

Abstract
The proliferation of all organisms depends on the coordination of enzymatic events within large multiprotein replisomes that duplicate chromosomes. Whereas the structure and function of many core replisome components have been clarified, the timing and order of molecular events during replication remains obscure. To better understand the replication mechanism, new methods must be developed that allow for the observation and characterization of short-lived states and dynamic events at single replication forks. Over the last decade, great progress has been made toward this goal with the development of novel DNA nanomanipulation and fluorescence imaging techniques allowing for the direct observation of replication-fork dynamics both reconstituted in vitro and in live cells. This article reviews these new single-molecule approaches and the revised understanding of replisome operation that has emerged.
DNA nanomanipulation and fluorescence imaging techniques allow for the direct observation of replication-fork dynamics (e.g., the formation and release of loops during Okazaki fragment synthesis).
The duplication of chromosomes in cells is conducted with high speed and accuracy by the concerted efforts of many cellular factors operating in concert within large replication complexes. A typical example is the replication machinery of Escherichia coli: It is composed of more than a dozen components forming an assembly with a molecular weight over 1 MDa that unwinds and copies DNA at a rate approaching 1000 base pairs per second (Chandler et al. 1975) while making less than one mistake per 109 nucleotide incorporations (Drake et al. 1969). What makes this achievement even more remarkable is that the process depends on the coordination of molecular events over a broad range of time and length scales, from the breaking and reformation of chemical bonds within DNA polymerases to the large-scale structural rearrangements of chromosomes. This high degree of efficiency is maintained even in eukaryotic cells where the process is more complex, involving additional regulation and a larger number of cellular components (Bell and Dutta 2002). Although decades of intense research has revealed the function and architecture of many core replisome subassemblies, the coordination of enzymatic events at the replication fork remains poorly understood because of the challenge of directly observing replication complexes in action.
Recent work has further complicated our view of the processes at the replication fork by revealing that replisome composition is highly dynamic, with components rapidly exchanging during synthesis. A partial explanation for these observations emerges when considering that robust replication in the cellular environment depends not only on the efficiency and accuracy of synthesis, but also on the ability of replisomes to overcome obstacles encountered on parental chromosomes, such as transcription complexes and DNA lesions (Cox et al. 2000). The recent discoveries of novel mechanisms for replication restart have provided insight into how replisomes cope with these obstacles (Heller and Marians 2006; Lopes et al. 2006; Pomerantz and O’Donnell 2008; Yeeles and Marians 2011) and revealed that replication-fork collapse and reassembly may be a common occurrence even within a single cell cycle (Cox et al. 2000; Indiani et al. 2009; Langston et al. 2009). These studies support the notion that replisomes do not operate as static assemblies but instead have a dynamic composition that is responsive to the cellular environment.
To construct a complete understanding of the replication cycle, we must develop and employ new methods that allow for the observation and characterization of short-lived states and dynamic events at the replication fork. The last decade has seen great progress toward these goals with the advent of new techniques for observing and characterizing single replisomes both reconstituted in vitro and in live cells. These efforts have started to provide a dynamic view of the events that occur during chromosome duplication. Flow-stretching nanomanipulation and fluorescence-based assays have revealed the formation and release of loops during Okazaki fragment synthesis and the dynamic exchange of polymerases during replication (Hamdan et al. 2009; Loparo et al. 2011; Tanner et al. 2011; Lia et al. 2012). The development of new fluorescence imaging techniques has allowed for the real-time observation of individual replisomes in vivo, which has led to a revised understanding of replisome composition in bacteria (Reyes-Lamothe et al. 2010). In this article, we review our current understanding of replisome function while highlighting a broad range of new methods and technologies that allow for the direct observation of events at single replication forks.
EXPERIMENTAL TOOLBOXES
Mechanical Stretching
The mechanical manipulation of individual DNA molecules by means of optical trapping, magnetic tweezing, and flow stretching has provided powerful tools for probing the function of nucleic-acid processing enzymes involved in DNA replication. Optical trapping and magnetic tweezing allow for the direct application of force on individual DNA molecules and the read out of enzymatic function using feedback control of force (Fig. 1A,B). These techniques have been used to study the mechanism used by helicases to unwind DNA (Johnson et al. 2007; Lionnet et al. 2007; Sun et al. 2011) and the forces at work within polymerases during nucleotide incorporation (Maier et al. 2000; Wuite et al. 2000). Although these methods provide the highest resolution of enzymatic function on DNA, in most cases (Ribeck and Saleh 2008), they are not compatible with multiplexing, which makes collection of statistically significant samples sizes difficult and time consuming. An alternative approach that can be multiplexed is a technique where individual DNA molecules are stretched by laminar flow, and their lengths are monitored by tracking the position of small beads attached to their ends (Fig. 1C) (van Oijen et al. 2003; Tanner and van Oijen 2009). Both leading-strand and coordinated synthesis by replisomes have been observed using this method. In the former case, bead movement reports on the conversion from an extended double-stranded state to a contracted single-stranded state (Lee et al. 2006; Tanner et al. 2008). In the latter case, bead movement reports on the formation and release of replication loops on the lagging strand (Hamdan et al. 2009).
Figure 1.
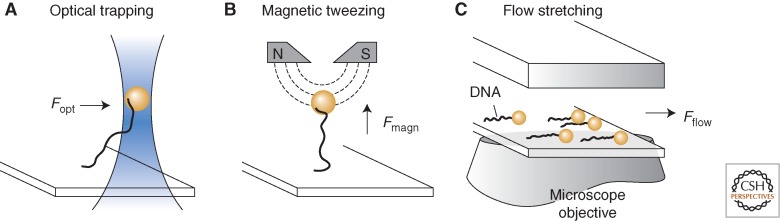
DNA stretching techniques. Three methods of DNA nanomanipulation are shown. (A) Optical trapping: A focused beam of light traps a bead that is tethered to DNA. Feedback control allows the position and force on the bead to be precisely monitored and controlled. (B) Magnetic tweezing: Permanent magnets located above the sample generate a vertical extending force on DNA through a magnetic bead. (C) Flow stretching: DNA-tethered beads are extended using laminar flow and imaged using wide-field optical microscopy. In all cases, the end of the DNA opposite the beads is attached to a functionalized glass surface.
Fluoresence Imaging
Although mechanical manipulation techniques provide detailed kinetic information about enzymatic function, they do not provide direct information about the composition of protein assemblies or conformational states adopted by single enzymes during operation. A complete understanding of the events at the replication fork will only be possible by simultaneously observing the composition and function of single replisomes in operation. Fortunately, many single-molecule fluorescence techniques have emerged that allow for the characterization of these properties. Single-molecule fluorescence resonance energy transfer (FRET) has been used to distinguish between different replisome assembly pathways (Xi et al. 2005a,b; Zhang et al. 2005; Smiley et al. 2006) and to observe the conformational states adopted by polymerases during substrate engagement (Luo et al. 2007). Single-molecule fluorescence techniques have also been used to visualize replication products and short-lived changes in DNA structure during synthesis (Hamdan et al. 2009; Pandey et al. 2009). Using these methods, the activity of fully assembled E. coli replisomes has been observed in real time on rolling-circle substrates by flow stretching the products and staining with a DNA-intercalating dye (Fig. 2A,B) (Georgescu et al. 2009; Tanner et al. 2009, 2011; Georgescu et al. 2012). Finally, both the composition and structure of the phage T7 replisome have been observed simultaneously by combining nanomanipulation techniques with the labeling of individual polymerases and replication substrates (Fig. 2C) (Loparo et al. 2011).
