

Abstract
Background
Transthyretin-associated Familial Amyloid Polyneuropathy (TTR-FAP) is an autosomal dominant disease caused by the deposition of abnormal transthyretin that results from a gene mutation. Although rare worldwide, there are descriptions of several endemic foci, such as in Majorca, Balearic Islands, Spain. We aimed at describing a contemporary series of TTR-FAP patients in Son Llàtzer Hospital in Majorca from an epidemiological point of view in order to report their main clinical and laboratory characteristics.
Methods
A retrospective, observational study was performed. Medical records from adult patients diagnosed with TTR-FAP from a single hospital were reviewed.
Results
Out of a total of 107 cases, 75 subjects were included: asymptomatic carriers (52.3%) and symptomatic patients (47.7%). Mean age was 58.3 years at the time of the study, and 50.7% were men. Mean age at diagnosis was 49.8 years. In addition, 42 patients (39.2%) had received a liver transplant, and time to liver transplantation was on average 29.56 months from the initial diagnosis. They all had the Val30Met mutation. The organs of the nervous system were those most frequently impaired (57.3%), and 83.9% of the symptomatic patients were fully ambulant (stage 1). Family history was reported in 69.3% of the cases, with the patient’s father being the most commonly affected relative. Comorbidities were frequent, with high blood pressure being the most common.
Conclusions
Our findings provide additional information on this condition and are useful for describing the demographic features, clinical presentation, diagnosis, and natural course of TTR-FAP in Majorca.
Keywords: Familial amyloid polyneuropathy, TTR, Amyloidosis, Transthyretin, Majorca, Asymptomatic carriers, Symptomatic patient, Liver transplant
Background
Familial amyloid polyneuropathies (FAP) are a group of systemic amyloidoses caused by the extracellular deposition of insoluble amyloid fibrils. Three main types of FAP have been described, based on the abnormal amyloid precursor protein resulting from a gene mutation (transthyretin [TTR], apolipoprotein A-1, or gelsolin) [1]. FAP caused by a mutated TTR is the most common form, with the Val30Met mutation in the TTR gene being by far the most common variant (accounting for about 50% of cases worldwide). It is virtually the only one in some countries, such as Portugal and Sweden [2], although at present 113 amyloidogenic mutations have been described in the TTR gene [1-3]. The abnormal TTR protein is mainly produced in the liver and to a lesser extent in the choroid plexus and retina.
Transthyretin-associated familial amyloid polyneuropathy (TTR-FAP) is a life-threatening disease transmitted as an autosomal dominant trait. The most typical presentation of TTR-FAP is a progressive nerve length-dependent, sensory-motor polyneuropathy, which usually begins with loss of thermal and pain sensation in the feet and slowly ascends up the limbs. This is then followed by autonomic manifestations and motor dysfunction or impairment. The progression of the disease is usually described as going through 3 stages according to the symptoms and signs shown by the patients. Stage 1 is characterized by mild motor dysfunction/impairment of the lower extremities, moderate autonomic manifestations, no or minimum impairment in daily activities, and full ambulation. Stage 2 is characterized by mild-to-moderate motor dysfunction/impairment of the lower or upper extremities, mild autonomic manifestations, significant impairment in daily activities, and the need for help in ambulation. Lastly, stage 3 is described as severe motor dysfunction/impairment of the lower or upper extremities, severe autonomic manifestations, profound impairment in daily activities, and confinement to a wheelchair [4]. Diagnosis is based on family history, neurographic evidence of polyneuropathy, detection of mutated TTR or DNA in the blood, and identification of amyloid deposits in the tissues (mainly in intestinal, nerve, salivary gland or abdominal fat biopsies). The disease is occasionally diagnosed in sporadic cases with no known family history. Immunolabeling or mass spectroscopy-based proteomic analysis can be useful in identifying the amyloid type, but a TTR/DNA test is mandatory [1]. Early- and late-onset presentations have been described, with late-onset presentations showing a slower progression and fewer autonomic symptoms. TTR-FAP is usually fatal within 10 to 13 years of the first symptoms [5-7].
