

Abstract
The zinc-dependent deacetylase LpxC catalyzes the committed step of lipid A biosynthesis in Gram-negative bacteria and is a validated target for development of novel antibiotics to combat multidrug-resistant Gram-negative infections. Many potent LpxC inhibitors contain an essential threonyl-hydroxamate head group for high-affinity interaction with LpxC. We report the synthesis, antibiotic activity, and structural and enzymatic characterization of novel LpxC inhibitors containing an additional aryl-group in the threonyl-hydroxamate moiety, which expands the inhibitor-binding surface in LpxC. These compounds display enhanced potency against LpxC in enzymatic assays and superior antibiotic activity against F. novicida in cell culture. Comparison of the antibiotic activities of these compounds against a leaky E. coli strain and the wild-type strain reveals the contribution of the formidable outer membrane permeability barrier that reduces the compound efficacy in cell culture and emphasizes the importance of maintaining a balanced hydrophobicity and hydrophilicity profile in developing effective LpxC-targeting antibiotics.
Keywords: LpxC, antibiotic, lipid A biosynthesis, Gram-negative bacteria
INTRODUCTION
Gram-negative bacterial infections remain as a serious risk factor to public health.1 As the rate of multidrug-resistant Gram-negative bacterial infections continues to rise, these infections are becoming more difficult to treat, resulting in increased mortality and morbidity.2, 3 Gram-negative bacteria differ from Gram-positive bacteria in that they possess a unique outer membrane, with the outer leaflet of the outer membrane enriched with lipid A, the membrane anchor of lipopolysaccharide (LPS) and the active component of bacterial endotoxin.4 Lipid A shields bacteria from the damage of antibiotics and detergent molecules, and its biosynthesis is required for the survival of nearly all Gram-negative organisms. Since the essential lipid A pathway has never been exploited by existing antibiotics, its biosynthetic enzymes, particularly LpxC that catalyzes the second and committed step of lipid A biosynthesis, have drawn increasing attention as promising novel antibiotic targets.5 LpxC is a zinc-dependent UDP-3-O-(acyl)-N-acetylglucosamine deacetylase. Early studies by Merck researchers have led to the discovery of the first nanomolar LpxC inhibitor 1 (L-161,240) containing a zinc-binding hydroxamate group attached to a phenyl oxazoline scaffold (Figure 1A).6 This compound shows impressive antibiotic activities against E. coli in vitro and in vivo, but it is inactive against P. aeruginosa. In the last decade, tremendous progress in medicinal chemistry has resulted in steady improvement of the potency and spectrum of inhibition for LpxC-targeting antibiotics. Among well-characterized LpxC inhibitors, the most potent compound reported to date is the biphenyl diacetylene-based inhibitor 2 (LPC-011), which inhibits E. coli LpxC with a KI of ~0.039 nM and exhibits significant antimicrobial activity against a wide range of Gram-negative bacteria (Figure 1A).7
Figure 1. Structure of representative LpxC inhibitors and PaLpxC in complex with 5 (PDB entry: 3P3E).

(A) LpxC inhibitors show distinct structural scaffolds. (B) A hydrophobic patch, proximal to the active site, is a conserved structural feature across LpxC orthologs. Insert I and Insert II are highlighted in orange and blue, respectively. Inhibitor and side chains of protein forming zinc coordination and the hydrophobic patch are shown as sticks, and zinc ion is shown as a sphere. An additional rotameric state of the compound 5 threonyl head group is colored in gray.
Our previous structural and biochemical characterization of the substrate analog 3 (TU-514) bound to Aquifex aeolicus LpxC has revealed three conserved features of LpxC-inhibitor interactions in addition to the essential hydroxamate-zinc interaction, including the acyl-chain binding hydrophobic passage, a hydrophobic patch consisting of three phenylalanine residues adjacent to the passage, and a basic patch located at the opposite side of the active site. 8, 9 Subsequent studies of the threonyl-hydroxamate-containing biphenyl-acetylene compound 4 (CHIR-090) and biphenyl diacetylene compounds 5 (LPC-009) and 2 (Figure 1A) have further validated the important contributions of these three areas for efficient inhibitor interaction with LpxC.7, 10, 11 In particular, the biphenyl acetylene and biphenyl diacetylene “tail” groups of 4, 5, and 2 all insert into the hydrophobic passage, whereas their threonyl methyl group forms vdW contact with the first phenylalanine (F191 of P. aeruginosa LpxC, PaLpxC) of the hydrophobic patch, and the hydroxyl group forms a hydrogen bond with a catalytically important lysine residue (K238 of PaLpxC) of the basic patch (Figure 1B). It is interesting to note that in the PaLpxC/5 complex, the threonyl group can adopt an additional rotameric state (Figure 1B).11 In this alternative conformation, the threonyl methyl group points toward the K238, whereas the hydroxyl group faces up to form a hydrogen bond with the backbone carbonyl group of F191 of LpxC, leaving the F191-contacting methyl position unoccupied. The observation of two rotameric states of the compound 5 threonyl head group reveals the existence of additional space in the LpxC active site that can be further exploited to expand the inhibitor-LpxC interaction (Figure 1B).
Here, we describe the synthesis and biochemical and structural characterization of compound 2 derivatives containing an aryl group in order to enhance the inhibitor interaction with the hydrophobic patch of LpxC. The best compound of this series 24c is significantly more effective than 2 against Francisella novicida, the bacterium closely related with the category A Gram-negative pathogen Francisella tularensis. Although 24c possesses an overall similar antibiotic activity as 2 against wild-type E. coli and P. aeruginosa, it displays much enhanced activity against a leaky E. coli strain, suggesting that the membrane permeability barrier negatively affects the penetration of 24c and thus its potency. Detailed enzymatic characterization reveals a KI value of ~0.024 nM of 24c toward E. coli LpxC (EcLpxC), ~1.6-fold improvement over 2. This success demonstrates the feasibility to enhance the LpxC-inhibitor binding by expanding the interaction of the inhibitor head group with the hydrophobic patch of LpxC.
CHEMISTRY
Synthesis of 8a began with amide coupling between 4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzoic acid 6 7 and L-histidine methyl ester hydrochloride (Scheme 1). Then the methyl ester was converted to the corresponding hydroxamic acid 8a by treatment with hydroxylamine under basic conditions. Compounds 8b, 8c and 8d were synthesized by employing the same procedure.
Scheme 1.
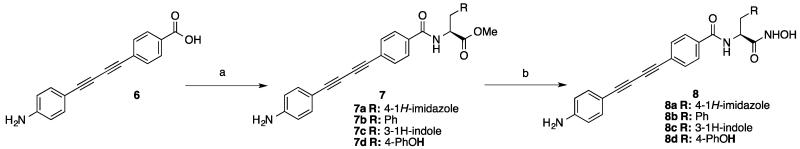
Synthesis of compound 8 a.
a Reagents and conditions: (a) EDCI, HOBt, DIPEA, DMF, Amino Acid, 0 °C-rt; (b) NH2OH.HCl, NaOMe, MeOH/THF, 0 °C-rt.
Intermediate serine aldehyde 14 (Scheme 2) 12, 13 was obtained from Cbz-L-serine 11. The oxetane tosylate 10 was prepared using standard conditions as a stable crystalline material with a 72% yield. Subsequent reaction of Cbz-L-serine with the oxetane tosylate 10, in the presence of 5% tetrabutylammonium iodide and triethylamine in anhydrous DMF afforded the desired L-serine oxetane ester 12. The formation of the ortho ester 13 from the oxetane ester 12 was performed in DCM with a catalytic amount of BF3.Et2O (3 mol%). Finally, oxidation of ortho ester 13, under Swern conditions, gave the intermediate serine aldehyde 14.
Scheme 2.

Synthesis of serine aldehyde 14a.
a Reagents and conditions: (a) TsCl, Pyridine, rt; (b) 10, tetrabutylammonium iodide , TEA, DMF, rt; (c) BF3•Et2O, TEA, 0 °C; (d) DMSO, (COCl)2, DIPEA,−78 °C.
Reaction of serine aldehyde 14 with different Grignard reagents led to the corresponding protected β-hydroxy amino acids 15a-15c (Scheme 3). The reaction was run at −78 °C in a mixture of DCM/THF or DCM/Et2O, resulting in reasonable yields. The β-hydroxy adducts were then oxidized under Swern conditions to afford the corresponding ketones 16a-16c in good yields. The oxidization products were purified by chromatography on silica gel without racemization. Reduction of the ketone 16a by LiBH4 at −78 °C regenerated the β-hydroxy amino acid 19, but with the opposite configuration at β-carbon.14, 15 Reaction of ketones 16a-16c with Grignard reagents afforded the corresponding dialkyl-β-hydroxy α-amino acid derivatives 17a-17c. Removal of the Cbz group from β-hydroxy amino acids 15a, 15b, 17a, 17b and 19 were accomplished by hydrogenolysis.
Scheme 3.

Synthesis of intermediate 18, 20, 21 a.
a Reagents and conditions: (a) R1MgBr, DCM, −78 °C; (b) DMSO, (COCl)2, DCM, −78 °C; (c) R2MgBr, DCM, −78 °C; (d) H2, Pd/C, MeOH, rt; (e) LiBH4, THF, −78 °C.
Under the standard amide coupling condition (EDC/HOBt/DIPEA), the β-hydroxy amino acids were reacted with compound 6 to give amide 22 (Scheme 4) in 70-90% yield. Then exposure of the oxa-bicyclic ortho-ester (OBO ester) 22 to a 1:1:1 mixture of dioxane/H2O/acetic acid allowed for quantitative opening of the OBO ester to give 23, which was then converted to the hydroxamic acid 24a, 24b, 24c and 24d, using hydroxylamine in the presence of KCN.
Scheme 4.

Synthesis of compound 24 a.
a Reagents and conditions: (a) EDCI, HOBt, DIPEA, DMF, Amino Acids 0 °C-rt; (b) 1:1:1:1 dioxane:HOAc:H2O, rt; (c) NH2OH, KCN, MeOH, 0 °C-rt.
As shown in Scheme 5, 29 was prepared starting from intermediate 17c, which was treated with TBAF to remove the TMS group. The terminal alkyne 25 was reacted with (azidomethyl)benzene under copper (I)-mediated conditions 16, 17 to give the triazole 26 regioselectively with concomitant ring-opening of the OBO ester to yield the diol. Amide coupling of compound 27 with compound 6 under basic conditions afforded the intermediate 28, which was then converted to the hydroxamic acid 29 as described above.
Scheme 5.

Synthesis of compound 29 a.
a Reagents and conditions: (a) TBAF, THF, 0 °C; (b) CuSO4•5H2O, Sodium L-ascorbate, tBuOH/H2O, rt; (c) H2, Pd/C, HCl, MeOH, rt; (d) EDCI, HOBt, DIPEA, DMF, 0 °C-rt ; (e) NH2OH, KCN, MeOH, 0 °C-rt.
