

Abstract
Significant advances in fluorescence microscopy tend be a balance between two competing qualities wherein improvements in resolution and low light detection are typically accompanied by losses in acquisition rate and signal-to-noise, respectively. These trade-offs are becoming less of a barrier to biomedical research as recent advances in optoelectronic microscopy and developments in fluorophore chemistry have enabled scientists to see beyond the diffraction barrier, image deeper into live specimens, and acquire images at unprecedented speed. Selective plane illumination microscopy has provided significant gains in the spatial and temporal acquisition of fluorescence specimens several mm in thickness. With commercial systems now available, this method promises to expand on recent advances in 2-photon deep-tissue imaging with improved speed and reduced photobleaching compared to laser scanning confocal microscopy. Superresolution microscopes are also available in several modalities and can be coupled with selective plane illumination techniques. The combination of methods to increase resolution, acquisition speed, and depth of collection are now being married to common microscope systems, enabling scientists to make significant advances in live cell and in situ imaging in real time. We show that light sheet microscopy provides significant advantages for imaging live zebrafish embryos compared to laser scanning confocal microscopy.
Keywords: confocal microscopy, photoactivatable GFP, 2-photon microscopy, superresolution microscopy, selective plane illumination microscopy, light sheet microscopy
Introduction
Ask any scientist what the most important part of a microscope is and he or she will invariably point to the numerical aperture (NA) of the objective lens. The logic to this answer, first published by Ernst Abbe in 1873 and later refined by Lord Rayleigh, lies in the well-known diffraction limit that defines the relationship between illuminating wavelength and resolution. Abbe was the first to define NA and develop simple formulas for measuring the distance between two Airy patterns capable of being distinguished as separate entities.
The Abbe limits for lateral and axial resolution in a light microscope are:
Abbe Resolution x,y = λ/2 NA
Abbe Resolution z = 2λ/NA2
These formulas show that shorter wavelengths and larger NA yield more detail, since spatial resolution is defined by the ratio of wavelength to NA or, in empirical terms, the ratio of diffraction to aperture opening. Substituting 488 nm light for excitation of green fluorescent protein (GFP) with a highly corrected 1.4 NA oil immersion objective lens into the above equation yields a theoretical lateral resolution of 174 nm and an axial resolution of 498 nm using a standard epifluorescence widefield microscope. In reality, these values are difficult to approximate, especially when looking into large samples such as whole embryos, where resolution is decreased due to diffraction, poor signal to noise, and imperfect illumination, resulting in a real world lateral resolution closer to 250 nm and axial resolution of ~ 650 nm. The gap separating theoretical resolution values from observed values limited microscopists for more than a century, and most scientists accepted as dogma that resolution in x, y is ~half the wavelength of incident light, a value immortalized as the Abbe diffraction limit. In this review, we highlight several notable advances that have dramatically altered our ability to see light microscope images with much better resolution than Abbe hypothesized and acquire deep tissue images at previously unimaginable acquisition speeds. Perhaps most surprisingly, all of the major breakthroughs discussed here do not require use of shorter λ and improvement in the NA of lenses. Abbe and Rayleigh weren’t wrong; diffraction is still rate-limiting, it’s just that the physical tools and fluorophores now available for fluorescence imaging have changed our way of thinking about microscopy.
Pinholes Increase Resolution
One of the major limitations to widefield fluorescence microscopy is the significant out-of-focus fluorescence that creates background noise and image degradation. This background fluorescence can be reduced with measurable gains in resolution using laser scanning confocal microscopy (LSCM), where a rapidly scanned excitation spot is used to build an image point-by-point [1]. The resulting fluorescence signal is regulated by adjustable apertures (confocal pinholes) placed in front of detectors and conjugate with the sample to remove out-of-focus emissions above and below the diffraction-limited focal volume (Figure 1). Using the Rayleigh criterion for measurements of resolution, which is defined by the minimum resolvable distance separating the intensity profiles or point spread functions (PSF) of two point sources, LSCM lateral resolution of 178 nm and axial resolution of 455 nm were observed when imaging isolated fluorescent microspheres [2]. While these values are within ~10 percent of the theoretical limit of the confocal microscope, they were obtained using fluorescent beads in the absence of background noise. Such impressive resolution is not possible using LSCM in biological specimens due to diffraction, light scatter, and absorption, problems that are particularly evident in thick samples. The primary benefit resulting from gains in axial resolution using confocal microscopy is the ability to acquire thin in-focus optical slices through entire samples, including live embryos. The resulting image z-stacks are then used for 3D or 4D (x, y, z and time) reconstructions.
Figure 1.