Figure 2.

Study of reconstituted replisomes. (A) Schematic of single-molecule rolling-circle replication assay. Leading-strand synthesis displaces DNA from the circle as the replisome rolls around the template. The displaced tail is then used for lagging-strand synthesis. The DNA that couples the M13 circle to the surface increases in length as replication proceeds. Synthesis is monitored by fluorescently staining and flow stretching the newly synthesized products. (B) Example image of rolling-circle replication products collected using total internal reflection fluorescence (TIRF) microscopy. (C) Schematic of single-molecule assay for simultaneously measuring replisome activity and composition. Leading-strand synthesis is performed using a replication fork introduced at one end of λ DNA. During synthesis, the template is unwound and a compact single-stranded product is generated on the lagging strand. The conversion from double-stranded to single-stranded DNA is monitored using a quantum dot (QD) attached to the template. Fluorescently labeled polymerases are monitored as they enter and exit the replisome during synthesis. All replication substrates are attached to functionalized glass surfaces for TIRF imaging.
Observing Replication-Fork Dynamics In Vivo
Understanding the action of molecular machines, like replisomes, in their natural cellular environment is the ultimate goal of biochemical research. The use of single-molecule fluorescence microscopy in live cells promises to reveal details of composition, architecture, and dynamics of active replisomes. The main limitation of this approach is the difficulty in detecting single fluorophores over a high background, but use of strategies to illuminate small sections of the cells can provide the desired sensitivity, as is the case for TIRF, HILO, and SPIM (Fig. 3A) (Tokunaga et al. 2008; Li and Xie 2011). Bacterial cells (with widths <1 µm) have much lower background fluorescence derived from autofluorescence and out-of-focus fluorophores, thereby permitting the detection of single molecules using standard epifluorescence (Li and Xie 2011). The small number of replication forks and range of genetic tools make bacteria perfect models for this approach. The use of single-molecule techniques has permitted the determination of stoichiometry and the study of dynamics of individual molecules in bacteria. Two strategies have been adopted for these studies. In the first technique, the sample is illuminated by short flashes of light at intervals limited by the camera reading time (strobing illumination) (Xie et al. 2008). Strobing illumination has the advantage of allowing large fields of view. The second technique used consists in concentrating the laser beam in the sample so it only illuminates a very small region of the slide, originally ∼6 µm diameter (slimfield microscopy) (Reyes-Lamothe et al. 2010). Slimfield microscopy can only be used for small cells but has the advantage of permitting capture rates of 3 msec and up (Fig. 3B).
Figure 3.

Study of replisomes in live cells. (A) Two examples of techniques used to enhance the contrast of fluorescence microscopy: TIRF and high inclined and laminated optical sheet (HILO) microscopy. Both techniques use a tilted light beam, as opposed to epifluorescence, where the beam is perpendicular to the slide. Depending on the angle used, the beam either results in the illumination of only the ∼100 nm closer to the slide (TIRF) or the creation of a sheet of light that illuminates a slice of the sample (HILO). By reducing the volume illuminated, the contribution of out-of-focus fluorescent or autofluorescent molecules is also reduced. (B) Two strategies to detect single molecules in cells: illuminating the sample using short pulses of light followed by intervals with a length no shorter than the camera’s processing time (strobing) and, alternatively, illuminating a small region of the sample with the signal captured continuously (slimfield). (C) Use of fluorescence recovery after photobleaching (FRAP) to study dynamics of molecular machines (enclosed in blue circles). Fluorescent molecules (yellow balls) are either relatively immobile, when part of a machine and generating fluorescent spots, or rapidly diffusing in the cytoplasm. Bleaching fluorescence of a region of the cell causes a fraction of molecules to lose their fluorescence (grey balls). The exchange between fluorescent and bleached molecules can be followed by changes in the intensity of spots.
Fluorescence recovery after photobleaching (FRAP) is widely used to probe molecular dynamics in cells (Lippincott-Schwartz et al. 2001). The technique consists of irreversibly bleaching a region of the sample and monitoring the diffusion and turnover of fluorescent molecules expressed as recovery of fluorescence (Fig. 3C). This technique has been used to study both the diffusion of free replisome components and turnover of active replisome components in bacteria and eukaryotes (Sporbert et al. 2002; Solovjeva et al. 2005; Su’etsugu and Errington 2011). Use of photoswitchable proteins (Patterson et al. 2010) promises to offer an alternative to FRAP in studies of component turnover, and as it helps to provide single-molecule data of densely packed replisomes like those of eukaryotic cells.
LIFE OF A REPLISOME
Given the importance of faithful replication, the assembly and activity of the replisome must be stringently orchestrated. The unit of replication, the replicon, is controlled through initiation by action of a specific initiator acting at the replicon’s origin of replication (Jacob et al. 1963). The key step during replication initiation is the helicase loader-mediated loading of the ring-shaped replicative helicase to an active replication origin bound by the initiator. The function of the helicase loader is to open the otherwise stable toroidal structure of the helicase, so it can entrap DNA at its center. Both initiator and helicase loader proteins are members of the ATPase associated with various activities (AAA+) superfamily (Duderstadt and Berger 2008), and their action is regulated by nucleotide binding. The initiator in bacteria, DnaA, destabilizes the duplex by binding and oligomerizing at oriC, permitting the recruitment of the DnaB-DnaC helicase-loader complex onto ssDNA (Mott and Berger 2007). In contrast, eukaryotes, and possibly archaea, load the MCM helicase onto dsDNA (Evrin et al. 2009; Remus et al. 2009).
Movement of the replicative helicase away from the origin occurs concomitantly with dissociation of the helicase loader and recruitment of primase, the protein responsible for the synthesis of RNA primers on the lagging-strand template. Recruitment of primase permits the loading of the remaining replisome components, including the replicative polymerase, processivity clamp, and clamp loader (Fig. 4). Replication initiation results in the assembly of two sister replisomes, each facing outward from the origin. Synchronous loading and firing of sister replisomes protects chromosomes from exposing recombinogenic replisome-free forked structures. Eukaryotes achieve coordination of these events through the activity of cell-cycle-regulated phosphorylation of replisome components, with an inactive MCM helicase loaded on the origin long before it gets activated (Masai et al. 2010). In bacteria, coordination is achieved by coupling cell-cycle-regulated origin opening by DnaA-ATP to immediate loading of two DnaB helicases, which in turn stimulate the assembly of two sister replisomes (Katayama et al. 2010).
Figure 4.

