CSF1R/Ems (Horiuchi et al. 2007b). These RTK pathways play important immune roles including regulating haematopoiesis (cKit and FLT3) (Ashman 1999; Naoe and Kiyoi 2004) or macrophage and osteoclast development (CSF1R) (Cecchini et al. 1997). The ligand for cKit is expressed in two alternative splice forms: Kit ligand 1 (KL-1) contains an extra exon upstream of the juxtamembrane region that renders it readily susceptible to proteolytic shedding, whereas KL-2 lacks this exon and is cleaved less efficiently (Huang et al. 1992). As for many other RTK ligands, the principal Kit ligand sheddase is unknown, and several enzymes including ADAMs have been suggested (Huang et al. 1992). At least in mouse embryonic fibroblasts, TACE is essential for phorbol ester-induced shedding of KL-1 and KL-2, as well as for the constitutive shedding of KL-2 (Kawaguchi et al. 2007). However, constitutive shedding of KL-1 is not impaired in TACE null fibroblasts and cannot be inhibited by the metalloprotease inhibitor BB94, implicating another sheddase. Despite the importance of ligand shedding to activate Kit signaling, it is important to note that expression of soluble Kit ligand in mice cannot fully rescue the KitL knockout phenotype, illustrating that juxtamembrane signaling is also important (Brannan et al. 1991).
DEREGULATED LIGAND SHEDDING AND CANCER
A common theme for all RTK families is that once activated, the receptors can drive tumor growth, promote cell survival, migration, and resistance to chemotherapy (Lemmon and Schlessinger 2010; Sastry and Elferink 2011). Because proteolysis is irreversible, shedding can be the commitment point for unleashing potent signals. Amplification or mutation of RTKs can render them more sensitive to growth factors. When combined with excessive ligand shedding, this establishes an autocrine loop that enables cells to proliferate independently of the requirement for external cues (Di Marco et al. 1989). This is one route to malignant transformation (Sporn and Roberts 1985). EGFR amplification combined with increased growth factor shedding has been observed in diverse human cancers including gliomas and malignancies of the lung, bladder, gastric tract, esophagus, breast, ovaries, and head and neck tumors (Gullick 1991; Salomon et al. 1995). Other deregulated feedback loops can also drive cancer. Activation of the EGFR drives Ras activity, which can promote increased ligand transcription (Baselga et al. 1996), and kinases downstream of Ras can regulate TACE; this in turn amplifies EGFR signaling (Diaz-Rodriguez et al. 2002; Soond et al. 2005).
Increased expression of sheddases, including TACE, occurs in many tumors, contributing to metastasis and correlating with reduced patient survival rates (Murphy 2008). Increased crosstalk between many GPCRs and the EGFR also plays a significant role in cancer development and metastasis (Lappano and Maggiolini 2011). Another important factor is the interplay in signals between tumor and stromal cells. Recruitment of inflammatory cells to the tumor microenvironment is known to result in secretion of RTK ligands that enhance tumor growth (Wyckoff et al. 2004). These examples all highlight the medical importance of the control mechanisms discussed in this chapter and imply the therapeutic rationale of targeting the proteases that lie at the heart of much RTK activation. The ADAM proteases, for example, with their central position in both growth factor and cytokine activation, have been the focus of much pharmaceutical interest (Saftig and Reiss 2011). Attempts to inhibit them, however, have so far been disappointing, possibly because metalloproteases share a common active site architecture, resulting in off-target effects and toxicity (Coussens et al. 2002; DasGupta et al. 2009).
CONCLUDING REMARKS
It is clear that ligand shedding is a pivotal step for RTK activation in many contexts, particularly for the EGFR, but also for other RTKs. Whereas there has been progress in identifying the major physiological ligand proteases, much remains to be understood about the diverse mechanisms that trigger and regulate shedding. The primary mechanistic theme that we have emphasized is the role of regulated trafficking and compartmentalization of ligands and shedding proteases as a powerful and versatile mode of control. This regulatory logic, which exploits the exquisite regulation of cellular membrane trafficking systems, occurs repeatedly, even when specific components differ. Particular challenges for the future include understanding how the multiple overlapping signals and their regulators are integrated in vivo, and how pathological stimuli, including those emanating from GPCRs, control protease activity. Clearly sheddases are important drug targets; however, the ability to inhibit them specifically and potently still eludes us. Understanding more about control of proteolysis and the requirement for trafficking regulators and cofactors will no doubt identify new drugs targets for the many important pathways they control.