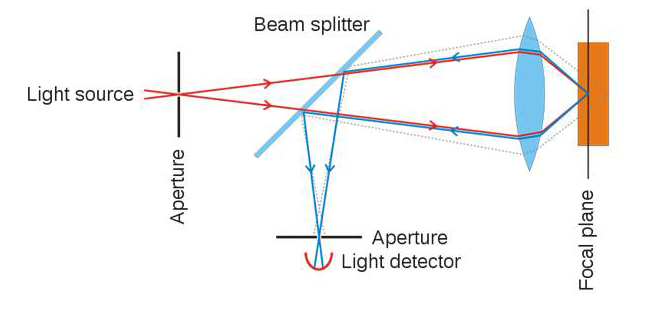
Confocal pinhole principle. Excitation light (red), fluorescence emission light passing through pinhole to detector (blue). Out of focus fluorescence (stippled line) is rejected from the detector by the adjustable pinhole aperture. ©Wikimedia Commons.
Two Photons Meet at the Focal Point
One limitation of LSCM is photobleaching of the sample, particularly during z-stack and time-lapse acquisitions. The confocal pinhole acts downstream of the sample and has no role in modulating excitation laser light. Hence, out-of-focus regions in the sample are repeatedly irradiated with excitation light for the duration of image stack acquisition, a period of minutes to hours depending on the sample and experimental design. To minimize out-of-focus photodamage, 2-photon excitation microscopy (2-P) has been adapted for use with LSCM. The photophysics of 2-P microscopy is based on the activation energy required to achieve an excited state (Figure 2a). In routine epifluorescence microscopy, one photon (1-P) is used to excite one fluorophore, and the energy of the incident light is commensurate with the absorption peak of the fluorophore. Short wavelengths with high energy are used for excitation of BFP and GFP, whereas longer wavelengths of lower energy are used for excitation of red-emitting probes (dsRed). In the early 1930s, Maria Göppert-Mayer [3] proposed that the energy required to excite an atom could be obtained by combining the lower energies of two (or more) longer wavelength photons (Figure 2a). This laid the foundation for the development of 2-P microscopy by Denk et al., who showed that a UV excitable fluorescent nucleic acid stain (Hoechst 33258) could be excited by 2-P absorption using 630 nm light [4]. This technique was subsequently adapted by biologists using the widely popular probe GFP, which can be excited by one photon of light at either 488 nm or simultaneously with 2 photons ranging from 800 to 950 nm delivering half the excitation energy. In both cases, the GFP emits green light with a peak emission at 516 nm.
Figure 2.
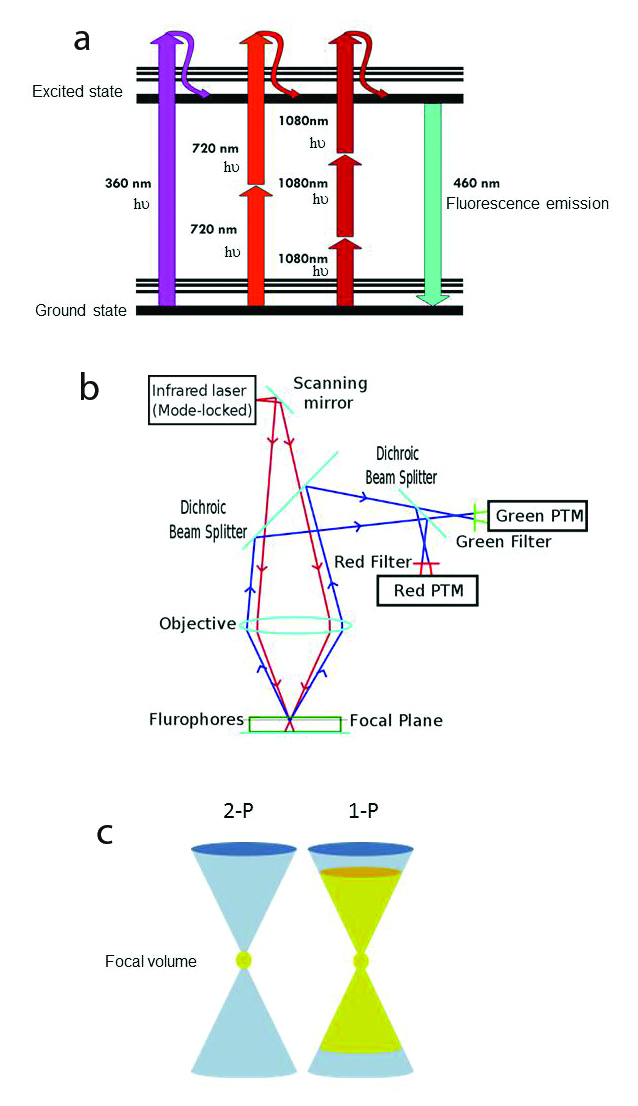
a) Jablonski fluorescence diagram showing excitation of atoms with 1PE: 360 nm (magenta), 2PE: 720 nm light (scarlet red) and 3PE: 1080 nm light (burgundy red. The thicker lines represent electronic energy levels, while the thinner lines indicate various vibrational energy states in the ground (lower) and excited (upper) states. In each case, the resulting fluorescence emission is 460 nm (cyan) visible light. © Wikimedia Commons/Alberto Diaspro, Paolo Bianchini, Giuseppe Vicidomini, Mario Faretta, Paola Ramoino and Cesare Usai. b) Scheme of 2-photon excitation microscopy. Incident excitation pulsed laser light (red). Fluorescence emission (blue). Photomultiplier tube (PTM). © Wikimedia Commons. c) Schematic illustration of the difference between 1- and 2-photon absorption. 2-photon absorption occurs only in the small focal volume (yellow) defined by the wavelength and NA of the objective. On the right, 1-photon excitation (yellow) by out-of-focus photons occurs above and below the focal plane, causing unnecessary photodamage to the sample. From: Magnus B. Lilledahl, Gary Chinga-Carrasco and Catharina de Lange Davies. Three-Dimensional Visualization and Quantification of Structural Fibres for Biomedical Applications. Available from: http://www.intechopen.com/books/confocal-laser-microscopy-principles-and-applications-in-medicine-biology-and-the-food-sciences/three-dimensional-visualization-and-quantification-of-structural-fibres-for-biomedical-applications.