RESULTS AND DISCUSSION
Aryl group substitution of the threonyl group
In our previous study of the LpxC/4 complex,10 we have noted that the compound 4 threonyl head group and UDP occupy adjacent, but non-overlapping conformational space in the LpxC active site, with the UDP pyrophosphate group located next to the Cβ atom of the threonyl head group of 4, suggesting that it is possible to enhance the compound 4-LpxC interaction by expanding the threonyl head group of 4 through the Cβ-position. Since nearly all LpxC inhibitors containing the threonyl head group share a similar binding mode, with the methyl group contacting F191 of the hydrophobic patch and the hydroxyl group forming a hydrogen bond with K238 of the basic patch, inclusion of an additional substitution would position such a functional group near the hydrophobic patch of LpxC. We reasoned that introduction of an additional aryl group at the Cβ position of the threonyl group could augment the inhibitor interaction with the hydrophobic patch of LpxC without disrupting existing compound-protein interactions, thus improving the potency of LpxC inhibitors. To search for a suitable aryl group, we first replaced the threonyl group of 2 with aromatic amino acids such as L-phenylalanine, L-tyrosine, L-tryptophan and L-histidine and evaluated their effectiveness as antibiotics against wild-type E. coli (W3110), E. coli (CMR300)—a Kdo-deficient strain with enhanced membrane permeability,18 P. aeruginosa (PAO1), and a modified E. coli strain with the native lpxC gene replaced by that of P. aeruginosa (W3110PA) (Table 1).7 In general, replacing the threonyl group with aromatic side chains reduces the antimicrobial activity. Among tested bacterial strains, the wild-type E. coli (W3110) is the most tolerant toward bulky aromatic functional groups, presumably due to a higher degree of conformational flexibility within the EcLpxC active site. Among the aromatic analogues, substitution of the threonyl group with a tryptophan group containing an indole ring at the Cβ position (8c) is most detrimental, resulting a >400-fold increase of the MIC (minimum inhibitory concentration) value for 8c (12.5 μg/mL) over 2 (0.03 μg/mL) against the wild-type E. coli (W3110). The phenylalanine substituent containing a benzene ring at the Cβ position (8b) similarly causes a significantly increase in MIC (6.3 μg/mL). In contrast, tyrosine and histidine analogs are much better tolerated, with MIC values of 0.8 μg/mL for the Tyr analog 8d and 0.4 μg/mL for the His analog 8a. It is interesting to note that while the MIC values of 8a are identical for the wild-type P. aeruginosa strain and the E. coli PaLpxC-knock-in strain W3110PA (6.3 μg/mL), 8d is ~3-fold less active against P. aeruginosa (12.5 μg/mL) than the W3110PA strain (4.7 μg/mL). Since the E. coli W3110PA strain also expresses PaLpxC instead of EcLpxC, the difference in the MIC values indicates that 8d is less efficient in penetrating the membrane of P. aeruginosa.
Table 1.
MICs of LpxC Inhibitors
| Code | Name | Structure | cLogD 7.4 | MIC (μg/mL) | ||||
|---|---|---|---|---|---|---|---|---|
|
E. coli
Wild type (W3110) |
E. coli
CMR300 |
E. coli
W3110PA |
P. aeruginosa
PAO1 |
Ratio
(W3110/ CMR300) |
||||
| 4 |
CHIR
-090 |

|
1.1 | 0.2 | 0.01 | 1.3 | 1.6 | 20 |
| 2 |
LPC
-011 |

|
2.61 | 0.03 | 0.001 | 0.32 | 0.32 | 30 |
| 8a |
LPC
-014 |

|
1.71 | 0.4 | 0.02 | 6.3 | 6.3 | 20 |
| 8b |
LPC
-025 |

|
4.19 | 6.3 | 0.014 | >50 | >50 | 450 |
| 8c |
LPC
-029 |

|
4.12 | 12.5 | 0.06 | >50 | >50 | 208 |
| 8d |
LPC
-033 |

|
3.45 | 0.8 | 0.01 | 4.7 | 12.5 | 80 |
| 24a |
LPC
-049 |
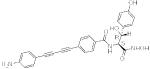
|
3.45 | 50 | 0.15 | >50 | >50 | 333 |
| 24b |
LPC
-050 |
|
3.45 | 0.13 | 0.003 | 0.9 | 1.0 | 43 |
| 24c |
LPC
-051 |
|
3.81 | 0.03 | 0.0001 | 0.5 | 0.7 | 300 |
| 24d |
LPC
-052 |

|
3.81 | 0.5 | 0.008 | 3.2 | 5.0 | 62 |
| 29 |
LPC
-072 |
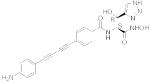
|
2.54 | 0.06 | 0.004 | 0.5 | 0.5 | 3.9 |
In order to determine whether the phenol group and imidazole ring at the Cβ position of the aromatic substituents occupy the previously noted UDP binding pocket of LpxC and could be used to augment the inhibitor interaction with the hydrophobic patch of LpxC, we determined the crystal structure of the PaLpxC/8a complex (Figure 2A). Our structure shows that the imidazole ring of 8a points toward and cuts across the phenyl ring of F191, forming a favorable π-π interaction. Additionally, the histidine side chain of 8a forms a hydrogen bond with the backbone carbonyl of F191and further enhances its interaction with LpxC. Most importantly, the histidine side chain of 8a occupies a conformational space not utilized by the threonyl group of 2; although we have not been able to capture a high-resolution structure of the LpxC/8d complex, the tyrosine analog is expected to adopt a similar side chain romateric position as the side chain of the histidine analog, suggesting that both the imidazole ring and the phenol group can be attached to the Cβ position of the threonyl head group to enhance its methyl interaction with the hydrophobic patch of LpxC, while maintaining a favorable hydrogen bond between its hydroxyl group and the catalytically important Lys residue of LpxC (K238 in PaLpxC) from the basic patch.
Figure 2. The aryl-substituted diacetylene LpxC Inhibitors 8a, 24b and 24c form specific interactions with the hydrophobic patch of LpxC.

(A–D) Shown are the binding modes of 8a (A), 24b (B) and 24c (C) around the hydrophobic patch proximal to the active site of LpxC (top panel). The bottom panels show multiple rotamer conformations of F193 in PaLpxC/8a and PaLpxC/24b complexes and lowest energy conformation of F193 in PaLpxC/24c. Purple and gray meshes represent the 2mFo – DFc electron densities for each inhibitor and protein residues, respectively. The contouring level of the electron map is 1.4 σ. Favorable protein-inhibitor interactions are denoted as dashed lines. Inhibitors and important protein residues are shown as sticks, and zinc ion is shown as a sphere.
Activity of Cβ-doubly and triply substituted compounds
To test this structure-based prediction, we modified the tyrosine analog 8d and histidine analog 8a by attaching a hydroxyl group, a methyl group, or both at the Cβ-position. The attachment of a hydroxyl group in the S-configuration to the tyrosine analog (24b) improves the antibiotic potency over the parental compound 8d by 5.2- to 12.5-folds against wild-type E. coli, P. aeruginosa (PAO1) and the E. coli W3110PA strain. The stereochemistry at the Cβ-position appears crucial for maintaining antibiotic activity of the tyrosine analogues. For example, 24a, of which the absolute configuration in the Cβ-position is inverted from S to R, displays significantly diminished activity compared to its S-diastereoisomer 24b (over 500-fold for E. coli). Such a strict chirality requirement is consistent with the inhibitor binding mode revealed in the crystal structure of the PaLpxC/24b complex (Figure 2B), where the appended hydroxyl group of 24b forms a hydrogen bond with the conserved K238, and the inhibitor’s phenol ring interacts with the F191. With the same overall binding mode, but opposite chirality, both of these favorable interactions are lost, resulting in a large energetic penalty. Inclusion of an additional methyl attachment to the Cβ-position also requires strict stereochemistry. 24c containing a Cβ-methyl substitution in the S-configuration displays further improved potency (1.5- to 3-folds) over the parent compound 24b against the wild-type E. coli, P. aeruginosa and the E. coli W3110PA strain. In contrast, the R-diastereomer 24d shows reduced antibiotic activity (3- to 5-folds) compared to the parent 24b.
In order to obtain direct evidence that inclusion of the phenol group indeed expands the interaction between the inhibitor and the hydrophobic patch of LpxC, we also determined the crystal structure of PaLpxC/24c. The complex structure (Figure 2C) reveals that the hydrogen bond between the inhibitor Cβ-hydroxyl group and K238 of LpxC remains intact, and the inhibitor Cβ-methyl group similarly makes a favorable vdW contact with the side chain of F191 of LpxC as previously observed in the 2 complex. The additional phenol ring of 24c points toward F191 with its surface crossing F191 at a nearly perpendicular angle, forming a favorable T-shaped π-π interaction. Furthermore, the phenolic hydroxyl group picks up an additional OH-π interaction with adjacent F193 located within the same hydrophobic patch. Indeed, the side chain of F193 is sufficiently flexible to display two rotameric states in the PaLpxC/8a, PaLpxC/2 and PaLpxC/24b complexes, whereas it is held in a low energy rotamer by the phenolic hydroxyl group of 24c. A similar attempt was made to attach the methyl and hydroxyl groups to the Cβ-position of the 8a. In this case, the imidazole of the 8a was replaced by a triazole ring in order to facilitate chemical synthesis. This modification (compound 29) also increases the antibiotic activity over 8a by 6.7- to 12.6-fold against the wild-type E. coli, P. aeruginosa, and E. coli (W3110PA). Of note, 29 shares the same stereo center as 24c at the Cβ position, despite an apparent inversion of nomenclature (S to R) at this position due to the change of IUPAC priority of the substituted groups (Table 1).
To examine the overall antibiotic profiles of the aryl analog 24c and 29, we measured their MICs against a panel of Gram-negative bacterial strains, including S. typhimurium, K. pneumoniae, V. cholerae and F. novicida, a non-virulent subspecies of F. tularensis (Table 2). While 24c and 29 have an overall antimicrobial potency comparable to that of 2 for many tested bacterial species, both compounds show enhanced activity (2- to 4-fold) over 2 against F. novicida, supporting the notion that expanding the inhibitor interaction with LpxC via the hydrophobic patch can be utilized to design more potent inhibitors.
Table 2.