ACKNOWLEDGMENTS
We thank Viorica Lastun and Kvido Strisovsky for helpful comments on the manuscript. C.A. was supported by a long-term fellowship from The International Human Frontier Science Program Organization and was the recipient of an EMBO Long-Term Fellowship. The Freeman group is supported by the Medical Research Council Programme number U105178780.
Footnotes
Editors: Joseph Schlessinger and Mark A. Lemmon
Additional Perspectives on Signaling by Receptor Tyrosine Kinases available at www.cshperspectives.org
REFERENCES
- Adrain C, Freeman M 2012. New lives for old: Evolution of pseudoenzyme function illustrated by iRhoms. Nat Rev Mol Cell Biol 13: 489–498 [DOI] [PubMed] [Google Scholar]
- Adrain C, Strisovsky K, Zettl M, Hu L, Lemberg MK, Freeman M 2011. Mammalian EGF receptor activation by the rhomboid protease RHBDL2. EMBO Rep 12: 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrain C, Zettl M, Christova Y, Taylor N, Freeman M 2012. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335: 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arduise C, Abache T, Li L, Billard M, Chabanon A, Ludwig A, Mauduit P, Boucheix C, Rubinstein E, Le Naour F 2008. Tetraspanins regulate ADAM10-mediated cleavage of TNF-α and epidermal growth factor. J Immunol 181: 7002–7013 [DOI] [PubMed] [Google Scholar]
- Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, Ishiguro H, et al. 2002. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: Metalloproteinase inhibitors as a new therapy. Nat Med 8: 35–40 [DOI] [PubMed] [Google Scholar]
- Ashman LK 1999. The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol 31: 1037–1051 [DOI] [PubMed] [Google Scholar]
- Baselga J, Mendelsohn J, Kim YM, Pandiella A 1996. Autocrine regulation of membrane transforming growth factor-α cleavage. J Biol Chem 271: 3279–3284 [DOI] [PubMed] [Google Scholar]
- Berditchevski F, Odintsova E 2007. Tetraspanins as regulators of protein trafficking. Traffic 8: 89–96 [DOI] [PubMed] [Google Scholar]
- Bier E, Jan LY, Jan YN 1990. Rhomboid, a gene required for dorsoventral axis establishment and peripheral nervous system development in Drosophila melanogaster. Genes Dev 4: 190–203 [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al. 1997. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 385: 729–733 [DOI] [PubMed] [Google Scholar]
- Blobel CP 2005. ADAMs: Key components in EGFR signalling and development. Nat Rev Mol Cell Biol 6: 32–43 [DOI] [PubMed] [Google Scholar]
- Blobel CP, White JM 1992. Structure, function and evolutionary relationship of proteins containing a disintegrin domain. Curr Opin Cell Biol 4: 760–765 [DOI] [PubMed] [Google Scholar]
- Brannan CI, Lyman SD, Williams DE, Eisenman J, Anderson DM, Cosman D, Bedell MA, Jenkins NA, Copeland NG 1991. Steel-Dickie mutation encodes a c-kit ligand lacking transmembrane and cytoplasmic domains. Proc Natl Acad Sci 88: 4671–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KE, Kerr M, Freeman M 2007. The EGFR ligands Spitz and Keren act cooperatively in the Drosophila eye. Dev Biol 307: 105–113 [DOI] [PubMed] [Google Scholar]
- Cecchini MG, Hofstetter W, Halasy J, Wetterwald A, Felix R 1997. Role of CSF-1 in bone and bone marrow development. Mol Reprod Dev 46: 75–83; discussion 83–4 [DOI] [PubMed] [Google Scholar]
- Citri A, Skaria KB, Yarden Y 2003. The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Exp Cell Res 284: 54–65 [DOI] [PubMed] [Google Scholar]
- Cook PW, Mattox PA, Keeble WW, Pittelkow MR, Plowman GD, Shoyab M, Adelman JP, Shipley GD 1991. A heparin sulfate-regulated human keratinocyte autocrine factor is similar or identical to amphiregulin. Mol Cell Biol 11: 2547–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM 2002. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 295: 2387–2392 [DOI] [PubMed] [Google Scholar]
- Cowan CA, Yokoyama N, Saxena A, Chumley MJ, Silvany RE, Baker LA, Srivastava D, Henkemeyer M 2004. Ephrin-B2 reverse signaling is required for axon pathfinding and cardiac valve formation but not early vascular development. Dev Biol 271: 263–271 [DOI] [PubMed] [Google Scholar]
- DasGupta S, Murumkar PR, Giridhar R, Yadav MR 2009. Current perspective of TACE inhibitors: A review. Bioorg Med Chem 17: 444–459 [DOI] [PubMed] [Google Scholar]