2-P microscopy of biological specimens involves simultaneous absorption of near infrared (NIR) laser light by visible fluorophores (Figure 2b). This is typically achieved using high NA objectives and tunable high power Ti:Sapphire lasers (680 – 1080 nm) that emit high frequency (80 MHz) short duration (~100 fs) pulses of excitation light. When properly aligned, the 2-P laser creates a very high instantaneous photon flux at the diffraction limited focal volume, and only those fluorophores localized within this spot absorb two photons simultaneously and emit fluorescence (Figure 2c). The long wavelength photons have insufficient energies to excite visible fluorophores by 1-P absorption. Consequently, fluorophores outside the diffraction-limited focal volume remain in the ground state and photodamage and phototoxicity are markedly reduced outside the image plane. 2-P absorption at the focal point increases quadratically (nonlinear microscopy) with excitation intensity. Since fluorescence emission occurs only from the vicinity of the focal point and there is no out of focus fluorescence to degrade the image, 2-P microscopy does not require a confocal pinhole.
Another advantage of 2-P microscopy is the ability to obtain optical sections deeper into living tissues. This is mostly a function of the wavelength of choice: long wavelengths used in 2-P (e.g., 680-1080 nm) diffract less and penetrate deeper into samples than visible light (405 to 630 nm) used in 1-P microscopy. When coupled with a temperature-controlled stage incubator, 2-P microscopy is preferable for imaging deep cell movements in live samples, albeit the relatively slow speed of LSCM acquisition precludes real-time 4D acquisitions of whole embryos. A common misperception is that resolution is improved with 2-P compared to 1-P confocal microscopy. It is not, but image contrast and signal vary considerably between these excitation techniques depending on the sample and imaging parameters. For thin samples with low background, 1-P confocal resolution is markedly superior to 2-P methods. This is easily explained using the above equations showing that resolution is inversely related to wavelength and consistent with reports showing 2-P microscopy resolution is decreased by 10 percent in x, y and 30 percent along the z axis due to the larger focal spot formed using near IR excitation light [5]. When imaging deep into tissues, visible light of 1-P illumination is more easily scattered, yielding a larger excitation volume (greater axial PSF) with increasing image depth compared to 2-P excitation using near IR light. The resulting 2-P image is of much higher contrast due to better signal-to-noise ratios, albeit the theoretical resolution is no better than an optimized 1-P confocal microscope. 2-P excitation microscopy is better for deep tissue observations of live samples where minimizing photodamage is a priority, but offers no major advantage when used for fixed thin samples such as monolayers of cultured cells grown on coverslips. Neither imaging technique offers details beyond the Abbe limits.
Exploiting Physical Properties of Fluorophores and Light to Achieve Superresolution Microscopy
The key to resolving objects with resolution better than the ~250 nm diffraction limit came primarily from thinking outside the box. For innovators like Stefan Hell, the barrier set by diffraction of light is not a rate-limiting law of physics, but rather a value established by far-field localization microscopy methods, and seeing beyond the diffraction barrier is no different than traveling faster than the speed of sound. Marked improvements in resolution were initially reported using 4Pi-confocal microscopy coupled with 2-P excitation using an optical setup consisting of two opposing objective lenses capable of ~100 nm resolution in three dimensions [6]. While impressive compared to conventional confocal techniques, 4Pi microscopy wasn’t widely embraced due to sample size restrictions resulting from the shallow working distance of two closely opposed high NA objectives (Figure 3).
Figure 3.
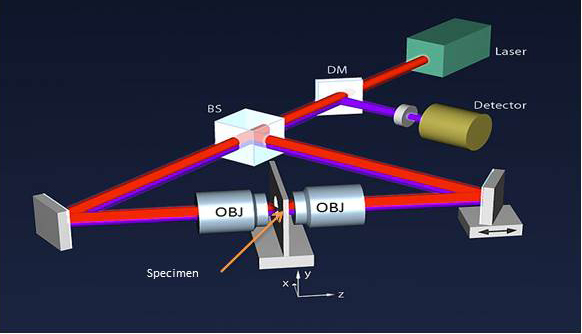
Schematic of a 4-Pi microscope. Incident excitation light (red). Emitted fluorescence (magenta). Objective lens (OBJ), Beamsplitter (BS), Dichroic mirror (DM). © Wikipedia/Hartmut Sebesse. CC-BY-SA-3.0; CC-BY-SA-3.0-DE; BILD-GFDL-NEU; Licensed under the GFDL by the author.