Liver transplantation provides a useful and specific therapy for TTR-FAP by allowing for the suppression of the main source of the mutant TTR; it prevents neuropathy progression in approximately 70% of cases with Val30Met mutation on a long-term basis [8,9].
In November 2011, the European Medicines Agency [10] approved tafamidis meglumine, a small molecule that stabilizes and inhibits the dissociation of circulating TTR tetramers, thus preventing the deposition and formation of the abnormal TTR fibrils, for TTR-FAP Val30Met patients with stage-1 polyneuropathy, and it seems to be very promising [11,12].
TTR-FAP was initially described by Andrade in 1952 in Póvoa de Varzim, Portugal [13], and has subsequently been reported in several other European countries such as Sweden [14,15], as well as in countries like Japan [16,17] and Brazil [18,19], among others. It is a relatively rare disease worldwide, although it has a relatively high prevalence in some specific areas. A prevalence of 151/100,000 inhabitants has been reported in some areas in northern Portugal (including Póvoa de Varzim, where the first cases were discovered) [20] and of 104/100,000 inhabitants in northern Sweden [14]. The reported prevalence in Japan is 1.5/100,000 [16], and data from Cyprus suggest a prevalence of 3.72/100,000 [21].
In Spain, a significant area was found to be Majorca in the Balearic Islands. According to Munar-Qués et al. [22,23], the rate of prevalence amounts to 5/100,000 inhabitants in Majorca and 1/100,000 in Minorca, also a Balearic Island. TTR-FAP has also been described in other areas in Spain, including Valverde del Camino (Huelva), the Safor area (Valencia), Barcelona, Cantabria [24-26], and Vigo (Galicia) [25]. However, the number of patients in these areas is low. In Majorca the first case was described in 1976; subsequently the diagnosis of TTR-FAP was also clinically detected in 15 additional patients [22]. The disease could have arrived in Majorca from Portugal as a founder mutation in the 13th or 14th centuries, or it could have appeared independently in Majorca, as well as in other parts of the world. In 2005, a series of 102 confirmed patients from Majorca was well described by Munar-Qués et al. [27]. According to their conclusions, TTR-FAP seems to now be a growing public health problem in the Balearic Islands owing to its relatively high prevalence, together with its significant social and clinical impact.
We aimed at describing a contemporary series of TTR-FAP patients followed at Son Llàtzer Hospital, Majorca, from an epidemiological point of view and at reporting their clinical and laboratory characteristics.
Methods
A retrospective, observational study was performed. Medical records from all adult (≥ 18 years old) patients who met the following inclusion criteria: being diagnosed with TTR-FAP at Son Llàtzer Hospital (a 350-bed community center located in Palma de Majorca, Spain). The study period ran from January 1, 2001 to January 13, 2012, at which point the recorded data were reviewed. We described both the symptomatic patients and the asymptomatic carriers at the time of diagnosis. The cohort of TTR-FAP patients that underwent liver transplantation was also described.
Demographic details (age, sex, occupational status) and clinical data (family history, type of mutation, and disease stage) were obtained. The results were analyzed by comparing asymptomatic carriers to symptomatic patients. As supplemental data we also recorded health care resource utilization from and at diagnosis.
Diagnostic criteria included: presence of TTR mutation (both in symptomatic patients and asymptomatic carriers).
Additional tests were performed in symptomatic patients: electromyogram to confirm the presence of mixed polyneuropathy; tissue biopsy to detect amyloid deposits; echocardiogram (ECG) or Holter monitoring (24-hour ECG) when cardiac involvement was suspected; and lastly, eye exam when appropriate.
Data confidentiality was respected when decoupling personal data. The study was classified by the Spanish Agency of Medicines and Medical Devices and was approved by the Independent Ethics Committee of Son Llàtzer Hospital.