Antibiotic Profile of Lead Compounds
| Pathogen | MIC (μg/mL) |
|||
|---|---|---|---|---|
| 4 | 2 | 24c | 29 | |
| Escherichia coli | 0.2 | 0.03 | 0.03 | 0.06 |
| W3110 | ||||
| Pseudomonas aeruginosa | 1.6 | 0.32 | 0.7 | 0.5 |
| PA01 | ||||
| Salmonella typhimurium | 0.16 | 0.024 | 0.05 | 0.1 |
| LT2 | ||||
| Klebsiella pneumoniae | 0.64 | 0.13 | 0.16 | 0.25 |
| 43816 | ||||
| Vibrio cholerae | ||||
|
P4 (P27459ΔctxAB::KmR,
SmR) |
0.16 | 0.01 | 0.016 | 0.03 |
| Francisella novicida | 3.1 | 2.5 | 0.63 | 1.0 |
| U112 | ||||
Effect of the LPS membrane barrier on antibiotic activities of LpxC inhibitors
In order to determine whether a lack of improvement in antibiotic activities of 24c and 29 over 2 against E. coli is due to a lack of enhanced inhibition of EcLpxC by these compounds or due to other factors, such as membrane impermeability, we determined the inhibition constants of these two inhibitors against EcLpxC using an enzymatic assay. Both 24c and 29 potently inhibit EcLpxC with a KI value of 0.024 ± 0.004 nM, corresponding to ~1.6-fold improvement of potency over 2 (KI =0.039 ± 0.003 nM). The more efficient inhibition of EcLpxC by 24c and 29 in vitro is consistent with the expanded interaction surface between the additional inhibitor aryl group and the hydrophobic patch of LpxC, indicating that a lack of improvement in antibiotic activities of these compounds in cell culture is likely caused by issues related to the membrane impermeability.
The outer monolayer of the outer membrane of Gram-negative bacteria is enriched with lipid A-anchored LPS containing O-antigen repeats, which acts as a predominant shield against hydrophobic molecules. In order to dissect the contribution of the LPS barrier to impeding membrane penetration of LpxC inhibitors, we tested our compounds against a leaky E. coli strain (CMR300) that lacks the core saccharides and O-antigen repeats of lipopolyssacharide due to a deletion of the kdtA gene. 18 Although all compounds display reduced MIC values against the leaky E. coli strain, the more hydrophobic compounds experience a greater effect (Table 1), consistent with the notion that LPS is a major shield against hydrophobic molecules. Since LpxC contains a hydrophobic substrate passage and a hydrophobic patch in the active site, potent LpxC inhibitors are typically more hydrophobic compared to antibiotics inhibiting other targets. However, our results suggest that despite enhanced potency against LpxC in vitro, a more hydrophobic compound may not necessarily translate its tighter inhibition in enzymatic assays into a better antibiotic in cell culture, as the formidable LPS barrier discriminates against the more hydrophobic compounds. Therefore, a balanced hydrophobic and hydrophilic profile is most efficient in translating in vitro potency of LpxC inhibitors to antibiotic activities in cell cultures.
CONCLUSION
In this study, we have designed and synthesized new series of diacetylene-based threonyl-hydroxamate derivatives containing an additional aryl head group in order to enhance the inhibitor interaction with the hydrophobic patch of LpxC, a conserved feature across distinct LpxC orthologs. Our structure-activity relationship study shows that attaching imidazole- (or triazole-) and tyrosyl-substituents at the Cβ-position of the threonyl group of 2 introduces additional interactions with the hydrophobic patch of LpxC and augments the existing threonyl methyl and hydroxyl group interactions with LpxC. Such an expanded inhibitor-LpxC interaction has resulted in a further reduction of KI values to ~24 pM in E. coli LpxC enzymatic assays and 4-fold improved antibiotic activity against F. novicida for the best compound 24c. The newly identified aryl derivative series show impressive antibiotic activity against a wide range of Gram-negative bacteria and highlight the Cβ-position of the threonyl group in 2 as an effective branch point to expand the binding interface of existing LpxC inhibitors. Our study additionally highlights the importance of balancing the hydrophilicity and hydrophobicity for development of effective antibiotics targeting Gram-negative bacteria. Such an observation is particularly important in future efforts to develop in vivo active antibiotics targeting LpxC based on in silico screening or structural insights. These compounds can be readily solubilized at concentrations of 10-40 mg/mL in 100 mM β-cyclodextrin, making them compatible with IV formulation for further evaluations of safety and efficacy in mouse infection models. Our compounds include both acetylene and hydroxamate moieties, residues that are sometime associated with metabolic toxicity.19, 20 However, both acetylene and hydroxamate groups occur in FDA-approved drugs (such as efavirenz,21 linagliptin,22 and vorinostat23) for chronic administration, and their presence in LpxC inhibitors intended for short-term treatment of Gram-negative infections should not prevent the deployment of this novel class of antibiotics.
EXPERIMENTAL SECTION
MIC tests
The MIC of each compound was tested in triplicate as described previously using a modified NCCLS protocol, which is adapted to 96-well plates and LB media in presence of 5% DMSO.7, 11 100 μL of various concentrations of compounds are prepared on a standard 96-well plate (Corning Costar 3596, flat bottomed with lid, polystyrene wells), in 2-fold dilution series with range of 0.0005 to 1 μg/mL, 0.0025 to 5 μg/mL or 0.024 to 50 μg/mL. Bacterial cells, grown to OD600 = 0.6, were diluted 100-fold with fresh LB medium, and 100 μL of the diluted cells was added into each well. The inoculated plates were incubated at 37 °C for 22 hours. After the incubation, 50 μL of 1 mg/mL [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide solution (MTT) was added and incubated at 37 °C for another 3 hr. The MIC was considered the lowest concentration of compound that showed no visible color change (yellow to black).
LpxC enzymatic inhibition assay
The substrate for the enzymatic assay, UDP-3-O-[(R)-3-hydroxymyristoyl]-N-acetylglucosamine and [α-32P] UDP-3-O-[(R)-3-hydroxymyristoyl]-N-acetylglucosamine, were prepared as previously described.24 The assays were performed in 25 mM HEPES buffer, pH 7.4, 100 mM KCl, 1 mg/mL BSA, 2 mM DTT, 0.5 μM substrate with 10% DMSO or 0.19 pM to 12.5 nM inhibitor in DMSO. The reactions were initiated with addition of 2 pM of purified E. coli LpxC. Aliquots were taken after 1 to 4 hours of incubation at 30 °C to quantify reaction progression (< 5% conversion of substrate to product). The kinetic parameters for enzyme activity in the presence of inhibitors were determined as reported previously. 7, 11 Briefly, K appI were determined by plotting fractional activity versus inhibitor concentration and fitting to Morrison’s quadratic equation. 25 KM and Vmax values were determined by varying substrate concentration from 0.5 to 50 μM. Finally, the KI values were calculated based on the relationship between K appI and KI for a tight binding competitive inhibitor. 25 All measurements were done in duplicates.
Crystallization and structure determination
Protein samples were prepared as described previously. 11 Purified PaLpxC (1-299, C40S) in a buffer of 25 mM HEPES, pH 7.1, 50 mM NaCl and 2 mM TCEP (tris(2-carboxyethyl)phosphine) was diluted to a final concentration of 12 mg/mL with the same buffer. A four-fold molar excess of individual compounds, dissolved in DMSO, was mixed with the diluted protein and then 10 mM zinc sulfate was added into the complex sample. Crystals of PaLpxC in complex with 8a, 24b or 24c were obtained using the sitting-drop vapor-diffusion method at 20 °C in drops containing 1 μL of the inhibitor-protein complex and 1 μL of well solution consisting of 0.1 M sodium acetate trihydrate (pH 4.7-5.2) and 2.4-2.7 M ammonium nitrate. The crystals were cryoprotected with perfluoropolyether (PFO-X175/08) before flash-freezing. Diffraction data were collected in-house at 100 K using a Rigaku MicroMax-007 HF rotating anode generator and R-Axis IV++ detector. The collected diffraction data were processed with HKL2000. 26 Molecular replacement using AUTOMR in the PHENIX package 27 was carried out by using the structure of the PaLpxC/5 complex (PDB entry 3P3E) as the search model. The final coordinate was completed by iterative cycles of model building (COOT) and refinement (PHENIX). 27, 28 The statistics for the LpxC-inhibitor complexes are shown in Table 3.
Table 3.
X-ray crystallography data collection and refinement statistics
| PaLpxC/8a | PaLpxC/24b | PaLpxC/24c | |
|---|---|---|---|
| Resolution range (Å) | 24.29 - 1.60 (1.66 - 1.60)a | 28.27 - 1.57 (1.62 - 1.57) | 19.86 - 1.60 (1.65 - 1.60) |
| Space group | P 21 21 21 | P 21 21 21 | P 21 21 21 |
| Cell dimensions | |||
| a, b, c (Å) | 53.19 73.82 88.18 | 53.53 73.62 88.29 | 53.20 73.53 88.04 |
| a, P, Y(°) | 90 90 90 | 90 90 90 | 90 90 90 |
|
Reflections
(unique/total) |
42841 / 237334 | 48969/141160 | 45214 / 275467 |
| Completeness (%) | 91.88 (98.87) | 98.72 (98.46) | 97.06 (93.07) |
| I/σ | 20.52 (4.27) | 18.92 (5.26) | 15.42 (2.78) |
| Wilson B-factor | 24.27 | 20.87 | 24.47 |
| R-merge (%) | 4.2 (45.3) | 3.7 (24.3) | 6.1 (45.3) |
| R-factor | 0.191 (0.215) | 0.179 (0.210) | 0.180 (0.248) |
| R-free | 0.220 (0.255) | 0.206 (0.262) | 0.215 (0.294) |
| Number of atoms | 2707 | 2849 | 2755 |
| macromolecules | 2333 | 2332 | 2321 |
| ligands | 78 | 100 | 102 |
| water | 296 | 417 | 332 |
| protein residues | 298 | 296 | 296 |
| RMS (bonds) | 0.004 | 0.004 | 0.009 |
| RMS (angles) | 0.90 | 0.89 | 1.24 |
|
Ramachandran
favored (%) |
98 | 98 | 97 |
|
Ramachandran
outliers (%) |
0 | 0 | 0 |
| Clashscore | 4.44 | 5.29 | 5.31 |
| Average B-factor | 24.20 | 23.30 | 26.10 |
| macromolecules | 22.80 | 20.80 | 24.00 |
| solvent | 33.70 | 35.90 | 37.80 |
Values in parentheses are for highest-resolution shell.
Chemical synthesis
LC/MS analysis was conducted on an Agilent 1200 HPLC with a quadrupole mass analyzer. LC chromatography used an Agilent XDB-C18 column (4.6×50 mm, 1.8 m) with a water/acetonitrile (each with 0.2% (v/v) formic acid) gradient at a flow rate of 0.5 mL/min. HRMS analyses were performed at the Duke MS Center. 1H and 13C spectra were recorded at 300 (400) and 75 (100) MHz, respectively, on a Varian Spectrometer. Column chromatography was conducted using either silica gel (Silicycle 40-64 m) or prepacked RediSep columns (Teledyne Isco Inc., Lincoln, NE) on an Isco CombiFlash Rf instrument. All moisture-sensitive reactions were carried out using dry solvents and under a slight pressure of ultra-pure quality argon. Glassware was dried in an oven at 140 °C for at least 12h prior to use, and then assembled quickly while hot, sealed with rubber septa, and allowed to cool under a stream of argon. Reactions were stirred magnetically using Teflon-coated magnetic stirring bars. Commercially available disposable syringes were used for transferring reagents and solvents. All reagents were used as received from commercial sources, unless specified otherwise, or prepared as described in the literature. The purities of all compounds were determined to be > 95% by NMR and LC-MS.