Rather than attempting to use shorter wavelengths (UV) of illuminating light, which isn’t transmitted well through glass, or design lenses with greater NAs (not practical due to refractive index limitations of glass), Hell and others [7,8] developed true superresolution microscopes by exploiting breakthroughs in fluorescence chemistry and advances in optoelectronics. Many of the visionary pioneers [7,9] responsible for superresolution microscopy use the phrase “breaking the diffraction barrier” when discussing their research. This parenthetical phrase is somewhat misleading and should not be interpreted literally, but rather viewed as a figurative definition that provides sufficient meaning but still requires further interpretation. Yes, superresolution microscopy enables visualization of objects closer than the diffraction limit (~250 nm), but this is accomplished using physical tricks. No laws of physics are broken when this barrier is passed, and superresolution microscopy image formation is still restricted by diffraction of light. Perhaps “seeing beyond the diffraction barrier” is more appropriate than “breaking the diffraction barrier,” as the former doesn’t infer any violation of nature.
How Do We See Beyond the Diffraction Barrier?
Superresolution microscopy is currently divided into two classes based on methodologies: 1) ensemble techniques that require shaping of the incident light and 2) single-molecules techniques that depend on precise localization of point emissions. Both classes are further subdivided based on method of excitation, detection, and use of specific optic elements. In each imaging modality, reports of ~20 to ~50 nm resolution are common, values ~10-fold better than achieved using standard widefield epifuorescence microscopy.
Ensemble Superresolution Microscopy
One of the initial ensemble-based superresolution systems designed by Hell in 1994 is stimulated emission depletion microscopy (STED), which uses stimulated emission to overcome the Abbe diffraction limit [9]. STED microscopy uses a short excitatory wavelength (typically 488 nm, blue) to energize fluorophores to an excited state coupled with a second laser pulse of longer wavelength (e.g., 592 nm, red) superimposed over the excitatory light as a donut-shaped ring. When properly aligned, the donut of red laser light suppresses the fluorescence at the margins of the emitting focal volume (Figure 4). Stimulated emission depletion is most effective when an electron in an excited state is stimulated by a photon with the same energy required for the transition back to the ground state. This is readily achieved using 1 ps STED laser pulses to deplete electrons from the excited state independent of spontaneous fluorescence emission.
Figure 4.

STED superresolution microscopy. Display of excitation focus (left), de-excitation focus (center), and remaining fluorescence distribution in a STED microscope. Note that the final fluorescent spot size is reduced to a diameter <250 nm. © Wikipedia/Marcel Lauterbach.
The thickness of the fluorescence-depleting donut can be expanded by increasing the laser intensity, thereby decreasing the diameter of the centrally located focal emission spot to very small values (Figure 4). The smaller emission spot is then scanned over the sample using a confocal microscope raster to build a superresolution image point-by-point. STED is unique for light microscopy in that this technique does not have a defined bandwidth limit. In theory, the emission spot size can be reduced infinitely, with corresponding sub-nm resolution. In practice, STED superresolution images with <70 nm resolution can be processed in real time using scaled-gradient-projection deconvolution [10]. When combined with 2-P excitation for whole organ imaging, STED superresolution microscopy achieves 60 nm resolution up to 30 um deep in live brain [11].
The major shortcoming of STED is that the system is rather difficult to design in-house. Commercial systems are available from Leica, albeit the utility of other ensemble superresolution systems based on structured illumination are available from both Nikon (N-SIM/N-STORM) and Zeiss (SR-SIM) and may be better suited for multi-user facilities. For detailed reviews on structured illumination see [12].
Single Molecule Superresolution
In contrast to STED, which excites multiple molecules in the focal volume, single molecule techniques depend on precise localization of fluorescence from point sources. Advances in fluorescent protein engineering coupled with manageable optical design make this method preferable for biologists and multi-user core facilities. Following the expression of GFP as a fluorescent protein marker by Chalfie et al. [13] and improvement of green fluorescence by Tsien and coworkers in 1995 [14], fluorescent protein chemistry developed into a recognized industry driven by genetic engineering of variants that excited and emitted over a range of visible colors and with increasing photostability (BFP: 402/547, CFP: 458/480, YFP: 508/524). One variant with properties ideal for superresolution microscopy is photoactivatable-GFP (PA-GFP). Unconverted PA-GFP absorbs well at 405 nm, but not at 488 nm. Following irradiation of PA-GFP with 405 nm laser light, the absorbance maximum shifts closer to 488 nm, but this converted form does not emit useful green fluorescence until exposed to light at 488 nm, a process that results in a 100-fold increase in green fluorescence emission compared to the unconverted forms. Converted PA-GFP provides a significant increase in signal-to-noise since the fluorescent region of interest is limited only by the size of the laser focal volume, and regions of the sample outside the focal spot of 405 nm light do not emit fluorescence during standard GFP excitation/emission (488/520 nm) image acquisition settings [15].