Replisome architecture. The arrangement of the different components of the E. coli replisome is shown. At the front of the replication fork, a hexameric helicase melts the dsDNA. Helicase binds primase, and it is connected to the three replicative polymerases (Pol III) via the τ subunit of the clamp loader. A leading-strand polymerase travels in the same direction of the fork, while the two remaining polymerases can potentially act on the lagging strand. Each active polymerase is bound to the homodimeric β-clamp, which acts as a processivity factor. The leading strand is synthesized continuously, whereas the lagging strand requires cycles of chain elongation. Progression of helicase causes the accumulation of ssDNA, which is covered by the homotetrameric SSB. The clamp loader (heteropentamer) loads β-clamp, mediates primase handover to polymerase, and connects the polymerase with helicase. The architecture of the replisome in other systems is thought to be similar to that of E. coli (Yao and O’Donnell 2010). Some differences with phage and eukaryote replisomes are listed.
An idea that has lingered for decades is that, after replisome assembly at origins, sister replisomes remain attached to each other as replication progresses, with their action potentially coordinated. This idea was born as part of a segregation model proposed before the concept of bidirectional elongation had been established (Jacob et al. 1963), and it was later formulated explicitly by Dingman (1974). This concept of a replication factory containing sister replisomes was expanded to accommodate the observation that each eukaryote replication fluorescent focus needed to contain up to 40 replisomes to accommodate the total number of replication forks per cell predicted by other means (Cook 1999). One could imagine that in such putative mega-factories, coordination of the activities of all origins could be coupled, or that any coordination would be solely between sister replisomes. Support for at least the sister-replisome factory model came from structural and biochemical data of SV40 helicase, T-ag (it is loaded onto the origin as double hexamer, and its action is dependent on the maintenance of this link between hexamers) (Weisshart et al. 1999; Valle et al. 2000; Alexandrov et al. 2002). Early fluorescence microscopy studies of replisomes and chromosomal loci within Bacillus subtillis and Saccharomyces cerevisiae also supported the existence of a replication factory that pulled in the unreplicated DNA and extruded the replicated DNA (Lemon and Grossman 1998, 2000; Kitamura et al. 2006).
Nevertheless, with better resolution assays, evidence for independent action and cellular positioning of replisomes has accumulated. Fluorescence microscopy of E. coli cells showed that single replisomes track independently on the chromosome during replication, although closely spaced sister replisomes are not always spatially separable in epifluorescence (Reyes-Lamothe et al. 2008). The situation may be the same in eukaryotes (e.g., in Xenous egg extracts, where sister replisomes separated from each other as replication proceeded on a stretched template without any visible effect on their rate) (Yardimci et al. 2010). Consistent with the independent progression of sister forks, their functional independence has been demonstrated in E. coli, B. subtillis, and S. cerevisiae (Breier et al. 2005; Wang et al. 2007; Doksani et al. 2009), where blocking or slowing down one of them does not prevent the normal progression of its sister. There is no a priori reason for sister replisomes to stay together, and if they were complexed into a fixed structure, it would seem difficult to generate the precatenane interlinks between newly replicated sisters that result from individual fork rotation as replication proceeds (Hearst et al. 1998; Wang et al. 2008). It seems likely that, in many cases, the proposed factories result from diffraction-limited light microscopy of densely replisome-populated chromosomal regions. Indeed, super-resolution microscopy of human cells showed the number of replisomes per fluorescent spot to be between two and three (Cseresnyes et al. 2009).
Elongation of replicating DNA strands occurs with a 5′ to 3′ directionality (Kornberg and Baker 1992) (Fig. 4). The anti-parallel nature of DNA permits continuous synthesis only in the strand elongated in the same direction as the migration of the replication fork, the leading strand. Meanwhile, the copying of the complementary strand, lagging strand, occurs discontinuously and requires cycles of priming, clamp loading, polymerase loading, and polymerase release at every Okazaki fragment (Fig. 5). Single-stranded DNA binding proteins have to cover and protect the stretches of ssDNA until they are displaced by polymerase. Other auxiliary activities, like removal of RNA primers and ligation of Okazaki fragments, mediated by proteins not generally thought of as part of the replisome, have to be accomplished to obtain an uninterrupted copy of DNA. The cytoplasmic pool of proteins involved in lagging-strand synthesis is limited, and this imposes their need to be recycled (Fig. 5).
Figure 5.

Replisome dynamics. The major source of molecular exchange during replication is produced during the synthesis of the lagging strand. Primase binds to ssDNA (1) and produces a primer forming a short loop in the process (2). The primase then dissociates from helicase, possibly remaining bound to the primer (3). Loading of β-clamp displaces the primase (4) and recruits one of the idle polymerase in the replisome (5). Polymerase elongates the chain causing the growth of a DNA loop (6). Finally, the lagging-strand polymerase detaches from the β-clamp and causes the release of the DNA loop, while the clamp remains bound to DNA (7). For simplicity, only the collision mechanism for loop release is depicted. SSB, β-clamp, primase, and likely polymerase (during loop release, not shown here) are exchanged with the diffusing pool of proteins in the cytoplasm at different stages of the cycle. For clarity, only a part of the replisome is represented in the different steps.
Once replication has initiated, the two diverging forks normally continue until they each meet a converging fork and replication terminates. Precisely what occurs mechanistically when replication forks converge, how the torsional tension is handled, and how overreplication is avoided still needs to be determined. Replicon size varies from a few kb to many Mb (Table 1), and is determined by the spacing of potentially active origins within a chromosome; in eukaryotes, replicon size can change dramatically during development (Méchali 2010). Replisomes are not always loaded in pairs. Replication of some plasmids, some of them ∼100 kb long (Nordstrom 2006), is unidirectional, presumably because only a single replisome is loaded by a single replicative helicase, showing that there is no physical impediment to a single replisome forming and acting at a given initiation.
Table 1.
Facts on DNA replication dynamics
| T4 | T7 | E. coli | Archaea | S. cerevisiae | Human | |
|---|---|---|---|---|---|---|
| Length of Okazaki fragment (kb)a | 1.5–2.2 | 2.3 | 1.5 | 0.09–0.16 | 0.16b | 0.1–0.2c |
| Primer length (nt)d | 4–5 | 4–5 | 10–12 | ∼10 | 8–10 | 11–14 |
| SSB/RPA binding (nt)e,f | 10 | 14 | 32 or 64 | 4.5–5 | 19 or 27 | 8 or 30 |
| Rate of fork progression (bp sec−1)g | 600 | 370 | 600–1000 | 88–330 | 26–48 | 25 |
| Replicon size (kb)h | 40 | 166 | 4600 | 1000, 700, 500 | 36 | ∼100 |
aEdenberg and Huberman 1975; Kornberg and Baker 1992; Debyser et al. 1994; Chastain et al. 2000; Matsunaga et al. 2003; Smith and Whitehouse 2012.
bOkazaki fragment length matches that of nucleosome binding in yeast. Multiples of 165 nt, 330 nt, and 495 nt were reported.
cExtrapolation from the study of SV40 system.
dFrick and Richardson 2001; Matsunaga et al. 2003.
eKim et al. 1992; Kornberg and Baker 1992; Blackwell and Borowiec 1994; Kumaran et al. 2006.
fExtent of binding by a functional unit; T4 gp32 is a monomer, T7 gp2.5 is a homodimer, bacterial SSB is a homotetramer, archaeal SSB is a monomer, and the eukaryotic RPAs are heterotrimers.
gEdenberg and Huberman 1975; Rabkin and Richardson 1990; Kornberg and Baker 1992; Myllykallio et al. 2000; Raghuraman et al. 2001; Kerr et al. 2003; Duggin et al. 2008.
hEdenberg and Huberman 1975; Kornberg and Baker 1992; Duggin et al. 2008; Liachko et al. 2010.