General Procedure for the amide coupling (Method A). To a stirred mixture of 4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzoic acid (100 mg, 0.39 mmol, 1.00 equiv) and amino acid hydrochloride (0.54 mmol, 1.40 equiv) in anhydrous DMF (5 mL) was added N-ethyl-N’-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC hydrochloride) (103 mg, 0.54 mmol, 1.40 equiv) and 1-hydroxybenzotriazole (HOBt) (73 mg, 0.54 mmol, 1.40 equiv) at room temperature. Then the mixture was cooled to 0 °C, and diisopropylethylamine (DIEA) (0.27 mL, 1.53 mmol, 4.00 equiv) was added. The whole reaction mixture was stirred at 0 °C under argon for 1h, then was allowed to warm to temperature with the stirring continued overnight (18h). The resulting yellow solution was concentrated to dryness with a rotary evaporator, and the residue was treated with water (20 mL), extracted with EtOAc (3×50 mL). The combined extracts were washed with water (20 mL), brine (20 mL), and dried over anhydrous Na2SO4. Evaporation of the solvent afforded the crude products, which was purified by flash chromatography.
General Procedure for the hydroxamic acid (Method B). To an ice-cold solution of ester (0.23 mmol, 1.00 equiv) dissolved in anhydrous MeOH (1 mL) and THF (1 mL) was added hydroxylamine hydrochloride (104 mg, 1.50 mmol, 5.00 equiv) followed by 25% sodium methoxide in methanol solution (0.54 mL, 2.25 mmol, 7.50 equiv). The reaction mixture was stirred at 0 °C under argon for 2h, then was allowed to warm to ambient temperature with the stirring continued overnight (16h). The resulting yellow suspension was condensed to dryness with a rotary evaporator, and the residue was treated water (20 mL), acidified by 0.5 N HCl to pH 6~7, and extracted with EtOAc (3×50 mL). The combined extracts were washed with water (20 mL), brine (20 mL), and dried over anhydrous Na2SO4. Evaporation of the solvent afforded the crude product, which was purified by flash chromatography.
General Procedure for the hydroxamic acid (Method C). Ester (0.10 mmol, 1.00 equiv) was dissolved in a mixture of THF/MeOH (1:1 volume) 2.0 mL. 50% aq hydroxylamine (0.20 mL, 3.0 mmol, 30.00 equiv) was added followed by potassium cyanide (6.5 mg, 0.10 mmol, 1.00 equiv) at room temperature under argon. The reaction mixture was stirred at room temperature for 14h. Then the yellow solution was concentrated to dryness. The residue was treated with water (30 mL), acidified by 0.5 N HCl to pH 6~7, extracted with EtOAc (3×50 mL). The combined organic layers were washed with water (30 mL), brine (30 mL), and dried (anhydrous Na2SO4). The crude product was purified by flash chromatography.
General Procedure for Swern Oxdiation (Method D). The alcohol (21.27 mmol, 1.00 equiv) was dissolved in anhydrous DCM (100 mL) under argon and cooled to −78 °C in flask I. Oxalyl chloride (17.00 mL, 2.0 M in DCM, 70.19 mmol, 1.60 equiv) was added to anhydrous DCM (100 mL) in a separate flask II under argon and cooled to −78°C. Anhydrous DMSO (5.36 mL, 70.19 mmol, 3.30 equiv) was added to the oxalyl chloride solution (flask II), and the mixture was stirred at −78 °C for 15 min. The alcohol solution was transferred by syringe at −78 °C to the flask II over a period of 20 min. The resulting cloudy white mixture was stirred for 1.5h at −78 °C. DIPEA (18.44 mL, 106.35 mmol, 5.0 equiv) was added, and the reaction mixture was stirred for 30 min at −78 °C and 10 min at 0 °C. Ice-cold DCM (200 mL) was added, and the solution was washed with ice-cold 3% NH4Cl (3×200 mL), brine (200 mL), dried (anhydrous Na2SO4). The crude product was crystallized from DCM/Hexane.
General Procedure for Grignard Addition (Method E). Aldehyde or ketone (5.00 mmol, 1.00 equiv) was dissolved in anhydrous DCM (20 mL) under argon. A solution of RMgBr in THF (20 mL, 20 mmol, 4.00 equiv) was added quickly by syringe at −78 °C or 0 °C and stirred vigorously. The reaction mixture was stirred at −78 °C for 30 min-2h. Then the reaction mixture was quenched by saturated NH4Cl (300 mL, pH~7) and was stirred for an additional 15 min. The mixture was extracted with DCM (3×80 mL). The combined organic layers were washed with water (50 mL), brine (50 mL), dried (anhydrous Na2SO4). The crude product was purified by flash chromatography or recrystallization.
General Procedure for hydrogenolysis (Method F). To a solution of Cbz protected compound (1.2 mmol) in anhydrous MeOH (10 mL) was added 10% Pd/C (10 w/w%) at room temperature under a balloon of hydrogen. The reaction mixture was stirred for 14h. Then the catalyst was removed by filtration through a pad of celite and washed with MeOH (50 mL). The filtrate was evaporated under reduced vacuum to obtain the crude product, which was utilized for further transformation without any purification.
General Procedure for ring-opening of OBO ester (Method G). OBO ester (0.286 mmol) was dissolved in dioxane (1.0 mL). The mixture was diluted with acetic acid (1.0 mL) and water (1.0 mL) at room temperature under argon and the resulting suspension was stirred for 70 min. Then the yellow clear solution was concentrated to dryness. The residue was treated with saturated NaHCO3 (20 mL) to pH~10, extracted with EtOAc (3×40 mL). The combined organic layers were washed with water (2×30 mL), brined (30 mL), and dried (anhydrous Na2SO4). The crude product was purified by flash chromatography.
(S)-Methyl 2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-(1H-imidazol-4-yl)propanoate (7a)
Method A, (0.97 g, 45% yield) as white solid. 1H NMR (300 Hz, CDCl3) δ 3.20 (s, br, 2H), 3.67 (s, 3H), 4.93 (s, br, 1H), 6.59 (d, J=8.7 Hz, 2H), 7.32 (d, J=11.1Hz, 2H), 7.54 (d, J=8.4 Hz, 2H), 7.80-7.83 (m, J=8.4 Hz, 2H), 8.35 (s, br, 1H); 13C NMR (75 Hz, CDCl3) δ 29.24, 52.65, 53.38, 72.07, 79.78, 80.10, 83.07, 84.44, 110.45, 114.84, 115.079, 125.99, 127.52, 132.49, 132.65, 134.41, 135.47, 148.06, 166.70, 172.17; LCMS: 413.1 [M+H+].
(S)-Methyl-2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-phenylpropanoate (7b)
Method A, (143 mg, 89% yield) as yellow solid. 1H NMR (300 Hz, DMSO-d6) δ 3.06-3.19 (m, J=39 Hz, 1H), 3.62 (s, 1H), 4.60-4.68 (m, J=5.846 Hz, 2H), 6.53 (d, J=8.7 Hz, 2H), 7.16-7.27 (m, J=34.2 Hz, 7H), 7.62 (d, J=8.1 Hz, 2H), 7.78 (d, J=8.4 Hz, 2H), 8.96 (d, J=7.8 Hz, 1H); 13C NMR (75 Hz, DMSO-d6) δ 36.86, 52.71, 55.05, 71.77, 77.40, 80.75, 86.45, 105.77, 114.26, 124.96, 127.21, 218.43, 128.95, 129.74, 132.70, 134.38, 134.71, 138.30, 144.23, 151.574, 166.23, 172.75; LCMS: m/s 424.2 [M+2H]+.
(S)-methyl-2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-(1H-indol-3-yl)propanoate (7c)
Method A, (173 mg, 98% yield) as yellow solid. 1H NMR (300 Hz, CD3OD) δ 3.29-3.40 (m, J=31.8 Hz, 2H), 3.41 (d, J=5.4 Hz, 1H), 3.70 (s, 3H), 6.60 (d, J=8.4 Hz, 2H), 6.99 (t, J=14.4 Hz, 1H), 7.06-7.11 (m, J=15.0 Hz, 2H), 7.23 (d, J=8.1 Hz, 2H), 7.32 (d, J=7.8 Hz, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.55 (d, J=7.8 Hz, 1H), 7.69 (d, J=8.1 Hz, 2H); 13C NMR (75 Hz, CD3OD) δ 27.05, 51.63, 54.44, 70.81, 76.61, 79.034, 84.55, 108.33, 109.78, 111.22, 114.23, 117.98, 118.73, 121.36, 123.23, 125.89, 127.49, 132.00, 133.83, 136.88, 150.19, 168.07, 172.86; LCMS: m/s 462.3 [M+H]+.
(S)-Methyl-2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-(4-hydroxyphenyl)propanoate (7d)
Method A, (148 mg, 90% yield) as yellow solid.1H NMR (300 Hz, CD3OD) δ 3.00 (q, J=24.3 Hz, 1H), 3.18 (q, J=19.5 Hz, 1H), 3.70 (s, 3 H), 4.81 (q, J=40.5 Hz, 1H), 6.61 (d, J=8.7 Hz), 6.70 (d, J=8.7 Hz, 2H), 7.06 (d, J=8.7 Hz, 2H), 7.24 (d, J=8.7 Hz, 2H), 7.52 (d, J=8.7 Hz, 2H), 7.71 (d, J=8.7 Hz, 2H); 13C NMR (75 Hz, CD3OD) δ 36.23, 51.61, 55.07, 70.81, 76.62, 79.05, 84.57, 108.32, 114.23, 115.12, 125.92, 127.48, 127.84, 130.06, 132.04, 133.84, 150.20, 156.22, 168.10, 172.56; LCMS: m/s 439.5 [M+H]+.
(S)-4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-(1-(hydroxyamino)-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl)benzamide (8a)
Method B, (24 mg, 48% yield) as yellow solid. 1H NMR (300 Hz, DMSO-d6) δ 3.12 (q, 2H, J=20.7 Hz, 4.74 (q, 1H, J=14.4 Hz), 6.62 (d, 2H, J=9.0 Hz), 6.93 (s, 1H), 7.22 (d, 2H, J=8.7 Hz), 7.51-7.56 (m, 3H, J=14.1 Hz), 7.68 (d, 1H, J=1.5 Hz), 7.76-7.80 (m, 3H, J=12.6 Hz); 13C NMR (75 Hz, DMSO-d6) δ 29.24, 52.06, 70.70, 76.59, 78.96, 80.01, 84.56, 108.28, 114.20, 116.74, 125.96, 127.54, 131.85, 131.99, 133.79, 135.09, 150.24, 167.80, 169.17; HRMS HRMS: m/z calc’d for C23H19N5O3[M]+: 413.1488; found: 413.1490.
