

Abstract
The differentiation of bone marrow–derived progenitors into monocytes, tissue macrophages and some dendritic cell (DC) subtypes requires the growth factor CSF1 and its receptor, CSF1R. Langerhans cells (LCs) and microglia develop from embryonic myeloid precursors that populate the epidermis and central nervous system (CNS) before birth. Notably, LCs and microglia are present in CSF1-deficient mice but absent from CSF1R-deficient mice. Here we investigated whether an alternative CSF1R ligand, interleukin 34 (IL-34), is responsible for this discrepancy. Through the use of IL-34-deficient (Il34LacZ/LacZ) reporter mice, we found that keratinocytes and neurons were the main sources of IL-34. Il34LacZ/LacZ mice selectively lacked LCs and microglia and responded poorly to skin antigens and viral infection of the CNS. Thus, IL-34 specifically directs differentiation of myeloid cells in the skin epidermis and CNS.
INTRUCTION
The mononuclear phagocyte system encompasses a group of myeloid cells with crucial functions in maintaining the homeostasis of multiple tissues as well as promoting inflammatory and repair responses to microbial, chemical and physical insults1–5. The development of many mononuclear phagocytes, including blood monocytes and tissue macrophages, requires colony-stimulating factor 1 (CSF1; also known as macrophage colony-stimulating factor (M-CSF)6,7. CSF1 binds to and activates the tyrosine kinase receptor CSF1R (also known as M-CSFR or CD115) on bone marrow progenitors, which results in their proliferation and differentiation into monocytes and macrophages. The CSF-1–CSF1R ligand-receptor pair also promotes the differentiation of multinucleated osteoclasts8, which are created by the fusion of mononuclear myeloid progenitors and participate in the resorption and remodeling of bone. Moreover, signaling via CSF1R contributes to the development of two dendritic cell (DC) subsets: Langerhans cells (LCs), which are the resident DCs of the epidermis; and monocyte-derived DCs, which constitute a major DC population in inflamed tissues1–5,9.
Although monocytes, tissue macrophages, osteoclasts, monocyte-derived DCs and LCs express CSF1R, their dependence on CSF1 varies considerably10. This heterogeneity is reflected in the disparate phenotypes of osteopetrotic mutant mice that lack CSF1 because of an inactivating mutation (op/op) in the gene encoding CSF1 (Csf1op/op mice)11 and mice deficient in CSF1R (Csf1r−/− mice)12. Both Csf1op/op and Csf1r−/− mice lack osteoclasts and thus are osteopetrotic. However, Csf1r−/− mice have more severe defects in several mononuclear phagocyte subsets than are Csf1op/op mice. Notably, microglia, which are the resident macrophages of the central nervous system (CNS)13, are present in Csf1op/op mice but absent from Csf1r−/− mice14,15. Similarly, LCs are present in Csf1op/op mice but absent from Csf1r−/− mice14,16. Such discrepancies suggest the existence of an alternative ligand for CSF1R that can partially compensate for the absence of CSF1 in Csf1op/op mice. Relevant to this, interleukin 34 (IL-34) has been identified as the second functional ligand for CSF1R17,18. IL-34 shares little aminoacid homology with CSF1 but can bind to and activate CSF1R, at least in vitro19,20. Furthermore, transgenic expression of IL-34 ‘rescues’ the bone, osteoclast and tissue macrophage defects of Csf1op/op mice19. Although IL-34 can compensate for the lack of CSF1, the expression patterns of Il34 mRNA and Csf1 mRNA are spatially and temporally distinct19, which suggests that they have complementary rather than redundant roles in CSF1R activation in vivo.
In this study, we generated IL-34-deficient reporter mice in which the Il34 coding sequence was replaced by the gene encoding β-galactosidase (Il34LacZ/LacZ mice). Analysis of these mice showed that IL-34 was produced mainly by keratinocytes and neurons. Consistent with that distribution pattern, we found that Il34LacZ/LacZ mice had a selective defect in the generation of LCs and microglia. The lack of LCs resulted in diminished contact hypersensitivity (CHS), whereas the deficiency in microglia resulted in greater susceptibility to viral infection of the CNS. In contrast to its effect on LCs and microglia, targeted deletion of Il34 had little effect on other myeloid-cell compartments, except for slightly fewer CD11b+ DCs in the lung. Blood monocytes, other tissue macrophages and DC subsets were largely unaffected, reflective of the predominant role of other growth factors, such as CSF1 and Flt3, for their maturation.
LCs and microglia are unique among myeloid cells in their development; they arise from embryonic myeloid progenitors that populate the skin and the CNS, respectively, before birth. LCs and microglia proliferate before birth and during the first days of postnatal life and then self-renew with a slow turnover rate13,16,21–23. Our results show that IL-34 promotes a tissue-specific differentiation pathway that is selectively required for myeloid cells that populate the epidermis and CNS.
RESULTS
IL-34 expression mainly in the skin and CNS
We generated IL-34-mutant mice from a genetically targeted embryonic stem cell line designed to create a null, lacZ reporter allele by Cre-mediated deletion (Supplementary Fig. 1). We bred mice carrying the targeted Il34 gene to transgenic mice expressing Cre driven bya ubiquitous cytomegalovirus promoter (CMV-Cre) to produce Il34LacZ/+ mice in which exons 3–5 were deleted and replaced by an internal ribosome entry site–lacZ cassette. We intercrossed offspring to generate homozygous Il34LacZ/LacZ mice. To identify β-galactosidase (LacZ)-producing cells, we stained frozen tissue sections of Il34LacZ/+ mice with the β-galactosidase substrate X-gal. We found strong and punctate X-gal staining in the skin, brain, kidney and testes (Fig. 1). At the cellular level, X-gal staining was evident in keratinocytes, hair follicles, neurons, proximal renal tubule cells and seminiferous tubule germ cells. In the brain, we detected X-gal staining in the cerebral cortex and hippocampus but not in the cerebellum (Supplementary Fig. 2a–d). We did not find X-gal staining in the meninges, but it was present in a fraction of ependymal cells lining the ventricular system and those of the choroid plexus (Supplementary Fig. 2a–d), which suggested that IL-34 was secreted into the cerebrospinal fluid. X-gal staining was weak or undetectable in other nonlymphoid organs as well as spleen and lymph nodes (Fig. 1 and data not shown). X-gal staining in the CNS and skin substantially overlapped the pattern of Il34 mRNA expression in the Biology Gene Portal System (BioGPS) microarray data set (http://biogps.org/#goto=genereport&id=76527) and its expression reported before19. In contrast, X-gal staining did not overlap the pattern of Csf1 mRNA expression (http://biogps.org/#goto=genereport&id=12977), except in bones. These data indicated a nonredundant function for IL-34 in the CNS and skin, but a possibly redundant or dispensable role for IL-34 in other tissues in which CSF-1 is produced. Il34 mRNA was almost undetectable in skin and brain of homozygous Il34LacZ/LacZ mice (Supplementary Fig. 2e, f), which confirmed that Il34LacZ/LacZ mice completely lacked IL-34.
Figure 1.

Tissue specific expression of IL-34. Staining for X-gal (blue) in skin (a), brain cerebral cortex (b), testis (c), kidney tubules (d), spleen (e) and lymph nodes (f) from Il34LacZ/LacZ mice. Tissues from wild-type mice did not show any X-gal staining (data not shown). HF, hair follicles; GL, kidney glomerulus. Arrows indicate X-gal+ cells. Original magnification, ×400 (a; scale bar, 50 μm) or ×600 (b–f; scale bars, 33 μm). Data are representative of 2 experiments with three mice (25 high-power fields per tissue per mouse).