Statistical analysis: Pearson’s chi-squared test or Fisher’s exact test were used for categorical data. Continuous variables were analyzed using Student’s t-test or one-way analysis of variance (ANOVA) (or Mann–Whitney U test for non-parametric variables). A p-value of < 0.05 was used to show statistically significant associations. All statistical analyses were based on SPSS ver. 17.0 or later.
Results
A total of 107 cases with heterozygote TTR-Val30Met pathogenic mutations were diagnosed with TTR-FAP at Son Llàtzer Hospital in the study period running from January 2001 to January 2012. We have epidemiological and clinical data from 75 cases of asymptomatic carriers and symptomatic patients that were included in this study, and the remaining 42 patients received a liver transplantation.
Description of asymptomatic carriers and symptomatic patients
All of the subjects had the Val30Met mutation. Mean age was 58.33 ± 16.13 years (range from 28 to 92), and 38 (50.7%) were men (Figure 1) with a mean age at diagnosis of 49.8 ± 17.33 years (range from 13 to 81). Mean life expectancy without liver transplantation was 7.4 years. When assessing for this analysis (2011), we did not report all data, but 36 of 57 subjects (63.2%) had an active employment status. The rest of the demographic data are shown in Table 1.
Figure 1.
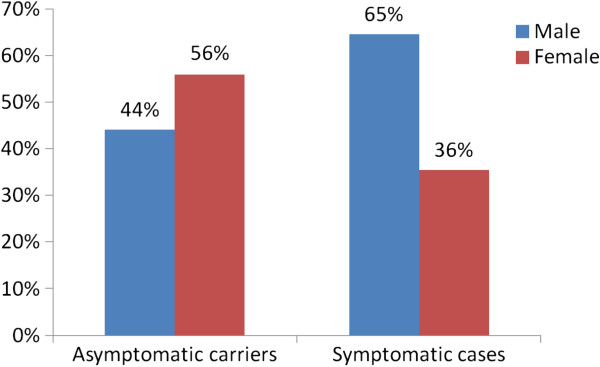
TTR-FAP related in Son Llàtzer Hospital (Majorca) comparison of asymptomatic carriers and symptomatic patients.
Table 1.
Demographic data of subjects included in the study at TTR-FAP diagnosis
| Variable | Number* | Percentage | Mean ± SD | Range |
|---|---|---|---|---|
|
Sex (n = 75) |
|
|
|
|
| Men |
38 |
50.7 |
|
|
| Women |
37 |
49.3 |
|
|
|
Age, years (n = 75) |
|
|
58.33 ± 16.13 |
28–92 |
|
Occupational status (n = 57) |
|
|
|
|
| Active |
36 |
63.2 |
|
|
| Retired |
16 |
28.1 |
|
|
| Other |
5 |
8.8 |
|
|
|
Weight, kg (n = 24) |
|
|
67.00 ± 13.05 |
35–87 |
| Mean age at diagnosis, years (n = 54) | 49.8 ± 17.33 | 13–81 |
*Total number of patients analyzed; some patients did not report all data.
At diagnosis (we have data for 65 subjects), there were 34 (52.3%) asymptomatic carriers and 31 (47.7%) symptomatic patients. Clinical stage for those symptomatic patients at diagnosis was as follows: stage 1 (fully ambulant) for 26 patients (83.9%); stage 2 (needing help with ambulation) for 4 patients (12.9%); and stage 3 (confined to wheelchair) for 1 patient (3.2%) (Figure 2).
Figure 2.
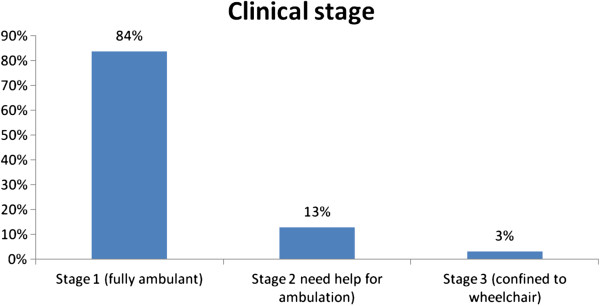
Clinical stage in symptomatic patients at diagnosis.