(S)-4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-(1-(hydroxyamino)-1-oxo-3-phenylpropan-2-yl)benzamide (8b)
Method B, (77 mg, 77% yield) as yellow solid.1H NMR (300 Hz, DMSO-d6) δ 3.00 (d, J=6.6 Hz 2H), 4.57 (d, J=7.2 Hz, 1H), 5.84 (s, 2H), 6.53 (d, J=8.7 Hz, 2H), 7.15 (d, J=7.2 Hz, 2H), 7.20-7.30 (m, J=30.6, 5H), 7.58 (d, J=8.1 Hz, 2H); 13C NMR (75 Hz, DMSO-d6) δ 38.13, 53.66, 71.80, 77.23, 80.84, 86.33, 105.82, 114.26, 124.63, 126.94, 128.50, 129.77, 132.52, 134.70, 138.94, 151.54, 165.91, 168.46; HRMS: m/z calc’d for C26H21N3O3 [M]+: 423.1583; found: 423.1585.
(S)-4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-(1-(hydroxyamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)benzamide (8c)
Method B, (77 mg, 70% yield) as yellow solid.1H NMR (300 Hz, DMSO-d6) δ 3.13 (d, J=6.9 Hz, 2H), 4.63 (q, J=15.6 Hz, 1H), 5.84 (s, 2H), 6.52 (d, J=8.4 Hz, 2H), 6.93-7.05 (m, J=35.7 Hz, 2H), 7.18 (s, 1H), 7.22-7.29 (m, J=21.9 Hz, 3H), 7.58 (d, J=8.1 Hz, 2H), 8.70 (d, J=8.1 Hz, 1H), 8.88 (s, 1H), 10.76 (s, 1H), 10.86 (s, 1H); 13C NMR (75 Hz, DMSO-d6) δ 28.31, 52.74, 71.80, 77.25, 80.84, 86.34, 105.81, 110.94, 112.00, 114.26, 118.94, 119.18, 121.58, 124.41, 124.64, 127.83, 128.55, 132.51, 134.71, 134.88, 136.72, 151.56, 165.92, 169.04; HRMS: m/z calc’d for C28H22N4O3[M]+: 462.1692; found: 462.1694.
(S)-4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-(1-(hydroxyamino)-3-(4-hydroxyphenyl)-1-oxopropan-2-yl)benzamide (8d)
Method B, (74 mg, 67% yield) as yellow solid.1H NMR (300 Hz, DMSO-d6) δ 2.87 (d, J=7.2 Hz, 2H), 4.47 (d, J=23.4 Hz, 1H), 5.83 (s, 2H), 6.53 (d, J=8.7 Hz, 2H), 6.60 (d, J=8.4 Hz, 2H), 7.08 (d, J=8.4 Hz, 2H), 7.24 (d, J=8.4 Hz, 2H), 7.59 (d, J=8.1 Hz, 2H), 7.80 (d, J=8.4 Hz, 2H), 8.68 (d, J=8.4 Hz, 1H), 8.86 (s, 1H), 9.14 (s, 1H), 10.75 (s, 1H); 13C NMR (300 Hz, DMSO-d6) δ 53.96, 71.81, 77.23, 80.85, 86.33, 105.82, 114.26, 115.59, 124.63, 128.53, 128.86, 130.74, 132.53, 134.70, 134.93, 151.55, 156.42, 165.96.168.75; HRMS: m/z calc’d for C26H21N3O4[M]+]: 439.1532; found: 439.1533.
(3-methyloxetan-3-yl)methyl 4-methylbenzenesulfonate (10)
To a stirred solution of p-toluenesulfonyl chloride (74.36 g, 0.39 mol, 1.5 equiv) in anhydrous pyridine (300 mL) was added dropwise 3-methyl-3-oxetane-methanol (26.52 g, 0.26 mol) over 10 min at 0°C under argon. After 5 min, the reaction mixture was allowed to warm to room temperature with the stirring continued for an additional 1.5h. The mixture was then slowly added to a vigorously stirred mixture of milliQ water (800 mL) and crushed ice (800 g) for 30 min. Then the white precipitate was collected on Whatman filter #1 and washed with cold water (300 mL). The product was dried under high vacuum to obtain a white power of oxetane tosylate 10 (47.88 g 72% yield). 1H NMR (300 MHz, CDCl3) δ 1.28 (s, 3H), 2.43 (s, 3H), 4.08 (s, 2H), 4.29-4.35 (m, 2H), 7.34 (d, J=7.8 Hz, 2H), 7.78 (d, J=7.5 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 20.85, 21.86, 39.45, 74.50, 79.13, 128.15, 130.22, 132.81, 145.34; LCMS: m/s 164.9 [M+H]+.
(S)-(3-methyloxetan-3-yl)methyl-2-(((benzyloxy)carbonyl)amino)-3-hydroxypropanoate (12)
A solution of Cbz-L-Ser 11 (29.60 g, 0.167 mol), 3-methyl-(3-tosylmethyl oxetane) 10 (29.95 g, 0.125 mol), tetrabutylammonium iodide (2.30 g, 0.06 mol, 0.05 equiv) and TEA (14.04 g, 0.138 mol, 1.1 equiv) in anhydrous DMF (100 mL) was slowly heated to 70°C for 36h under argon. The reaction mixture was allowed to cool to room temperature, and the DMF was removed under reduced pressure. The remaining residue was dissolved in 1.0 L of EtOAc, washed with 1.0 N HCl (2×250 mL), saturated NaHCO3 (2×250 mL), brine (250 mL), and dried (anhydrous Na2SO4). The solvent was removed under reduced pressure, and the remaining residue was purified on a CombiFlash system (eluting with 0-50% EtOAc in hexane) to give 12 (25.20 g, 64% yield) as pale yellow oil. 1H NMR (300 MHz, CDCl3) δ 1.26 (s, 3H), 3.34 (br, s, 1H), 3.84-3.87 (m, 1H), 4.02-4.07 (m, 2H), 4.35-4.52 (m, 6H), 5.11 (s, 2H), 5.94 (d, J=7.5 Hz, 1H), 7.26-7.33 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 20.98, 39.70, 56.57, 63.39, 67.34, 69.18, 79.66, 128.33, 128.44, 128.75, 136.38, 156.51, 170.98; LCMS: m/s 324.1 [M+H]+.
(S)-benzyl-(2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)ethyl)carbamate (13)
Cbz-L-Ser-oxetane ester 12 (11.00 g, 34.00 mmol) was dissolved in anhydrous DCM (100 mL) and was cooled to 0°C under argon. BF3.Et2O (0.13 mL, 0.92 mmol, 0.03 equiv) was diluted in DCM (5.0 mL) and added to the reaction flask. The reaction mixture was allowed to warm to room temperature. After 5h, Et3N (1.40 mL, 9.86 mmol, 0.3 equiv) was added with the stirring continued for an additional 30 min. Then the result solution was concentrated to dryness to thick oil. The crude product was purified on CombiFlash (eluting with 0-5% MeOH in DCM) to afford 13 (6.90 g, 62.7% yield) as white foam. 1H NMR (300 MHz, CDCl3) δ 0.78 (s, 3H), 2.64 (q, J=12.6 Hz, 1H), 3.62-3.95 (m, 9H), 5.03-5.14 (m, 2H), 5.37 (d, J=9.0 Hz, 1H), 7.26-7.34 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 14.48, 30.76, 55.60, 62.14, 67.12, 72.90, 108.68, 128.28, 128.36, 128.69, 136.69, 156.67; LCMS: m/s 324.1 [M+H]+.
(S)-benzyl-(1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)-2-oxoethyl)carbamate (14)
Method D, (4.3 g, 63% yield) as yellow solid. 1H NMR (300 MHz, CDCl3) δ 0.81 (s, 3H), 3.93 (s, 6H), 4.60 (d, J=8.7 Hz, 1H), 5.12 (s, 2H), 5.36 (d, J=8.7 Hz, 1H), 7.26-7.40 (m, 5H), 9.68 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 14.47, 31.09, 63.50, 67.43, 73.12, 107.40, 128.36, 128.70, 136.40, 156.42, 159.90; LCMS: m/s 307.4 [M+H]+.
Benzyl((1S,2R)-2-(4-(benzyloxy)phenyl)-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)ethyl)carbamate (15a)
Method E, (0.272 g, 54% yield) as foam solid. 1H NMR (300 MHz, CDCl3) δ 0.83 (s, 3H), 3.36 (s, 1H), 3.98 (s, 6H), 4.07 (d, J=10.5 Hz, 1H), 4.88-5.08 (m, 5H), 5.48 (d, J=10.5 Hz, 1H), 6.91 (d, J=9.0 Hz, 2H), 7.22-7.46 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 14.56, 30.93, 58.87, 66.76, 70.16, 70.65, 73.06, 109.04, 114.72, 127.36, 127.77, 127.99, 128.14, 128.63, 128.80, 132.84, 136.99, 137.414, 156.61, 158.33; LCMS: m/s 506.2 [M+H]+.
Benzyl((1S,2R)-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)propyl)carbamate (15b)
Method E, (1.52 g, 50% yield) as white solid. 1H NMR (300 MHz, CDCl3) δ 0.80 (s, 3H), 1.11 (d, J=6.3 Hz, 3H), 2.87 (s, 3H), 3.75 (d, J=10.5 Hz, 1H), 3.92 (s, 6H), 4.35 (q, J=19.5 Hz, 1H), 5.08-5.18 (m, 2H), 5.34 (d, J=10.5 Hz, 1H), 7.26-7.36 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 14.53, 19.22, 30.84, 57.95, 65.43, 67.06, 72.93, 108.99, 127.77, 128.19, 128.67, 136.85, 157.19; LCMS: m/s 378.1 [M+H]+.
Benzyl((1S,2R)-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)-4-(trimethylsilyl)but-3-yn-1-yl)carbamate (15c)
Method E, (2.20 g, 70% yield) as white foam. 1H NMR (300 MHz, CDCl3) δ 0.12 (s, 9H), 0.81 (s, 3H), 3.09 (s, 1H), 3.92 (s, 6H), 4.12 (d, J=10.2 Hz, 1H), 4.90 (s, 1H), 5.13 (dd, J1=21.0 Hz, J2=12.3 Hz, 2H), 5.45 (d, J=10.5 Hz, 1H), 7.32-7.36 (m, 5H); 13C NMR (75MHz, CDCl3) δ 0.27, 14.25, 30.65, 57.62, 62.11, 66.91, 72.74, 72.89, 89.99, 102.66, 108.31, 127.98, 128.06, 128.43, 136.53. 156.43; LCMS: m/s 420.2 [M+H]+.
(S)-benzyl (2-(4-(benzyloxy)phenyl)-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)-2-oxoethyl)carbamate (16a)
Method D, (0. 62 g, 84% yield) as white foam. 1H NMR (300 MHz, CDCl3) δ 0.75 (s, 3H), 3.86 (s, 6H), 5.10 (s, 2H), 5.13 (s, 2H), 5.55 (d, J=9.3 Hz, 1H), 5.95 (d, J=9.3 Hz, 1H), 7.00 (d, J=9.0 Hz, 2H), 7.27-7.45 (m, 10H), 8.06 (d, J=9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 14.48, 30.90, 57.35, 67.23, 70.37, 73.17, 107.60, 114.60, 127.76, 128.30, 128.48, 128.67, 128.93, 129.91, 132.120, 136.43, 136.60, 156.16, 163.22, 193.49; LCMS: m/s 504.1 [M+H]+.