IL-34 deficiency impairs epidermal LCs
Because we found that keratinocytes produced IL-34 (Fig. 1a), and LC development depends on CSF1R but not CSF1 (ref. 16), we hypothesized that IL-34 may be essential for the development of LCs. To test our hypothesis, we isolated cells from the ears of wild-type and Il34LacZ/LacZ mice and analyzed their content of LCs and dermal DCs by flow cytometry. We identified LCs as CD11c+ major histocompatibility complex class II (MHCII)+ cells that express CD207 (langerin) but not CD103, which is a marker of some dermal DCs2–4,22. We categorized dermal DCs into two subsets. A larger subset included CD11c+MHCII+ cells that lacked CD103 and CD207 (refs. 2–4,22). These cells also expressed the common myeloid marker CD11b (data not shown). A smaller subset included CD11c+MHCII+ cells that expressed both CD103 and CD207 (refs. 24–28). We found considerably fewer CD11c+ MHCII+ CD103− CD207+ LCs in the skin of Il34LacZ/LacZ mice than in the skin of wild-type mice (Fig. 2a, b). In contrast, CD103+CD207+ and CD103− CD207−dermal DCs were equally abundant in Il34LacZ/LacZ and wild-type mice (Fig. 2a, b).
Figure 2.

IL-34 deficiency leads to the absence of LCs. (a, b) Flow cytometry of single-cell suspensions of wild-type (WT) and Il34LacZ/LacZ (KO) skin digests stained for CD45, CD11c, MHCII, CD103 and intracellular CD207 (langerin). (a) Gating of CD11c+MHCII+ cells among live CD45+ cells (left), followed by further discrimination (right) of LCs (CD103−CD207+) and dermal DCs (CD103+CD207+ and CD103−CD207−). (b) Frequency of total CD11c+MHCII+ cells (among live CD45+ cells), LCs and dermal DCs (among CD11c+MHCII+ cells) in the skin of wild-type and Il34LacZ/LacZ mice (n = 6 per group). *P < 0.0001, (unpaired Student’s t-test). (c–e) Immunohistochemical analysis of langerin (brown; c), MHCII (brown; d) and CD11b (brown; e) in the skin of wild-type and Il34LacZ/LacZ mice. Arrows indicate positive cells in the epidermis. Original magnification, ×400 (scale bars, 50 μm). (f) Flow cytometry of single-cell suspensions of wild-type and Il34LacZ/LacZ skin-draining lymph nodes stained for CD11c and MHCII to identify DC subsets (left), followed by further analysis (right) of CD11c+MHCIIhi cells (I) to determine the frequency of migrating LCs and CD103+ dermal DCs; of CD11chiMCHIIint cells (II) to determine the frequency of CD8α+ DCs; and of CD11cloMHCIIlo cells (III) to determine the frequency of pDCs marked by expression of Siglec-H. (g) Frequency of CD11c+MCHII+ cells in gates I, II, and III. (h) Frequency of CD103−CD207+ LCs, CD103+CD207+ dermal DCs (in gate I), CD8α+ DCs (in gate II) and pDCs (in gate III) in the skin-draining lymph nodes of wild-type and Il34LacZ/LacZ mice (n = 6 or 7 per group). (i) Immunohistochemistry analysis of langerinin cervical lymph nodes of wild-type and Il34LacZ/LacZ mice. Original magnification, ×600 (scale bars, 33 μm). Numbers adjacent to outlined areas (a, f) indicate percent cells in each gate. Each symbol (b, g, h) represents an individual mouse; small horizontal lines indicate the mean. *P < 0.0001 (unpaired Student’s t-test). Data represent at least two independent experiments.
To confirm the defect in LCs and the maintenance of dermal DCs in the skin of Il34LacZ/LacZ mice, we isolated epidermal sheets from the ears of wild-type and Il34LacZ/LacZ mice and looked for LCs identified as CD11c+ and MHCII+ cells. The epidermis of Il34LacZ/LacZ mice had considerably fewer CD11c+MHCII+ cells than did the epidermis of wild-type mice, as shown by both confocal microscopy (Supplementary Fig. 3a, b) and flow cytometry (Supplementary Fig. 3c, d). We also examined dermal DCs, identified as CD11c+MHCII+ cells in the dermal sheets of the ears. Wild-type and Il34LacZ/LacZ mice had a similar frequency of dermal CD11c+MHCII+ cells (Supplementary Fig. 3e, f). Dermal DC subsets, identified as CD103−CD11b+ and CD103+CD11b− cells, were present in equal abundance in wild-type and Il34LacZ/LacZ mice (Supplementary Fig. 3g–j). We also evaluated LCs and dermal DCs in the body-wall skin by assessing the expression of CD207, MHCII and CD11b by immunohistochemistry. Il34LacZ/LacZ mice had almost no cells that expressed CD207, MHCII and CD11b in the epidermis, whereas these cells were present in the dermis (Fig. 2c–e). Wild-type mice had cells that expressed CD207, MHCII and CD11b in both the epidermis and dermis.
Because LCs migrate from the skin to the lymph nodes4,22, we analyzed skin-draining lymph nodes by flow cytometry. To distinguish among DC subsets, we costained lymph node cells for CD11c and MHCII (Fig. 2f–h). CD11c+MHCIIhi cells (gate I) include migrating DCs; CD11c+MHCIIint cells (gate II) include CD8α+ DCs; CD11c+MHCIIlo cells (gate III) include plasmacytoid DCs. To further distinguish between LCs and dermal DCs, we also stained cells for CD103 and CD207, as described above. We noted considerably fewer migrating CD103−CD207+ LCs in Il34LacZ/LacZ mice than in wild-type mice (Fig. 2f–h). In contrast, migrating dermal DCs, including CD103+CD207+ and CD103−CD207−CD11b+ subsets, were present in equal abundance in the lymph nodes of Il34LacZ/LacZ and wild-type mice (Fig. 2f–h). IL-34 deficiency had no effect on lymph node CD8α+ DCs or Siglec-H+ plasmacytoid DCs (Fig. 2f). Immunohistochemical analysis of lymph nodes showed considerably fewer CD207 cells in Il34LacZ/LacZ mice than in wild-type mice (Fig. 2i). Although some dermal DCs also express CD207 (refs. 24–28), the flow cytometry analysis indicated that the lower abundance of CD207+ cells in the lymph nodes solely reflected a lower abundance of migrating epidermal LCs. Moreover, Il34LacZ/LacZ mice and wild-type mice had similar numbers of CD207+ cells in the spleen and thymus (Supplementary Fig. 3k), consistent with the idea that these cells are not derived from epidermal LCs24,25.
IL-34 deficiency affects T cell priming in CHS
LCs are antigen-presenting cells of the epidermis29 that prime T cell responses to skin allergens30–33. Given that, we assessed the effect of IL-34 and LC deficiency on CHS. We sensitized wild-type and Il34LacZ/LacZ mice to the hapten DNFB by cutaneous application of DNFB to the shaved abdomen on day 0. After 5 d, we challenged mice by painting the left ear with DNFB and the right ear with vehicle only, as a control. We calculated ear swelling for each mouse by subtracting the thickness of the vehicle-treated ear from that of the DNFB-treated ear. Ear swelling in wild-type mice peaked between days 1 and 2 after challenge and decreased at day 3 (Fig. 3a). At all times after challenge, Il34LacZ/LacZ mice were less sensitive than were wild-type mice to DNFB-mediated CHS.
Figure 3.