A family tree following the disease was performed for all patients and we found a family history of TTR-FAP in 52 cases (69.3%), with subjects’ fathers being the most commonly affected relative. Comorbidities at initial diagnosis were frequent 23 (30.7%), with high blood pressure being the most common, found in 18 subjects (24.0%). The rest of clinical data, including specific family history, comorbidities, and initial organ involvement, are shown in Table 2.
Table 2.
Clinical data of patients included in the study at TTR-FAP diagnosis
| Variable | Number n = 75 | Percentage |
|---|---|---|
|
Family history |
|
|
| No |
8 |
10.7 |
| Yes |
52 |
69.3 |
| Father |
22 |
42.3 |
| Mother |
12 |
23.1 |
| Grandfather/grandmother |
3 |
5.8 |
| Uncle/aunt |
16 |
30.8 |
| Sibling |
23 |
44.2 |
| Other (cousin, niece, nephew, etc.) |
26 |
50.0 |
| Unknown |
15 |
20 |
|
Comorbidities |
|
|
| Any comorbidity |
23 |
30.7 |
| High blood pressure |
18 |
24.0 |
| Dyslipidemia |
7 |
9.3 |
| Diabetes mellitus |
3 |
4.0 |
| Kidney failure |
3 |
4.0 |
| Smoking |
3 |
4.0 |
| Obesity |
2 |
2.7 |
| Heart failure |
1 |
1.3 |
|
Initial organ involvement |
|
|
| Nervous system |
43 |
57.3 |
| Gastrointestinal tract |
11 |
14.7 |
| Erectile dysfunction |
4 |
5.3 |
| Cardiovascular |
3 |
4.0 |
| Kidneys | 3 | 4.0 |
All subjects were diagnosed with a TTR/DNA/genetic test. Moreover, additional tests were performed in symptomatic patients: electromyogram to rule out the presence of mixed polyneuropathy; biopsy to try to detect tissue amyloid deposits; echocardiogram or Holter monitoring (24-hour ECG) when cardiac involvement was suspected; and ophthalmic exam when appropriate (Figure 3).
Figure 3.
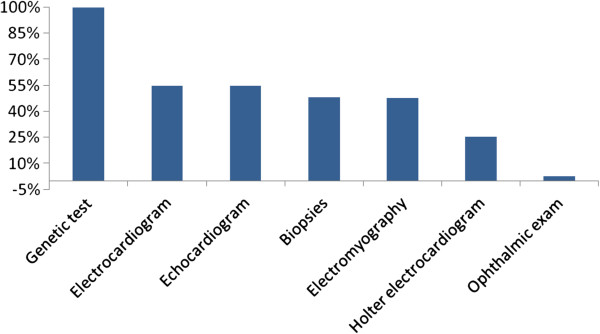
Additional tests performed.
In the comparison between asymptomatic carriers and symptomatic patients, we found statistically significant differences in mean age at diagnosis (54 vs. 44 years; p < 0.05) and also for active employment status (75.8% vs. 50.0%; p < 0.01). In contrast, we found no statistically significant differences in mean age. Comorbid disease profile was similar in both groups.
Patients with liver transplantation
During the data analysis, liver transplantation had already been performed in 42 patients (39.2%). Not all the patients could undergo liver transplantation, mainly owing to advanced age, contraindication (comorbidities), or because they were at stage 3 of the disease progression. The time to inclusion on the transplantation waiting list was 29.56 months (range from 16.72 to 37.88).
After transplantation, 34 patients (80.9%) showed no disease progression; 23 (54.7%) needed no help with ambulation; and most patients (35, or 83.3%) stated that they were satisfied with their therapy (transplantation). The post-transplantation course was generally good, with a relatively high proportion of patients being stable and with many subjects having an active employment status (77.8% of patients were able to resume their usual working activity).