(S)-benzyl (1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)-2-oxopropyl)carbamate (16b)
Method D, (1.06 g, 82% yield) as white solid.1H NMR (300 Hz, CDCl3) δ 0.79 (s, 3H), 2.30 (s, 3H), 3.90 (s, 6H), 4.58 (d, J=8.4 Hz, 1H), 5.09 (s, 2H), 5.62 (d, J=8.1 Hz, 1H), 7.26-7.34 (m, 5H); 13C NMR (300 Hz, CDCl3) δ 14.44, 29.94, 30.87, 63.38, 67.26, 73.17, 107.13, 128.30, 128.68, 136.50, 156.21, 202.86; LCMS: m/s 356.2 [M+H]+.
(S)-benzyl(1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)-2-oxo-4-(trimethylsilyl)but-3-yn-1-yl)carbamate (16c)
Method D, (0. 62 g, 80% yield) as white foam. 1H NMR (300 MHz, CDCl3) δ 0.01 (s, 9H), 0.58 (s, 3H) 3.69 (s, 6H), 4.45 (d, J=9.3 Hz, 1H), 4.89 (s, 2H), 5.40 (d, J=9.0 Hz, 1H), 7.04-7.13 (m, 5H); 13C NMR (75MHz, CDCl3) δ 0.84, 14.21, 30.80, 63.88, 67.17, 72.93, 101.23, 102.11, 106.88, 128.12, 128.47, 136.16, 155.72, 181.35; LCMS: m/s 418.1 [M+H]+.
Benzyl((1S,2S)-2-(4-(benzyloxy)phenyl)-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)propyl)carbamate(17a)
Method E, (0.81 g, 98% yield) as white solid. 1H NMR (300 MHz, CDCl3) δ 0.73 (s, 3H), 1.50 (s, 3H), 3.73 (s, 3H), 3.71-3.80 (m, 6H), 4.22 (d, J=10.2 Hz, 1H), 4.98-5.14 (m, 4H), 5.33 (d, J=10.5 Hz, 1H), 6.90 (d, J=8.7 Hz, 2H), 7.28-7.43(m, 12H); 13C NMR (75 MHz, CDCl3) δ 14.45, 27.17, 30.62, 61.09, 66.96, 70.13, 72.60, 76.04, 109.50, 113.93, 126.76, 127.11, 127.76, 128.12, 128.68, 128.75, 136.90, 137.51, 138.83, 156.60, 157.49; LCMS: m/s 520.2 [M+H]+.
Benzyl((1S,2R)-2-(4-(benzyloxy)phenyl)-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)propyl)carbamate(17b)
Method E, (0.62 g, 40% yield) as white solid. 1H NMR (300 MHz, CDCl3) δ 0.82 (s, 3H), 1.66 (s, 3H), 3.48 (s, 1H), 3.87-3.99 (m, 6H), 4.19 (d, J=10.2 Hz, 1H), 4.87-5.06 (m, 4H), 5.31-5.33 (m, 2H), 7.26-7.46 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 14.56, 29.71, 30.74, 60.71, 66.50, 70.11, 72.72, 76.03, 109.78, 114.27, 126.23, 127.64, 127.77, 127.92, 128.12, 128.55, 128.78, 137.08, 137.45, 138.45, 156.52, 157.60; LCMS: m/s 520.2 [M+H]+.
Benzyl((1S,2S)-2-hydroxy-2-methyl-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)-4-(trimethylsilyl)but-3-yn-1-yl)carbamate (17c)
Method E, (0.1 g, 74% yield) as white solid. 1H NMR (300 MHz, CDCl3) δ 0.16 (s, 9H), 0.81 (s, 3H), 1.47 (s, 3H), 3.72 (s, 1H), 3.90 (s, 6H), 4.05 (d, J=10.5 Hz, 1H), 5.13 (s, 2H), 5.27 (d, J=9.6 Hz, 1H), 7.27-7.35 (m, 5H); 13C NMR (75MHz, CDCl3) δ 0.03, 14.30, 27.38, 30.57, 60.23, 66.92, 68.45, 72.40, 88.53, 107.31, 108.83, 127.97, 128.02, 128.46, 136.50, 156.55; LCMS: m/s 434.2 [M+H]+.
4-((1R,2S)-2-amino-1-hydroxy-2-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)ethyl)phenol (18)
Method F, (0.277 g, 99% yield) as white solid. 1H NMR (300 MHz, CD3OD) δ 0.81 (s, 3H), 3.94 (s, 6H), 5.02 (d, J=2.4 Hz, 1H), 6.73 (d, J=8.7 Hz, 2H), 7.17 (d, J=8.7 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 13.05, 30.36, 60.33, 70.65, 72.57, 108.90, 114.70, 127.11, 133.26, 156.48; LCMS: m/s 282.2 [M+H]+.
Benzyl((1S,2S)-2-(4-(benzyloxy)phenyl)-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)ethyl)carbamate (19)
Intermediate 18 (0.50 g, 1.00 mmol) was dissolved in anhydrous DCM (15 mL) and MeOH (15 mL) under argon, and then cooled to −78 °C. Then a solution of 2.0 M LiBH4 in THF (4.80 mL, 8.00 mmol, 8.0 equiv) was added dropwise. The reaction mixture was stirred at −78 °C for 10h, and then slowly warm to room temperature. The mixture was diluted to with DCM (100 mL) and then quenched with saturated NH4Cl (50 mL). The organic layer was separated. And the aqueous layer was extracted with DCM (2×50 mL). The organic layers were combined, washed with saturated NH4Cl (50 mL), brine (50 mL), and dried (anhydrous Na2SO4). The solvent was reduced in vacuum to dryness. The crude product was purified on CombiFlash (eluting with 0-60% EtOAc in hexane) to obtain 19 (0.44g, 88% yield) as white solid. 1H NMR (300 MHz, CD3Cl) δ 0.83 (s, 3H), 3.96 (s, 6H), 4.11 (d, J=1.5 Hz, 1H), 4.14-4.21 (m, 1H), 4.78-4.87 (m, 3H), 5.00 (s, 1H), 5.02 (s, 2H), 6.91 (d, J=8.4 Hz, 2H), 7.11 (d, J=6.6 Hz, 2H), 7.25-7.45 (m, 10H); 13C NMR (75 MHz, CD3Cl) δ 14.51, 30.92, 58.94, 66.72, 70.10, 73.03, 74.02, 108.96, 114.51, 127.71, 127.83, 128.11, 128.60, 128.78, 129.12, 132.95, 136.83, 137.38, 156.04, 158.62; LCMS: m/s 505.2 [M+H]+.
4-((1S,2S)-2-amino-1-hydroxy-2-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)ethyl)phenol (20)
Method F, (0.280 g, 96% yield) as white solid. 1H NMR (300 MHz, CD3OD) δ 0.82 (s, 3H), 3.02 (d, J=7.2 Hz, 1H), 3.95 (s, 6H), 4.72 (d, J=7.2 Hz, 1H), 6.75 (d, J=8.4 Hz, 2H), 7.19 (d, J=8.7 HZ, 2H); 13C NMR (75 MHz, CD3OD) δ 12.97, 30.32, 59.78, 72.52, 73.42, 108.66, 114.71, 128.85, 157.18; LCMS: m/s 282.2 [M+H]+.
4-((1S,2S)-1-amino-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)propan-2-yl)phenol (21a)
Method F, (0.343 g, 97% yield) as white solid. 1H NMR (300 MHz, CD3OD) δ 0.77 (s, 3H), 1.60 (s, 3H), 2.96 (s, 1H), 3.86 (s, 6H), 6.73 (d, J=8.7 Hz, 2H), 7.26 (d, J=8.7 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 12.98, 24.07, 30.11, 62.99, 72.29, 75.52, 109.59, 114.24, 127.21, 136.78, 16.25; LCMS: m/s 296.1 [M+H]+.
4-((1S,2R)-1-amino-2-hydroxy-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)propan-2-yl)phenol (21b)
Method F, (0.339 g, 99 % yield) as white solid. 1H NMR (300 MHz, CDCl3) δ 0.82 (s, 3H), 1.57 (s, 3H), 3.93 (s, 6H), 4.06 (br, s, 2H), 5.29 (s, 1H), 6.54 (d, J=8.1 Hz, 2H), 7.13 (d, J=7.8 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 14.51,29.91, 30.67, 61.76, 72.74, 74.88, 109.85, 115.72, 125.63, 137.17, 155.57; LCMS: m/s 296.1 [M+H]+.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((1S,2S)-2-hydroxy-2-(4-hydroxyphenyl)-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)ethyl)benzamide (22a)
Method A, (165 mg, 87% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 0.82 (s, 3H), 3.98 (s, 6H), 4.54 (d, J=8.4 Hz, 1H), 4.95 (d, J=8.4 Hz, 1H), 6.61 (d, J=8.7 Hz, 2H), 6.68 (d, J=8.7 Hz, 2H), 7.21 (d, J=6.6 Hz, 2H), 7.23 (d, J=6.6 Hz, 2H), 7.43 (s, 4H); 13C NMR (75 MHz, CD3OD) δ 12.99, 30.460, 57.49, 70.77, 72.67, 73.07, 76.29, 79.06, 84.38, 108.35, 108.66, 114.21, 114.47, 125.41, 127.26, 128.85, 131.68, 131.79, 133.79, 134.74, 150.19, 156.97, 167.68; LCMS: m/s 525.1 [M+H]+.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)ethyl)benzamide (22b)
Method A, (337 mg, 98% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 0.79 (s, 3H), 3.96 (s, 6H), 4.41 (d, J=1.5 Hz, 1H), 5.27 (s, 1H), 6.61 (d, J=9.0 Hz, 2H), 6.67 (d, J=8.7 Hz, 2H), 7.17 (d, J=8.4 Hz, 2H), 7.25 (d, J=8.7 HZ, 2H), 7.52 (d, J=8.7 Hz, 2H), 7.70 (d, J=8.7 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 13.08, 30.46, 58.04, 70.50, 7.91, 72.68, 76.58, 79.16, 84.58, 108.38, 108.51, 114.28, 114.68, 125.72, 127.08, 127.36, 132.07, 132.68, 133.87, 134.41, 150.159, 156.47, 167.73; LCMS: m/s 525.2 [M+H]+.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((1S,2S)-2-hydroxy-2-(4-hydroxyphenyl)-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)propyl)benzamide (22c)
Method A, (350 mg, 78% yield) as yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 0.70 (s, 3H), 1.50 (s, 3H), 3.78 (s, 6H), 4.42 (d, J=8.7 Hz, 1H), 4.53 (s, 1H), 5.82 (s, 1H), 6.53 (d, J=8.4 Hz, 4H), 7.18 (d, J=8.7 Hz, 2H), 7.24 (d, J=8.4 Hz, 2H), 7.56 (d, J=8.4 Hz, 2H), 7.68-7.73 (m, 3H), 9.00 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 14.29, 27.15, 30.57, 49.29, 60.06, 71.80, 72.32, 75.56, 77.08, 80.89, 86.26, 105.86, 109.45, 114.27, 124.39, 127.54, 132.44, 134.68, 135.48, 137.60, 151.54, 155.96, 165.50; LCMS: m/s 539.4 [M+H]+.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((1S,2R)-2-hydroxy-2-(4-hydroxyphenyl)-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)propyl)benzamide (22d)
Method A, (353 mg, 79.1% yield) as yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 0.74 (s, 3H), 1.56 (s, 3H), 3.84 (s, 6H), 4.47 (s, 1H), 5.82 (s, 2H), 6.51 (d, J=2.7 Hz, 2H), 6.54 (d, J=2.7 Hz, 2H), 7.16 (d, J=8.7 Hz, 2H), 7.23 (d, J=8.4 Hz, 2H), 7.49-7.57 (m, 4H), 9.00 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 14.38, 29.55, 30.62, 59.47, 71.79, 72.33, 75.58, 77.08, 80.81, 86.25, 105.85, 109.40, 114.26, 114.65, 124.35, 126.62, 128.19, 132.53, 134.68, 135.42, 138.53, 151.53, 155.90, 165.04; LCMS: m/s 539.2 [M+H]+.
