

Abstract
B cells play a major role in the pathogenesis of many autoimmune disorders including rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis and type I diabetes mellitus, as indicated by the efficacy of B cell-targeted therapies in these diseases. Therapeutic effects of the most commonly used B cell-targeted therapy, anti-CD20 monoclonal antibody, are contingent upon long-term depletion of peripheral B cells. Here, we describe an alternative approach involving the targeting of CD79, the transducer subunit of the B cell antigen receptor. Unlike anti-CD20 mAbs, the protective effects of CD79-targeted mAb are do not require cell depletion, but rather act by inducing an anergic-like state. Thus, we describe a novel B cell-targeted approach predicated on the induction of B cell anergy.
INTRODUCTION
B cells have proven to be effective targets for the treatment of multiple autoimmune disorders and B-lineage cancers (1, 2). The most widely used B cell-targeted drug is rituximab, which has been approved in the United States since 1997 for treatment of B cell lymphoma and since 2006 for treatment of rheumatoid arthritis (RA). Therapeutic utility of rituximab has recently been shown in multiple other autoimmune diseases, such as multiple sclerosis (MS) and Type I diabetes mellitus (T1DM) (3, 4). Despite inconclusive data from Phase III clinical trials in SLE, rituximab continues to see significant off-label use for treatment of this disease (5).
Rituximab is a chimeric human/mouse IgG1 mAb that targets CD20 and mediates long-lasting depletion of peripheral B cells (6). CD20 is a surface protein that is abundantly expressed on B-lineage cells from the pre-B cell stage to the plasmablast stage (7). As CD20 is not expressed on plasma cells, rituximab does not impair established antibody-mediated immunity gained from past infections and vaccinations (8). Empirical evidence supports at least three direct modes of B cell depletion by rituximab: antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cellular cytotoxicity (CDC) and the direct induction of apoptosis via CD20 cross-linking (9-11). The primacy of these mechanisms in rituximab-induced B cell loss in humans is unclear.
Rituximab is not consistently efficacious even among autoimmunities known to be antibody mediated. For example, in mouse models of lupus in which B cells express human CD20, rituximab was unable to efficiently deplete B cells from secondary lymphoid tissues or affect the course of disease despite depletion of peripheral blood B cells (12). Indeed, the very applicability of rituximab in SLE remains controversial. Two large, double-blinded, placebo-controlled studies in SLE patients found that rituximab does not have any benefit over placebo (5, 13). However, results of a number of non-blinded clinical trials and off-label use of rituximab suggest that it does has clinical efficacy in SLE, although perhaps less than seen in RA (14-16)
CD79 (Ig-α/β) may emerge as an alternative target for the treatment of B cell-dependent autoimmunity (17). CD79 is a disulphide-linked heterodimer of CD79a (Ig-α) and CD79b (Ig-β), and is associated with membrane immunoglobulin (mIg) on the surface of B-lineage cells. Together, these components constitute the B cell antigen receptor (BCR). Upon an antigen-induced BCR aggregation, CD79 is phosphorylated and initiates a cascade of down-stream signaling events. B cells are thus activated and ready to receive further co-activating signals that drive proliferation and differentiation, ultimately delivering a memory cell pool and an appropriate humoral response. During this process, B cells become robust antigen presenting cells and release cytokines that can influence the quality of the immune response.
Work in our laboratory and others has defined and characterized an alternate mode of BCR signaling that is induced by chronic antigen receptor stimulation and maintains a state of B cell unresponsiveness termed, ‘anergy’ (18-23). Anergic B cells are characterized by the partial down-regulation of surface BCR and impaired propagation of activating signals that normally emanate from CD79, including activation of the SYK tyrosine kinase and extracellular Ca2+ influx; and have a life-span that is reduced from ~40 days of a typical naïve B cell to ~5 days (19, 21, 24-26). We hypothesized that the mechanism of B cell anergy might be harnessed for therapeutic inactivation of B cells.
Recently, the therapeutic effectiveness of anti-CD79b mAb in the MRL/lpr mouse model of lupus was demonstrated (17). In the present study, we addressed the mechanism of anti-CD79b mAb-mediated immune suppression. We report here that anti-CD79b mAb induces a polyclonal B cell anergy that is capable of preventing collagen-induced arthritis (CIA). These findings introduce a new strategy for therapeutic targeting of B cells that does not require B cell depletion, but instead acts by disabling antigen receptor function.
MATERIALS AND METHODS
Mice
Unless otherwise noted, female mice were used at 2-6 months of age. C57BL/6 mice purchased from Jackson Laboratories were used as wildtype controls. FcRγ-/- mice, were a kind gift from the laboratory of Dr. E. Gelfand. FcγRIIB-/- mice were purchased from Taconic Laboratories. These mice were bred and housed at the animal facility at NJH and the experiments were performed under approved IACUC protocols. CIA experiments were undertaken using adult eight-week-old male DBA/1J.
Induction of collagen-induced arthritis (CIA)
CIA was induced in male DBA/1J as described in (27). Briefly, mice were immunized with bovine Collagen Type II (CII) emulsified in Complete Freund Adjuvant (CFA) at day 0. After 21 days, mice received a secondary immunization with CII emulsified in Incomplete Freund Adjuvant (IFA).
Anti-CD79b, m-anti-CD79b, m-anti-CD79b-D/A, anti-CD20, anti-CD20-D/A and control hamster IgG and control mouse IgG2a were administered subcutaneously (s.c.) on day 0. Two hours after mAb injection the mice were immunized with bovine CII. Clinical scores were assessed after the secondary immunization on individual paws, applying a scale ranging from 0 to 4, as previously described (28).
BrdU Labeling
For analysis of newly-generated B cells, mice were given drinking water containing 0.8 mg/ml of BrdU for three weeks. Fresh BrdU+ drinking water was protected from light and refreshed every day. For the first week the BrdU+ drinking water was supplemented with 1.0% glucose to avoid drinking aversion. Intracellular staining was conducted with BD Cytofix/Cytoperm (BD Biosciences) and the FITC-conjugated anti-BrdU antibody, B44 (BD Biosciences)
Histological assessment of arthritis
Animals were sacrificed at experimental end point on day 41. The knee joints of 4 representative animals per group were collected and processed as previously described and embedded in paraffin blocks. Serial midsagital sections (8μm of thickness) of the whole knee joint were either stained with Hematoxylin and Eosin (H&E) or Safranin O/fast green counterstaining (28). Histological sections were then graded as previously described (28).
Measurement of free serum anti-CD79 mAb
One million C57BL/6 splenocytes/well were distributed in a V-bottom 96-well plate and blocked with anti-CD16/CD32 (BD Biosciences) for 20 min at 4°C. After washing the cells, 5 μl of whole mouse serum (or a dilution thereof) was diluted to a final volume of 50 μl and was added to each well, followed by incubation for 30 min at 4°C. After subsequent washing, cells were stained with a secondary fluorescent antibody to mouse IgG2a and analyzed by flow cytometry to quantitate B cell staining by the anti-CD79 mAb remaining in the original serum.
Tissue harvesting
Spleens, lymph nodes (LN), bone marrow (BM) and peripheral blood lymphocytes (PBL) were prepared for analysis by a variety of methods. To harvest spleens and lymph nodes a single cell suspension was prepared by mechanical disruption in IMDM medium supplemented with 5% FCS, HEPES, Pen/Strep and Gentamycin. Red blood cells (RBC) were lysed using ACK (150mM NH4Cl, 10mM KHCO3, 100mM Na2 EDTA) for 2 minutes at RT. The cells were then resuspended in 10X volume of sterile PBS and washed in FACS buffer (1% BSA, 0.02% NaN3 in PBS). To harvest PBL, peripheral blood was collected in tubes containing 5μl of heparin. PBL were washed twice with ACK to lyse the RBCs and then resuspended in FACS buffer. To collect bone marrow, the femurs were flushed with the same medium as above using a syringe. Collected bone marrow cells were then washed once in ACK and resuspended in FACs buffer. For CIA-related experiments, spleens or draining lymph nodes were digested individually with collagenase (2.4 mg/ml; Gibco/Invitrogen) and DNase (1 mg/ml; Sigma-Aldrich) for 30 minutes at RT. When appropriate, B cells were negatively selected using a CD43 B cell isolation kit (Miltenyi).