The photochemistry of PA-GFP is based on conversion of a neutral molecule to a stable anionic (fluorescent) species. Strategies for engineering novel fluorescent proteins include using either directed evolution or error-prone PCR. The current trend has emphasized identification and modification of fluorescent proteins from marine invertebrates, particularly corals, that emit in the red spectrum (mFruit series) [16] and has extended beyond metazoans to include archaeal proteins. Recently, Kralj et al. developed a pH-sensitive fluorescent voltage sensor using archaerhdopsin 3 from Halorurum, enabling neuroscientists to study action potentials in mammalian neurons [17]. Collectively termed optical highlighters, these photoconvertible proteins are being used as novel FRET sensors of protein-protein interactions and Ca2+ indicators and essential probes for select methods of superresolution microscopy.
The simplest strategy for superresolution microscopy involves visualization of single fluorescent molecules with high spatial precision. This is not practical using routine immunofluorescence microscopy methods in which the fluorophore density can easily exceed 100,000 molecules/um2, but through use of PA-GFP or the red emitting PA-mCherry, which can be incorporated into cells at sufficiently high densities and excited stochastically and repeatedly over time. This ladder point is essential, since single fluorophores must be able to undergo repeated rounds of excitation sufficient for detection by a sensor. For low level expressing fluorescent proteins, the period of excitation necessary for useful analysis can last seconds or tens of seconds.
Like all point sources, emission of a photoactivated fluorescent protein generates a three-dimensional pattern of diffracted light (PSF) that can be assigned a centroid position. The trick is to use a relatively low laser power to stochastically photoactivate only a very small population of fluorescent proteins in the sample at any given time. With repeated rounds of stochastic photoactivation, a sufficient signal can be recorded against a low background, since for each excitation pulse, most of the fluorescent proteins remain unactivated. Fluorescence emission from objects closer than one Airy disc can then be assigned centroids with subpixel resolution and diffraction-limited objects spaced closer than the 250 nm barrier detected. This strategy has resulted in the development of three nearly identical methods of single molecule detection superesolution microscopy: 1) photoactivated localization microscopy (PALM); 2) fluorescence photoactivation localization microscopy (FPALM); and 3) stochastic optical reconstruction microscopy (STORM) [15]. In spite of the differing acronyms, they all share the ability to temporally separate spatially overlapping fluorescence signals.
PALM microscopy has the inherent advantage of being the easiest and least expensive form of home-built superresolution microscopy, requiring relatively straightforward modifications to an existing TIRF microscope mated with available free software for image acquisition and processing (https://code.google.com/p/quickpalm/). Depending on the sample, sensor, and processor, PALM can achieve resolutions of ~40 nm in real time [18], making this method ideal for studies of subcellular dynamics. When coupled with 2-P excitation, Vaziri et al. report sub 50 nm resolution in deep image planes of biological samples [19].
STORM provides an advantage over PALM due to improved fluorophore localization accuracy, a direct result of introducing astigmatism using a cylindrical lens placed between the imaging lens and the objective lens [7]. Huang and coworker found that use of a cylindrical lens in the light path did not markedly alter resolution in x and y compared to previous 2D STORM observations made without a cylindrical lens. By contrast, their accuracy in z using the 3D STORM system was approximately twice the localization accuracy measure in x and y [7].
Imaging with a Plane of Light
When imaging large samples such as live fish or fly embryos, developmental biologists have made significant advances in elucidating mechanisms of tissue morphogenesis using 2-P microscopy coupled with superresolution and laser scanning confocal microscopy. The trade-off for the spatial resolution available using a scanning microscope is slow acquisition rate. A single high quality scan can take >10 sec, and depending on optical slice thickness and axial step size, 100-400 slices may be needed to build a single z-stack, making high resolution 4D tracking of single cells in live specimens difficult. To better meet the need for rapid whole embryo imaging, Huisken et al. developed selective plane illumination microscopy (SPIM) using two objective lenses: one for illumination and one placed orthogonally to the illuminating light sheet used for detection of fluorescence emission [20,21]. This technique is also referred to as single plane illumination microscopy and light sheet microscopy, names that reflect the shape of the illuminating beam (Figure 5). In SPIM, optical sections are created using a cylindrical lens placed in front of a projection objective to focus the excitation laser beam into a thin light sheet of uniform thickness (2-6 um) across the field of view. Since the light sheet excites only a narrow plane of the sample at a given time, phototoxicity and photobleaching are minimized (Table 1). Another significant advantage, and perhaps most useful for studies of cellular and subcellular events in real time in toto, is that SPIM images can be collected very rapidly, often limited by only by the transfer rate of the CCD or CMOS sensors.
