

Abstract
Background
Although technical advances in genomics and proteomics research have yielded a better understanding of the coding capacity of a genome, one major challenge remaining is the identification of all expressed proteins, especially those less than 100 amino acids in length. Such information can be particularly relevant to human pathogens, such as Trypanosoma brucei, the causative agent of African trypanosomiasis, since it will provide further insight into the parasite biology and life cycle.
Results
Starting with 993 T. brucei transcripts, previously shown by RNA-Sequencing not to coincide with annotated coding sequences (CDS), homology searches revealed that 173 predicted short open reading frames in these transcripts are conserved across kinetoplastids with 13 also conserved in representative eukaryotes. Mining mass spectrometry data sets revealed 42 transcripts encoding at least one matching peptide. RNAi-induced down-regulation of these 42 transcripts revealed seven to be essential in insect-form trypanosomes with two also required for the bloodstream life cycle stage. To validate the specificity of the RNAi results, each lethal phenotype was rescued by co-expressing an RNAi-resistant construct of each corresponding CDS. These previously non-annotated essential small proteins localized to a variety of cell compartments, including the cell surface, mitochondria, nucleus and cytoplasm, inferring the diverse biological roles they are likely to play in T. brucei. We also provide evidence that one of these small proteins is required for replicating the kinetoplast (mitochondrial) DNA.
Conclusions
Our studies highlight the presence and significance of small proteins in a protist and expose potential new targets to block the survival of trypanosomes in the insect vector and/or the mammalian host.
Keywords: Genomics, Proteomics, Mass spectrometry data, Non-coding RNA, Mitochondria
Background
Recent advances in high-throughput sequencing technologies have led to the discovery of a large number of transcripts originating from regions of the genome previously thought to be silent [1]. One major challenge arising from these observations is to determine whether these transcripts code for a protein or should be classified as non-coding RNAs. This task is rather overwhelming, since a majority of these transcripts only have the potential to encode small proteins, generally less than 100 amino acids (aa) [2,3]. Historically, an arbitrary cutoff for open reading frames of 100 aa was applied in genome annotation projects [4,5] and thus the extent and functional significance of small open reading frames (sORFs) remains a largely unexplored territory in many organisms. Nevertheless, copious reports clearly indicate that they play crucial biological roles, including protection against pathogens [6,7], signal transduction [8], serving as molecular chaperones [9], developmental regulation [10-13] and even calcium transport in cardiac muscle contraction [14].
Several proteins encoded by sORFs have been identified serendipitously by biochemical methods as part of a complex or the product of a processed precursor protein. One example is the Drosophila tarsal-less (tal) gene, originally annotated as non-coding, but later shown to encode three small proteins with a crucial role in fly development [13]. Several studies have used genome-wide approaches to gauge the prevalence of sORFs. When examining potential small proteins in Drosophila melanogaster, Ladoukakis et al. identified 4,561 sORFs that were conserved in a closely related species, Drosophila pseudoobscura[15]. Synteny, evidence of transcription and nucleotide substitution, narrowed the 4,561 to a more conservative estimate of 401 sORFs. A study on the Arabidopsis small proteome assessed evolutionary conservation and examined evidence of transcription to predict the expression of as many as 3,241 sORFs [16]. A report on the mammalian small proteome by Frith et al. used FANTOM cDNA data to identify a potential 1,240 sORFs using a CRITICA gene-detection program [17]. Additionally, 25 sORFs were GFP-tagged and, following transfection into cells, 14 of the fusion proteins were detected, providing evidence of translation [17]. More recently, using a novel combination of peptidomics and RNA-Sequencing (RNA-Seq), Slavoff et al. identified 86 novel small proteins in humans and two were tagged and shown to localize to the mitochondria and cytoplasm [18]. Nevertheless, to date few functional studies of proteins encoded by sORFs have been performed. In yeast, 140 small proteins were tested by generating gene deletions and 22 had an effect on Saccharomyces cerevisiae growth under various conditions [19], whereas overexpression of 473 small proteins in Arabidopsis resulted in 49 recognizable phenotypes [20].
Mass spectrometry, a powerful technique in proteomics to validate the existence of putative protein candidates, has been applied in several studies [18,21-25]. High-resolution mass spectrometry provides very accurate precursor ion masses and combined with stringent statistical methods enhances the certainty of peptide identification [26]. This is a key issue in the validation of newly identified sORFs. In general, a protein database derived from the genome is used in shotgun proteomics to identify peptides and proteins from mass spectrometric raw data, but six frame translation of the genome is also frequently employed [24,25]. In either case, the certainty of the existence of any protein can be increased by an observed corresponding RNA transcript. Recently, we used a combination of stringent methods, that is, ribosome footprinting, next generation sequencing and advanced mass spectrometric technology, to discover a plethora of novel sORFs in cytomegalovirus, many of which we determined to exist at the protein level [23].