- Davis CG 1990. The many faces of epidermal growth factor repeats. New Biol 2: 410–419 [PubMed] [Google Scholar]
- Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Goldfarb M, Yancopoulos GD 1994. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 266: 816–819 [DOI] [PubMed] [Google Scholar]
- Delwig A, Rand MD 2008. Kuz and TACE can activate Notch independent of ligand. Cell Mol Life Sci 65: 2232–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey PJ, Meise KS, Yoshitake Y, Nishikawa K, Coffey RJ 1997. Apical enrichment of human EGF precursor in Madin-Darby canine kidney cells involves preferential basolateral ectodomain cleavage sensitive to a metalloprotease inhibitor. J Cell Biol 138: 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Rodriguez E, Montero JC, Esparis-Ogando A, Yuste L, Pandiella A 2002. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: A potential role in regulated shedding. Mol Biol Cell 13: 2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco E, Pierce JH, Fleming TP, Kraus MH, Molloy CJ, Aaronson SA, Di Fiore PP 1989. Autocrine interaction between TGF-α and the EGF-receptor: Quantitative requirements for induction of the malignant phenotype. Oncogene 4: 831–838 [PubMed] [Google Scholar]
- Doedens JR, Mahimkar RM, Black RA 2003. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem Biophys Res Commun 308: 331–338 [DOI] [PubMed] [Google Scholar]
- Dong J, Opresko LK, Dempsey PJ, Lauffenburger DA, Coffey RJ, Wiley HS 1999. Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc Natl Acad Sci 96: 6235–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DR, Handsley MM, Pennington CJ 2008. The ADAM metalloproteinases. Mol Aspects Med 29: 258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falls DL 2003. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res 284: 14–30 [DOI] [PubMed] [Google Scholar]
- Fambrough D, Pan D, Rubin GM, Goodman CS 1996. The cell surface metalloprotease/disintegrin Kuzbanian is required for axonal extension in Drosophila. Proc Natl Acad Sci 93: 13233–13238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Turck CW, Derynck R 2003. Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-α converting enzyme and of an alternatively translated polypeptide. J Biol Chem 278: 18617–18627 [DOI] [PubMed] [Google Scholar]
- Foltenyi K, Greenspan RJ, Newport JW 2007. Activation of EGFR and ERK by rhomboid signaling regulates the consolidation and maintenance of sleep in Drosophila. Nat Neurosci 10: 1160–1167 [DOI] [PubMed] [Google Scholar]
- Freeman M 1994. The Spitz gene is required for photoreceptor determination in the Drosophila eye where it interacts with the EGF receptor. Mech Dev 48: 25–33 [DOI] [PubMed] [Google Scholar]
- Freeman M 1998. Complexity of EGF receptor signalling revealed in Drosophila. Curr Opin Genet Dev 8: 407–411 [DOI] [PubMed] [Google Scholar]
- Freeman M 2009. Rhomboids: 7 years of a new protease family. Semin Cell Dev Biol 20: 231–239 [DOI] [PubMed] [Google Scholar]
- Georgakopoulos A, Litterst C, Ghersi E, Baki L, Xu C, Serban G, Robakis NK 2006. Metalloproteinase/Presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J 25: 1242–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales PE, Galli JD, Milla ME 2008. Identification of key sequence determinants for the inhibitory function of the prodomain of TACE. Biochemistry 47: 9911–9919 [DOI] [PubMed] [Google Scholar]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A 2001. Cell communication networks: Epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 20: 1594–1600 [DOI] [PubMed] [Google Scholar]
- Guinea-Viniegra J, Zenz R, Scheuch H, Hnisz D, Holcmann M, Bakiri L, Schonthaler HB, Sibilia M, Wagner EF 2009. TNFα shedding and epidermal inflammation are controlled by Jun proteins. Genes Dev 23: 2663–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullick WJ 1991. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull 47: 87–98 [DOI] [PubMed] [Google Scholar]
- Harris RC, Chung E, Coffey RJ 2003. EGF receptor ligands. Exp Cell Res 284: 2–13 [DOI] [PubMed] [Google Scholar]
- Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena Illert A, von Figura K, et al. 2002. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for α-secretase activity in fibroblasts. Hum Mol Genet 11: 2615–2624 [DOI] [PubMed] [Google Scholar]