Flow cytometric analysis
For cell surface staining the following mAbs were used (from BD Biosciences unless otherwise stated): Anti-CD3-FITC (145-2C11), anti-CD4-APC (RM4-5), anti-CD19-APC (1D3), anti-CD19-Pacific Blue (1D3, eBioscience), anti-CD43-Biotin (S7) detected with SA-Alexa-647 (Invitrogen), anti-CD43-FITC (S7), anti-CD44-FITC (IM7), anti-CD45-PercP (30F11), anti-CD69-PE (H1.2F3), anti-CD79 (polyclonal rabbit) detected with goat-anti-rabbit IgG-Alexa-649 (Molecular Probes), anti-CD79b-DyLight-488 (HM79), anti-CD79b-Dylight-649 (HM79), anti-CD80-PE (1610A1), anti-CD86-PE (GL-1), anti-B220-PerCP (RA3-6B2), anti-FcγRIIB-Dylight-488 (2.4G2), anti-Hamster IgG FITC (G70-204/G94.56), anti-IA/IE FITC (2G9), anti-IgD-PE (11-26, Southern Biotech), anti-IgM-Dylight-649 (b-7-6), anti-MHC-II-Dylight-488 (D3.137), and anti-PNA FITC (Sigma). Cells were incubated with Fc block (BD Biosciences) and stained in FACS buffer (PBS+1% BSA + 0.02% NaN3) to reduce non-specific staining. Flow cytometry was performed on cells using a CYAN flow cytometer (Beckman Coulter) or FACS Calibur flow cytometer (BD Biosciences) and analyzed using FlowJo (Treestar), CellQuestPro software (BD Biosciences) and PRISM (Graphpad Software).
Intracellular Ca2+ mobilization
Primary B cells were prepped for real-time intracellular calcium analysis as previously described (19). For stimulation in vitro, B cells were stimulated with 3.0μg of b-7-6 (anti-μ) or 1.5μg of 1-3-5 (anti-δ) anti-immunoglobulin heavy chain antibodies, 15.0μg of polyclonal rabbit-anti-mouse-CD79 or ionomycin (1.0μM) and analyzed by cytometry using an LSR-II (B.D. Sciences) and FloJo software (Treestar). Experiments using B cells from CIA mice were conducted as previously described; cells were resuspended in RPMI1640 supplemented with 0.1% BSA and 25 mM HEPES and loaded with Fura-2AM (1μM, Invitrogen) for 30 minutes at 37°C. After washing, the cells were transferred to a 96-well plate. Stimulation was performed with 7.5μg of polyclonal goat-anti-mouse-IgM using the Flexstation II device (Molecular Devices) and analyzed by SoftMax Pro software. Following stimulation, the response was measured based on area under the curve of the relative intracellular calcium concentration, minus that of the unstimulated background.
Immunization with NP4Ova
To prepare NP4Ova immunogen, a 1:1 volume mixture of 1mg/mL NP4Ova and 10mg/ml Alum (Brenntag) was incubated at RT on a rotator for 3 hours. 200 μl of the mixture was injected i.p. per mouse, to achieve a final immunization with 100μg of NP4Ova and 1mg of Alum.
NP-specific ELISPOT assay
For ELISPOT analysis, serial two-fold dilutions of 1×106/ml splenocytes per sample were resuspended and plated, in triplicate, in 96-well plates. Cells were incubated at 37°C for 6 hours. The plates were then washed 3 times with PBS + 0.05% Tween letting each wash sit for 10 minutes at RT. The plates were then washed twice with PBS. Next, 50μl of secondary goat-anti-mouse IgG (1:4000) with 2% BSA in PBS was added and incubated at 4°C overnight. The plates were then washed 3 times with PBS + 0.05% Tween. Next, the plates were incubated with ESA buffer (25 μM BCIP, 100mM NaCl, 100mM Tris, 10mM MgCl2 at pH9.5) for 1 hour. The reaction was stopped by washing the plate 3 times with ddH20. The number of spots at a cell dilution in the linear range was determined and the number of antibody secreting cells was calculated.
HEL-specific ELISPOT assay
For ELISPOT analysis, serial two-fold dilutions of 1×106/ml splenocytes per sample were resuspended and plated, in triplicate, in 96-well plates. Plates were coated with 10ug/ml of HEL per well and incubated at RT for 2 hours and then blocked with 2% BSA in PBS for 2 hours. Next, splenocytes from harvested mice were incubated at 37°C for 6 hours. The plates were then washed 3 times with PBS + 0.05% Tween letting each wash sit for 10 minutes at RT. Finally plates were washed twice with PBS. Next, 50μl of RS3.1-bt (anti-IgMa) (1:2000) with 2% BSA in PBS was added and incubated at 4°C overnight. The plates were then washed 3 times with PBS + 0.05% Tween and then incubated with Streptavidin-AP (1:2000) with 2% BSA in PBS and incubated at 4 C overnight. The plates were again washed 3 times with PBS + 0.05% Tween Next, the plates were incubated with ESA buffer (25 μM BCIP, 100mM NaCl, 100mM Tris, 10mM MgCl2 pH9.5) for 1 hour. The reaction was stopped by washing the plate 3 times with ddH20. The number of spots at a cell dilution in the linear range was determined and the number of antibody secreting cells was calculated.
HEL Immunization and B cell Adoptive Transfer
B cells from MD4 or MD4/ML5 mice treated with anti-CD79b mAb or control IgG were enriched by depletion of CD43+ cells with magnetic beads (MACS anti-mouse CD43; Miltenyi Biotec). A total of 106 B cells in 200 ml PBS was adoptively transferred by i.v. injection. One hour later the recipient mice were immunized i.p. with HEL-conjugated SRBCs as described previously (29).
Measurement of IgG and NP-specific IgG
EIA/RIA 96-well plates (Costar) were coated with 50μl of 20μg/mL of NP20BSA or NP2BSA or 1μg/mL of goat anti-mouse IgG or goat ant-mouse IgM in ELISA capture buffer (PBS + 150mM NaCl). Plates were allowed to incubate overnight at 4°C. The next day, wells were aspirated and then blocked with 2% BSA in PBS and allowed to incubate overnight at 4°C. Serum was then added and serially diluted in three-fold increments across the plate. The first dilutions were: 1:500 for total IgM, 1:1000 for total IgG, 1:200 for NP2BSA or NP20BSA specific detection. Plates were incubated overnight at 4°C, then washed three times with PBS + 0.05% Tween. Depending upon the desired mode of detection, 100μl of secondary detection antibody was then added. These reagents included goat-anti-mouse IgG1-Alkaline Phosphatase (1:2000), goat-anti-mouse IgG-Alkaline Phosphatase (1:2000) or goat-anti-mouse IgM-Alkaline Phosphatase (1:1000) in 1% BSA in PBS + 0.05% Tween.
Cloning and expression of recombinant IgG2a chimeric antibody HM79 and 18B12
18B12 VH and VL nucleotide sequences were synthesized by DNA2.0 according to the sequences described in patent application US 2007/0136826 A1. For expression in mammalian cells, 18B12 VH and VL sequences were sub-cloned in frame of an IL-2 signal sequence into pFUSE vectors (Invivogen) containing wild-type mouse IgG2a backbone (pFUSEss-CHIg-mG2a) and mouse constant IgK (pFUSE2ss-CLIg-mK) respectively. The pFUSEss-CHIg-mG2a was then subjected to site directed mutagenesis using Quikchange site-directed mutagenesis kit (Agilent) to introduce D265A mutation with the following primers: 5’-CATGTGTGGTGGTGGCTGTGAGCGAGGATGAC-3’ and 5’-GTCATCCTCGCTCACAGCCACCACCACACATG-3’.