The question of whether functional small proteins exist is particularly relevant in organisms with a tightly organized genome, such as the parasitic protozoan Trypanosoma brucei. Protein-coding genes are arranged in long unidirectional clusters with intergenic regions only a few hundred nucleotides in length, thus leaving little space for sORFs or non-coding RNAs. The initial sequencing and annotation of the 11 megabase-sized chromosomes, published in 2005, predicted 9,068 protein-coding genes [27]. As of November 2013, this number has increased to 10,574 (TriTrypDB); however, a major challenge remains to identify all expressed proteins. This quest was addressed by several RNA-Seq studies using Illumina high-throughput cDNA sequencing [28-31]. In particular, we provided evidence that the coding potential of the T. brucei genome was larger than originally anticipated by identifying 1,114 transcripts mapping to regions of the genome with no annotated ORFs [28]. A total of 993 of these transcripts have the potential to contain a coding sequence (CDS) of at least 25 amino acids and the remaining 121 transcripts either have no coding potential at all or no ORF larger than 75 nucleotides. However, it remains to be established whether these transcripts encode functional proteins.
Founded on the set of transcripts identified by our transcriptome analysis [28], we applied bioinformatics approaches to identify small proteins conserved across kinetoplastid species and representative eukaryotes. Combined with mass spectrometry data, we pinpointed 42 high-confidence small proteins ranging in size from 49 to 219 amino acids. RNAi-knockdown revealed seven essential proteins in the insect-stage of the life cycle and their diverse subcellular localizations suggested involvement in many aspects of T. brucei biology.
Results
T. brucei transcripts encoding evolutionarily conserved potential small proteins
We previously published a single-nucleotide resolution genomic map of the T. brucei transcriptome, which included 1,114 transcripts not originating from annotated CDS ( [28]; original RNA-Seq data have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive - SRA at [32] - under accession no. SRA012290 and the 1,114 transcripts are accessible through a community file, Tbrucei_novel_transcripts.fasta, on TriTrypDB at [33]). After a reexamination of this data set using the latest T. brucei genome annotation (GeneDB version 5, [34]), we excluded 39 and 10 transcripts coding for snoRNAs and annotated proteins larger than 300 amino acids, respectively, and added two novel transcripts coding for proteins identified by mass spectrometry (MS) data (Figure 1). Setting a lower limit of 25 aa, 987 of the remaining transcripts contain between one (112 transcripts) and 31 (1 transcript) ORFs for a total of 4,699 ORFs [see Additional file 1]. Eighty transcripts were classified as non-coding RNAs, since the predicted ORFs were less than 75 nucleotides. However, we cannot exclude the possibility that the latter category has coding potential by using alternative initiation codons or encoding proteins smaller than 25 aa.
Figure 1.

Flowchart of the strategy used to analyze T. brucei transcripts not coinciding with annotated coding sequences (CDS).
The selected 4,699 ORFs were highly enriched in short ORFs (sORFs), that is, less than 100 amino acids, with 4,499 ORFs (96%) falling into this category [see Additional file 2: Figure S1]. Since proteins encoded by sORFs largely escape standard genome annotations, we examined evolutionary conservation in combination with computational approaches to screen for ORFs conserved in kinetoplastidae and representative eukaryotes as a benchmark for protein expression. Kinetoplastid protists belong to the phylum Euglenozoa and include a significant number of disease-causing parasites, such as T. brucei and T. cruzi, the causative agent of African trypanosomiasis and Chagas disease, respectively, and the Old and New World Leishmania parasites, which cause various forms of leishmaniasis worldwide. First, we conducted Basic Local Alignment Search Tool (BLAST) analyses [35] of kinetoplastid genomes and annotated proteins, excluding the T. brucei subspecies (see Methods for details). Of the 987 transcripts, 157 encoded one ORF that was conserved in at least one kinetoplastid organism and four transcripts (Tb4.NT.51, Tb5.NT.84, Tb6.NT.58 and Tb8.NT.142) encoded between two and twelve conserved ORFs for a total of 173 conserved ORFs [see Additional file 3]. Second, we compared the selected 4,699 ORFs to the annotated proteins from representative eukaryotes, namely S. cerevisiae, Caenorhabditis elegans, Arabidopsis thaliana, D. melanogaster, Mus musculus and Homo sapiens. We found that 13 ORFs had significant alignments with BLAST bit scores ranging from 34 to 227, with 6 coding for ribosomal proteins [see Additional file 4]. It is worth noting that these 13 ORFs were part of the set conserved in kinetoplastids. We next surveyed the 173 conserved ORFs for known protein domain(s) using the CD-Search Tool (cdsearch/cdd v3.10 [36]) and detected domains in 61 ORFs covering a broad spectrum [see Additional file 5]. However, the ribosomal protein superfamily (six hits), various Zn finger domains (five hits) and the RNA recognition motif (RRM) superfamily (three hits) were overrepresented. Finally, our analysis of SignalP [37] and TMHMM [38] predictions revealed that 5 of the 173 potential small proteins have a predicted signal peptide and that a considerable number (43 or 25%) have a predicted trans-membrane domain with seven having more than one predicted domain.