COORDINATION OF LEADING- AND LAGGING-STRAND SYNTHESIS
Processive replication depends on the spatial and temporal coordination of enzymatic events at the replication fork. An essential characteristic of this coordination is the reorientation of the lagging-strand DNA through the formation of a loop during Okazaki-fragment synthesis, which allows the leading- and lagging-strand polymerases to move coordinately in parallel. Electron-microscopy studies have visualized the formation of these lagging-strand loops and characterized their length distributions (Park et al. 1998; Lee et al. 2002; Chastain et al. 2003), but the timing and mechanisms underlying the transitions that occur during Okazaki-fragment synthesis have remained less clear. There are at least three enzymatic events that must occur during each cycle: the synthesis of a primer, its handoff to the lagging-strand polymerase, and loop release (Fig. 5). Several recent studies have begun to elucidate the kinetics of this process, providing experimental observations that discriminate between different mechanisms of regulation for each stage of the cycle.
Two mechanisms have been proposed for ensuring efficient loop release and a fluid transition from the synthesis of one Okazaki fragment to the next. In the signaling mechanism, primer synthesis triggers loop release even if nascent Okazaki-fragment synthesis is unfinished. Supporting this model is the observation that Okazaki-fragment size is dependent on primase activity in the E. coli, T7, and T4 systems (Li and Marians 2000; Lee et al. 2002; Yang et al. 2006) and sensitive to helicase-primase interactions in E. coli (Tougu and Marians 1996). Additional support comes from the observation of ssDNA gaps on the lagging strands in E. coli and T4 (Yang et al. 2006; Georgescu et al. 2012). An alternative model, the collision mechanism, proposes that the lagging-strand polymerase dissociates once it has encountered the 5′ terminus of the previously synthesized Okazaki fragment. This model is supported by the observation that the T4 DNA polymerase and E. coli DNA polymerase III holoenzyme dissociate rapidly when they encounter a 5′ terminus (Hacker and Alberts 1994; Carver et al. 1997; Li and Marians 2000; Lopez de Saro et al. 2003; Yao et al. 2009). However, a recent E. coli study suggests the collision mechanism is too slow to explain the observed rate of replication (Dohrmann et al. 2011).
Direct observation of coordinated synthesis using single-molecule methods has provided compelling evidence that reconciles the seemingly contradictory findings of past work. The formation and release of single replication loops has been observed by fully reconstituted T7 replisomes by tracking length changes in flow-stretched DNA substrates (Hamdan et al. 2009). This work shows that loop formation occurs only half the time during replication, a finding consistent with previous electron microscopy studies (Park et al. 1998; Lee et al. 2002; Chastain et al. 2003). Analysis of loop sizes and delay times between loops, a step unobservable in bulk-phase assays, shows that both the initiation of primer synthesis and the completion of an Okazaki fragment serve as triggers for loop release. Single-molecule studies conducted with E. coli replisomes have also revealed a mixture of loop-release mechanisms (Tanner et al. 2011; Georgescu et al. 2012). Furthermore, E. coli replisomes composed of two polymerases prematurely terminate Okazaki synthesis, whereas replisomes composed of three polymerases almost always complete synthesis (Georgescu et al. 2012). These studies show that replisomes use both signaling and collision mechanisms to regulate lagging-strand synthesis. The combination of these two mechanisms is likely critical for highly processive and coordinated synthesis. While the signaling mechanism allows for rapid transitions from Okazaki-fragment synthesis to the site of a new primer, the collision mechanism ensures that polymerases do not stall if a new primer has not been synthesized before they encounter the previous Okazaki fragment.
Although lagging-strand synthesis involves many more enzymatic steps than leading-strand synthesis does, the duplication of DNA on both strands is coordinated by mechanisms that are not completely understood. Single-molecule techniques have allowed for detailed investigations of this issue, which have provided new evidence in support of several explanations. Flow-stretching experiments, conducted using the T7 replisome, have shown that activation of primase, by the addition of required ribonucleotides, causes transient pauses in leading-strand synthesis (Lee et al. 2006; Hamdan et al. 2009) with an average duration consistent with the kinetics of primer synthesis (Kusakabe and Richardson 1997; Frick et al. 1999). These observations reveal a possible mechanism for ensuring that the slow enzymatic events on the lagging strand do not cause leading-strand synthesis to proceed faster than lagging-strand synthesis. An alternative explanation is supported by recent single-molecule experiments investigating the T7 leading-strand synthesis reaction (Pandey et al. 2009) and T4 helicase-primase complex (Manosas et al. 2009). These studies demonstrate the formation of a small ssDNA loop between the helicase and primase that may allow the replisome to continue during primer synthesis. In this case, a faster rate of lagging-strand synthesis has been suggested as a mechanism to ensure efficient coupling of daughter-strand synthesis.
A complete understanding of how primer synthesis influences coordinated replication depends on careful consideration of the steps involved in RNA polymerization. Several studies have demonstrated that primer synthesis proceeds in two distinct stages (Kusakabe and Richardson 1997; Frick et al. 1999; Qimron et al. 2006): the formation of a dinucleotide and the slow extension of that dinucleotide to form a full-length primer. During replication, this is a stochastic process in which only a fraction of priming sequences are recognized and utilized by primase (Lee et al. 2006). In between these actual primer-synthesis events, primase scans the lagging strand for initiation sites. Whereas it has been demonstrated that dinucleotide formation and primer synthesis can trigger replication loop release and cause pausing (Lee et al. 2006; Hamdan et al. 2009), the molecular events that cause the primase to utilize one priming site and bypass others are not understood. The formation of a small ssDNA priming loop may allow the primase to efficiently utilize the primase-recognition sequences for dinucleotide formation while allowing the helicase to continue unwinding (Fig. 5). Then, once a ribodinucleotide has formed, slow extension into a full-length primer may occur, a process that could cause the replisome to pause. This dynamic balance between priming-loop formation and replisome pausing could provide an optimal compromise between speed and coordination and would provide an explanation for the current experimental evidence. Work conducted with E. coli and T7 systems has shown that primase activity depends on in trans interactions between adjacent primases bound to the helicase (Lee and Richardson 2002; Corn et al. 2005), providing a structural framework that could explain the observed pausing behavior.