Figure 5.
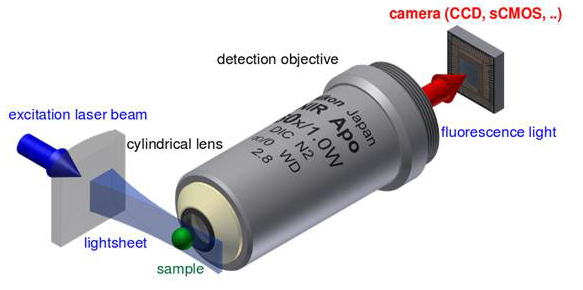
Principle setup of a light sheet fluorescence microscope using a single excitation laser (blue) and a single detection objective lens. ©Wikipedia/Jan Krieger
Table 1. Properties of single plane illumination microscopy.
| Optimized for speed and light sensitivity |
| Useful for whole live embryos and other large samples |
| Bleaching reduced compared to confocal and widefield microscopy |
| Scatter in illumination plane does not degrade image |
| Scattered fluorescence light contributes to image formation |
| Light sheet thickness determines lateral and axial resolution |
| Better resolution than 2-P and confocal microscopy at NA < ~0.8 [28] |
| 3D multiview stacks recorded by rotation of sample or using cameras placed at offset angles |
Embryos are prepared by embedding them in low melt agarose using capillary tubes as molds, and the sample is mounted as a partially extruded cylinder suspended in the optical axis of a temperature-controlled chamber filled with water or physiological buffer (Figure 6). Water immersion objectives with 1.0 NAs are used for live sample imaging to minimize refractive changes with biological specimens. Since the immersion lens, and often the illuminating objective, are fixed in position, the sample is orientated in the light sheet by rotation around the capillary long axis and movement in x, y, and z.
Figure 6.

Single plane illumination microscopy (SPIM) light path in the Zeiss Lightsheet Z.1 microscope (light sheet fluorescence microscopy: LSFM) using a whole zebrafish embryo. Illumination lenses (black), detection objective: W Plan-APHCHORMAT 20x/1.0 (white), lightsheet (blue). LSFM separates fluorescence excitation and detection into two separate light paths, with the axis of sheet illumination orthogonal to the detection axis (green). Laser light is focused into a thin light sheet by cylindrical lenses (not shown) upstream of the illuminating objectives. Only the in-focus light sheet excites fluorescence, significantly reducing phototoxicity. Use of two oppositional illuminating objectives reduces shadowing artifacts and increases acquisition speed. Fluorescence is collected on ultrafast CMOS cameras enabling z-stack acquisition speeds >100-fold faster than laser scanning confocal microscopy. Image courtesy of Carl Zeiss Microscopy GmbH. Used with permission.
A downside to illumination with a light sheet from only one side of the embryo is that image quality is hindered by light scatting and absorption, especially at deeper tissues, leading to formation of a shadowing effect. Shadow artifacts typically are visible as darker areas that are formed when light interacts with opaque structures in the sample. Reduction in shadowing artifacts can be reduced by rotation of the sample around an optical axis, but deep tissue scatting remains unchanged. To address this problem, Huisken and Stainier advanced their earlier SPIM system with the addition of a third lens, enabling the sample to be illuminated from both sides [22]. Termed multidirectional SPIM (mSPIM), resolution is improved in deep tissues since two separate light sheets enter from opposite sides of the embryo, effectively reducing the path length of excitation by 50 percent. Software registration of the oppositional beams allows reconstruction of multiple views in real time, enabling stunning 3D and 4D images of embryogenesis. Shadowing effects in the sample are reduced further by pivoting the light sheet several degrees during acquisitions [22].
A commercial selective plane illumination system is available from Applied Scientific Instrumentation (http://asiimaging.com/products/light-sheet-microscopy/selective-plane-illumination-microscopy-ispimdispim) and sold in three configurations: 1) The fixed sheet system; 2) single–sided; and 3) double-sided selective plane illumination microscopy systems. The fixed sheet system is the least expensive but is limited in that the light sheet is stationary. The single-sided system involves a scanned light sheet from one side, and the double-sided system involves scanned light sheets entering from both sides of the specimen. This flexible design stems from collaborations with Shroff and coworkers at the Section on High Resolution Optical Imaging, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health [23]. These light sheet configurations require precise alignment of objectives (either stationary or piezo-positioned) mounted to a steel housing that bolts onto an inverted microscope base. The Applied Scientific Instrumentation SPIMs offer considerable flexibility for relatively low cost and seem most appropriate for laboratories with considerable microscopy expertise.