IL-34 deficiency leads to diminished DNFB-mediated CHS. (a) Ear swelling in wild-type mice (n = 6) and Il34LacZ/LacZ mice (n = 7) at 24, 48 and 72 h after challenge with DNFB. *P = 0.015, **P < 0.0001 and ***P = 0.0034 (unpaired Student’s t-test). (b) Flow cytometry of cells from the ear-draining lymph nodes of wild-type and Il34LacZ/LacZ mice after painting of ears with CellTracker green. Left panel, DCs were separated as CD11cintCD40int cells (population I), CD11chiCD40hi cells (population II) and CD11chiCD40lo cells (population III). Right, flow cytometry of cells in the populations gated at left (black lines, wild-type; gray lines, Il34LacZ/LacZ). Numbers above bracketed lines (colors match line colors) indicate percent cells positive for CellTracker green (CTG). (c) Flow cytometry of MHCIIhiCD11c+CTG+ cells at 48 h after the ear painting in b, to assess the migration of LCs and dermal DCs. Numbers adjacent to outlined areas indicate percent CD103+CD207+ cells (bottom) or CD103+CD207+ cells (top). (d) IFN-γ in skin-draining (inguinal) lymph nodes isolated from wild-type and Il34LacZ/LacZ mice without sensitization (naive) or 6 d after DNFB sensitization (DNFB). Lymph node cells (2 × 105 cells/well) were cultured for 24 or 48 h in the presence of plate-bound anti-CD3 and cytometric bead array analysis of culture supernatants. *P = 0.04 (unpaired Student’s t-test). Data are representative of three independent experiments (a), represent two independent experiments with two mice per group (b, c) or represent one of two independent experiments with three mice per group in each (d).
Because LCs deliver skin antigens to draining lymph nodes34, we assessed whether the diminished CHS in Il34LacZ/LacZ mice depended on defective antigen delivery. We painted the ears of wild-type and Il34LacZ/LacZ mice with the dye CellTracker Green and monitored CellTracker+ cells in the draining cervical lymph nodes 40–48 h later. To distinguish between skin-derived DCs and resident DCs, we costained lymph node cells for CD40 and CD11c35. The frequency of CellTracker+ cells among CD40hiCD11chi DCs was much lower in Il34LacZ/LacZ mice than wild-type mice (Fig. 3b, population II), which suggested that CD40hiCD11chi DCs included migrating LCs. In contrast, the frequency of CellTracker+ cells among CD40intCD11cint cells was similar in the draining lymph nodes of wild-type and Il34LacZ/LacZ mice (Fig. 3b, population I), which suggested that the CD40intCD11cint cells corresponded to migrating dermal DCs. Resident DCs, identified as CD40loCD11chi, were negative for CellTracker (Fig. 3b, population III) and were present in equal abundance in wild-type and Il34LacZ/LacZ mice. Thus, deficiency of IL-34 resulted in selectively fewer CellTracker+ cells of epidermal origin in the draining lymph nodes. To confirm those results, we evaluated migrating LCs and dermal DCs in the lymph nodes after painting with CellTracker Green, using CD207 and CD103 as markers. The draining lymph nodes of Il34LacZ/LacZ mice had considerably fewer CellTracker+CD207+CD103− LCs than did those of wild-type mice, whereas we observed a similar proportion of CellTracker+CD207+CD103+ dermal DCs in Il34LacZ/LacZ and wild-type mice (Fig. 3c).
CD8+ and CD4+ T cells that produce interferon-γ (IFN-γ) mediate CHS to DNFB36. Thus, we examined the effect of deficiency in IL-34 and LCs on the priming of T cells in draining lymph nodes after sensitization with DNFB. We collected inguinal lymph nodes 6 d after sensitizing mice with DNFB, then stimulated the lymph nodes with plate-bound antibody to CD3 (anti-CD3) and assessed IFN-γ production 24 and 48 h later. Lymph node cells from Il34LacZ/LacZ mice produced less IFN-γ than did cells isolated from wild-type mice (Fig. 3d). Lower IFN-γ production was paralleled with fewer total cells in the lymph nodes of Il34LacZ/LacZ mice than in the lymph nodes of wild-type mice, consistent with the lower inflammation (Supplementary Fig. 4a). The lack of IL-34, however, had no effect on the normal development of CD8+ or CD4+ T cells in the thymus or the number of CD8+ or CD4+ T cells in lymph nodes of naive mice (Supplementary Fig. 4b). Together these results demonstrated that the paucity of epidermal LCs due to lack of IL-34 impaired antigen delivery and priming of allergen-specific T cells in local draining lymph nodes.
It has been shown that LCs are necessary and sufficient for direct antigen presentation and the generation of an antigen-specific response by the TH17 subset of helper T cells in a model of infection of the skin with Candida albicans37. To see whether IL-34 and LC deficiency affected the skin response to C. albicans, we sensitized Il34LacZ/LacZ and wild-type mice by applying C. albicans to shaved dorsal skin. One week after sensitization, we challenged mice with C. albicans in the footpad and measured swelling the next day. Il34LacZ/LacZ mice had modestly but significantly (P = 0.0237) less swelling than did wild-type mice (Supplementary Fig. 4c). We found fewer cells in the draining lymph nodes of Il34LacZ/LacZ mice than in those of wild-type mice (Supplementary Fig. 4d), which provided further evidence of an attenuated inflammatory skin response to C. albicans. This result confirmed the CHS data and indicated that the lack of LCs did have at least some effect on delayed-type hypersensitivity responses to pathogens in the skin.
IL-34 deficiency impairs microglia cell numbers
Because we found that neurons produced IL-34 (Fig. 1b and Supplementary Fig. 2a, c) and microglia are present in Csf1op/op mice but absent from Csf1r−/− mice14,15, we hypothesized that IL-34 contributes to the development of microglia. We purified myeloid cells from the brains of Il34LacZ/LacZ and wild-type mice with anti-CD11b magnetic beads and identified microglia (CD11b+CD45int) by flow cytometry13. The number of CD11b+CD45int cells isolated from brains of Il34LacZ/LacZ mice was <20% of that in wild-type mice (Fig. 4a). Staining of CNS sections with IBA-1, a marker of myeloid cells, confirmed that the cerebral cortex, corpus callosum, hippocampus and basal ganglia of Il34LacZ/LacZ mice had considerably fewer microglia than did those of wild-type mice (Fig. 4b–e and Table 1). Notably, the cerebellum of Il34LacZ/LacZ mice and wild-type mice had a similar abundance of microglia (Fig. 4f and Table 1), perhaps because of local production of CSF1. We purified residual microglia of Il34LacZ/LacZ mice and stimulated them in vitro with ligands of Toll-like receptors (TLRs). Purified microglia from Il34LacZ/LacZ mice produced inflammatory cytokines in amounts similar to those produced by equal numbers of wild-type microglia (Supplementary Fig. 5a), which indicated that IL-34 deficiency affected mainly development but not the ability of microglia to produce inflammatory cytokines.
Figure 4.

Impaired microglia development in IL-34-deficient mice. (a) Total CD45intCD11b+ microglial cells in the brains of wild-type and Il34LacZ/LacZ mice (n = 4 per group). Each symbol represents an individual mouse; small horizontal lines indicate the mean. *P = 0.0004 (unpaired Student’s t-test). (b–f) Immunohistochemistry of wild-type and Il34LacZ/LacZ mouse cerebral cortex (b), corpus callosum (c), hippocampus (d), striatum (e) and cerebellum (f) in fixed brain tissues stained with the myeloid marker IBA-1. Original magnification, ×200 (scale bars, 100 μm). Data are representative of three experiments (a) or represent two independent experiments (b–f).
Table 1.