Identification of small protein candidates through mass spectrometry data
For an alternative approach based on peptide evidence to recognize transcripts coding for small proteins, we surveyed MS data ( [21,22] and this study) for peptides matching the 4,699 selected ORFs described above. As reported previously [28], searching the proteome data of Panigrahi et al. [21] provided evidence for the expression of 16 small proteins, with all 16 being part of the 173 small protein candidates identified bioinformatically. In addition, MS data from Butter et al. [22] and this study revealed 63 hits. As well as providing validation for 58 of the 173 small protein candidates, our data also predicted five small proteins specific to T. brucei with no recognizable homologues in other kinetoplastids. We also performed a search against hexatranslations of the trypanosome genome, which revealed the same set of newly identified proteins (data not shown). Taken together, we were able to provide supporting MS data for 63 predicted small proteins with 22 being represented in more than one MS data set, and, except for the T. brucei-specific hits, all the other matches were among the evolutionarily conserved 173 small protein candidates [see Additional file 3].
The 63 small protein candidates with supporting MS data were filtered further by removing predicted ribosomal proteins or annotated proteins with a predicted function and CDS with multiple copies in the genome, leaving us with 42 small proteins for further analysis [see Additional file 3]. This final group of selected proteins ranges in size from 49 to 219 amino acids and 35 qualify as small proteins. Transcript lengths vary from 333 to 4,100 nucleotides and the average 5’ UTR length is 119 nucleotides with a median of 110 nucleotides. This is similar to the global analysis of the transcriptome [28,30], where a median length of 128 to 130 nucleotides was reported. On the other hand, the 3’ UTR is on average 390 nucleotides long with a median of 237 nucleotides, with the latter being notably smaller than the medians reported in the aforementioned transcriptome studies, namely 400 nucleotides [30] and 388 nucleotides [28].
Noteworthy characteristics of this collection of 42 small proteins are as follows: three have putative homologues in representative eukaryotes (Tb7.NT.49, Tb11.NT.47 and Tb11.NT.220); predicted domains include two RRMs and two Zn-finger domains; sixteen have a predicted trans-membrane domain; and one has a predicted signal peptide [see Additional file 3].
RNAi screen of the 42 small proteins revealed 7 to be essential in the insect life-cycle stage
RNAi knock-down strategies have revolutionized the functional analysis of genes in T. brucei[39]. mRNA degradation is triggered most efficiently by double-stranded RNA (dsRNA) produced in vivo as a hairpin RNA transcribed from a tetracycline-inducible promoter. Thus, we generated a hairpin construct for each of the 42 ORFs using the pTrypRNAiGate plasmid [40]. Each construct was stably integrated in the non-transcribed rRNA spacer region of a special procyclic-form recipient strain, named 29.13.6, expressing the tet repressor and T7 RNA polymerase [41], and clonal cell lines were established. Upon RNAi induction with tetracycline, 12 had a growth phenotype that differed from un-induced control cells. Three of the knockdowns resulted in a slow-growth phenotype. For example, RNAi of Tb5.NT.58 resulted in a cell division time of 16 hours as compared to 8.5 hours for un-induced cells and this phenotype was not accompanied by noticeable changes in cell morphology [see Additional file 2: Figure S2]. In addition, knockdown of two small proteins (Tb11.NT.222 and Tb11.NT.66) resulted in faster growth with no obvious changes in cell morphology [see Additional file 2: Figure S2]. Monitoring cell growth after RNAi induction of the remaining seven revealed that all cell lines stopped dividing and eventually died demonstrating that Tb3.NT.18, Tb10.NT.86, Tb10.NT.87, Tb10.NT.90, Tb11.NT.28, Tb11.NT.29 and Tb11.NT.108 are essential genes (Table 1; Additional file 2: Figure S3). For subsequent analyses we focused on the seven essential predicted small proteins and the RNAi knockdowns revealing a change in the doubling time were not pursued further.