In a recent hands-on trial, we were able to evaluate the Zeiss Lightsheet Z.1 microscope, a commercial mSPIM system (Figure 6), for imaging live zebrafish embryos. Using a full frame CMOS sensor and a water immersion objective, we obtained high contrast images with resolutions comparable to those of embryos obtained using our Zeiss LSM 510 LSCM (Figure 7). The parfocal length of the Zeiss 20x/1.0 NA W Plan-APOCHROMAT used in the Lightsheet Z1 is 75mm, or 30 mm longer than the standard 45 mm length for the Plan-APOCHROMAT objectives mounted on our Zeiss LSM 510 confocal microscope. So it was not possible for us to swap objectives on both microscopes and perform direct image comparisons using a single lens. For comparison, we used a high quality 20x Plan APOCHROMAT / 0.8 NA dry objective on our Zeiss LSM 510 LSCM. For typical whole-embryo imaging of zebrafish, we find that publication-quality full frame (512 x 512) scans (two frame averages) take ~15 sec. In contrast, comparable quality images using the Lightsheet Z.1 microscope were obtained in ~120 to 150 ms, and we were able to acquire 400 optical slices/min for 4D reconstructions of live zebrafish embryos over a period of 3 to 4 hours, yielding a ~100-fold increase in z-stack acquisition speed using the Lightsheet Z.1 microscope compared to the LSM 510 (Figure 7). Based on our limited access to the Z.1 microscope, we are quite confident light sheet microscopy is the best available method for single cell tracking in live embryos over time (hours to days), enabling better understanding of cellular and subcellular morphogenesis during development.
Figure 7.

Comparison of LSM 510 laser scanning confocal (A, C) and Lightsheet Z.1 (B, D) images of zebrafish embryos stained with nuclear localization protein tags (red) and fibronectin (green). Optical slices of live embryos imaged at depths of 15 um (A, B) and 50 um (C, D) from the dorsal surface. The signal to noise is markedly improved in deep tissue images using lightsheet microscopy (D) compared to laser scanning confocal microscopy (C). Lights sheet images obtained using a 20x W Plan APOCHROMAT /1.0 NA water immersion objective. LSM 510 confocal images obtained at 512 x 512 pixel resolution using a 20x Plan–APOCHROMAT/0.8 NA dry objective with 2 line averaging. Acquisition time for images: A, C (15 sec), for B, D (125 ms). Scale bar: A, B: 50 um, C, D 100 um.
Conclusion and Perspective
One of the greatest challenges now facing microscopists is data processing and management. Rather than developing massive storage and computing systems, Schmid et al. engineered a four-lens SPIM system with ultrafast cameras and software capable of real-time image processing for generation of radial maximum intensity projections [24]. Rapid movement of the specimen past the focused light sheet enables fluorescence emission of every cell in a zebrafish embryo to be recorded on a CMOS sensor in <10s, or >50-fold faster than conventional laser scanning confocal microscopy [24].
Krzic et al. developed a four-lens, multview SPIM (MuVi SPIM) that allows views of the front and back of an embryo without moving the sample [25], a process that eliminates the time required for sample rotation around the optical axis. Light sheets aligned using fluorescent beads illuminate both sides of the sample, and two detection objectives gather emitted fluorescence. This arrangement increases the speed of acquisition and improves resolution in deep tissue. To eliminate computational bottlenecks associated with 4D acquisitions, which can easily exceed100GB, these authors used dedicated software for image acquisition and to fuse data in real time [25].
Taking SPIM one step further, Friedrich et al. showed that SPIM can be combined with STED superresolution (STED-SPIM), demonstrating for the first time that superresolution can be achieved in a light sheet [26]. Perhaps the most innovative breakthrough comes from the work of Zanacchi et al., who used 2-P excitation of photoactivatable individual molecules to obtain superresolution in the single plane illumination microscope (IML-SPIM) [27]. By combining the deep tissue penetration of 2-P excitation of single PA-fluorophores in a light sheet, these authors have built a microscopy system devoid of any Achilles’ heel. Biologists no longer need to compromise resolution for speed when imaging subcellular events in whole embryos.