200μg of purified hamster HM79 (IgG2/λ) antibody were subjected to SDS-PAGE and transferred to PVDF membrane. The membrane was stained using Ponceau Red. Higher and lower molecular bands, corresponding to heavy and light chains, respectively, were then submitted to N-terminal Edman sequencing. The EVRLLES sequence was obtained after 7 cycles for the hamster mAb higher molecular band. No data were obtained with hamster mAb lower molecular band suggesting that the sample was N-terminally blocked. After total RNA extraction from the HM79 hybridoma cell line and cDNA synthesis, the VH was PCR amplified using a degenerate forward primer, based on the EVRLLES sequence described in human and rodent VHs 5’-GAGGTGCGGCTKTTGGARTCTGG-3’ and reverse primer 5’-CTACGTTGCAG-GTGATGGGCTGCTTG-3’. Purified PCR product was further cloned into pCR4-TOPO vector (Invitrogen) before sequencing. Then, the HM79 VH was sub-cloned in frame of an IL-2 signal sequence into pFUSEss-CHIg-mG2a vectors containing mouse IgG2a wt and mouse IgG2a D265A backbones (mouse IgG2a D265A was generated by site directed mutagenesis as described above). For VL, part of the hamster constant lambda chain was cloned using degenerate forward primers 5’-CAARGCYAC-MYTGGTGTGTMYG-3’ and reverse primer, 5’-GRVRCABTCWGCASGRGMC-ARRCTC-3’ on the basis of hamster H57 Fab protein sequence (1NFD_E, GI: 2914251). Then, a specific reverse primer annealing in the hamster constant Lambda chain was designed, 5’-GTTTCCCCTTCATGGGTAACTTGGCAGG-3’. Finally, HM79 VL nucleotide sequence was amplified by RACE PCR using the SMART™ RACE cDNA Amplification Kit (Clontech) according to manufacturer’s instruction and cloned into pCR4-TOPO vector for sequencing. The VL sequence was further sub-cloned in frame of an IL-2 signal sequence into pFUSE2ss-CLIg-mL1 vector containing mouse constant Igλ1 for expression in mammalian cells.
Full-length 18B12 and chimeric HM79 heavy and light chain constructs were co-transfected in CHO cells and stable cell lines were obtained by selection using Blasticidin and Zeocin (Invivogen). Conditioned supernatant of Ig expressing CHO cells were collected and recombinant antibodies were purified by protein G affinity column chromatography (GE Healthcare).
CD79a and CD79b cloning and expression in PEAK cells
The cDNAs encoding mouse CD79a (BC027633) in pCMV6-AC-GFP vector and mouse CD79b (NM_008339) in pCMV6 vector were purchased from Origene Technologies. PEAK cells were cultured and transfected with this constructs as previously described (30). The cells were used for FACS staining with anti-CD79b mAbs 48 hours post-transfection.
Statistics
Because of the non-normal distributions of both the arthritis score and the B-cell counts, we used a nonparametric permutation testing approach to analyze the data in Figure 1A, Figure 7A, and Figure 7B (31). Additionally, we adjusted the p-values from these tests to compensate for the multiple significance testing. This adjustment used the permutation approach described by Westfall and Young (32), a procedure which has the further advantage of taking into account the correlation structure of the data (multiple assessments recorded for each animal). All statistical analyses were done using SAS 9.2.
Figure 1. Anti-CD79b mAb Inhibits Development of Collagen-Induced Arthritis.
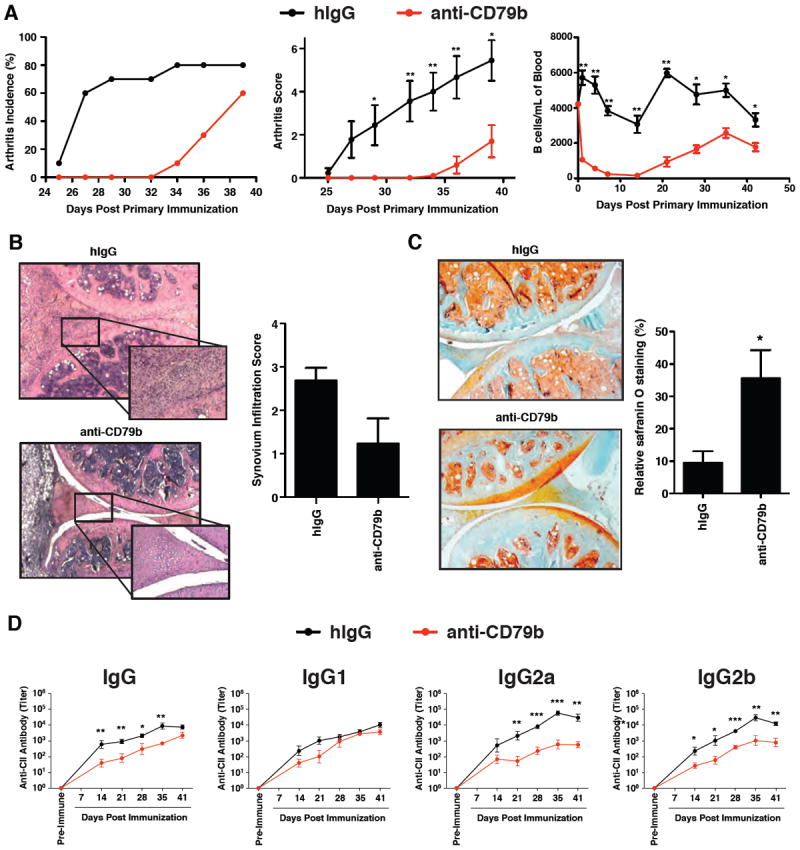
DBA/1 mice (n=10 per group) were treated with 1.0 mg of anti-CD79b mAb or hamster IgG (hIgG) as control 2 hours before primary immunization with C-II. Twenty-one days later, the mice received a second immunization with C-II. A. The arthritis incidence (left panel), arthritis score (middle panel) and B cell (CD19+) count in peripheral blood (right panel) were assessed. This experiment is representative of three independent experiments. B. Representative H&E stained joint sections at day 41 (original magnification X5, enlarged magnification X20). C. Representative Safranin O/fast green stained joint sections at day 41(original magnification X5, enlarged magnification X20). D. Serum was collected from five mice in each group once a week from day 14 to day 41 and the serum titers of anti-C-II IgG, IgG1, IgG2a and IgG2b was determined.
Figure 7. Loss of B cells Following anti-CD79b mAb Treatment Reflects Transient Relocalization of Cells Rather Than B Cell Death.
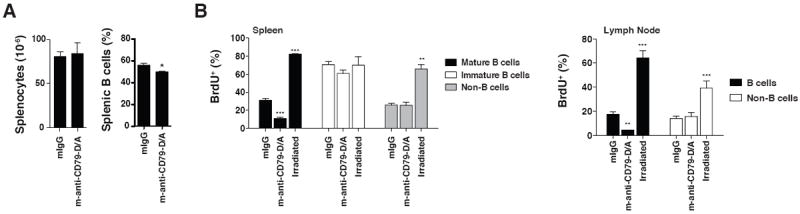
A. C57BL/6 wild-type mice (n=5) were injected with 1.0 mg of control mouse IgG or m-anti-CD79b-D/A mAb. 3 weeks later, they were harvested for analysis and splenocytes were enumerated and the proportion B cells determined by FACS. B. C57BL/6 wild-type mice (n=4) were injected with 1.0 mg of control mouse IgG or m-anti-CD79b-D/A mAb or were sublethally irradiated. The mice were then fed BrdU+ drinking water (0.8mg/ml) for three weeks after which the mice were sacrificed and harvested as described in the methods. The percentage of BrdU+ mature B cells (CD19+/CD93-), immature B cells (CD19+/CD93+) and non-B cells (CD19) were determined by intracellular BrdU staining and FACS analysis.
All other data are depicted as means with standard error of the mean. Data was graphed using PRISM software. Excluding the statistics described above, p-values were calculated using PRISM using 1-tailed or 2-tailed t-tests, where appropriate. Abbreviations used in the figures: * = p-value < 0.05, ** = p-value <0.01, *** = p-value <0.001, n.s. = not significant.
Study Approval
All animal studies were conducted under the procedures approved by the IACUC at National Jewish Health or according to license from the Swiss veterinary office for animal experimentation (OVC).