Primase action not only regulates the length of Okazaki fragments, but also the transfer of the replicative polymerase to the primer. A single Toprim-family protein comprises bacterial and phage primases, whereas two subunits (which share no homology to bacteria primases) comprise archaeal and eukaryotic primases (Kuchta and Stengel 2010). Nevertheless, in all cases, the result of their function is a primer that is just a few bp long (Table 1). What determines the distribution of Okazaki-fragment sizes (Table 1) is not known, but it is likely that the interaction between primase and helicase plays a role. Binding of bacterial DnaG primase to DnaB helicase stimulates primase ssDNA binding and priming (Corn and Berger 2006). In turn, association of up to three primase molecules increases the processivity of helicase, probably by stabilizing its structure (Bailey et al. 2007). E. coli primase, present at 50–100 copies per cell (Kornberg and Baker 1992), remains bound to DNA after primer synthesis every cycle (Yuzhakov et al. 1999), implying the need for fast turnover of the primase. Finally, two primase proteins may need to interact to function (Corn and Berger 2006). Considering all these constraints, the cellular primase pool only accounts for the elongation of less than 75 kb of DNA. It is therefore possible that the activity of the otherwise idle primase is fine-tuned by the frequency of its association to helicase (Corn and Berger 2006). Such a scenario is supported by the observation that priming frequency increases with primase concentration, this holding true for T4 and even T7 phage systems, the latter having primase and helicase as parts of a single peptide. The situation is less clear for archaea and eukaryotes. However, primase interacts with the MCM helicase through GINS, mediated by Ctf4 in S. cerevisiae, suggesting a role of this interaction for the coordination of their respective activities (Marinsek et al. 2006; Gambus et al. 2009; Tanaka et al. 2009).
Primase remains bound to the primer-template to stabilize, protect, and selectively transfer it to the replicative polymerase (Yuzhakov et al. 1999). Handing off the primer to the polymerase seems to occur in a number of ways according to the different replisome architectures. In E. coli, as with other bacteria, the handing off of primer seems to occur simultaneously with the loading of the clamp onto the primer. After synthesizing the primer, primase forms a ternary complex with DNA, and binding of primase to SSB stabilizes this interaction (Yuzhakov et al. 1999). The nonessential component of the clamp loader, χ, was suggested to compete for the interaction with SSB, releasing primase from the template (Yuzhakov et al. 1999). It is nevertheless likely this model is incomplete given that χ is not essential for viability (Kelman and O’Donnell 1995) and because genetic and structural evidence suggests rather a structural role for χ maintaining processivity of leading-strand synthesis (Marceau et al. 2011). The question of how the switch from primase to polymerase is regulated in bacteria remains open. In eukaryotes, primase is part of a heterotetramer that also includes DNA polymerase Pol α. In this system, the length of primer and the handover to the DNA polymerase subunit is regulated by the noncatalytic subunit of primase (p58) (Kuchta and Stengel 2010). A typical RNA primer would be ∼10 nt in length, onto which Pol α adds ∼20 nt more before it is replaced by Pol ε via RFC clamp-loader-mediated handoff (Waga and Stillman 1998). For binding to RPA, RFC competes with Pol α; for binding to the primer, PCNA gets loaded, and then Pol ε displaces RFC again through a competition for RPA (Yuzhakov et al. 1999; Maga et al. 2000). The theme of a stable interaction between primase and polymerase facilitating primer transfer has also been proposed in the replisome of phage T7 (Kato et al. 2004).
PERSPECTIVES
Although great progress has been made in understanding the coordination of enzymatic events at the replication fork, many mechanistic questions remain unanswered. In particular, how conformational and structural changes within replisomes are coupled to replication-fork progression remains largely unknown. The studies discussed in this article demonstrate that a complex network of interactions between different replisome components regulates each stage of lagging-strand synthesis. However, detailed kinetic studies are needed to clarify the timing and importance of each interaction within active replication complexes. For example, although primer synthesis has been shown to cause polymerase pausing events and the formation of small priming loops, the importance of these events and the conformational states involved still need to be determined. Likewise, the mechanism by which newly synthesized primers are handed off to polymerases on the lagging strand remains obscure.
Until recently, the replisome was thought to operate as a stable multiprotein machine with a defined structure throughout replication. Now it is clear that replisome architecture is actually highly dynamic with individual components rapidly exchanging with their counterparts in solution. In particular, during lagging-strand synthesis, primase, SSB/RPA, the lagging-strand replicative polymerase, and the enzymes that remove the RNA primers and ligate the new Okazaki fragments are known to dissociate and turnover locally or to be recycled into the cellular pool. The turnover frequency of clamp-loader components is not known, but recent work has shown that leading-strand polymerases frequently exchange in contrast to previously proposed models (Loparo et al. 2011). Sliding clamps are recruited continuously during lagging-strand synthesis, with single molecule in vitro analysis showing that they can be recycled locally (Tanner et al. 2011), but the situation in vivo remains unclear. The influence of these turnover events on replication-fork progression is not fully understood. However, subunit exchange is emerging as a general feature of the action of many large multiprotein complexes, as evidenced by the demonstration that flagellar motor components are continually turned over as the bacterial flagella rotates (Delalez et al. 2010; Li and Xie 2011).
Although the importance of continual subunit exchange for efficient replisome operation is not known in all cases, these events appear to play a critical role when the replisome encounters obstacles. During replication, DNA lesions and other barriers to fork progression lead to replication stalling or breaking of the template. In response to these events, a range of different machineries can be recruited specifically to the replisome through association with the sliding clamp, SSB/RPA, or single-stranded DNA or though recognition of other structures at the stalled or broken fork. Recently, many novel mechanisms of replication restart have been discovered, revealing a web of possible reaction pathways to overcome these obstacles (Heller and Marians 2006; Lopes et al. 2006; Pomerantz and O’Donnell 2008; Yeeles and Marians 2011). In particular, specialized translesion polymerases, which are capable of synthesizing across damaged DNA bases (Fujii and Fuchs 2004), enter the replisome through exchange events and allow for lesion bypass. Even though these specialized polymerases are capable of bypassing DNA damage, they synthesize at slower rates and introduce more errors making them poorly suited for normal replication. To alleviate this problem, these specialized polymerases exchange after bypass, returning the replisome to normal operation.
The single-molecule in vivo and in vitro techniques highlighted in this article provide a powerful set of tools to address complex mechanistic questions about replisome function. In vivo imaging allows for the direct visualization of single replication complexes during operation under the most physiologically relevant conditions. These techniques have been used to determine replisome composition in E. coli cells, revealing for the first time that the replisome operates with three polymerases, one more than is required to synthesize the two daughter strands. In vivo imaging methods have also been used to visualize lagging-strand polymerase exchange events within active replisome complexes, demonstrating that highly dynamic processes can be probed (Lia et al. 2012). However, many remaining questions about replisome operation cannot be addressed using in vivo techniques alone because of the difficulty of directly monitoring synthesis rate, the introduction of components with severe mutations or truncations, and the lack of control over reaction conditions. In vitro single-molecule approaches are ideal for these types of situations because both the replication rate and replisome composition can be probed simultaneously and a wide range of reaction conditions can be tested (Loparo et al. 2011; Tanner et al. 2011). The combination of both in vivo and in vitro techniques discussed in this article promise to address a wide range of complex mechanistic questions about DNA replication previously inaccessible using traditional approaches.