Numbers of microglial cells in various regions of the brains of wild-type and Il34LacZ/LacZ mice
| Region | Cell count | P | |
|---|---|---|---|
| WT | KO | ||
| Cerebral cortex | 10.3 (2.6) | 3.7 (1.8) | <0.0001 |
| Corpus callosum | 5.6 (1.8) | 2.8 (1.5) | <0.001 |
| Hippocampus | 7.3 (2.9) | 3.0 (1.7) | <0.001 |
| Striatum | 7.4 (2.4) | 4.2 (2.2) | <0.01 |
| Cerebellum | 3.5 (1.5) | 2.9 (1.3) | 0.055 |
Quantification of microglial cells in wild-type and Il34LacZ/LacZ mice cell, assessed by microscopy (×400) of paired regions of the brain stained with anti-IBA-1. Data are representative of one experiment with at least 25 high-power fields per region per mouse (mean and s.d.; 3 mice/group).
IL-34 deficiency affects antiviral resistance in the CNS
Microglia are thought to protect the CNS from viral infection through several mechanisms, including the production of cytokines that inhibit viral replication, the phagocytosis of virus-infected and dying neurons and the induction of neuronal repair and homeostasis13. We examined the effect of deficiency in IL-34 and microglia in vivo during infection with a neurotropic virus. We infected wild-type and Il34LacZ/LacZ mice with West Nile virus (WNV) via an intracranial route and monitored survival. Infection with wild-type virulent WNV resulted in rapid mortality in both Il34LacZ/LacZ mice and wild-type mice, as 100% of both groups of mice succumbed to infection by 8 d after infection (Fig. 5a). In contrast, when we infected mice with an attenuated WNV strain (WNV-E218A), we observed more death of Il34LacZ/LacZ mice than of wild-type mice by 10–12 d after infection. This result suggested that a lack of IL-34 and consequent lower abundance of microglia impaired CNS defenses against WNV infection. Notably, we detected similar viral burdens in various regions of the brain of wild-type and Il34LacZ/LacZ mice on both day 3 and day 6 after infection (Supplementary Fig. 5b), which suggested that the protective effect of microglia may have been due to their ability to limit neuronal death and/or promote CNS repair rather than their ability to directly inhibit viral replication.
Figure 5.

IL-34-deficient mice are more susceptible to infection after intracranial inoculation of attenuated WNV. (a) Survival of 9-week-old wild-type mice (n = 27) and Il34LacZ/LacZ mice (n = 18) inoculated via an intracranial route with wild-type WNV (10 plaque-forming units) or attenuated WNV (WNV-E218A; 1 × 105 plaque-forming units) and monitored for 21 d. *P = 0.0155 (Gehan-Breslow-Wilcoxon test). (b–e) Microscopy of fixed, frozen sections of the cerebral cortex (b), hippocampus (c), brainstem (d) and cerebellum (e) of wild-type and Il34LacZ/LacZ mice infected by intracranial injection of attenuated WNV (1 × 105 PFU), assessed at day 6 after infection by staining for the neuronal antigen MAP-2 (green), TUNEL analysis (red) and nuclear staining with ToPro-3 (blue). Arrows indicate TUNEL+ neurons. Original magnification, ×63 (scale bars, 20 μm). (f) Quantification of TUNEL+ cells in b–e (n = 4 mice; three high-power fields (HPF) per brain region per mouse). Each symbol represents an individual mouse; small horizontal lines indicate the mean. *P = 0.0268, **P < 0.0001, ***P = 0.0005 and **** P = 0.0077 (unpaired Student’s t-test). Data represent three independent experiments (a) or one experiment with four to six mice per group (b–f).
To test that hypothesis, we assessed neuronal apoptosis in various regions of the brains of wild-type and Il34LacZ/LacZ mice on day 6 after infection by TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling) assay. Il34LacZ/LacZ mice had more neuronal apoptosis in all brain areas analyzed (Fig. 5b–f). Analysis of WNV-infected brains by flow cytometry showed that Il34LacZ/LacZ mice had fewer microglia than did wild-type mice (Supplementary Fig. 5c). Thus, although WNV infection causes inflammation and the recruitment of bone marrow–derived myeloid cells, such as inflammatory monocytes38, such recruitment was not sufficient to compensate for the defect in resident microglia in Il34LacZ/LacZ mice. WNV-infected Il34LacZ/LacZ and wild-type mice had a similar abundance of brain macrophages and total CD4+ T cells and CD8+ T cells, as well as WNV-specific CD8+ T cells (Supplementary 5c–e), which further suggested that susceptibility of Il34LacZ/LacZ mice to WNV was due to insufficient resident microglia and a consequent lack of the protective and trophic factors provided by these cells.
Limited effect of IL-34 on other macrophage and DC subsets
In vitro studies have suggested that IL-34 can promote differentiation of bone marrow–derived macrophages (BMDMs)19. Furthermore, transgenic expression of IL-34 ‘rescues’ the bone, osteoclast and tissue macrophage defects of Csf1op/op mice19. Whether IL-34 is necessary for the development of macrophages in vivo in tissues other than the skin and the brain remains unknown. To assess this, we examined whether IL-34 deficiency affected myeloid cell development in lymphoid organs. Notably, Il34LacZ/LacZ and wild-type mice had similar frequencies of CD11b+, F4/80+, Ly6C+ and Ly6G+ cell subsets in the peripheral blood and bone marrow (Supplementary Fig. 6a). Analysis of DC subsets in the spleens of Il34LacZ/LacZ and wild-type mice also showed a similar frequency of CD8α+, CD11b+ and CD4+CD11c+ DCs as well as Siglec-H+ plasmacytoid DCs (Supplementary Fig. 6b).
We next assessed whether IL-34 deficiency also affected the development of tissue-resident macrophages in nonlymphoid organs. Kupffer cells are liver-resident macrophages39 that include two subsets identified by expression of the macrophage marker F4/80 and CD11b40. The frequency of F4/80+CD11b− and F4/80+CD11b+ Kupffer cells was similar in Il34LacZ/LacZ and wild-type mice (Fig. 6a). IL-34 deficiency also had no effect on lung alveolar macrophages, identified as CD11c+Siglec-F+F4/80+ cells (Fig. 6b), or total DCs, identified as CD11c+Siglec-F− cells (Fig. 6c). However, among lung DC subsets, Il34LacZ/LacZ mice had fewer CD11c+CD11b+ DCs (Fig. 6b, c). These results suggested a smaller effect of IL-34 on the development of macrophages and DCs than on the development of LCs and microglia, probably because of redundancy with CSF-1.
Figure 6.
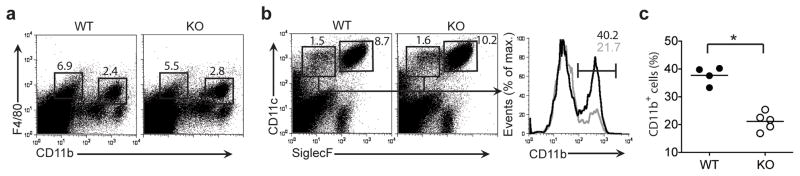
IL-34 deficiency does not affect the development of myeloid cells in the liver but partially affects the frequency of CD11b+ DCs in the lungs. (a) Flow cytometry of single-cell suspensions of wild-type and Il34LacZ/LacZ liver cells, to identify two subsets of Kupffer cells. Numbers above outlined areas indicate percent F4/80+CD11b− cells (left) and F4/80+CD11b+ cells (right). (b) Flow cytometry of cells from the lungs of wild-type and Il34LacZ/LacZ mice (left). Numbers adjacent to outlined areas indicate percent CD11c+Siglec-F+ cells (lung alveolar macrophages; right) and CD11c+Siglec-F− cells (lung DCs; left). Right, frequency (numbers above brackets) of CD11b+ lung DCs in wild-type mice (black lines) and Il34LacZ/LacZ mice (gray lines). (c) Frequency of CD11b+ DCs among CD11c+Siglec-F− DCs in wild-type and Il34LacZ/LacZ mouse lungd. Each symbol represents an individual mouse; small horizontal lines indicate the mean. *P = 0.0001 (unpaired Student’s t-test). Data represent two independent experiments with at least two mice per group.