RESULTS
Anti-CD79b mAb Inhibits Development of Collagen-Induced Arthritis (CIA)
CIA has been shown to require B cells and has been used to define the therapeutic actions of B cell targeted mAb (33). To study the efficacy of anti-CD79b mAb in this model, 8-week old DBA/1 mice were pretreated with control polyclonal hamster IgG (hIgG) or anti-CD79b mAb prior to induction of CIA. Mice treated with a single dose of anti-CD79b mAb exhibited a delay in arthritis onset as well as reduced disease incidence and severity (Figure 1A, left and middle panel). Reduced B cell numbers were seen in peripheral blood for two weeks, followed by a gradual, but not entirely complete, recovery for the duration of the experiment (Figure 1A, right panel). There was reduced synovial lymphocytic infiltrate in anti-CD79b mAb-treated mice although this did not reach a significant level (p=0.054) (Figure 1B). Reduced infiltration was correlated with increased preservation of cartilage in anti-CD79b mAb-treated mice as indicated by histological analysis (Figure 1C). The reduction in disease anti-CD79b mAb treated mice was also correlated with decreased serum C-II-specific IgG, in particular IgG2a and IgG2b subclasses (Figure 1D). Interestingly, not all subclasses were equally affected by anti-CD79b mAb treatment, as IgG1 antibody levels were not significantly reduced.
Anti-CD79b mAb Treatment Leads to a Reduction in peripheral blood, lymph node and splenic B cells
Anti-CD79b mAb treatment reduced B cell numbers in peripheral blood, as has been reported by studies using other B cell-targeted treatments in both mice and humans (1-6, 17). To determine if anti-CD79b mAb treatment affected B cell numbers in other organs we treated 8-12 week-old C57BL/6 mice with 0.5 mg of control hIgG, anti-CD79b mAb or anti-CD20 mAb and analyzed spleens, lymph nodes, bone marrow and peripheral blood. One week postadministration, anti-CD79b mAb treatment reduced B cell numbers in the spleen by approximately 50% (Figure 2A, 2B). No concomitant increase in the number of peripheral T cells was observed (Supplemental Figure 1). However, loss of B cells was much less complete than that induced by an anti-CD20 mAb (18B12), a mouse IgG2a specific for mouse CD20 (Figure 2A). Consistent with previously published reports, anti-CD20 mAb treatment led to loss of > 95% of B cells from spleen (34, 35). B cell loss in the lymph nodes and peripheral blood was more efficient although still not approaching the depletion previously reported with anti-CD20 mAb treatment (Figure 2B). Genetic ablation of genes whose products are required for antibody-dependent cellular cytotoxicity (FcRγ and FcγRIIb) or disabling complementdependent cytotoxicity did not impair the ability of anti-CD79b mAb to induce B cell loss from these organs, suggesting that the depression in B cell numbers may be due to effects of the antibody on B cells other than Fc engagement of cytotoxic effectors (Supplemental Figure 2).
Figure 2. Anti-CD79b mAb Treatment Leads to a Reduction in Peripheral B cells.
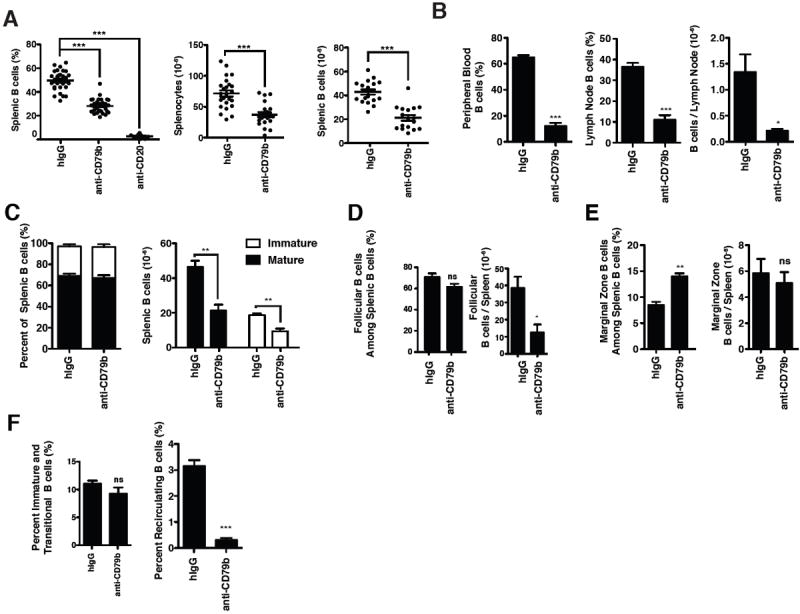
Wild-type C57BL/6 mice were injected with 0.5 mg of control polyclonal hamster IgG (hIgG), anti-CD79b mAb (HM79) or 0.25 mg of anti-CD20 mAb (18B12). Tissues were harvested for analysis one week later. A. The percentage of B cells (CD19+) among splenocytes (left), absolute number of splenocytes (middle) and absolute number of splenic B cells (CD19+) (right). Data are the total of 5-7 independent experiments as and consist of 30 to 10 mice. B. The percentage of B cells (CD19+) in peripheral blood (left) and lymph node (middle). The absolute number of B cells (CD19+) in the lymph node (right panel). C. The frequency (left) and absolute numbers (right) of immature (CD93+) and mature (CD93-) B cells among the total splenic B cells (CD19+). D. The frequency (left) and absolute number (right) of follicular B cells (CD23+, CD21+) B cells among splenic B cells (CD19+). E. The frequency (left) and absolute number (right) of marginal zone (CD21+, CD1dhi) B cells among splenic B cells (CD19+) F. The frequency of immature B cells (CD23-/CD24hi/CD93+/CD19+) (left) and recirculating mature B cells (CD23+/CD24int/CD93-/CD19+/B220+) (right panel) among total bone marrow cells. B-F. These experiments are representative of three independent experiments of 3-5 mice per group.
We observed no differential effect of anti-CD79 treatment on splenic immature (CD93+), mature (CD93-) or follicular B cell numbers (Figure 2C, 2D). Surprisingly, however, anti-CD79b mAb treatment had no effect on the number of marginal zone B cells recovered (CD21+/CD1dhi) (Figure 2E). No change was observed in developing B cell populations (CD23-/CD24hi/CD93+/B220+) in the bone marrow despite loss of the mature recirculating B cell populations (CD23+/CD24int/CD93-/B220+/CD19+) (Figure 2F).
Targeting CD79 with mAb Does Not Induce B cell Activation
B cell activation is also a potential consequence of BCR ligation in vivo. To explore the status of B cells following anti-CD79b mAb treatment, we assessed expression of B cell activation markers ex vivo 12-48 hours following administration of anti-CD79b mAb. B cells in mice treated with anti-CD79b mAb exhibited a slight up-regulation of CD69 and CD86, but not CD44, at 12 hours (Figure 3A). However, by 24 hours, expression of these markers returned to baseline. Analysis of cells one week following treatment revealed no significant up-regulation of MHCII, CD80, CD44 or CD86 (Figure 3B). As an additional assay of B cell activation and differentiation to immunoglobulin-secreting cells, we analyzed serum levels of total IgM and IgG in anti-CD79b mAb treated mice. We detected no change in serum IgM or IgG levels relative to controls (Figure 3C). Thus, BCR signaling induced in vivo by anti-CD79b does not cause significant B cell activation.
Figure 3. Targeting CD79 with mAb Does Not Induce B cell Activation.
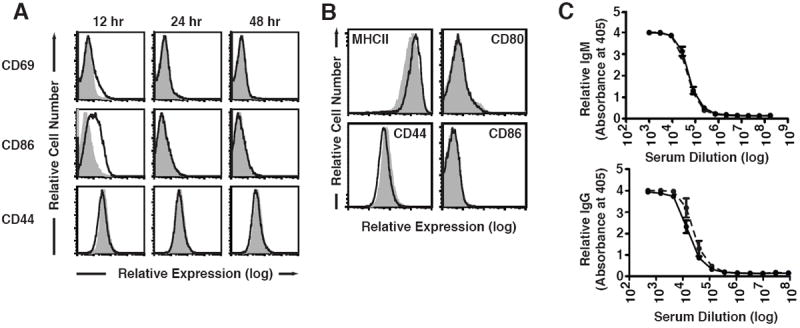
Wild-type, C57BL/6 mice (2-3 per group) were injected with 0.5 mg of control polyclonal hamster IgG (shaded trace) or anti-CD79b mAb (HM79) (red trace). A. Tissues were harvested from mice at 12, 24 and 48 hours post injection with antibody and splenic B cells (CD19+) were stained for surface expression of CD69, CD86 and CD44, as described in Methods. B. Tissues were harvested one week after injection with mAb and splenic B cells (CD19+) were stained for surface expression of MHC-II, CD44, CD80 and CD86. C. Serum was collected one week after injection with 0.5 mg of control polyclonal hamster IgG (solid line) or anti-CD79b mAb (HM79) (dashed line) and analyzed for the levels of serum IgM and IgG.