Discussion
Our findings, which are based on a relatively large series from a local community hospital in Majorca, provide additional information on this condition and are useful for describing the demographic features, clinical presentation, diagnosis, and natural course of TTR-FAP in the Majorca area, a significant endemic focus.
Age of onset has not been consistent in all reported series (Figure 4). Our findings (mean age at diagnosis of 49.8 years) are similar to previously reported data from Majorca (age of onset of 45.7 years) [27], as well as to those from some other Mediterranean areas, like Cyprus (46 years) [21]. An earlier age of onset has been found in the main endemic areas in Portugal (33.5 years) [20], Japan (35.6 years) [16], and Brazil (32 years) [18], whereas a later age of onset has been reported in northern Sweden (56.7 years) [15]. In contrast, an even later age of onset (61 years) has been reported in sporadic cases in a series from a non-endemic area [28]. Taking into account that the causal mutation is probably the same in Portugal and Majorca, and that patients who are not aware of relatives who had the disease (sporadic cases) in other areas show a later age of onset, the difference in age of onset between Portuguese and Majorcan cases could possibly be owing to environmental or epigenetic factors.
Figure 4.

Mean age of onset or diagnosis of TTR-FAP in several series [14,16,20-22].
Although a male predominance has been previously reported in Majorca (62% versus 38%) [27], our results show a ratio of 51% to 49%. Similar findings have been reported in some other areas, including Brazil (45% to 55%) [18] and Cyprus (44% to 56%) [21]. A later age of onset in women has been described in Portugal [20] and Brazil [18], whereas no differences based on gender were found in Sweden [15], Cyprus [21], or the previous Majorcan series [27]. Our findings confirm a similar age of onset in both men and women.
Clinical data from our series show a high proportion of patients with a positive family history, mostly owing to the father. As expected, the existence of a family history was more common in asymptomatic carriers. Such individuals had also an earlier diagnosis. A large number of symptomatic patients received a liver transplant, with mean time to receiving a liver transplant being just slightly longer than 2.5 years from the patient’s diagnosis. Data on employment activity are scarce in previous studies, and our findings are particularly relevant for demonstrating the real impact of the disease on a patient’s life.
Weaknesses
This is the first TTR-FAP epidemiological study to include both clinical and work productivity data during disease progression. The isolated nature of TTR-FAP and its being limited to a specific geographical area make it difficult to extrapolate results. In addition, limitations stemming from the retrospective nature of this study are also applicable.
Conclusions
TTR-FAP is a life-threatening neurodegenerative disease that constitutes a public health problem in Majorca, with a disease focus growing mainly due to TTR-FAP is being better understood so patients could be diagnosed earlier. The clinical characteristics of our series are similar to those previously reported in the literature, especially in Mediterranean areas, but with a slightly older age at onset and relatively higher neurological and digestive involvement at presentation. Because TTR-FAP is a multi-pathological disease, in Majorca a multidisciplinary group has been created that includes experts in internal medicine, neurology, cardiology, gastroenterology, ophthalmology and pathology, thereby enabling a quicker assessment of disease involvement at presentation. This multidisciplinary approach will probably decrease the age of onset in Majorca in further studies. The proportion of symptomatic TTR-FAP patients in our series who underwent liver transplantation seems relatively high (56%) compared with other countries.
Abbreviations
FAP: Familial amyloid polyneuropathy; TTR: Transthyretin; TTR-FAP: Transthyretin-associated familial amyloid polyneuropathy; ECG: Echocardiogram; ANOVA: Analysis of variance.
Competing interests
This study was sponsored by Pfizer Spain. Editorial Support (medical writing) was provided by Content Ed Net Communications S.L. and was funded by Pfizer Spain.
Authors’ contributions
All authors had complete access to the data, participated in the analysis and/or interpretation of results, and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Juan Buades Reinés, Email: doctorjuanbuades@gmail.com.