ACKNOWLEDGMENTS
Work in the Sherratt laboratory is funded by a Wellcome Trust Programme grant to D.J.S. and by a Todd-Bird Junior Research Fellowship of New College (Oxford) to R.R.-L. The research on DNA replication in the van Oijen group is funded by an ERC Starting grant (281098; SINGLEREPLISOME), an NWO Vici grant (680-47-607) to A.M.vO. and a Human Frontier Science Program Fellowship to K.E.D.
Footnotes
Editors: Stephen D. Bell, Marcel Méchali, and Melvin L. DePamphilis
Additional Perspectives on DNA Replication available at www.cshperspectives.org
REFERENCES
- Alexandrov AI, Botchan MR, Cozzarelli NR 2002. Characterization of simian virus 40 T-antigen double hexamers bound to a replication fork: The active form of the helicase. J Biol Chem 277: 44886–44897 [DOI] [PubMed] [Google Scholar]
- Bailey S, Eliason WK, Steitz TA 2007. Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 318: 459–463 [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A 2002. DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Blackwell LJ, Borowiec JA 1994. Human replication protein A binds single-stranded DNA in two distinct complexes. Mol Cell Biol 14: 3993–4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier AM, Weier H-UG, Cozzarelli NR 2005. Independence of replisomes in Escherichia coli chromosomal replication. Proc Natl Acad Sci 102: 3942–3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver TE Jr, Sexton DJ, Benkovic SJ 1997. Dissociation of bacteriophage T4 DNA polymerase and its processivity clamp after completion of Okazaki fragment synthesis. Biochemistry 36: 14409–14417 [DOI] [PubMed] [Google Scholar]
- Chandler M, Bird RE, Caro L 1975. The replication time of the Escherichia coli K12 chromosome as a function of cell doubling time. J Mol Biol 94: 127–132 [DOI] [PubMed] [Google Scholar]
- Chastain PD II, Makhov AM, Nossal NG, Griffith J 2000. Analysis of the Okazaki fragment distributions along single long DNAs replicated by the bacteriophage T4 proteins. Mol Cell 6: 803–814 [DOI] [PubMed] [Google Scholar]
- Chastain PD II, Makhov AM, Nossal NG, Griffith J 2003. Architecture of the replication complex and DNA loops at the fork generated by the bacteriophage t4 proteins. J Biol Chem 278: 21276–21285 [DOI] [PubMed] [Google Scholar]
- Cook PR 1999. The organization of replication and transcription. Science 284: 1790–1795 [DOI] [PubMed] [Google Scholar]
- Corn JE, Berger JM 2006. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res 34: 4082–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corn JE, Pease PJ, Hura GL, Berger JM 2005. Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol Cell 20: 391–401 [DOI] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ 2000. The importance of repairing stalled replication forks. Nature 404: 37–41 [DOI] [PubMed] [Google Scholar]
- Cseresnyes Z, Schwarz U, Green CM 2009. Analysis of replication factories in human cells by super-resolution light microscopy. BMC Cell Biol 10: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debyser Z, Tabor S, Richardson CC 1994. Coordination of leading and lagging strand DNA synthesis at the replication fork of bacteriophage T7. Cell 77: 157–166 [DOI] [PubMed] [Google Scholar]
- Delalez NJ, Wadhams GH, Rosser G, Xue Q, Brown MT, Dobbie IM, Berry RM, Leake MC, Armitage JP 2010. Signal-dependent turnover of the bacterial flagellar switch protein FliM. Proc Natl Acad Sci 107: 11347–11351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingman CW 1974. Bidirectional chromosome replication: Some topological considerations. J Theor Biol 43: 187–195 [DOI] [PubMed] [Google Scholar]
- Dohrmann PR, Manhart CM, Downey CD, McHenry CS 2011. The rate of polymerase release upon filling the gap between Okazaki fragments is inadequate to support cycling during lagging strand synthesis. J Mol Biol 414: 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, Bermejo R, Fiorani S, Haber JE, Foiani M 2009. Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell 137: 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Allen EF, Forsberg SA, Preparata RM, Greening EO 1969. Genetic control of mutation rates in bacteriophageT4. Nature 221: 1128–1132 [PubMed] [Google Scholar]
- Duderstadt KE, Berger JM 2008. AAA+ ATPases in the initiation of DNA replication. Crit Rev Biochem Mol Biol 43: 163–187 [DOI] [PubMed] [Google Scholar]
- Duggin IG, McCallum SA, Bell SD 2008. Chromosome replication dynamics in the archaeon Sulfolobus acidocaldarius. Proc Natl Acad Sci 105: 16737–16742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Huberman JA 1975. Eukaryotic chromosome replication. Annu Rev Genet 9: 245–284 [DOI] [PubMed] [Google Scholar]
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C 2009. A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci 106: 20240–20245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick DN, Richardson CC 2001. DNA primases. Annu Rev Biochem 70: 39–80 [DOI] [PubMed] [Google Scholar]
- Frick DN, Kumar S, Richardson CC 1999. Interaction of ribonucleoside triphosphates with the gene 4 primase of bacteriophage T7. J Biol Chem 274: 35899–35907 [DOI] [PubMed] [Google Scholar]
- Fujii S, Fuchs RP 2004. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J 23: 4342–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A, Labib K 2009. A key role for Ctf4 in coupling the MCM2–7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J 28: 2992–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Kurth I, Yao NY, Stewart J, Yurieva O, O’Donnell M 2009. Mechanism of polymerase collision release from sliding clamps on the lagging strand. EMBO J 28: 2981–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Kurth I, O’Donnell ME 2012. Single-molecule studies reveal the function of a third polymerase in the replisome. Nat Struct Mol Biol 19: 113–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker KJ, Alberts BM 1994. The rapid dissociation of the T4 DNA polymerase holoenzyme when stopped by a DNA hairpin helix: A model for polymerase release following the termination of each Okazaki fragment. J Biol Chem 269: 24221–24228 [PubMed] [Google Scholar]
- Hamdan SM, Loparo JJ, Takahashi M, Richardson CC, van Oijen AM 2009. Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 457: 336–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearst JE, Kauffman L, McClain WM 1998. A simple mechanism for the avoidance of entanglement during chromosome replication. Trends Genet 14: 244–247 [DOI] [PubMed] [Google Scholar]
- Heller RC, Marians KJ 2006. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439: 557–562 [DOI] [PubMed] [Google Scholar]
- Indiani C, Langston LD, Yurieva O, Goodman MF, O’Donnell M 2009. Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc Natl Acad Sci U S A 106: 6031–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Brenner S, Cuzin F 1963. On the regulation of DNA replication in bacteria. Cold Spring Harbor Symp Quant Biol 28: 329–347 [Google Scholar]
- Johnson DS, Bai L, Smith BY, Patel SS, Wang MD 2007. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell 129: 1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T, Ozaki S, Keyamura K, Fujimitsu K 2010. Regulation of the replication cycle: Conserved and diverse regulatory systems for DnaA and oriC. Nat Rev Microbiol 8: 163–170 [DOI] [PubMed] [Google Scholar]