Protection from Autoimmunity is Not Dependent on Anti-CD79b-induced Reduction of Peripheral B cells
To determine whether the therapeutic activity of anti-CD79b mAb is dependent on Fc domain function we engineered anti-CD79b and anti-CD20 mAbs to eliminate ADCC and CDC activity, and tested their effectiveness in preventing disease. Variable regions of the hamster anti-CD79b antibody, HM79, were cloned into mouse lambda chain and IgG2a heavy chain constant region backbones to create a chimeric hamster/mouse anti-CD79b mAb (m-anti-CD79b). We then engineered an alanine substitution at aspartic acid 265 in the chimeric anti-CD79b mAb to generate m-anti-CD79b-D/A and, as a control, into the anti-CD20 mAb generating anti-CD20-D/A, 18B12. The 265 aspartic acid residue has been shown to be required for the functional engagement of IgG Fc with FcγR and complement (36). Importantly, both mutant and non-mutant m-anti-CD79b mAbs retained ability to bind CD79b-expressing cells ex vivo and reduced B cell populations similarly to the parental hamster mAbs in vivo (Supplemental Figures 3)
To test the efficacy of these antibodies, we examined their ability to modulate arthritis induced in DBA/1 mice as described in Figure 1. Mice were injected once at the beginning of the experiment with control mouse IgG2a (mIgG2a), m-anti-CD20 mAb, m-anti-CD20-D/A mAb, m-anti-CD79b mAb, or m-anti-CD79b-D/A mAb. B cell numbers in peripheral blood and disease activity were assessed at regular intervals thereafter. As previously reported, mice treated with m-anti-CD20 mAb did not develop arthritis and had remarkably reduced peripheral B cells for the duration of the experiment (Figure 4A). Importantly, introduction of the D265A mutation eliminated the protective effects of m-anti-CD20 mAb, while its ability to deplete peripheral B cell was partially retained (Figure 4A). These data suggest that the protective effects of anti- CD20 mAb are Fc-dependent and predicated upon efficient and complete peripheral B cell clearance.
Figure 4. Protection from Autoimmunity is Not Dependent on Anti-CD79b-induced Reduction of Peripheral B cells.
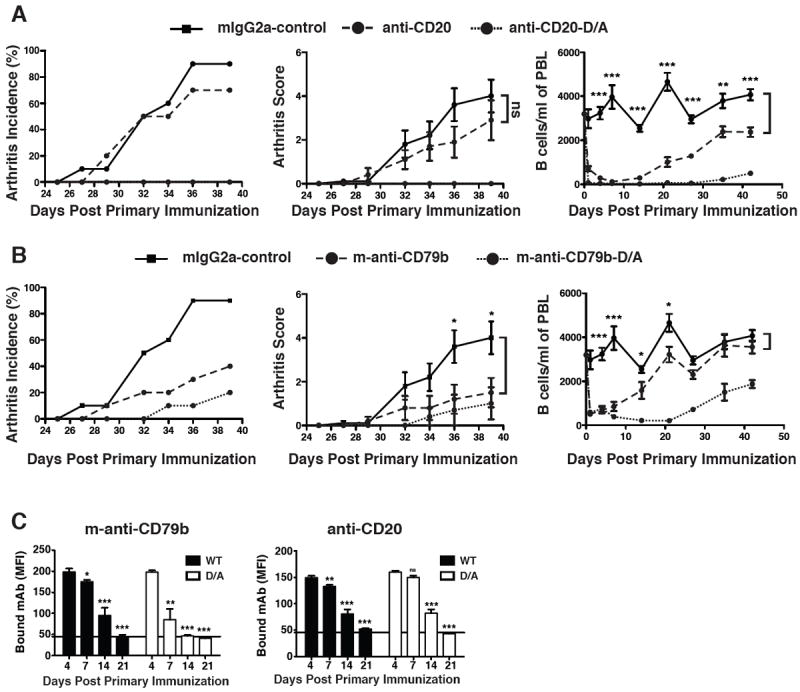
Arthritis was initiated in DBA/1 mice as described in methods. A&B. On the same day as the first C-II immunization, mice (n=10) were treated with 1.0 mg of mouse IgG2a, anti-CD20 mAb, anti-CD20-D/A mAb, m-anti-CD79b mAb, or m-anti-CD79b-D/A mAb. The arthritis incidence (left panels), arthritis score (middle panels) and B cell count in peripheral blood (right panels) were assessed. Statistical significance between the isotype control and the D/A mAb is as indicated. This experiment is representative of two independent experiments. C. Serum was collected from five mice in each group from day 4 to day 41 and used to stain freshly isolated target B cells.
Similar to the results shown in Figure 1, incidence and clinical score of collagen-induced arthritis was reduced in mice treated with m-anti-CD79b-D/A mAb despite the fact that this treatment caused less B cell loss than the functionally ineffective m-anti-CD20-D/A antibody (Figure 4B).
Consistent with a non-depleting mode of action of m-anti-CD79b-D/A antibody, peripheral blood B cell numbers returned to near normal levels within two weeks of treatment (Figure 4B). Despite complete recovery of the peripheral B cell pool prior to the secondary collagen challenge on day 21, m-anti-CD79b-D/A mAb treatment impaired the onset of collagen-induced arthritis and reduced disease severity. These observations are in contrast to those seen with anti-CD20-D/A mAb, which, despite more efficient peripheral B cell depletion, did not suppress arthritis development. Thus, depletion of peripheral B cells cannot account for the therapeutic efficacy of anti-CD79b mAb.
To asses the effect of D/Z mutations on mAb half-life, we determined the serum levels of free mAb in mice from Figure 4A and 4B. As expected, the levels of free serum m-anti-CD20 mAb were not altered by the D265A mutation which lies outside of the FcRn binding site (Figure 4C, right panel) (37). Unexpectedly, the D265A mutation significantly reduced the half-life of free serum m-anti-CD79b (Figure 4D, left panel). Like m-anti-CD20 and m-anti-CD20-D/A, m-anti-CD79b mAb was detectable in the serum of treated mice for more than two weeks following injection, while m-anti-CD79b-D/A was cleared about a week earlier. Thus m-anti-CD79-D/A retains protective activity despite having a reduced half-life and truncated biological activity relative to m-anti-CD20-D/A.
Anti-CD79b mAb Induces B cell Antigen Receptor Desensitization
The effectiveness of m-anti-CD79b-D/A mAb in CIA suggests that therapeutic benefit may derive solely from the mAbs’ interaction with its antigen, CD79b. Thus we hypothesized that exposure of B cells to a chronic antigen receptor stimulus derived from the anti-CD79b mAb may mimic the tolerizing signals that occur naturally and drive autoreactive cells into a silenced state termed ‘B cell anergy’ (20, 23, 24). Consistent with this hypothesis, anti-CD79b mAb was able to productively engage BCR-signaling as demonstrated by Ca2+ mobilization in B cells, while anti-CD20 mAb was not (Figure 5A). To further explore the possible role of anergy in mediating the effects of anti-CD79b mAb, we determined whether in vivo treatment affects the ability of B cells to signal through their antigen receptors. B cells recovered from mice 7 days following a single administration of anti-CD79b mAb expressed reduced surface levels of IgM, IgD and CD79b consistent with ligand encounter (Figure 5B). These recovered B cells were coated with anti-CD79b mAb as indicated by staining with anti-hamster Ig antibody (Figure 5B). Control experiments indicated that binding of staining reagents, including the affinity purified rabbit anti-CD79 used to measure CD79 levels, was not significantly hindered by monoclonal anti-CD79 adsorbed to the B cells.
Figure 5. Anti-CD79b mAb Induces B cell Antigen Receptor Desensitization.