(2S,3S)-3-hydroxy-2-(hydroxymethyl)-2-methylpropyl 2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-hydroxy-3-(4-hydroxyphenyl)propanoate (23a)
Method G, (126 mg, 88% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 0.86 (s, 3H), 3.45-3.43 (m, 4H), 4.09 (q, J=32,4 Hz, 2H), 4.89 (d, J=7.5 Hz, 1H), 5.05 (d, J=7.5 Hz, 1H), 6.60 (d, J=8.7 Hz, 2H), 6.78 (d, J=8.4 Hz, 2H), 7.24 (d, J=8.7 Hz, 2H), 7.26 (d, J=8.7 Hz, 2H), 7.48 (d, J=8.4 Hz, 2H), 7.60 (d, J=8.7 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 15.75, 40.83, 59.21, 64.58, 67.31, 70.92, 73.33, 76.75, 79.10, 84.65, 108.44, 114.32, 115.03, 125.98, 127.42, 127.96, 131.62, 132.07, 133.76, 133.88, 150.10, 157.23, 167.84, 171.21; LCMS: m/s 543.2 [M+H]+.
(2S,3R)-3-hydroxy-2-(hydroxymethyl)-2-methylpropyl 2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-hydroxy-3-(4-hydroxyphenyl)propanoate (23b)
Method G, (120 mg, 90% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 0.87 (s, 3H), 3.43-3.44 (m, 4H), 4.07 (d, J=1.2 Hz, 2H), 4.91 (d, J=4.2 Hz, 1H), 5.28 (d, J=3.9 Hz, 1H), 6.60 (d, J=8.7 Hz, 2H), 6.75 (d, J=8.7 Hz, 2H), 7.24 (d, J=5.4 Hz, 2H) 7.27 (d, J=5.4 Hz, 2H), 7.53 (d, J=8.4 Hz, 2H), 7.72 (d, J=8.7 Hz, 2H); 1H NMR (75 MHz, CD3OD) δ 15.68, 40.87, 59.83, 64.44, 67.43, 70.84, 72.84, 76.68, 79.05, 84.60, 108.39, 114.26, 114.94, 126.00, 127.29, 127.46, 131.73, 132.09, 133.84, 150.16, 157.03, 168.30, 170.73; LCMS: m/s 565.1 [M+Na]+.
(2S,3S)-3-hydroxy-2-(hydroxymethyl)-2-methylpropyl 2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-hydroxy-3-(4-hydroxyphenyl)butanoate (23c)
Method G, (61 mg, 99% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 0.65 (s, 3H), 1.57 (s, 3H), 3.17-3.26 (m, 4H), 3.70 (d, J=10.8 Hz, 1H), 3.93 (d, J=11.1 Hz, 1H), 5.06 (s, 1H), 6.62 (d, J=8.7 Hz, 2H), 6.79 (d, J=8.7 Hz, 2H), 7.25 (d, J=9.0 Hz, 2H), 7.36 (d, J=8.7 Hz, 2H), 7.59 (d, J=8.7 Hz, 2H), 7.83 (d, J=8.7 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 15.52, 27.12, 40.38, 61.79, 64.47, 67.39, 70.80, 75.32, 76.80, 78.98, 84.68, 108.33, 114.24, 114.82, 126.20, 126.35, 127.51, 132.19, 133.84, 135.88, 150.20, 156.50, 168.00, 170.56; LCMS: m/s 557.2 [M+H]+.
(2S,3R)-3-hydroxy-2-(hydroxymethyl)-2-methylpropyl 2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-hydroxy-3-(4-hydroxyphenyl)butanoate (23d)
Method G, (320 mg, 97% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 0.91 (s, 3H), 1.69 (s, 3H), 3.42-3.51 (m, 4H), 4.11 (q, J=31.8 Hz, 2H), 4.95 (s, 1H), 6.61 (d, J=6.9 Hz, 2H), 6.74 (d, J=9.0 Hz, 2H), 7.32 (d, J=8.7 Hz, 2H), 7.34 (d, J=8.7 Hz, 2H), 7.49 (d, J=8.7 Hz, 2H), 7.54 (d, J=8.7 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 15.83, 27.46, 40.77, 62.13, 64.47, 64.61, 67.39, 70.78, 75.10, 76.67, 78.95, 84.60, 108.33, 114.23, 114.77, 125.98, 126.13, 127.21, 132.05, 133.81, 136.31, 150.18, 156.36, 167.65, 170.50; LCMS: m/s 557.2 [M+H]+.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((1R,2S)-1-hydroxy-3-(hydroxyamino)-1-(4-hydroxyphenyl)-3-oxopropan-2-yl)benzamide (24a)
Method C, (36 mg, 79% yield) as yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 4.55 (q, J=13.5 Hz, 1H), 4.97 (t, J=9.9 Hz, 1H), 5.54 (d, J=6.0 Hz, 1H), 5.83 (s, 2H), 6.54 (d, J=8.7 Hz, 2H), 6.64 (d, J=8.7 HZ, 2H), 7.14 (d, J=8.7 Hz, 2H), 7.25 (d, J=8.4 HZ, 2H), 7.60 (d, J=8.4 Hz, 2H), 7.80 (d, J=8.7 Hz, 2H), 8.12 (d, J=8.7 Hz, 1H), 8.79 (s, 1H), 9.22 (s, 1H), 10.60 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 58.86, 71.81, 72.90, 77.29, 80.83, 86.37, 105.85, 114.28, 115.30, 124.72, 128.01, 128.47, 132.60, 133.06, 134.71, 134.98, 151.56, 157.03, 165.99, 167.23; HRMS: m/z calc’d for C26H21N3O5 [M]+: 455.1481; found: 455.1483.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((1S,2S)-1-hydroxy-3-(hydroxyamino)-1-(4-hydroxyphenyl)-3-oxopropan-2-yl)benzamide (24b)
Method C, (35 mg, 77% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 4.69 (d, J=8.7 Hz, 1H), 4.91 (d, J=9.0 Hz, 1H), 6.61 (d, J=8.4 Hz, 2H), 6.72 (d, J=8.4 Hz, 2H), 7.23 (d, J=9.0 Hz, 2H), 7.27 (d, J=8.4 Hz, 2H), 7.48 (d, J=8.1 Hz, 2H), 7.56 (d, J=8.1 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 56.73, 70.72, 73.14, 76.49, 78.96, 84.48, 108.32, 114.20, 114.81, 125.80, 127.34, 128.26, 131.91, 132.01, 133.78, 134.04, 150.21, 157.21, 167.37, 168.52; HRMS: m/z calc’d for C26H21N3O5 [M]+: 455.1481; found: 455.1483.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((2S,3S)-3-hydroxy-1-(hydroxyamino)-3-(4-hydroxyphenyl)-1-oxobutan-2-yl)benzamide (24c)
Method C, (28 mg, 59 % yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 1.50 (s, 3H), 4.86 (s, 1H), 6.62 (d, J=8.7 Hz, 2H), 6.76 (d, J=8.7 Hz, 2H), 7.24 (d, J=8.1 Hz, 2H), 7.34 (d, J=8.4 Hz, 2H), 7.57 (d, J=8.1 Hz, 2H), 7.82 (d, J=8.4 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 25.86, 57.98, 70.73, 75.68, 76.64, 78.96, 84.58, 108.30, 114.21, 114.73, 126.06, 126.45, 127.57, 132.08, 133.81, 133.94, 135.69, 150.24, 156.48, 167.89; HRMS: m/z calc’d for C27H23N3O5 [M]+: 469.1636; found: 469.1638.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((2S,3R)-3-hydroxy-1-(hydroxyamino)-3-(4-hydroxyphenyl)-1-oxobutan-2-yl)benzamide (24d)
Method C, (22 mg, 46% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 1.63 (s, 3H), 4.75 (s, 1H), 6.61 (d, J=8.4 Hz, 2H), 6.71 (d, J=8.7 Hz, 2H), 7.23 (d, J=8.4 Hz, 2H), 7.33 (d, J=8.7 Hz, 2H), 7.49 (d, J=8.4 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ 26.93, 59.36, 70.71, 74.83, 76.57, 78.92, 84.53, 108.29, 114.20, 114.60, 125.89, 126.21, 127.24, 131.98, 133.78, 136.00, 150.22, 156.28, 167.28, 168.09; HRMS: m/z calc’d for C27H23N3O [M]+: 469.1636; found: 469.1638.
Benzyl ((1S,2S)-2-hydroxy-2-methyl-1-(4-methyl-2,6,7-trioxabicyclo[2.2.2]octan-1-yl)but-3-yn-1-yl)carbamate (25)
To a solution of 17c (50 mg, 0.115 mmol, 1.00 equiv) in THF (5 mL) was added TBAF (30.0 mg, 0.115 mmol, 1.00 equiv) at 0 °C under argon. The reaction mixture was stirred at 0 °C for 5 min. Then it was concentrated to dryness. The crude product was purified on CombiFlash (eluting with 0-50% EtOAc in hexane) to give 25 (38 mg, 90% yield) as white solid. 1H NMR (300 MHz, CDCl3) δ 0.81 (s, 3H), 1.50 (s, 3H), 2.48 (s, 1H), 3.81 (s, 1H), 3.92 (s, br, 6H), 4.07 (d, J=11.1 Hz, 1H), 5.19 (dd, J1=18.6 Hz, J2=12.3 Hz, 2H), 5.29 (d, J=10.5 Hz, 1H), 7.28-7.35 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 14.27, 27.36, 30.57, 60.23, 66.97, 68.31, 72.47, 85.77, 108.75, 127.97, 128.74, 128.46, 136.48, 156.65; LCMS: m/s 362.2 [M+H]+.