The development of microglia and LCs require IL-34
Microglia and LCs originate during embryogenesis from primitive precursors that populate the skin and CNS before birth. Precursor cell populations expand before and soon after birth. In the adults, microglia and LCs self-renew with a slow turnover rate under homeostatic conditions13,16,21–23. We sought to determine whether the defects in LCs and microglia observed in adult Il34LacZ/LacZ mice was due to impaired development or maintenance in the adult once these cells developed. To address this, we examined microglia and LCs in neonates. Il34LacZ/LacZ neonates had considerably fewer microglia than did wild-type neonates (Fig. 7a, b). Similarly, although they were present in wild-type neonates, LCs were absent from the skin of Il34LacZ/LacZ mice (Fig. 7c, d). Thus, IL-34 was crucial for the development of both cell subsets.
Figure 7.

IL-34 is required for the development of microglia and LCs in neonates but is dispensable for recolonization of LCs in adult mice after exposure to ultraviolet light. (a–d) Flow cytometry of cells from the brain (a, b) and epidermis (c, d) of neonatal (2-day-old) wild-type and Il34LacZ/LacZ mice (n = 5–6 per group). Numbers adjacent to outlined areas (a, c) indicate percent CD45intCD11b+ cells (microglia; a) or CD11c+MHCII+ cells (LCs; c). (b, d) Frequency of microglia (CD45intCD11b+; b) and LCs (CD11c+CD207+; d) in the CD45+ gate. *P = 0.0025 and **P < 0.0001 (unpaired Student’s t-test). (e, f) Flow cytometry of cells from wild-type and Il34LacZ/LacZ mice (n = 3–6 per group) left untreated (Naive) or irradiated with ultraviolet light (UV; assessed on day 21). Numbers above outlined areas indicate percent CD207+CD11c+ cells. (f) Quantification of the analysis in e. *P = 0.0083 (unpaired Student’s t-test). Each symbol (b, d, f) represents an individual mouse; small horizontal lines indicate the mean. Data represent two independent experiments.
LC recovery after inflammation is IL-34 independent
We next assessed whether IL-34 was necessary for the turnover of LCs during skin inflammation. It has been shown that after prolonged exposure to ultraviolet light, LCs rapidly disappear and are replaced by circulating monocytic precursors within 3 weeks41. We depleted wild-type and Il34LacZ/LacZ mice of LCs by exposing them to ultraviolet light and analyzed the epidermis of the ears 21 d after treatment. In wild-type mice, after the initial depletion caused by exposure to ultraviolet light (Supplementary Fig. 7), LCs were partially replenished by day 21 (Fig. 7e, f). Notably, Il34LacZ/LacZ mice, which lacked LCs before the treatment with ultraviolet light, had a detectable LC population at day 21 (Fig. 7e, f), which suggested that IL-34 was dispensable for the regeneration of LCs from monocytic progenitors in adult mice in response to inflammation.
DISCUSSION
In this study, we showed that IL-34 was produced mainly by keratinocytes and neurons and that a lack of this cytokine resulted in considerably fewer LCs in the epidermis and microglia in the CNS. These results explain the well-described difference in the phenotypes of Csf1op/op mice and Csf1r−/− mice12: LCs and microglia are present in Csf1op/op mice but absent from Csf1r−/− mice14–16. Thus, IL-34 selectively directs a developmental program specific for these unique subsets of myeloid cells. Both LCs and microglia develop from embryonic progenitors and populate their correspondent tissues during embryogenesis. There, they proliferate and differentiate before and soon after birth15,16,21. We envision that IL-34 stimulates the population expansion and/or differentiation of the LC and microglia progenitors once they reach the epidermis and CNS. Consistent with such a model, we found that Il34LacZ/LacZ mice lacked LCs and microglia at birth. It is possible that IL-34 also contributes to the maintenance of LCs and microglia in the steady state in the adult. However, as Csf1op/op mice have LCs at birth12 but are deficient in LCs as adults42, CSF1 also may contribute to the self-renewal of LCs. Notably, we found IL-34 in ependymal cells of the choroid plexus, which suggests that it may promote the development of myeloid cells at this site, although DCs in the choroid plexus are dependent on the ligand for Flt3 (ref. 43).
After epidermal damage and inflammation, LCs are replaced rapidly by monocytic precursors16. Similarly, during CNS injury, bone marrow–derived progenitors infiltrate the CNS to promote inflammation and repair38,44. In these situations, the breakdown of the epidermis and altered brain-blood barrier probably facilitates the infiltration of bone marrow–derived myeloid progenitors and additional cellular sources of CSF1 that may contribute to the regeneration of LCs and microglia. In support of that model, we found that after ultraviolet light–induced inflammation, the defect of LCs in Il34LacZ/LacZ mice was partially compensated for by LCs presumably derived from monocytic precursors41. Although brain infection with an attenuated WNV virus did not lead to the complete replenishment of microglia by incoming monocytes, this may occur in mice infected with virulent strains that cause more severe inflammation.
In addition to finding IL-34 production in the epidermis and neurons, we detected IL-34 production in other tissue compartments, particularly the kidney and testis, but also in a scattered pattern in the spleen. Given those findings, it is likely that IL-34 also contributes to the development of myeloid cells in other anatomic regions. However, Il34LacZ/LacZ mice were fertile and lacked apparent renal deficiency. Moreover, Il34LacZ/LacZ mice had no apparent defects in blood monocytes, tissue macrophages or DCs, with the exception of fewer CD11c+CD11b+ DCs in the lung. Thus, beyond the epidermis and CNS, cellular sources of CSF1 may compensate for an absence of IL-34. In some compartments, such as the skeletal system, CSF1 may act as the main agonist of CSF1R, as Csf1op/op mice develop osteopetrosis despite producing normal amounts of IL-34.
Because they selectively lacked LCs and microglia, Il34LacZ/LacZ mice provided a unique opportunity for assessing the function of these cells. LCs are antigen-presenting cells that intercalate with keratinocytes and establish tight contacts via E-cadherin-mediated adhesion29,45. Given the production of IL-34 by keratinocytes, such contacts may be essential for the differentiation and maintenance of LCs. LCs slowly detach from keratinocytes to cross the epidermal basement membrane and migrate into the draining lymph nodes and thus regulate skin-adaptive immune responses to physical injury, chemical allergens and infection46. Whether LCs elicit activation or tolerance of T cells in response to chemical allergens remains unclear46. One study has reported that mice with transgenic expression of the gene encoding diphtheria toxin under control of the human langerin promoter have constitutive deletion of LCs that results in enhanced CHS47,48. Thus, LCs seemed to have a tolerogenic function. However, conflicting results have been obtained with mice in which the gene encoding the receptor for diphtheria toxin is knocked into the langerin locus, so that LCs can be ablated conditionally by injection of diphtheria toxin. In these mice, deletion of LCs results in attenuated CHS30,31 or has a negligible effect34, which suggests that LCs have either a stimulatory function or no influence on T cell responses. CHS was attenuated in our Il34LacZ/LacZ mice relative to that in wild-type mice, which supports the proposal that LCs promote T cell stimulation, at least in response to a chemical hapten. The discrepancies among the studies may reflect differences in the extent and timing of LC defects as well as the type and amount of haptens and adjuvants used to induce CHS. Our study also showed that deficiency of IL-34 diminished the delayed-type hypersensitivity response to C. albicans, which supports the idea that LCs can promote the generation of T cell responses to pathogens37.