Abbreviations
- 1-P
1 photon
- 2-P
2-photon excitation microscopy
- LSCM
laser scanning confocal microscopy
- NA
numerical aperture
- SPIM
selective plane illumination microscopy
- IML-SPIM
individual molecule localization single plane illumination microscope
- mSPIM
multidirectional SPIM
- MuVi SPIM
multview SPIM
- STED
Stimulated Emission Depletion Microscopy
- CMOS
complementary metal-oxide-semiconductor
- CCD
charge-coupled device
- TIRF
total internal reflection fluorescence microscope
- PALM
photoactivated localization microscopy
- FPALM
fluorescence photoactivation localization microscopy
- STORM
stochastic optical reconstruction microscopy
- PSF
point spread function
- GFP
green fluorescence protein
References
- Amos WB, White JG. How the confocal laser scanning microscope entered biological research. Biol Cell. 2003;95(6):335–342. doi: 10.1016/s0248-4900(03)00078-9. [DOI] [PubMed] [Google Scholar]
- Cole RW, Jinadasa T, Brown CM. Measuring and interpreting point spread functions to determine confocal microscope resolution and ensure quality control. Nat Protoc. 2011;6(12):1929–1941. doi: 10.1038/nprot.2011.407. [DOI] [PubMed] [Google Scholar]
- Göppert-Mayer M. Über Elementarakte mit zwei Quantensprüngen. Annalen der Physik. 1931;9:273–294. German. [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248(6):73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Sako Y, Sekihata A, Yanagisawa Y, Yamamoto M, Shimada Y, Ozaki K. et al. Comparison of two-photon excitation laser scanning microscopy with UV-confocal laser scanning microscopy in three-dimensional calcium imaging using the fluorescence indicator Indo-1. J Microsc. 1997;185(Pt 1):9–20. doi: 10.1046/j.1365-2818.1997.1480707.x. [DOI] [PubMed] [Google Scholar]
- Hell SW, Stelzer EHK. Fundamental improvement of resolution with 4Pi-confocal fluorescence microscope using 2-photon excitation. Opt Commun. 1992;93:277–282. [Google Scholar]
- Huang B, Babcock H, Zhuang X. Breaking the Diffraction Barrier: Super-Resolution Imaging of Cells. Cell. 2010;143(7):1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith CG, Galbraith JA. Super-resolution microscopy at a glance. J Cell Sci. 2011;124(Pt 10):1607–1611. doi: 10.1242/jcs.080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19(11):780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Zanella R, Zanghirati G, Cavicchioli R, Zanni L, Boccacci P, Bertero M. et al. Towards real-time image deconvolution: application to confocal and STED microscopy. Sci Rep. 2013;3:2523. doi: 10.1038/srep02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki KT, Ding JB, Sabatini BL. Live-cell superresolution imaging by pulsed STED two-photon excitation microscopy. Biophys J. 2013;104(4):770–777. doi: 10.1016/j.bpj.2012.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost A, Heintzmann R. Superresolution Multidimensional Imaging with Structured Illumination Microscopy. Annu Rev Mater Res. 2013;43:261–282. [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263(5148):802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt A, Tsien RY. Improved green fluorescence. Nature. 1995;373(6516):663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Patterson G, Davidson M, Manley S, Lippincott-Schwart J. Superresolution Imaging using Single-Molecule Localization. Annu Rev Phys Chem. 2010;61:345–367. doi: 10.1146/annurev.physchem.012809.103444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MW, Campbell RE. Engineered fluorescent proteins: innovations and applications. Nat Methods. 2009;6(10):713–717. doi: 10.1038/nmeth1009-713. [DOI] [PubMed] [Google Scholar]
- Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D, Cohen AE. Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat Methods. 2011;9(1):90–95. doi: 10.1038/nmeth.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques R, Griffiths C, Hesper Rego E, Mhlanga MM. PALM and STORM: unlocking live-cell super-resolution. Biopolymers. 2011;95(5):322–331. doi: 10.1002/bip.21586. [DOI] [PubMed] [Google Scholar]
- Vaziri A, Tang J, Shroff H, Shank CV. Multilayer three-dimensional super resolution imaging of thick biological samples. Proc Natl Acad Sci USA. 2008;105(51):20221–20226. doi: 10.1073/pnas.0810636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J, Stainier DY. Selective plane illumination microscopy techniques in developmental biology. Development. 2009;136(12):1963–1975. doi: 10.1242/dev.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EHK. Optical Sectioning Deep Inside Live Embryos by Selective Plane Illumination Microscopy. Science. 2004;305(5686):1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- Huisken J, Stainier DYR. Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM) Opt Lett. 2007;32(17):2608–2610. doi: 10.1364/ol.32.002608. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wawrzusin P, Senseney J, Fischer RS, Christensen R, Santella A. et al. Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nat Biotechnol. 2013;31(11):1032–1038. doi: 10.1038/nbt.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid B, Shah G, Scherf N, Weber M, Thierbach K, Pérez Campos C. et al. High-speed panoramic light-sheet microscopy reveals global endodermal cell dynamics. Nat Commun. 2013;4:2207. doi: 10.1038/ncomms3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzic U, Gunther S, Saunders TE, Streichan SJ, Hufnagel L. Multiview light-sheet microscope for rapid in toto imaging. Nat Methods. 2012;9(7):730–733. doi: 10.1038/nmeth.2064. [DOI] [PubMed] [Google Scholar]
- Friedrich M, Gan Q, Ermolayev V, Harms GS. STED-SPIM: Stimulated Emission Depletion Improves Sheet Illumination Microscopy Resolution. Biophys J. 2011;100(8):L43–L45. doi: 10.1016/j.bpj.2010.12.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanacchi FC, Lavagnino Z, Faretta M, Furia L, Diaspro A. Light-sheet confined super-resolution using two-photon photoactivation. PLoS One. 2013;8(7):e67667. doi: 10.1371/journal.pone.0067667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbrecht CJ, Stelzer EH. Resolution enhancement in a light-sheet-based microscope (SPIM) Opt Lett. 2006;31(10):1477–1490. doi: 10.1364/ol.31.001477. [DOI] [PubMed] [Google Scholar]