Tomás Ripoll Vera, Email: tripoll@hsll.es.
Mercedes Uson Martín, Email: muson@hsll.es.
Hernán Andréu Serra, Email: handreu@hsll.es.
Ma Margarita Company Campins, Email: mmcompany@hsll.es.
José Miguel Diéguez Millán, Email: jmdieguez@hsll.es.
Cristina Gallego Lezaun, Email: vbenito@hsll.es.
Manuel Raya Cruz, Email: manuelraya@hotmail.com.
Acknowledgements
The authors would like to thank Marta Iglesias for her assistance in preparing the draft of this manuscript, and Vanessa Benito and Antonia Taura for their contribution during data collection. The authors would also like to thank Mercedes García-Vargas and Marina De Salas for their assistance in data analysis and manuscript review. Special acknowledgement to Isabel Fernandez-Arias and Joao Fonseca for their help in reviewing the manuscript.
References
- Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–1097. doi: 10.1016/S1474-4422(11)70246-0. [DOI] [PubMed] [Google Scholar]
- Saraiva MJ. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat. 2001;17:493–503. doi: 10.1002/humu.1132. [DOI] [PubMed] [Google Scholar]
- Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants. Amyloid. 2003;10:160–184. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]
- Coutinho P, Silva AM, Lima LJ, In: Amyloid and Amyloidosis. Glenner GG, editor. Amsterdam: Excerpta Medica; 1980. Forty years of Eexperience with type I amyloid neuropathy. review of 483 cases; pp. 88–98. [Google Scholar]
- Said G, Plante-Bordeneuve V. Familial amyloid polyneuropathy: a clinico-pathologic study. J Neurol Sci. 2009;284:149–154. doi: 10.1016/j.jns.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Plante-Bordeneuve V, Said G. Transthyretin related familial amyloid polyneuropathy. Curr Opin Neurol. 2000;13:569–573. doi: 10.1097/00019052-200010000-00011. [DOI] [PubMed] [Google Scholar]
- Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J Intern Med. 1994;235:479–485. doi: 10.1111/j.1365-2796.1994.tb01106.x. [DOI] [PubMed] [Google Scholar]
- Sharma P, Perri RE, Sirven JE, Zeldenrust SR, Brandhagen DJ, Rosen CB, Douglas DD, Mulligan DC, Rakela J, Wiesner RH, Balan V. Outcome of liver transplantation for familial amyloidotic polyneuropathy. Liver Transpl. 2003;9:1273–1280. doi: 10.1016/j.lts.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Adams D, Samuel D, Slama M. Treatment of familial amyloid polyneuropathy. Presse Med. 2012;41:793–806. doi: 10.1016/j.lpm.2011.11.027. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency. Vyndaqel. Summary of product characteristics. [ http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002294/WC500117862.pdf]
- Said G, Grippon S, Kirkpatrick P. Tafamidis. Nat Rev Drug Discov. 2012;11:185. doi: 10.1038/nrd3675. [DOI] [PubMed] [Google Scholar]
- Russo M, Stancanelli C, Gentile L, Toscano A, Vita G, Mazzeo A. Safety and tolerability of orally administered tafamidis meglumine in TTR FAP: preliminary data at 3-month follow-up. J Peripher Nerv Syst. 2012;17:S48–S49. [Google Scholar]
- Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–427. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- Andersson R. Familial amyloidosis with polyneuropathy. A clinical study based on patients living in northern Sweden. Acta Med Scand Suppl. 1976;590:1–64. [PubMed] [Google Scholar]