It was unexpected that the considerably lower abundance of microglia in Il34LacZ/LacZ mice did not cause a substantially altered CNS phenotypes under steady-state conditions, although we did no detailed neurological testing. However, it is possible that loss of the neuroprotective functions of microglia may expedite neurodegenerative pathology49. Alternatively, microglia may have a role in synaptic pruning during postnatal development, which would influence brain plasticity50. Thus, it will be important in future studies to use IL-34-deficient mice to assess whether a lack of microglia induces functional changes in mouse behavior. The lack of microglia did negatively affect the survival of Il34LacZ/LacZ mice infected intracranially with an attenuated strain of WNV. However, microglia did not directly affect viral replication, as Il34LacZ/LacZ and wild-type mice had similar viral burdens in many brain regions, which suggests that in these settings, microglia may act by preserving CNS integrity, presumably by removing virus-infected, dying cells, secreting neurotrophic factors and limiting neuronal injury and death13. In summary, our studies of Il34LacZ/LacZ mice have demonstrated an alternative pathway for the development of myeloid cells that is essential for LCs and microglia but redundant or dispensable for other myeloid-cell subsets. Functional analysis of Il34LacZ/LacZ mice has defined an important role for LCs in promoting CHS and for microglia in protecting the CNS from injury associated with viral infection.
METHODS
Generation of Il34LacZ/LacZ mice
Of three clones carrying the targeted allele Il34tm1a(EUCOMM)Wtsi (European Conditional Mouse Mutagenesis Consortium), two were confirmed by Southern blot analysis as being correctly targeted and were introduced into C57BL/6J-Tyrc-2J/J (B6-albino) eight-cell embryos by laser-assisted injection. Male chimeras (n = 18; most >80% derived from embryonic stem cells, as assessed by coat color) were obtained from clone EPD0146_4_D03 (embryonic stem line JM8.N4; C57BL/6). Chimeras were initially bred to B6-albino mice for analysis of germline transmission; those transmitting 100% were bred to CMV-Cre mice (>99% C57BL/6)51 for deletion of exons 3–5 of Il34 and the neomycin-resistance cassette; Il34LacZ/+ offspring were intercrossed to generate IL-34-null LacZ+ mice (Il34LacZ/LacZ). To delete the IRES-lacZ–neomycin-resistance cassette and generate mice carrying a loxP-flanked Il34 allele (Il34f/+), chimeras were also bred to CAG-FLPe C57BL/6 transgenic mice2, which express an enhanced form of the FLP recombinase driven by a chicken β-actin/rabbit β-globin hybrid promoter (AG) and the human CMV-IE enhancer. Il34f/+ mice were intercrossed to generate Il34f/f mice (Supplementary Fig. 1). Homozygous Il34LacZ/LacZ mice were used for analysis; wild-type controls were age- and sex-matched C57BL/6 or Il34f/f mice bred in the same room of the facility. All animal studies were approved by the Washington University Animal Studies Committee. Mice were genotyped by PCR with the following oligonucleotides: for Il34LacZ/LacZ, Il34 305959 (5′-TCAGATACAAATATGAAATTAGAG-3′), Il34 306329 (5′-TGCTGGCAAAGGGCTAAGAA-3′) and Il34 306539 (5′-GTCAGTATCGGCGGAATT-3′; wild-type product, 421 base pairs (bp); Editor: this PCR is deisigned to distinguish 3 possibilites: the endogenous allele, the external lox P site (5′ on our map, but not relevant otherwise) that is present in both the intitial targeted allele and in the floxed allele (post FLP), and the LacZ reporter (post Cre, exons 3–5 deleted) allele; have tried to make this more clear] Il34 external LoxP product, 371 bp; Il34LacZ reporter product, 581 bp); for Il34f/f, Il34 307868 (5′-CCATGGTCAGAGTCCCCAGG-3′) and Il34 315341 (5′-CCCTGGTCGGCTTTGCATGT-3′; wild-type product, 468 bp; Il34f product, 570 bp).
Immunohistochemistry and immunofluorescence
Sections for immunohistochemistry were obtained from formalin-fixed, paraffin-embedded tissues or frozen tissues. For morphology, slides were stained with hematoxylin and eosin. Cryostat sections 5 μm in thickness were air-dried overnight, fixed in acetone for 10 min and stained by with a β-Galactosidase Reporter Gene Staining Kit (Sigma). X-gal Stock Solution (Sigma) was prepared in N,N-dimethylformamide (Sigma). An X-gal working solution was prepared by dilution of the X-gal stock solution 1:40 in prewarmed dilution buffer. Slides were washed for 5 min at 25 °C in 1× PBS (Sigma) with three changes of the solution and were incubated for 4 h at 37 °C in X-gal working solution, in a humidified chamber. Sections were rinsed in 1× PBS (twice for 5 min each) and in distilled water (5 min). Sections were counterstained with hematoxylin or fast red. The β-galactosidase activity was visualized as a light blue signal. Sections (4 μm in thickness) from fixed tissues were stained with anti-IBA1 (rabbit; 1:300 dilution; Cat. No. 019-19741; Wako Chemicals) and reactivity was detected with an EnVision HRP-linked Rabbit kit (Dako) followed by diaminobenzidine. Nuclei were counterstained with haematoxylin. Cryostat sections (5 μm in thickness) from frozen tissues were stained with rat monoclonal antibody to langerin (1:100 dilution; 929F3.01; Dendritics), F4/80 (1:50 dilution; BM8; Caltag Laboratories), CD11b (1:100 dilution; M1/70; BD Pharmingen) or MHC class II I-A and I-E (1:50 dilution; 2G9; BD Pharmingen). Reactivity was detected with a Rat-on-mouse HRP-Polymer Kit (Biocare Medical) followed by diaminobenzidine as the chromogen. For microphotographs, immunostained sections were photographed with a DP-70 Olympus camera mounted on an Olympus BX60 microscope. Microglia in wild-type and mutant mice were quantified by staining with anti-IBA-1 (Cat. No. 019-19741; Wako Chemicals) in paired regions of the brain. At least 25 high-power-fields (magnification, ×400) per region were analyzed for each mouse with a Nikon Eclipse 50i. Freshly isolated epidermal sheets were fixed and rehydrated and the receptor FcγR was blocked with monoclonal antibody 2.4G2 (ATCC). Epidermal sheets were stained with biotin-conjugated antibody to MHC class II (M5/114.15.2; BD Pharmingen) followed by streptavidin–Texas Red before being mounted on slides and analyzed with a Zeiss LSM 510 laser-scanning confocal microscope. At least eight high-power fields (magnification, ×400) per dermal sheet were analyzed. After infection with WNV, brains were collected and perfused sequentially with 20 ml PBS and 20 ml 4% paraformaldehyde in PBS, then fixed overnight at 4 °C in 4% paraformaldehyde in PBS. Tissues were cryoprotected in 30% sucrose and embedded, then frozen sections were cut. Tissues were prepared and stained as described3. After saturation of nonspecific binding sites and permeabilization of cells, sections were incubated overnight at 4 °C with anti-MAP2 (AP-20Chemicon). Primary antibodies were detected with secondary Alexa Fluor 488–conjugated goat anti-mouse IgG (Cat. No. A11017Molecular Probes). Nuclei were counterstained with To-Pro3 (Molecular Probes). TUNEL staining was done with an in situ cell death detection kit according to the manufacturer’s instructions (Roche), with some modifications53,54. Fluorescence staining was visualized and quantified with a Zeiss 510 Meta LSM confocal microscope.