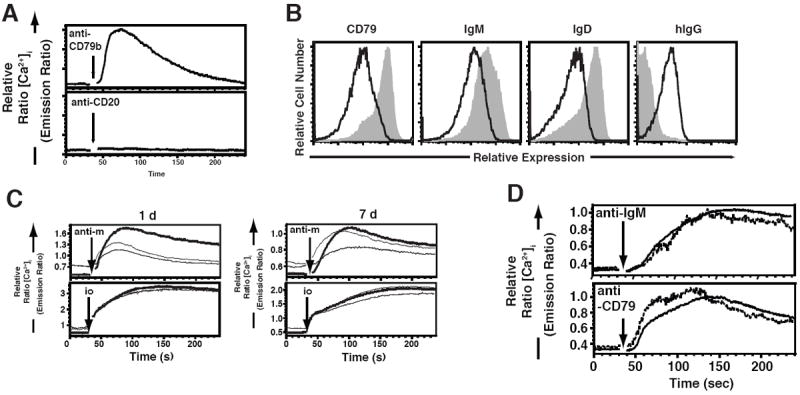
A. Analysis of [Ca2+]i of wild-type, splenic B cells (B220+) in response to 10μg/ml of anti-CD79b mAb or anti-CD20 mAb. Splenocytes were prepared as described in Methods. This experiment is representative of two independent experiments. B. Wild-type C57BL/6 mice (n=3 per group) were injected with 0.5 mg of hIgG (shaded trace) or anti-CD79b antibody (solid line). One week later, tissues were harvested for analysis. Splenic B cells (CD19+) were assessed for surface expression of bound hamster IgG, IgM, IgD and CD79 as described in Methods. This experiment is representative of three independent experiments. C. Wild-type C57BL/6 mice were injected with 1.0 mg of control mouse IgG (heavy line), m-anti-CD79b mAb (solid line) or m-anti- CD79b-D/A mAb (light line). One week later, tissues were harvested and intracellular Ca2+ influx of B cells (B220+) in response to BCR cross-linking was determined by flow cytometry. D. Wild-type C57BL/6 mice were injected with 0.5 mg of control polyclonal hamster IgG (black trace) or 0.25 mg of anti-CD20 antibody (dashed trace). One week later, tissues were harvested and intracellular Ca2+ influx of B cells (B220+) in response to BCR cross-linking was determined by flow cytometry.
Previous work from our laboratory has demonstrated that chronic engagement of the BCR in anergic B cells results in elevated basal Ca2+ and dampened BCR-mediated Ca2+ influx (19). Analysis of B cells recovered from m-anti-CD79b mAb and m-anti-CD79-D/A-treated mice one day following treatment revealed elevated basal Ca2+ and suppressed BCR-mediated Ca2+ influx (Fig 5C). One week later, B cells from anti-CD79b treated animals remained hyporesponsive, while those from m-anti-CD79-D/A-treated animals had partially recovered responsiveness (Fig 5C). This is likely a consequence of the reduced half-life of the m-anti-CD79-D/A mAb relative to the intact m-anti-CD79b mAb. Finally, B cells recovered following anti-CD20 mAb treatment did not display impaired BCR-mediated Ca2+ mobilization or increased basal [Ca2+]i (Figure 5D). Thus, treatment of mice with anti-CD79b mAb, but not anti-CD20 mAb, leads to B cell unresponsiveness to subsequent BCR stimulation, as is characteristic of anergic B cells.
Anti-CD79b mAb induces B cell Anergy
B cells from mice treated with anti-CD79b mAb share many features with anergic B cells including phenotypic markers and blunted B cell antigen receptor-mediated Ca2+ signaling. To determine if these characteristics extend to impairment of their ability to mount antibody responses in vivo, we treated mice with anti-CD79b mAb or control polyclonal hamster IgG and immunized i.p. with nitrophenyl-conjugated ovalbumin (NP4Ova) precipitated in Alum. On day 15 following immunization, serum levels of NP-specific IgG were measured. Antibody responses were reduced by 75% in anti-CD79b mAb treated mice (Figure 6A, top panel). Interestingly, high affinity NP-specific IgG was reduced by more than 90% by anti-CD79b mAb treatment, suggesting selective impairment of affinity matured responses (Figure 6A, bottom panel). ELISPOT enumeration of total and high-affinity NP-specific IgG antibody-secreting cells (ASC) revealed a similar anti-CD79-induced suppression. Control IgG treated mice had a mean of 117 ASC/106 splenocytes and approximately 43% of these were secreting high affinity antibody as indicated by detection with low-valency antigen. In contrast, anti-CD79b treated mice produced half as many total IgG+ anti-NP ASC and of these only 12% were high affinity (Figure 6B).
Figure 6. Anti-CD79b mAb Treatment Induces B cell anergy.
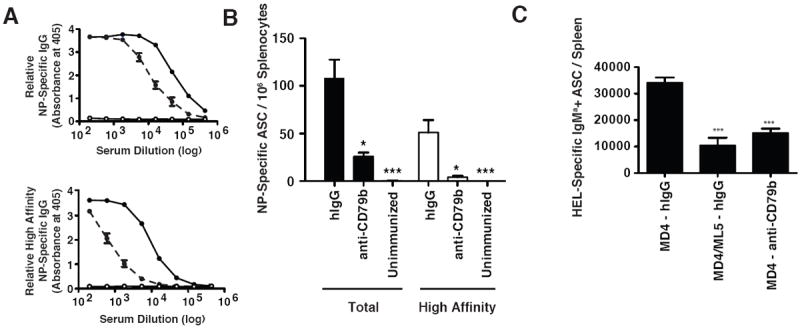
Wild-type, C57BL/6 mice (n=3) were injected on with 0.5 mg of hamster IgG or anti-CD79b mAb and immunized i.p. with 100μg of NP4Ova in Alum. On day 15, serum was collected and tissues were harvested for analysis. A. The relative amounts of total (top panel) and high-affinity (bottom panel) NP-specific IgG from mice treated with control polyclonal hamster IgG (black trace) or anti-CD79b mAb (dashed line). B. The number of the total and high-affinity NP-specific IgG+ antibody secreting cells per 106 splenocytes. The experiment is representative of two independent experiments. C. MD4 mice were injected with hIgG or anti-CD79b and MD4/ML5 mice were injected with hIgG as previously described. One day later B cells were purified and 106 B cells were adoptively transferred into recipient wild-type C57BL/6 mice (n=5). An hour post transfer 100 ul of 10% SRBC conjugated to HEL was injected I.P. 5 days later tissues were harvested for analysis and an the number of splenic HEL-specific IgMa+ antibody secreting cells were enumerated.
B cell anergy is a cell intrinsic phenomena due in part to altered BCR signaling. To determine whether anti-CD79b mAb-induced B cell suppression was also cell intrinsic we utilized the MD4/ML5 mouse model of B cell tolerance. MD4 transgenic mice express a hen-egg lysozyme (HEL) specific BCR and contain a mostly naïve peripheral B cell population. ML5 transgenic mice express soluble HEL and when crossed to the MD4 transgene results in the expression of HEL as a neo-cognate self-antigen (20). B cells from MD4/ML5 mice are rendered anergic as evidenced by reduced peripheral B cell numbers, lower surface IgM expression and fail to mobilize calcium efficiently upon BCR crosslinking (20). We treated MD4 mice with anti-CD79b mAb and one day later purified and transferred 1×106 splenic B cells from these donor mice into wild-type C57BL/6 (IgMb+) recipients. As control we transferred anergic B cells isolated from an anti-HEL/sHEL (MD4/ML5) transgenic mice. A naïve B cell control was provided by B cells from untreated anti-HEL (MD4) tg mice. One hour after transfer recipient mice were immunized by i.p. injection of SRBC-HEL. Five days after cell transfer, spleens from recipient mice were harvested and HEL-specific IgMa+ antibody secreting cells were measured by ELISPOT assay. Anergic B cells from MD4/ML5 mice and B cells from MD4 mice treated with anti-CD79b mAb prior to transfer exhibited a similarly impaired immune response relative to control treated MD4 B cells suggesting that the anti-CD79b mAb treatment led to a B cell intrinsic hyporesponsiveness functionally equivalent to anergy (Figure 6C).