- Kato M, Ito T, Wagner G, Ellenberger T 2004. A molecular handoff between bacteriophage T7 DNA primase and T7 DNA polymerase initiates DNA synthesis. J Biol Chem 279: 30554–30562 [DOI] [PubMed] [Google Scholar]
- Kelman Z, O’Donnell M 1995. DNA polymerase III holoenzyme: Structure and function of a chromosomal replicating machine. Annu Rev Biochem 64: 171–200 [DOI] [PubMed] [Google Scholar]
- Kerr ID, Wadsworth RI, Cubeddu L, Blankenfeldt W, Naismith JH, White MF 2003. Insights into ssDNA recognition by the OB fold from a structural and thermodynamic study of Sulfolobus SSB protein. EMBO J 22: 2561–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YT, Tabor S, Bortner C, Griffith JD, Richardson CC 1992. Purification and characterization of the bacteriophage T7 gene 2.5 protein: A single-stranded DNA-binding protein. J Biol Chem 267: 15022–15031 [PubMed] [Google Scholar]
- Kitamura E, Blow JJ, Tanaka TU 2006. Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell 125: 1297–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg A, Baker T 1992. DNA replication. Freeman, New York [Google Scholar]
- Kuchta RD, Stengel G 2010. Mechanism and evolution of DNA primases. Biochim Biophys Acta 1804: 1180–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran S, Kozlov AG, Lohman TM 2006. Saccharomyces cerevisiae replication protein A binds to single-stranded DNA in multiple salt-dependent modes. Biochemistry 45: 11958–11973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe T, Richardson CC 1997. Template recognition and ribonucleotide specificity of the DNA primase of bacteriophage T7. J Biol Chem 272: 5943–5951 [DOI] [PubMed] [Google Scholar]
- Langston LD, Indiani C, O’Donnell M 2009. Whither the replisome: Emerging perspectives on the dynamic nature of the DNA replication machinery. Cell Cycle 8: 2686–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Richardson CC 2002. Interaction of adjacent primase domains within the hexameric gene 4 helicase-primase of bacteriophage T7. Proc Natl Acad Sci 99: 12703–12708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Chastain PD II, Griffith JD, Richardson CC 2002. Lagging strand synthesis in coordinated DNA synthesis by bacteriophage T7 replication proteins. J Mol Biol 316: 19–34 [DOI] [PubMed] [Google Scholar]
- Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM 2006. DNA primase acts as a molecular brake in DNA replication. Nature 439: 621–624 [DOI] [PubMed] [Google Scholar]
- Lemon KP, Grossman AD 1998. Localization of bacterial DNA polymerase: Evidence for a factory model of replication. Science 282: 1516–1519 [DOI] [PubMed] [Google Scholar]
- Lemon KP, Grossman AD 2000. Movement of replicating DNA through a stationary replisome. Mol Cell 6: 1321–1330 [DOI] [PubMed] [Google Scholar]
- Li X, Marians KJ 2000. Two distinct triggers for cycling of the lagging strand polymerase at the replication fork. J Biol Chem 275: 34757–34765 [DOI] [PubMed] [Google Scholar]
- Li GW, Xie XS 2011. Central dogma at the single-molecule level in living cells. Nature 475: 308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lia G, Michel B, Allemand JF 2012. Polymerase exchange during Okazaki fragment synthesis observed in living cells. Science 335: 328–331 [DOI] [PubMed] [Google Scholar]
- Liachko I, Bhaskar A, Lee C, Chung SC, Tye BK, Keich U 2010. A comprehensive genome-wide map of autonomously replicating sequences in a naive genome. PLoS Genet 6: e1000946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T, Spiering MM, Benkovic SJ, Bensimon D, Croquette V 2007. Real-time observation of bacteriophage T4 gp41 helicase reveals an unwinding mechanism. Proc Natl Acad Sci 104: 19790–19795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A 2001. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol 2: 444–456 [DOI] [PubMed] [Google Scholar]
- Loparo JJ, Kulczyk AW, Richardson CC, van Oijen AM 2011. Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc Natl Acad Sci 108: 3584–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, Sogo JM 2006. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21: 15–27 [DOI] [PubMed] [Google Scholar]
- Lopez de Saro FJ, Georgescu RE, O’Donnell M 2003. A peptide switch regulates DNA polymerase processivity. Proc Natl Acad Sci 100: 14689–14694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, Wang M, Konigsberg WH, Xie XS 2007. Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc Natl Acad Sci 104: 12610–12615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maga G, Stucki M, Spadari S, Hubscher U 2000. DNA polymerase switching: I. Replication factor C displaces DNA polymerase alpha prior to PCNA loading. J Mol Biol 295: 791–801 [DOI] [PubMed] [Google Scholar]
- Maier B, Bensimon D, Croquette V 2000. Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc Natl Acad Sci 97: 12002–12007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manosas M, Spiering MM, Zhuang Z, Benkovic SJ, Croquette V 2009. Coupling DNA unwinding activity with primer synthesis in the bacteriophage T4 primosome. Nat Chem Biol 5: 904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau AH, Bahng S, Massoni SC, George NP, Sandler SJ, Marians KJ, Keck JL 2011. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J 30: 4236–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinsek N, Barry ER, Makarova KS, Dionne I, Koonin EV, Bell SD 2006. GINS, a central nexus in the archaeal DNA replication fork. EMBO Rep 7: 539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M 2010. Eukaryotic chromosome DNA replication: Where, when, and how? Annu Rev Biochem 79: 89–130 [DOI] [PubMed] [Google Scholar]
- Matsunaga F, Norais C, Forterre P, Myllykallio H 2003. Identification of short “eukaryotic” Okazaki fragments synthesized from a prokaryotic replication origin. EMBO Rep 4: 154–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méchali M 2010. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat Rev Mol Cell Biol 11: 728–738 [DOI] [PubMed] [Google Scholar]
- Mott ML, Berger JM 2007. DNA replication initiation: Mechanisms and regulation in bacteria. Nat Rev Microbiol 5: 343–354 [DOI] [PubMed] [Google Scholar]
- Myllykallio H, Lopez P, Lopez-Garcia P, Heilig R, Saurin W, Zivanovic Y, Philippe H, Forterre P 2000. Bacterial mode of replication with eukaryotic-like machinery in a hyperthermophilic archaeon. Science 288: 2212–2215 [DOI] [PubMed] [Google Scholar]
- Nordstrom K 2006. Plasmid R1—replication and its control. Plasmid 55: 1–26 [DOI] [PubMed] [Google Scholar]
- Pandey M, Syed S, Donmez I, Patel G, Ha T, Patel SS 2009. Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature 462: 940–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Debyser Z, Tabor S, Richardson CC, Griffith JD 1998. Formation of a DNA loop at the replication fork generated by bacteriophage T7 replication proteins. J Biol Chem 273: 5260–5270 [DOI] [PubMed] [Google Scholar]
- Patterson G, Davidson M, Manley S, Lippincott-Schwartz J 2010. Superresolution imaging using single-molecule localization. Annu Rev Phys Chem 61: 345–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, O’Donnell M 2008. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature 456: 762–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qimron U, Lee SJ, Hamdan SM, Richardson CC 2006. Primer initiation and extension by T7 DNA primase. EMBO J 25: 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabkin SD, Richardson CC 1990. In vivo analysis of the initiation of bacteriophage T7 DNA replication. Virology 174: 585–592 [DOI] [PubMed] [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL 2001. Replication dynamics of the yeast genome. Science 294: 115–121 [DOI] [PubMed] [Google Scholar]
- Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF 2009. Concerted loading of Mcm2–7 double hexamers around DNA during DNA replication origin licensing. Cell 139: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Possoz C, Danilova O, Sherratt DJ 2008. Independent positioning and action of Escherichia coli replisomes in live cells. Cell 133: 90–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Sherratt DJ, Leake MC 2010. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328: 498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeck N, Saleh OA 2008. Multiplexed single-molecule measurements with magnetic tweezers. Rev Sci Instrum 79: 094301. [DOI] [PubMed] [Google Scholar]
- Smiley RD, Zhuang Z, Benkovic SJ, Hammes GG 2006. Single-molecule investigation of the T4 bacteriophage DNA polymerase holoenzyme: Multiple pathways of holoenzyme formation. Biochemistry 45: 7990–7997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Whitehouse I 2012. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483: 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovjeva L, Svetlova M, Sasina L, Tanaka K, Saijo M, Nazarov I, Bradbury M, 2005. High mobility of flap endonuclease 1 and DNA polymerase eta associated with replication foci in mammalian S-phase nucleus. Mol Biol Cell 16: 2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC 2002. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol Cell 10: 1355–1365 [DOI] [PubMed] [Google Scholar]
- Su’etsugu M, Errington J 2011. The replicase sliding clamp dynamically accumulates behind progressing replication forks in Bacillus subtilis cells. Mol Cell 41: 720–732 [DOI] [PubMed] [Google Scholar]
- Sun B, Johnson DS, Patel G, Smith BY, Pandey M, Patel SS, Wang MD 2011. ATP-induced helicase slippage reveals highly coordinated subunits. Nature 478: 132–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Katou Y, Yagura M, Saitoh K, Itoh T, Araki H, Bando M, Shirahige K 2009. Ctf4 coordinates the progression of helicase and DNA polymerase alpha. Genes Cells 14: 807–820 [DOI] [PubMed] [Google Scholar]
- Tanner NA, van Oijen AM 2009. Single-molecule observation of prokaryotic DNA replication. Methods Mol Biol 521: 397–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NA, Hamdan SM, Jergic S, Loscha KV, Schaeffer PM, Dixon NE, van Oijen AM 2008. Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat Struct Mol Biol 15: 998. [DOI] [PubMed] [Google Scholar]
- Tanner NA, Loparo JJ, Hamdan SM, Jergic S, Dixon NE, van Oijen AM 2009. Real-time single-molecule observation of rolling-circle DNA replication. Nucleic Acids Res 37: e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NA, Tolun G, Loparo JJ, Jergic S, Griffith JD, Dixon NE, van Oijen AM 2011. E. coli DNA replication in the absence of free beta clamps. EMBO J 30: 1830–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata-Sogawa K 2008. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5: 159–161 [DOI] [PubMed] [Google Scholar]
- Tougu K, Marians KJ 1996. The interaction between helicase and primase sets the replication fork clock. J Biol Chem 271: 21398–21405 [DOI] [PubMed] [Google Scholar]
- Valle M, Gruss C, Halmer L, Carazo JM, Donate LE 2000. Large T-antigen double hexamers imaged at the simian virus 40 origin of replication. Mol Cell Biol 20: 34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oijen AM, Blainey PC, Crampton DJ, Richardson CC, Ellenberger T, Xie XS 2003. Single-molecule kinetics of λ exonuclease reveal base dependence and dynamic disorder. Science 301: 1235–1238 [DOI] [PubMed] [Google Scholar]
- Waga S, Stillman B 1998. The DNA replication fork in eukaryotic cells. Annu Rev Biochem 67: 721–751 [DOI] [PubMed] [Google Scholar]
- Wang JD, Berkmen MB, Grossman AD 2007. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc Natl Acad Sci 104: 5608–5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Reyes-Lamothe R, Sherratt DJ 2008. Modulation of Escherichia coli sister chromosome cohesion by topoisomerase IV. Genes Dev 22: 2426–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisshart K, Taneja P, Jenne A, Herbig U, Simmons DT, Fanning E 1999. Two regions of simian virus 40 T antigen determine cooperativity of double-hexamer assembly on the viral origin of DNA replication and promote hexamer interactions during bidirectional origin DNA unwinding. J Virol 73: 2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuite GJ, Smith SB, Young M, Keller D, Bustamante C 2000. Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature 404: 103–106 [DOI] [PubMed] [Google Scholar]
- Xi J, Zhang Z, Zhuang Z, Yang J, Spiering MM, Hammes GG, Benkovic SJ 2005a. Interaction between the T4 helicase loading protein (gp59) and the DNA polymerase (gp43): Unlocking of the gp59-gp43-DNA complex to initiate assembly of a fully functional replisome. Biochemistry 44: 7747–7756 [DOI] [PubMed] [Google Scholar]
- Xi J, Zhuang Z, Zhang Z, Selzer T, Spiering MM, Hammes GG, Benkovic SJ 2005b. Interaction between the T4 helicase-loading protein (gp59) and the DNA polymerase (gp43): A locking mechanism to delay replication during replisome assembly (erratum Biochemistry [2005] 44: 12264; 44: 4600). Biochemistry 44: 2305–2318 [DOI] [PubMed] [Google Scholar]
- Xie XS, Choi PJ, Li GW, Lee NK, Lia G 2008. Single-molecule approach to molecular biology in living bacterial cells. Annu Rev Biophys 37: 417–444 [DOI] [PubMed] [Google Scholar]
- Yang J, Nelson SW, Benkovic SJ 2006. The control mechanism for lagging strand polymerase recycling during bacteriophage T4 DNA replication. Mol Cell 21: 153–164 [DOI] [PubMed] [Google Scholar]
- Yao NY, O’Donnell M 2010. SnapShot: The replisome. Cell 141: 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao NY, Georgescu RE, Finkelstein J, O’Donnell ME 2009. Single-molecule analysis reveals that the lagging strand increases replisome processivity but slows replication fork progression. Proc Natl Acad Sci 106: 13236–13241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardimci H, Loveland AB, Habuchi S, van Oijen AM, Walter JC 2010. Uncoupling of sister replisomes during eukaryotic DNA replication. Mol Cell 40: 834–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, Marians KJ 2011. The Escherichia coli replisome is inherently DNA damage tolerant. Science 334: 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzhakov A, Kelman Z, Hurwitz J, O’Donnell M 1999a. Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. EMBO J 18: 6189–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzhakov A, Kelman Z, O’Donnell M 1999b. Trading places on DNA—a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 96: 153–163 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Spiering MM, Trakselis MA, Ishmael FT, Xi J, Benkovic SJ, Hammes GG 2005. Assembly of the bacteriophage T4 primosome: Single-molecule and ensemble studies. Proc Natl Acad Sci 102: 3254–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]