(2S,3R)-3-hydroxy-2-(hydroxymethyl)-2-methylpropyl 3-(1-benzyl-1H-1,2,3-triazol-4-yl)-2-(((benzyloxy)carbonyl)amino)-3-hydroxybutanoate (26)
To a solution of 25 (410 mg, 1.14 mmol, 1.00 equiv) and benzyl azide (151 mg, 1.14 mmol, 1.00 equiv) in a mixture of t-BuOH/H2O (1:1) (4 mL) was added a solution of sodium ascorbate (22.6 mg, 0.114 mmol, 0.10 equiv) in 0.5 mL of water and a solution of CuSO4 (14.23 mg, 0.057 mmol, 0.05 equiv) in 0.5 mL of water. The reaction mixture was stirred at room temperature under argon for 24 h. Removal of the volatiles afforded the crude product, which was purified on CombiFlash (eluting with MeOH in DCM 0-5%) to give 26 (520 mg, 92%) as white foam. 1H NMR (300 MHz, CDCl3) δ 0.71 (s, 3H), 1.62 (s, 3H), 1.81 (s, 1H), 3.07 (s, 1H), 3.15 (s, 1H), 3.43 (s, br, 4H), 4.02 (s, 2H), 4.16 (s, 1H), 4.60 (d, J=9.3 Hz, 1H), 5.10 (s, 2H), 5.46 (s, 2H), 5.92 (d, J=9.0 Hz, 1H), 7.23-7.45 (m, 10H); 13C NMR (75MHz, CDCl3) δ 16.85, 26.09, 40.27, 54.33, 61.79, 67.00, 67.27, 67.41, 68.10, 77.81, 121.02, 128.15, 128.19, 128.30, 128.56, 128.93, 129.20, 134.13, 135.92, 151.70, 156.59, 170.97; LCMS: m/s 513.2 [M+H]+.
(2S,3R)-3-hydroxy-2-(hydroxymethyl)-2-methylpropyl 2-amino-3-hydroxy-3-(1H-1,2,3-triazol-4-yl)butanoate (27)
Method F, (185 mg, 98%) as white solid. 1H NMR (300 MHz, CD3OD) δ 0.83 (s, 3H), 1.91 (s, 3H), 3.37-3.42 (m, 4H), 4.11 (dd, J1=75.6 Hz, J2=10.8 Hz, 2H), 4.51 (s, 1H), 8.39 (s, 1H); 13C NMR (75MHz, CD3OD) δ 15.56, 26.08, 40.35, 61.44, 64.00, 68.64, 69.40, 125.68, 146.41, 165.99; LCMS: m/s 289.2 [M+H]+.
(2S,3R)-3-hydroxy-2-(hydroxymethyl)-2-methylpropyl 2-(4-((4-aminophenyl)buta-1,3-diyn-1-yl)benzamido)-3-hydroxy-3-(1H-1,2,3-triazol-4-yl)butanoate (28)
Method A, (130 mg, 44% yield) as yellow solid. 1H NMR (300 MHz, CD3OD) δ 0.70 (s, 3H), 1.68 (s, 3H), 3.26-3.31 (m, 4H), 3.90 (dd, J1-64.4 Hz, J2=10.8 Hz, 2H), 5.13 (s, 1H), 5.46 (s, 1H), 6.60 (d, J=8.4Hz, 2H), 7.23 (d, J=8.8 Hz, 2H), 7.57 (d, J=8.8 Hz, 2H), 7.77 (s, 1H), 7.84 (d, J=8.4Hz, 2H); 13C NMR (75MHz, CD3OD) δ 15.31, 25.68, 40.26, 53.42, 61.43, 64.01, 64.12, 67.18, 70.56, 71.43, 76.58, 78.75, 84.46, 108.05, 110.25, 114.00, 117.18, 127.32, 131.97, 133.46, 133.61, 149.98, 167.83, 169.65; LCMS: m/s 532.2 [M+H]+.
4-((4-aminophenyl)buta-1,3-diyn-1-yl)-N-((2S,3R)-3-hydroxy-1-(hydroxyamino)-1-oxo-3-(1H-1,2,3-triazol-4-yl)butan-2-yl)benzamide (29)
Method C, (55 mg, 66% yield) as yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 1.52 (s, 3H), 4.79 (d, J=9.3 Hz, 1H), 5.84 (s, 2H), 6.53 (d, J=8.4 Hz, 2H), 7.25 (d, J=8.4 Hz, 2H), 7.61 (s, 1H), 7.65 (d, J=5.4 Hz, 2H), 7.83 (d, J=8.4 Hz, 2H), 8.45 (d, J=9.3 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 25.60, 58.47, 71.56, 71.65, 77.07, 80.58, 86.16, 105.56, 114.03, 124.55, 128.41, 132.34, 134.47, 134.73, 151.33, 165.92, 166.33; HRMS: m/z calc’d for C23H20N6O4 [M]+: 444.1546; found: 444.1548.
Acknowledgement
This research was supported by National Institutes of Health Grants AI055588 and GM051310 to P.Z. X-ray data collection was performed at the Duke University Macromolecular Crystallography Shared Resource Facility. We would like to acknowledge Dr. Meta J. Kuehn for providing Klebsiella pneumoniae and Vibrio cholerae strains.
Abbreviations Used
- MIC
minimum inhibitory concentration
- LpxC
UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase
- LPS
lipopolysaccharide
- BSA
bovine serum albumin
- TCEP
tris(2-carboxy-ethyl)phosphine
- DTT
dithiothreitol
Footnotes
ANCILLARY INFORMATION
Accession numbers
Structure factors and coordinates for the PaLpxC/8a (LPC-014), PaLpxC/24b (LPC-050) and PaLpxC/24c (LPC-051) complexes have been deposited in the RCSB Protein Data Bank with the accession codes 4LCF, 4LCG, and 4LCH, respectively.
REFERENCES AND NOTES
- 1.Diekema DJ, BootsMiller BJ, Vaughn TE, Woolson RF, Yankey JW, Ernst EJ, Flach SD, Ward MM, Franciscus CL, Pfaller MA, Doebbeling BN. Antimicrobial resistance trends and outbreak frequency in United States hospitals. Clin Infect Dis. 2004;38:78–85. doi: 10.1086/380457. [DOI] [PubMed] [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Rice LB. Emerging issues in the management of infections caused by multidrug-resistant gram-negative bacteria. Cleve Clin J Med. 2007;74(Suppl 4):S12–20. doi: 10.3949/ccjm.74.suppl_4.s12. [DOI] [PubMed] [Google Scholar]
- 4.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barb AW, Zhou P. Mechanism and inhibition of LpxC: an essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr Pharm Biotechnol. 2008;9:9–15. doi: 10.2174/138920108783497668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 7.Liang X, Lee CJ, Chen X, Chung HS, Zeng D, Raetz CR, Li Y, Zhou P, Toone EJ. Syntheses, structures and antibiotic activities of LpxC inhibitors based on the diacetylene scaffold. Bioorg Med Chem. 2011;19:852–860. doi: 10.1016/j.bmc.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coggins BE, Li X, McClerren AL, Hindsgaul O, Raetz CRH, Zhou P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat Struct Biol. 2003;10:645–651. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coggins BE, McClerren AL, Jiang L, Li X, Rudolph J, Hindsgaul O, Raetz CRH, Zhou P. Refined solution structure of the LpxC-TU-514 complex and pKa analysis of an active site histidine: insights into the mechanism and inhibitor design. Biochemistry. 2005;44:1114–1126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- 10.Barb AW, Jiang L, Raetz CR, Zhou P. Structure of the deacetylase LpxC bound to the antibiotic CHIR-090: Time-dependent inhibition and specificity in ligand binding. Proc Natl Acad Sci U S A. 2007;104:18433–18438. doi: 10.1073/pnas.0709412104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CJ, Liang X, Chen X, Zeng D, Joo SH, Chung HS, Barb AW, Swanson SM, Nicholas RA, Li Y, Toone EJ, Raetz CR, Zhou P. Species-specific and inhibitor-dependent conformations of LpxC: implications for antibiotic design. Chem Biol. 2011;18:38–47. doi: 10.1016/j.chembiol.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blaskovich MA, Evindar G, Rose NGW, Wilkinson S, Luo Y, Lajoie GA. Stereoselective Synthesis of Threo and Erythro β-Hydroxy and β-Disubstituted-β-Hydroxy α-Amino Acids. J. Org. Chem. 1998;63:3631–3646. [Google Scholar]
- 13.Hansen DB, Wan X, Carroll PJ, Joullie MM. Stereoselective Synthesis of Four Stereoisomers of β-Methoxytyrosine, a Component of Callipeltin A. J. Org. Chem. 2005;70:3120–3126. doi: 10.1021/jo047876z. [DOI] [PubMed] [Google Scholar]
- 14.Blaskovich MA, Lajoie GA. Synthesis of a Chiral Serine Aldehyde Equivalent and Its Conversion to Chiral a-Amino Acid Derivatives. J Am Chem Soc. 1993;115:5021–5030. [Google Scholar]
- 15.Blaskovich MA, Lajoie GA. Stereoselective Synthesis of allo-Threonine and β-2H-allo-Threonine from Threonine. Tetrahedron Letters. 1993;34:3837–3840. [Google Scholar]
- 16.Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 17.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds CM, Raetz CR. Replacement of lipopolysaccharide with free lipid A molecules in Escherichia coli mutants lacking all core sugars. Biochemistry. 2009;48:9627–9640. doi: 10.1021/bi901391g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalgutkar AS, Obach RS, Maurer TS. Mechanism-based inactivation of cytochrome P450 enzymes: chemical mechanisms, structure-activity relationships and relationship to clinical drug-drug interactions and idiosyncratic adverse drug reactions. Curr Drug Metab. 2007;8:407–447. doi: 10.2174/138920007780866807. [DOI] [PubMed] [Google Scholar]
- 20.Ritter JK. Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chem Biol Interact. 2000;129:171–193. doi: 10.1016/s0009-2797(00)00198-8. [DOI] [PubMed] [Google Scholar]
- 21.Ren J, Nichols C, Bird L, Chamberlain P, Weaver K, Short S, Stuart DI, Stammers DK. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J Mol Biol. 2001;312:795–805. doi: 10.1006/jmbi.2001.4988. [DOI] [PubMed] [Google Scholar]
- 22.Gallwitz B. Emerging DPP-4 inhibitors: focus on linagliptin for type 2 diabetes. Diabetes Metab Syndr Obes. 2013;6:1–9. doi: 10.2147/DMSO.S23166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 24.Jackman JE, Raetz CRH, Fierke CA. Site-directed mutagenesis of the bacterial metalloamidase UDP-(3-O-acyl)-N-acetylglucosamine deacetylase (LpxC). Identification of the zinc binding site. Biochemistry. 2001;40:514–523. doi: 10.1021/bi001872g. [DOI] [PubMed] [Google Scholar]
- 25.Copeland RA. Evaluation of enzyme inhibitors in drug discovery. John Wiley & Sons, Inc.; Hoboken, NJ: 2005. [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]