Reduced recovery of B cells following anti-CD79b mAb treatment reflects transient relocalization of cells
Repopulation of the B cell compartment following treatment with anti-CD20 requires weeks in mice and months in humans (38, 39). However, in mice treated with m-anti-CD79b-D/A mAb splenic B cells returned to near-normal levels within 14 days of maximal B cell reduction suggesting that treatment does not cause B cell death (Figure 7A). To determine if this repopulation was due to de novo generation of new B cells, mice were treated with antibodies then continuously fed bromodeoxyuridine (BrdU)-containing drinking water for three weeks to label B cells produced during the recovery period. Approximately 31.5% of the mature (CD93-) splenic B cells from control mice had incorporated BrdU, consistent with generation due to normal B cell turnover (Figure 7B). In mice that had been sublethally irradiated to kill most mature B cells, >80% of B cells incorporated BrdU during de novo generation in the bone marrow. Surprisingly, only 10.9% of mature B cells from m-anti-CD79b-treated mice incorporated BrdU despite near-complete restoration of the B cell compartment (Figure 7B). No differences were observed in BrdU incorporation in immature B cells, or in non-B cells. In treated and control animals, BrdU incorporation in the lymph node B cells was similarly affected by the respective treatments (Figure 7B). These data formally demonstrate that B cell “depleting” effects of m-anti-CD79 D/A mAb treatment does not reflect B cell death. Rather, the treatment must reflect transient relocation of some B cells to other niches from which they return following the clearance of m-anti-CD79b-D/A mAb from the animal.
DISCUSSION
Previous work by collaborators has demonstrated that anti-CD79b mAbs are therapeutic in the MRL/lpr mouse model of lupus (17). Here we report analysis of the mechanisms that underlie the efficacy of anti-CD79b mAb treatment. Unlike other B cell-targeted biologics that induce B cells death by ADCC, Complement fixation or survival factor starvation mediated cell death, anti-CD79b mAb appears to exert its therapeutic effect by inducing a transient state of B cell anergy. Analogous to naturally occurring anergic B cells, B cell exposure to anti-CD79b mAb in vivo leads to reduced surface immunoglobulin, impaired BCR-triggered calcium mobilization, and failure to mount a complete and robust response to antigen challenge. Further, we demonstrate that this polyclonal B cell anergy inhibits pathogenic immune responses in the absence of complete or long-term B cell depletion.
The unique mechanism of action of anti-CD79b mAb relative to anti-CD20 mAb derives from the distinct biologic function of its target antigen, CD79. CD20 is a membrane tetraspanner that was originally thought to have ion channel activity (40). Mice deficient in CD20 have relatively intact B cell development and humoral immunity (7). In contrast, CD79 is an integral component of the B cell antigen receptor complex and is required for B cell development and survival in the periphery (41-44). Signals emanating from CD79 are critical determinants of B cell fate decisions, among them peripheral B cell anergy.
In mice, peripheral B cell depletion by CD20-targeted mAb is primarily mediated by the interaction of the mAbs Fc domain with host Fcγ receptors and clearance of mAb coated B cells by ADCC (34). When the concentration of anti-CD20 mAb falls below the threshold necessary to maintain clearance, B cells newly produced in bone marrow repopulate the periphery, followed soon by a return of pathology (27, 38, 39). Some reports suggest that certain anti-CD20 mAbs can induce tolerance or apoptosis directly by cross-linking of CD20 (9, 11). However, considering the compelling evidence that ADCC is still critical for B cell depletion by these anti-CD20 mAbs, the direct induction of apoptosis is likely to be of only minor significance in anti-CD20 mAb action (45).
If the activity of anti-CD79b mAb does not require B cell depletion, then how is the antibody protective in CIA? Experiments reported here shed additional light on this question. The introduction of the D265A mutation, which ablates mAb ADCC and CDC activity, virtually eliminates the ability of anti-CD20 mAb to modify the course of CIA. In contrast to the anti-CD20 mAb, anti-CD79b mAb possessing the same mutation retain the ability to impair development of CIA despite return of B cells to peripheral lymphoid organs prior to disease onset. Experiments addressing the contribution of newly generated B cells to peripheral pools indicate that the disease preventive effects of m-anti-CD79b-D/A mAb are associated with transient B cell relocalization in the animal, rather than B cell death. Additional studies are required to elucidate the basis of this induced peripheral B cell redistribution and the contributions of this phenomena to the disease preventive activity of anti-CD79b mAb. It is interesting that FcR binding incompetent anti-CD3 mAbs induce a similarly transient redistribution of T cells in treated animals, and this is associated with cell localization in the gut and is apparently the consequence of TCR signaling (46). Whether a similar response is activated in B cells by anti-CD79 antibodies is currently under study.
It is not clear how similar the molecular underpinnings of anti-CD79b mAb -induced unresponsiveness is to naturally occurring anergy. Some minor differences do exist. For example, treatment with anti-CD79b mAb causes down-regulation of all components of the BCR, while in most cases anergic B cells display down-regulation of IgM, with IgD remaining at near normal levels (24). Additionally, preliminary results suggest that one of the apparently redundant regulatory enzymes that is important in maintaining anergy, the inositol 5-posphate phosphatase SHIP-1, is dispensable for anti-CD79b mAb-induced desensitization, at least in vitro (Hardy and Cambier, unpublished observations, (18). It is important to note that anergy is a rapidly reversible phenomenon, the maintenance of which requires chronic antigen receptor occupancy (19). The anergic-like state induced by anti-CD79b therapy appears similarly reversible and this would be advantageous when patients develop disadvantageous side effects or opportunistic infections.
We suggest that with development of additional strategies for suppression of pathogenic B cell function, may come a situation in multiple B cell-targeted therapies gain acceptance in the clinical market. As we better understand mode of action of B cell-targeted therapies in autoimmunity, and thus determinants of efficacy, it may be possible to predict an optimal course of B cell-targeted therapy based on the integrated calculus of disease state and the genetics of the patient. One clear aim of next-generation B cell-targeted therapy is achievement of greater specificity, thus improving upon the current paradigm of disease control at the expense of global immune suppression. Anti-CD79b mAb therapy, with its potential to allow rapid immune recovery, may represent a step toward this goal.
In conclusion, our studies define a novel, potentially therapeutic approach that silences B cells en masse by inducing an antigen unresponsive state reminiscent of anergy. These findings have far reaching implications for the design of therapeutics targeting the many other receptors that are subject to ligand-induced desensitization.
Supplementary Material
Acknowledgments
We thank Robert Dunn, Marilyn Kehry, and Biogen Idec, for generous provision of 18B12 mAb used in these experiments. We thank Les Huson for assistance in the statistical analyses of several experiments, and Sandra Duran for assistance in preparing the manuscript.