Quantitative PCR
RNA was extracted from skin and brain with an RNeasy Fibrous Tissue Mini kit as recommended by the manufacturer (Qiagen). After cDNA was synthesized from RNA with the Superscript III first-strand synthesis system for RT-PCR (Invitrogen), RNA expression was analyzed by quantitative PCR. SYBR Green PCR Master Mix (Bio-Rad Laboratories) and an ABI7000 (Applied Biosystems) were used according to the manufacturers’ instructions. IL-34 expression was detected with oligonucleotides that specifically amplify a 171-bp fragment from cDNA encoding exon 6 (forward primer, 5′-TACAGCCACCTCTGCTTGTG-3′; reverse primer, 5′-GCAAGATACGGCATTTGGTT-3′).
Cell isolation
Epidermal sheets were separated by treatment of mouse ears for 1 h at 37 °C with Dispase I (Sigma) at a concentration of 2 U/ml. For single-cell suspensions, epidermis or dermis was minced into fine pieces and incubated for 1 h at 37 °C in collagenase IV (4830 U/ml; Sigma) and DNAse I (0.1 mg/ml; Sigma). Kupffer cells were isolated as described55. Livers were minced and then treated for 1 h at 37 °C with collagenase D (100 μg/ml, Sigma). Cells in single-cell suspension were spun through 33.8% Percoll (Sigma) for 12 min at 693g. Red blood cells in recovered cell pellets were removed with red blood cell lysis buffer (Sigma) before analysis. Lung cells were extracted after digestion of minced lungs for 1 h at 37 °C with DNAse I (50 U/ml), hyaluronidase (100 U/ml, Sigma) and liberase (0.28 U/ml; Roche). For microglia purification, brains were minced into fine pieces and incubated for 90 min at 37 °C with 100 μg/ml collagenase D. For microglia purification, brains were minced into fine pieces and incubated for 90 min at 37 °C with 100 μg/ml collagenase D. Cells were dissociated into single-cell suspensions by a combination of pipetting and vortexing and were then spun through 50% percoll (Sigma) for 45 min at 200g. Microglia were identified as CD11b+CD45int by flow cytometry. In some experiments, microglia were further enriched to a purity of over 95% with CD11b magnetic beads (Miltenyi Biotech) and were stimulated with various TLR ligands (Invivogen) in vitro at the appropriate concentration. DCs from spleens and lymph nodes were dissociated into single-cell suspensions by incubation for 60 min with 100 μg/ml collagenase D before red blood cell lysis. In some experiment, DCs were further purified with CD11c magnetic beads (Miltenyi).
Viral infection
Wild-type WNV and WNV strain E218A were generated from an infectious cDNA clone of the New York 1999 strain and were propagated in C6/36 Aedes albopictus or BHK21-15 baby hamster kidney cells as described56,57. Vero African green monkey cells were used for measurement of viral titers of infected tissues by focus-forming assay58. Sex- and age-matched wild-type and Il34LacZ/LacZ mice 8–10 weeks of age were inoculated by intracranial injection with specified doses of WNV diluted in Hank’s balanced-salt solution supplemented with 1% heat-inactivated FBS (1 × 105 PFU in 10 μl). On specific days after infection, mice were killed and extensively perfused with iced PBS; organs were collected, weighed and processed for analysis of infiltrating CNS lymphocytes by a published protocol53 or were stored at −80 °C until further processing. Alternatively, survival of groups of mice was monitored for 21 d after infection.
Ear painting, analysis of CHS and delayed-type hypersensitivity and treatment with ultraviolet light
The migration of DCs and LCs was asssessed by painting of mouse ears with 100 μl 0.5% CellTracker Green (invitrogen) in acetone and dibutylphthalate (1:1) at 40–48 h before collection of cervical lymph nodes. CHS was induced in mice as described59. Mice were sensitized on day 0 by topical application, on the shaved abdomen, of 25 μl of 0.5% DNFB (2,4 dinitroflurobenzene; Sigma) in a mixture of acetone and olive oil (4:1). On day 5, mice were challenged with 10 μl of 0.2% DNFB on the left ear and 10 μl of vehicle on the right ear. Ear swelling was measured with an engineer’s micrometer (Fowler) 24 h after challenge. T cell priming during CHS was assessed as described60. Mice were sensitized by two daily paintings (days 0 and 1), on the shaved abdomen, of 25 μl of 0.5% DNFB. Inguinal lymph node cells from sensitized and control mice were cultured for 2 d in the presence of plate bound anti-CD3 (1 μg/ml; eBio500A2; eBioscience). IFN-γ in the supernatant was measured by cytometric bead array with a mouse inflammation kit (BD Bioscience). Induction of delayed-type hypersensitivity by C. albicans has been described61. Mice were shaved and chemically depilated with Nair hair remover. The stratum corneum was removed with 15 strokes with 220-grit sandpaper (3M). C. albicans was grown at 30 °C in yeast-peptone-adenine-dextrose medium until the absorbance at 600 nm reached 1.5–2.0. C. albicans (2 × 108 in 50 μl sterile PBS) was applied on to the skin. At 8 d after skin infection, mice were challenged with footpad injection of 1 × 107 heat-killed C. albicans. The specific delayed-type hypersensitivity was determined on the basis of the degree of footpad swelling 24 h after challenge. Treatment with ultraviolet light was done as described16.
Flow cytometry
DCs and other cell types in single-cell suspensions were incubated with supernatant of the hybridoma HB-197 (American Type Culture COllection) for blockade of Fc receptors and stained with anti-CD3 (145-2c11), anti-CD4 (GK1.5), anti-CD8α (53–6.7), anti-CD44 (IM7), anti-Ly6C (AL-21), anti-MHC-II (I-A/I-E, 2G9 or M5/114.15.2), anti-Siglec-F (E50-2440), anti-CD11b (M1/70; all from BD Pharmingen); anti-Ly6G (1A8), anti-CD62L (MEL-14), anti-Siglec-H (551; all from Biolegend); anti-CD11c (N418), anti-CD45 (30-F11), anti-CD103 (2E7; all from eBioscience); and anti body to the H-2Db–NS4B tetramer (US National Institutes of Health tetramer core facility). For intracellular staining with anti-CD207 (L31; eBioscience) and anti–granzyme B (GB12; Invitrogen), surfaces of cells were stained, then cells were fixed and permeabilized with Cytofix/Cytoperm buffer (BD Bioscience). Cells were processed on a FACSCalibur or FACSCanto II (BD Bioscience) and analyzed with FlowJo software (Tree Star).
Statistics
GraphPad Prism (GraphPad software) was used for all statistics analyses.
Supplementary Material
Acknowledgments
We thank M. Swiecki for help with flow cytometry and confocal microscopy; the European Conditional Mouse Mutagenesis Consortium for generating embryonic stem clones with the targeted allele Il34tm1a(EUCOMM)Wtsi; M. White for injection of Il34tm1a(EUCOMM)Wtsi embryonic stem cells; and T. Doering (Department of Molecular Microbiology, Washington University School of Medicine) for C. albicans SC5314. Supported by the National Heart, Lung and Blood Institute (2T32HL007317-31 to Y.W.), Fondazione Beretta (C.R.), Programma di Ricerca di interesse Nazionale of the Ministero dell’Istruzione dell’Università e della Ricerca, 2009 (W.V.), the European Commission Seventh Framework Programme (A.D.B.) and the US National Institutes of Health (GM77279 and HL097805 to M.Co.).