- Hattori M, Osterfield M, Flanagan JG 2000. Regulated cleavage of a contact-mediated axon repellent. Science 289: 1360–1365 [DOI] [PubMed] [Google Scholar]
- Hemler ME 2005. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 6: 801–811 [DOI] [PubMed] [Google Scholar]
- Higashiyama S, Abraham JA, Miller J, Fiddes JC, Klagsbrun M 1991. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science 251: 936–939 [DOI] [PubMed] [Google Scholar]
- Higashiyama S, Iwamoto R, Goishi K, Raab G, Taniguchi N, Klagsbrun M, Mekada E 1995. The membrane protein CD9/DRAP 27 potentiates the juxtacrine growth factor activity of the membrane-anchored heparin-binding EGF-like growth factor. J Cell Biol 128: 929–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP 2007a. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell 18: 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Miyamoto T, Takaishi H, Hakozaki A, Kosaki N, Miyauchi Y, Furukawa M, Takito J, Kaneko H, Matsuzaki K, et al. 2007b. Cell surface colony-stimulating factor 1 can be cleaved by TNF-α converting enzyme or endocytosed in a clathrin-dependent manner. J Immunol 179: 6715–6724 [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Morioka H, Takaishi H, Akiyama H, Blobel CP, Toyama Y 2009. Ectodomain shedding of FLT3 ligand is mediated by TNF-α converting enzyme. J Immunol 182: 7408–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Nocka KH, Buck J, Besmer P 1992. Differential expression and processing of two cell associated forms of the kit-ligand: KL-1 and KL-2. Mol Biol Cell 3: 349–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof I, Gasper WJ, Derynck R 2008. Association of tetraspanin CD9 with transmembrane TGF-α confers alterations in cell-surface presentation of TGF-α and cytoskeletal organization. J Cell Sci 121: 2265–2274 [DOI] [PubMed] [Google Scholar]
- Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, Raab G, et al. 2003. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci 100: 3221–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Hirata M, Hasuwa H, Iwamoto R, Umata T, Miyado K, Tamai Y, Kurisaki T, Sehara-Fujisawa A, Ohno S, et al. 1998. A metalloprotease-disintegrin, MDC9/meltrin-γ/ADAM9 and PKCδ are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J 17: 7260–7272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB 2005. Adam meets Eph: An ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell 123: 291–304 [DOI] [PubMed] [Google Scholar]
- Jansens A, van Duijn E, Braakman I 2002. Coordinated nonvectorial folding in a newly synthesized multidomain protein. Science 298: 2401–2403 [DOI] [PubMed] [Google Scholar]
- Kawaguchi N, Horiuchi K, Becherer JD, Toyama Y, Besmer P, Blobel CP 2007. Different ADAMs have distinct influences on Kit ligand processing: Phorbol-ester-stimulated ectodomain shedding of Kitl1 by ADAM17 is reduced by ADAM19. J Cell Sci 120: 943–952 [DOI] [PubMed] [Google Scholar]
- Lappano R, Maggiolini M 2011. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat Rev Drug Discov 10: 47–60 [DOI] [PubMed] [Google Scholar]
- Lee JR, Urban S, Garvey CF, Freeman M 2001. Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell 107: 161–171 [DOI] [PubMed] [Google Scholar]
- Le Gall SM, Meneton P, Mauduit P, Dreux C 2004. The sequential cleavage of membrane anchored pro-EGF requires a membrane serine protease other than kallikrein in rat kidney. Regul Pept 122: 119–129 [DOI] [PubMed] [Google Scholar]
- Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP 2009. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor α, L-selectin, and tumor necrosis factor α. Mol Biol Cell 20: 1785–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gall SM, Maretzky T, Issuree PD, Niu XD, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP 2010. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci 123: 3913–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M 2005. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J 24: 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemjabbar H, Basbaum C 2002. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat Med 8: 41–46 [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J 2010. Cell signaling by receptor tyrosine kinases. Cell 141: 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JD, Lin F, Milla ME 2005. Chaperone-like properties of the prodomain of TNFα-converting enzyme (TACE) and the functional role of its cysteine switch. Biochem J 387: 797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman RL, Wolfe MS 2007. From rhomboid function to structure and back again. Proc Natl Acad Sci 104: 8199–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohi O, Urban S, Freeman M 2004. Diverse substrate recognition mechanisms for rhomboids; thrombomodulin is cleaved by Mammalian rhomboids. Curr Biol 14: 236–241 [DOI] [PubMed] [Google Scholar]
- Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC 1993. TGF-α deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell 73: 263–278 [DOI] [PubMed] [Google Scholar]
- Mann GB, Fowler KJ, Gabriel A, Nice EC, Williams RL, Dunn AR 1993. Mice with a null mutation of the TGF-α gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell 73: 249–261 [DOI] [PubMed] [Google Scholar]
- Massague J, Pandiella A 1993. Membrane-anchored growth factors. Annu Rev Biochem 62: 515–541 [DOI] [PubMed] [Google Scholar]
- McIlwain DR, Lang PA, Maretzky T, Hamada K, Kazuhito O, Kumar Maney S, Berger T, Murthy A, Duncan G, Xu HC, et al. 2012. iRhom2 regulates innate immunity via TACE/ADAM17. Science 335: 229–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlos-Suarez A, Ruiz-Paz S, Baselga J, Arribas J 2001. Metalloprotease-dependent protransforming growth factor-α ectodomain shedding in the absence of tumor necrosis factor-α-converting enzyme. J Biol Chem 276: 48510–48517 [DOI] [PubMed] [Google Scholar]
- Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, English JL, Matrisian LM, Au B, Yeh WC, et al. 2004. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet 36: 969–977 [DOI] [PubMed] [Google Scholar]
- Murayama Y, Shinomura Y, Oritani K, Miyagawa J, Yoshida H, Nishida M, Katsube F, Shiraga M, Miyazaki T, Nakamoto T, et al. 2008. The tetraspanin CD9 modulates epidermal growth factor receptor signaling in cancer cells. J Cell Physiol 216: 135–143 [DOI] [PubMed] [Google Scholar]
- Murphy G 2008. The ADAMs: Signalling scissors in the tumour microenvironment. Nat Rev Cancer 8: 929–941 [DOI] [PubMed] [Google Scholar]
- Murphy G 2009. Regulation of the proteolytic disintegrin metalloproteinases, the “Sheddases.” Semin Cell Dev Biol 20: 138–145 [DOI] [PubMed] [Google Scholar]
- Murphy G 2011. Tissue inhibitors of metalloproteinases. Genome Biol 12: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy A, Defamie V, Smookler DS, Di Grappa MA, Horiuchi K, Federici M, Sibilia M, Blobel CP, Khokha R 2010. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J Clin Invest 120: 2731–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Guichard A, Castro CP, Xiao Y, Rizen M, Zhang HZ, Hu D, Bang A, Helms J, Bier E, et al. 2005. Characterization of a human rhomboid homolog, p100hRho/RHBDF1, which interacts with TGF-α family ligands. Dev Dyn 233: 1315–1331 [DOI] [PubMed] [Google Scholar]
- Naoe T, Kiyoi H 2004. Normal and oncogenic FLT3. Cell Mol Life Sci 61: 2932–2938 [DOI] [PubMed] [Google Scholar]
- Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL 2003. Anti-TNF-α therapies: The next generation. Nat Rev Drug Discov 2: 736–746 [DOI] [PubMed] [Google Scholar]
- Pandiella A, Bosenberg MW, Huang EJ, Besmer P, Massague J 1992. Cleavage of membrane-anchored growth factors involves distinct protease activities regulated through common mechanisms. J Biol Chem 267: 24028–24033 [PubMed] [Google Scholar]
- Pascall JC, Brown KD 2004. Intramembrane cleavage of ephrinB3 by the human rhomboid family protease, RHBDL2. Biochem Biophys Res Commun 317: 244–252 [DOI] [PubMed] [Google Scholar]
- Peiretti F, Canault M, Deprez-Beauclair P, Berthet V, Bonardo B, Juhan-Vague I, Nalbone G 2003a. Intracellular maturation and transport of tumor necrosis factor α converting enzyme. Exp Cell Res 285: 278–285 [DOI] [PubMed] [Google Scholar]
- Peiretti F, Deprez-Beauclair P, Bonardo B, Aubert H, Juhan-Vague I, Nalbone G 2003b. Identification of SAP97 as an intracellular binding partner of TACE. J Cell Sci 116: 1949–1957 [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, et al. 1998. An essential role for ectodomain shedding in mammalian development. Science 282: 1281–1284 [DOI] [PubMed] [Google Scholar]
- Piepkorn M, Pittelkow MR, Cook PW 1998. Autocrine regulation of keratinocytes: The emerging role of heparin-binding, epidermal growth factor-related growth factors. J Invest Dermatol 111: 715–721 [DOI] [PubMed] [Google Scholar]