This work was supported by the Colorado Bioscience Discovery Evaluation Grant Program, and National Institute of Health grants P01 AI022295 and R01 AI077597
References
- 1.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 2.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, Wey K, Royston I, Davis T, Levy R. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90:2188–2195. [PubMed] [Google Scholar]
- 3.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 4.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, Raskin P, Rodriguez H, Schatz DA, Wherrett D, Wilson DM, Lachin JM, Skyler JS. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, Zhang D, Brunetta PG. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, Hanna N, Anderson DR. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–445. [PubMed] [Google Scholar]
- 7.Uchida J, Lee Y, Hasegawa M, Liang Y, Bradney A, Oliver JA, Bowen K, Steeber DA, Haas KM, Poe JC, Tedder TF. Mouse CD20 expression and function. Int Immunol. 2004;16:119–129. doi: 10.1093/intimm/dxh009. [DOI] [PubMed] [Google Scholar]
- 8.Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, Webster AD. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003;48:2146–2154. doi: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 9.Byrd JC, Kitada S, Flinn IW, Aron JL, Pearson M, Lucas D, Reed JC. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood. 2002;99:1038–1043. doi: 10.1182/blood.v99.3.1038. [DOI] [PubMed] [Google Scholar]
- 10.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 11.Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91:1644–1652. [PubMed] [Google Scholar]
- 12.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 2007;179:3351–3361. doi: 10.4049/jimmunol.179.5.3351. [DOI] [PubMed] [Google Scholar]
- 13.Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, Maciuca R, Zhang D, Garg JP, Brunetta P, Appel G. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64:1215–1226. doi: 10.1002/art.34359. [DOI] [PubMed] [Google Scholar]
- 14.Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum. 2002;46:2673–2677. doi: 10.1002/art.10541. [DOI] [PubMed] [Google Scholar]
- 15.Perrotta S, Locatelli F, La Manna A, Cennamo L, De Stefano P, Nobili B. Anti-CD20 monoclonal antibody (Rituximab) for life-threatening autoimmune haemolytic anaemia in a patient with systemic lupus erythematosus. Br J Haematol. 2002;116:465–467. [PubMed] [Google Scholar]
- 16.Weide R, Heymanns J, Pandorf A, Koppler H. Successful long-term treatment of systemic lupus erythematosus with rituximab maintenance therapy. Lupus. 2003;12:779–782. doi: 10.1191/0961203303lu449cr. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Chen F, Putt M, Koo YK, Madaio M, Cambier JC, Cohen PL, Eisenberg RA. B cell depletion with anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr mice. J Immunol. 2008;181:2961–2972. doi: 10.4049/jimmunol.181.5.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity. 2009;31:749–760. doi: 10.1016/j.immuni.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol. 2005;6:1160–1167. doi: 10.1038/ni1256. [DOI] [PubMed] [Google Scholar]
- 20.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 21.Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, Lewis RS, Goodnow CC. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–428. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- 22.O’Neill SK, Getahun A, Gauld SB, Merrell KT, Tamir I, Smith MJ, Dal Porto JM, Li QZ, Cambier JC. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity. 2011;35:746–756. doi: 10.1016/j.immuni.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nossal GJ, Pike BL. Clonal anergy: persistence in tolerant mice of antigen-binding B lymphocytes incapable of responding to antigen or mitogen. Proc Natl Acad Sci U S A. 1980;77:1602–1606. doi: 10.1073/pnas.77.3.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benschop RJ, Aviszus K, Zhang X, Manser T, Cambier JC, Wysocki LJ. Activation and anergy in bone marrow B cells of a novel immunoglobulin transgenic mouse that is both hapten specific and autoreactive. Immunity. 2001;14:33–43. doi: 10.1016/s1074-7613(01)00087-5. [DOI] [PubMed] [Google Scholar]
- 25.Healy JI, Dolmetsch RE, Lewis RS, Goodnow CC. Quantitative and qualitative control of antigen receptor signalling in tolerant B lymphocytes. Novartis Found Symp. 1998;215:137–144. doi: 10.1002/9780470515525.ch10. discussion 144-135, 186-190. [DOI] [PubMed] [Google Scholar]
- 26.Vilen BJ, Famiglietti SJ, Carbone AM, Kay BK, Cambier JC. B cell antigen receptor desensitization: disruption of receptor coupling to tyrosine kinase activation. J Immunol. 1997;159:231–243. [PMC free article] [PubMed] [Google Scholar]
- 27.Anolik JH, Campbell D, Felgar RE, Young F, Sanz I, Rosenblatt J, Looney RJ. The relationship of FcgammaRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheum. 2003;48:455–459. doi: 10.1002/art.10764. [DOI] [PubMed] [Google Scholar]
- 28.Lissilaa R, Buatois V, Magistrelli G, Williams AS, Jones GW, Herren S, Shang L, Malinge P, Guilhot F, Chatel L, Hatterer E, Jones SA, Kosco-Vilbois MH, Ferlin WG. Although IL-6 trans-signaling is sufficient to drive local immune responses, classical IL-6 signaling is obligate for the induction of T cell-mediated autoimmunity. J Immunol. 2010;185:5512–5521. doi: 10.4049/jimmunol.1002015. [DOI] [PubMed] [Google Scholar]
- 29.Getahun A, Smith MJ, Kogut I, van Dyk LF, Cambier JC. Retention of anergy and inhibition of antibody responses during acute gamma herpesvirus 68 infection. J Immunol. 2012;189:2965–2974. doi: 10.4049/jimmunol.1201407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muraki M, Honda S. Efficient production of human Fas receptor extracellular domain-human IgG1 heavy chain Fc domain fusion protein using baculovirus/silkworm expression system. Protein Expr Purif. 2010;73:209–216. doi: 10.1016/j.pep.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Good P. Permutation Tests: A practical guide to resampling methods for testing hypothesis. Springer Series in Statistics 1994 [Google Scholar]
- 32.Westfall P, Y S. Resampling-Based Multiple Testing: Examples and Methods for p-Vallue Adjustment. Wiley Series in Probability and Statistics 1993 [Google Scholar]
- 33.Yanaba K, Hamaguchi Y, Venturi GM, Steeber DA, St Clair EW, Tedder TF. B cell depletion delays collagen-induced arthritis in mice: arthritis induction requires synergy between humoral and cell-mediated immunity. J Immunol. 2007;179:1369–1380. doi: 10.4049/jimmunol.179.2.1369. [DOI] [PubMed] [Google Scholar]
- 34.Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–1669. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu S, Dunn R, Kehry MR, Braley-Mullen H. B cell depletion inhibits spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2008;180:7706–7713. doi: 10.4049/jimmunol.180.11.7706. [DOI] [PubMed] [Google Scholar]
- 36.Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, Petry F, Ravetch JV, Nishimura S, Izui S. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol. 2008;181:6664–6669. doi: 10.4049/jimmunol.181.9.6664. [DOI] [PubMed] [Google Scholar]
- 37.Carver DJ, Aman MJ, Ravichandran KS. SHIP inhibits Akt activation in B cells through regulation of Akt membrane localization. Blood. 2000;96:1449–1456. [PubMed] [Google Scholar]
- 38.Beers SA, French RR, Chan HT, Lim SH, Jarrett TC, Vidal RM, Wijayaweera SS, Dixon SV, Kim H, Cox KL, Kerr JP, Johnston DA, Johnson PW, Verbeek JS, Glennie MJ, Cragg MS. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood. 2010;115:5191–5201. doi: 10.1182/blood-2010-01-263533. [DOI] [PubMed] [Google Scholar]
- 39.Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum. 2006;54:2377–2386. doi: 10.1002/art.22019. [DOI] [PubMed] [Google Scholar]
- 40.Bubien JK, Zhou LJ, Bell PD, Frizzell RA, Tedder TF. Transfection of the CD20 cell surface molecule into ectopic cell types generates a Ca2+ conductance found constitutively in B lymphocytes. J Cell Biol. 1993;121:1121–1132. doi: 10.1083/jcb.121.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong S, Nussenzweig MC. Regulation of an early developmental checkpoint in the B cell pathway by Ig beta. Science. 1996;272:411–414. doi: 10.1126/science.272.5260.411. [DOI] [PubMed] [Google Scholar]
- 42.Pelanda R, Braun U, Hobeika E, Nussenzweig MC, Reth M. B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-alpha and Ig-beta. J Immunol. 2002;169:865–872. doi: 10.4049/jimmunol.169.2.865. [DOI] [PubMed] [Google Scholar]
- 43.Bannish G, Fuentes-Panana EM, Cambier JC, Pear WS, Monroe JG. Ligand-independent signaling functions for the B lymphocyte antigen receptor and their role in positive selection during B lymphopoiesis. J Exp Med. 2001;194:1583–1596. doi: 10.1084/jem.194.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bannish G, Fuentes-Panana EM, Cambier JC, Pear WS, Monroe JG. Ligand-independent signaling functions for the B lymphocyte antigen receptor and their role in positive selection during B lymphopoiesis. J Exp Med. 2001;194:1583–1596. doi: 10.1084/jem.194.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Haij S, Jansen JH, Boross P, Beurskens FJ, Bakema JE, Bos DL, Martens A, Verbeek JS, Parren PW, van de Winkel JG, Leusen JH. In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer Res. 2010;70:3209–3217. doi: 10.1158/0008-5472.CAN-09-4109. [DOI] [PubMed] [Google Scholar]
- 46.Waldron-Lynch F, Henegariu O, Deng S, Preston-Hurlburt P, Tooley J, Flavell R, Herold KC. Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci Transl Med. 2012;4:118ra112. doi: 10.1126/scitranslmed.3003401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.