Footnotes
AUTHOR CONTRIBUTIONS
Y.W., K.J.S. and M.Ce. designed, did and analyzed experiments and wrote the manuscript; W.V. and C.R. did immunohistochemical analysis; M.S.D. supervised CNS-infection experiments; S.G. and A.D.B. generated Il34LacZ/LacZ mice; and M.Co. supervised research and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Gordon S, Mantovani A. Diversity and plasticity of mononuclear phagocytes. Eur J Immunol. 2011;41:2470–2472. doi: 10.1002/eji.201141988. [DOI] [PubMed] [Google Scholar]
- 2.Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234:45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 4.Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113:3418–3427. doi: 10.1182/blood-2008-12-180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 6.Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 8.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- 9.Choi JH, et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity. 2011;35:819–831. doi: 10.1016/j.immuni.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Cecchini MG, et al. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development. 1994;120:1357–1372. doi: 10.1242/dev.120.6.1357. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida H, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 12.Dai XM, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99:111–120. doi: 10.1182/blood.v99.1.111. [DOI] [PubMed] [Google Scholar]
- 13.Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- 14.Witmer-Pack MD, et al. Identification of macrophages and dendritic cells in the osteopetrotic (op/op) mouse. J Cell Sci. 1993;104:1021–1029. doi: 10.1242/jcs.104.4.1021. [DOI] [PubMed] [Google Scholar]
- 15.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginhoux F, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006;7:265–273. doi: 10.1038/ni1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin H, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320:807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 18.Ma X, et al. Structural basis for the dual recognition of helical cytokines IL-34 and CSF-1 by CSF-1R. Structure. 2012;20:676–687. doi: 10.1016/j.str.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Wei S, et al. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. 2010;88:495–505. doi: 10.1189/jlb.1209822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chihara T, et al. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010;17:1917–1927. doi: 10.1038/cdd.2010.60. [DOI] [PubMed] [Google Scholar]
- 21.Chorro L, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med. 2009;206:3089–3100. doi: 10.1084/jem.20091586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol. 2008;8:935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 23.Ginhoux F, Merad M. Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol. 2010;88:387–392. doi: 10.1038/icb.2010.38. [DOI] [PubMed] [Google Scholar]
- 24.Valladeau J, et al. Identification of mouse langerin/CD207 in Langerhans cells and some dendritic cells of lymphoid tissues. J Immunol. 2002;168:782–792. doi: 10.4049/jimmunol.168.2.782. [DOI] [PubMed] [Google Scholar]
- 25.Takahara K, et al. Identification and expression of mouse Langerin (CD207) in dendritic cells. Int Immunol. 2002;14:433–444. doi: 10.1093/intimm/14.5.433. [DOI] [PubMed] [Google Scholar]
- 26.Poulin LF, et al. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med. 2007;204:3119–3131. doi: 10.1084/jem.20071724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ginhoux F, et al. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med. 2007;204:3133–3146. doi: 10.1084/jem.20071733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bursch LS, et al. Identification of a novel population of Langerin+ dendritic cells. J Exp Med. 2007;204:3147–3156. doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunol Rev. 2010;234:120–141. doi: 10.1111/j.0105-2896.2009.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett CL, Noordegraaf M, Martina CA, Clausen BE. Langerhans cells are required for efficient presentation of topically applied hapten to T cells. J Immunol. 2007;179:6830–6835. doi: 10.4049/jimmunol.179.10.6830. [DOI] [PubMed] [Google Scholar]
- 31.Bennett CL, et al. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J Cell Biol. 2005;169:569–576. doi: 10.1083/jcb.200501071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bacci S, Alard P, Dai R, Nakamura T, Streilein JW. High and low doses of haptens dictate whether dermal or epidermal antigen-presenting cells promote contact hypersensitivity. Eur J Immunol. 1997;27:442–448. doi: 10.1002/eji.1830270214. [DOI] [PubMed] [Google Scholar]
- 33.Enk AH, Katz SI. Early events in the induction phase of contact sensitivity. J Invest Dermatol. 1992;99:39S–41S. doi: 10.1111/1523-1747.ep12668608. [DOI] [PubMed] [Google Scholar]
- 34.Kissenpfennig A, et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Ruedl C, Koebel P, Bachmann M, Hess M, Karjalainen K. Anatomical origin of dendritic cells determines their life span in peripheral lymph nodes. J Immunol. 2000;165:4910–4916. doi: 10.4049/jimmunol.165.9.4910. [DOI] [PubMed] [Google Scholar]
- 36.Christensen AD, Haase C. Immunological mechanisms of contact hypersensitivity in mice. APMIS. 2012;120:1–27. doi: 10.1111/j.1600-0463.2011.02832.x. [DOI] [PubMed] [Google Scholar]
- 37.Igyártó BZ, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. 2011;35:260–272. doi: 10.1016/j.immuni.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Getts DR, et al. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–163. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 40.Kinoshita M, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol. 2010;53:903–910. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 41.Merad M, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. 2002;3:1135–1141. doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi K, Naito M, Shultz LD. Differentiation of epidermal Langerhans cells in macrophage colony-stimulating-factor-deficient mice homozygous for the osteopetrosis (op) mutation. J Invest Dermatol. 1992;99:46S–47S. doi: 10.1111/1523-1747.ep12668982. [DOI] [PubMed] [Google Scholar]
- 43.Anandasabapathy N, et al. Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. J Exp Med. 2011;208:1695–1705. doi: 10.1084/jem.20102657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 45.Jiang A, et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity. 2007;27:610–624. doi: 10.1016/j.immuni.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romani N, Brunner PM, Stingl G. Changing views of the role of langerhans cells. J Invest Dermatol. 2012;132:872–881. doi: 10.1038/jid.2011.437. [DOI] [PubMed] [Google Scholar]
- 47.Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23:611–620. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 48.Kaplan DH. In vivo function of Langerhans cells and dermal dendritic cells. Trends Immunol. 2010;31:446–451. doi: 10.1016/j.it.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14:1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- 50.Paolicelli RC, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 51.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanki H, Suzuki H, Itohara S. High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp Anim. 2006;55:137–141. doi: 10.1538/expanim.55.137. [DOI] [PubMed] [Google Scholar]
- 53.Szretter KJ, et al. The immune adaptor molecule SARM modulates tumor necrosis factor α production and microglia activation in the brainstem and restricts West Nile Virus pathogenesis. J Virol. 2009;83:9329–9338. doi: 10.1128/JVI.00836-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuel MA, Morrey JD, Diamond MS. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J Virol. 2007;81:2614–2623. doi: 10.1128/JVI.02311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kinoshita M, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol. 2010;53:903–910. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 56.Daffis S, et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vogt MR, et al. Human monoclonal antibodies against West Nile virus induced by natural infection neutralize at a postattachment step. J Virol. 2009;83:6494–6507. doi: 10.1128/JVI.00286-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fuchs A, Pinto AK, Schwaeble WJ, Diamond MS. The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology. 2011;412:101–109. doi: 10.1016/j.virol.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Girardi M, et al. Resident skin-specific gammadelta T cells provide local, nonredundant regulation of cutaneous inflammation. J Exp Med. 2002;195:855–867. doi: 10.1084/jem.20012000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu H, DiIulio NA, Fairchild RL. T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production: interferon gamma-producing (Tc1) effector CD8+ T cells and interleukin (Il) 4/Il-10-producing (Th2) negative regulatory CD4+ T cells. J Exp Med. 1996;183:1001–1012. doi: 10.1084/jem.183.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Igyártó BZ, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. 2011;35:260–272. doi: 10.1016/j.immuni.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.