- Pitulescu ME, Adams RH 2010. Eph/ephrin molecules—A hub for signaling and endocytosis. Genes Dev 24: 2480–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente XS, Lopez-Otin C 2004. A genomic analysis of rat proteases and protease inhibitors. Genome Res 14: 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovida E, Paccagnini A, Del Rosso M, Peschon J, Dello Sbarba P 2001. TNF-α-converting enzyme cleaves the macrophage colony-stimulating factor receptor in macrophages undergoing activation. J Immunol 166: 1583–1589 [DOI] [PubMed] [Google Scholar]
- Ruohola-Baker H, Grell E, Chou TB, Baker D, Jan LY, Jan YN 1993. Spatially localized rhomboid is required for establishment of the dorsal-ventral axis in Drosophila oogenesis. Cell 73: 953–965 [DOI] [PubMed] [Google Scholar]
- Saftig P, Reiss K 2011. The “A disintegrin and metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur J Cell Biol 90: 527–535 [DOI] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP 2004. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164: 769–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon DS, Brandt R, Ciardiello F, Normanno N 1995. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 19: 183–232 [DOI] [PubMed] [Google Scholar]
- Sanderson MP, Erickson SN, Gough PJ, Garton KJ, Wille PT, Raines EW, Dunbar AJ, Dempsey PJ 2005. ADAM10 mediates ectodomain shedding of the betacellulin precursor activated by p-aminophenylmercuric acetate and extracellular calcium influx. J Biol Chem 280: 1826–1837 [DOI] [PubMed] [Google Scholar]
- Sastry SK, Elferink LA 2011. Checks and balances: Interplay of RTKs and PTPs in cancer progression. Biochem Pharmacol 82: 435–440 [DOI] [PubMed] [Google Scholar]
- Schafer B, Marg B, Gschwind A, Ullrich A 2004. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem 279: 47929–47938 [DOI] [PubMed] [Google Scholar]
- Schlesinger A, Kiger A, Perrimon N, Shilo BZ 2004. Small wing PLCγ is required for ER retention of cleaved Spitz during eye development in Drosophila. Dev Cell 7: 535–545 [DOI] [PubMed] [Google Scholar]
- Schlessinger J 2000. Cell signaling by receptor tyrosine kinases. Cell 103: 211–225 [DOI] [PubMed] [Google Scholar]
- Schlondorff J, Becherer JD, Blobel CP 2000. Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem J 347: 131–138 [PMC free article] [PubMed] [Google Scholar]
- Schneider MR, Wolf E 2009. The epidermal growth factor receptor ligands at a glance. J Cell Physiol 218: 460–466 [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA 2003. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev 17: 7–30 [DOI] [PubMed] [Google Scholar]
- Shilo BZ 2003. Signaling by the Drosophila epidermal growth factor receptor pathway during development. Exp Cell Res 284: 140–149 [DOI] [PubMed] [Google Scholar]
- Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M 2007. The epidermal growth factor receptor: From development to tumorigenesis. Differentiation 75: 770–787 [DOI] [PubMed] [Google Scholar]
- Siggs OM, Xiao N, Wang Y, Shi H, Tomisato W, Li X, Xia Y, Beutler B 2012. iRhom2 is required for the secretion of mouse TNF α. Blood 119: 5769–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AB, Harris RC 2005. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal 17: 1183–1193 [DOI] [PubMed] [Google Scholar]
- Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, DeSimone DW 2002. The cysteine-rich domain regulates ADAM protease function in vivo. J Cell Biol 159: 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS 2011. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 334: 1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soond SM, Everson B, Riches DW, Murphy G 2005. ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci 118: 2371–2380 [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB 1985. Autocrine growth factors and cancer. Nature 313: 745–747 [DOI] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, Freeman M 2009. Sequence-specific intramembrane proteolysis: Identification of a recognition motif in rhomboid substrates. Mol Cell 36: 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtevant MA, Roark M, Bier E 1993. The Drosophila rhomboid gene mediates the localized formation of wing veins and interacts genetically with components of the EGF-R signaling pathway. Genes Dev 7: 961–973 [DOI] [PubMed] [Google Scholar]
- Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, et al. 2002. Tumor necrosis factor-α converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem 277: 12838–12845 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Nanba D, Mori S, Shiba F, Ishiguro H, Yoshino K, Matsuura N, Higashiyama S 2004. ADAM binding protein Eve-1 is required for ectodomain shedding of epidermal growth factor receptor ligands. J Biol Chem 279: 41950–41959 [DOI] [PubMed] [Google Scholar]
- Tsruya R, Wojtalla A, Carmon S, Yogev S, Reich A, Bibi E, Merdes G, Schejter E, Shilo BZ 2007. Rhomboid cleaves Star to regulate the levels of secreted Spitz. EMBO J 26: 1211–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A, Schlessinger J 1990. Signal transduction by receptors with tyrosine kinase activity. Cell 61: 203–212 [DOI] [PubMed] [Google Scholar]
- Urban S, Dickey SW 2011. The rhomboid protease family: A decade of progress on function and mechanism. Genome Biol 12: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Freeman M 2003. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell 11: 1425–1434 [DOI] [PubMed] [Google Scholar]
- Urban S, Lee JR, Freeman M 2001. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell 107: 173–182 [DOI] [PubMed] [Google Scholar]
- Urban S, Lee JR, Freeman M 2002. A family of rhomboid intramembrane proteases activates all Drosophila membrane-tethered EGF ligands. EMBO J 21: 4277–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Ha Y 2006. Crystal structure of a rhomboid family intramembrane protease. Nature 444: 179–180 [DOI] [PubMed] [Google Scholar]
- Ward CL, Kopito RR 1994. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem 269: 25710–25718 [PubMed] [Google Scholar]
- Wasserman JD, Urban S, Freeman M 2000. A family of rhomboid-like genes: Drosophila rhomboid-1 and roughoid/rhomboid-3 cooperate to activate EGF receptor signaling. Genes Dev 14: 1651–1663 [PMC free article] [PubMed] [Google Scholar]
- White JM 2003. ADAMs: Modulators of cell–cell and cell–matrix interactions. Curr Opin Cell Biol 15: 598–606 [DOI] [PubMed] [Google Scholar]
- Wisniewska M, Goettig P, Maskos K, Belouski E, Winters D, Hecht R, Black R, Bode W 2008. Structural determinants of the ADAM inhibition by TIMP-3: Crystal structure of the TACE-N-TIMP-3 complex. J Mol Biol 381: 1307–1319 [DOI] [PubMed] [Google Scholar]
- Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J 2004. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64: 7022–7029 [DOI] [PubMed] [Google Scholar]
- Xu P, Derynck R 2010. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell 37: 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Sharma C, Hemler ME 2009. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J 23: 3674–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Iwamoto R, Saeki K, Asakura M, Takashima S, Yamazaki A, Kimura R, Mizushima H, Moribe H, Higashiyama S, et al. 2003. Mice with defects in HB-EGF ectodomain shedding show severe developmental abnormalities. J Cell Biol 163: 469–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zou H, Tian F, Grandis JR, Mixson AJ, Lu PY, Li LY 2008. Human rhomboid family-1 gene silencing causes apoptosis or autophagy to epithelial cancer cells and inhibits xenograft tumor growth. Mol Cancer Ther 7: 1355–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Mo M, Gutierrez-Lopez MD, Cabanas C 2011. Functional interplay between tetraspanins and proteases. Cell Mol Life Sci 68: 3323–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX 2001. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127–137 [DOI] [PubMed] [Google Scholar]
- Yogev S, Schejter ED, Shilo BZ 2008. Drosophila EGFR signalling is modulated by differential compartmentalization of rhomboid intramembrane proteases. EMBO J 27: 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M 2011. Rhomboid family pseudoproteases use the ER quality control machinery to regulate intercellular signaling. Cell 145: 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Schlondorff J, Blobel CP 2002. Evidence for regulation of the tumor necrosis factor α-convertase (TACE) by protein-tyrosine phosphatase PTPH1. J Biol Chem 277: 42463–42470 [DOI] [PubMed] [Google Scholar]