- Sousa A, Andersson R, Drugge U, Holmgren G, Sandgren O. Familial amyloidotic polyneuropathy in Sweden: geographical distribution, age of onset, and prevalence. Hum Hered. 1993;43:288–294. doi: 10.1159/000154146. [DOI] [PubMed] [Google Scholar]
- Araki S, Mawatari S, Ohta M, Nakajima A, Kuroiwa Y. Polyneuritic amyloidosis in a Japanese family. Arch Neurol. 1968;18:593–602. doi: 10.1001/archneur.1968.00470360015001. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology. 2002;58:1001–1007. doi: 10.1212/WNL.58.7.1001. [DOI] [PubMed] [Google Scholar]
- Bittencourt PL, Couto CA, Clemente C, Farias AQ, Palacios SA, Mies S, Goldberg AC. Phenotypic expression of familial amyloid polyneuropathy in Brazil. Eur J Neurol. 2005;12:289–293. doi: 10.1111/j.1468-1331.2004.00941.x. [DOI] [PubMed] [Google Scholar]
- Saporta MA, Zaros C, Cruz MW, Andre C, Misrahi M, Bonaiti-Pellie C, Plante-Bordeneuve V. Penetrance estimation of TTR familial amyloid polyneuropathy (type I) in Brazilian families. Eur J Neurol. 2009;16:337–341. doi: 10.1111/j.1468-1331.2008.02429.x. [DOI] [PubMed] [Google Scholar]
- Sousa A, Coelho T, Barros J, Sequeiros J. Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Povoa do Varzim and Vila do Conde (north of Portugal) Am J Med Genet. 1995;60:512–521. doi: 10.1002/ajmg.1320600606. [DOI] [PubMed] [Google Scholar]
- Dardiotis E, Koutsou P, Papanicolaou EZ, Vonta I, Kladi A, Vassilopoulos D, Hadjigeorgiou G, Christodoulou K, Kyriakides T. Epidemiological, clinical and genetic study of familial amyloidotic polyneuropathy in Cyprus. Amyloid. 2009;16:32–37. doi: 10.1080/13506120802676948. [DOI] [PubMed] [Google Scholar]
- Munar-Ques M, Costa PP, Saraiva MJ, Viader-Farre C, Munar-Bernat C. Familial type I (Portuguese form) amyloidotic polyneuropathy in Majorca. Study using the TTR (Met30) genetic marker. Med Clin (Barc) 1988;91:441–444. [PubMed] [Google Scholar]
- Munar-Ques M. Familial amyloid polyneuropathy 2003. Med Clin (Barc) 2003;121:100–101. doi: 10.1016/S0025-7753(03)73869-X. [DOI] [PubMed] [Google Scholar]
- Munar-Ques M. Up-date on amyloidosis. Hereditary amyloidosis. Med Clin (Barc) 1994;103:109–115. [PubMed] [Google Scholar]
- Tornero Estebanez C, Soriano Soriano C, Gimenez Escrich A, Rull Segura S. Late-onset familial amyloid polyneuropathy in the Safor (Valencia) area: four case reports. Rev Clin Esp. 2007;207:75–76. doi: 10.1157/13100199. [DOI] [PubMed] [Google Scholar]
- Gómez H, Araujo-Fernández S, Arca-Blanco A, Novoa-Lamazares L, González-Vázquez L, Sánchez-Conde P, Durán Muñoz O, De La Fuente Aguado J. Servicio de Medicina Interna. Hospital Povisa. Vigo. Polineuropatía amiloidótica familiar tipo 1 de Andrade: serie de 4 casos. Ferrol, Spain: In XXVIII Reunión de la Sociedad Gallega de Medicina Interna; 2011. 13-14 May. [Google Scholar]
- Munar-Ques M, Saraiva MJ, Viader-Farre C, Zabay-Becerril JM, Mulet-Ferrer J. Genetic epidemiology of familial amyloid polyneuropathy in the Balearic Islands (Spain) Amyloid. 2005;12:54–61. doi: 10.1080/13506120500032741. [DOI] [PubMed] [Google Scholar]
- Plante-Bordeneuve V, Ferreira A, Lalu T, Zaros C, Lacroix C, Adams D, Said G. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP) Neurology. 2007;13:693–698. doi: 10.1212/01.wnl.0000267338.45673.f4. [DOI] [PubMed] [Google Scholar]