Table 1.
Characteristics of the seven essential proteins in procyclics
|
ORF ID |
Transcript |
AA/MW |
pI |
Predicted |
Predicted |
Localization |
|---|---|---|---|---|---|---|
| size (nt) | signal peptide | TM domain | ||||
| Tb10.NT.87 |
478 |
64/8.2 |
11.46 |
No |
No |
Mitochondria |
| Tb11.NT.28 |
1,285 |
56/6.3 |
9.3 |
No |
Yes |
Mitochondria |
| Tb11.NT.29 |
796 |
62/7.6 |
6.96 |
No |
Yes |
Surface |
| Tb3.NT.18 |
709 |
85/9.3 |
7.91 |
No |
Yes |
Cytoplasm |
| Tb10.NT.86 |
605 |
93/10.5 |
9.67 |
No |
Yes |
Cytoplasm |
| Tb11.NT.108 |
461 |
96/10.5 |
6.49 |
No |
No |
Cytoplasm |
| Tb10.NT.90 | 529 | 67/7.3 | 4.49 | No | Yes | Nucleus |
AA, number of amino acids; MW, molecular weight; ORF, open reading frame; pI, isoelectric point; TM, transmembrane.
To confirm that the observed essentiality of the seven small proteins was specific to RNAi knockdown of the predicted transcript, we performed the following experiments. First, we verified the transcript length expected by the RNA-Seq data [28] using Northern blot analysis. In six of the seven cases a single predominant hybridizing band was detected and the observed size matched the predicted size within the limits of resolution of Northern blotting [see Additional file 2: Figure S4]. The seventh transcript, Tb3.NT.18, had two bands detected by Northern blot. One band corresponded to the size of the predicted novel transcript of 709 nucleotides. Further interrogation by Northern blotting and RT-PCR with probes specific for the upstream (Tb927.3.1080) and downstream annotated gene (Tb927.3.1090) led us to conclude that the longer RNA contained both Tb3.NT.18 and the downstream transcript encoding a component of the T. brucei U1 small nuclear ribonucleoprotein (snRNP). This finding was reminiscent of the presence of an upstream open reading frame (uORF) described in organisms from fungi to humans [42,43]. uORFs are defined as predominantly short ORFs found in the 5’ UTR of a previously annotated gene and experiments are ongoing to investigate whether Tb3.NT.18 qualifies as an uORF.
Second, semi-quantitative RT-PCR verified that the knockdown of the seven essential transcripts was efficient [see Additional file 2: Figure S5]. Third, to confirm the specificity of the RNAi knockdown, we set out to rescue each lethal phenotype with the expression of an RNAi-resistant construct. To do this, the CDS targeted by RNAi was assembled as a synthetic sequence bearing at least one silent mutation per 12 contiguous base-pairs [44] and flanked by heterologous UTR sequences (see Methods). In addition, an HA-TEV-FLAG epitope tag or a GFP tag was added to the C-terminus [see Additional file 2: Figure S6]. Upon co-expression of the hairpin targeting the endogenous transcript and the corresponding modified CDS in a stable cell line, the endogenous transcript was destroyed, as shown by RT-PCR, and in all seven cases the cells survived on the RNAi-resistant transcript encoding the same small protein [see Additional file 2: Figure S3]. These results led us to conclude that the essential phenotype was a direct consequence of the knockdown of the targeted CDS.
Initial characterization of the essential small proteins
The RNAi rescue experiments described above established that the epitope-tagged small proteins were functional and, thus, could be used for biochemical and cell biological experiments. Using fluorescence microscopy we detected expression of all seven small proteins (Figure 2) and Western blot analysis confirmed that the proteins had the predicted relative molecular mass [see Additional file 2: Figure S7]. Tb11.NT.28 and Tb10.NT.87 revealed a fluorescence pattern typical of the procyclic trypanosome branched tubular mitochondrion (Figure 2). Tb11.NT.29 appeared to be a surface protein, an observation supported by subsequent experiments (see below). By immunofluorescence, three proteins (Tb3.NT.18, Tb10.NT.86 and Tb11.NT.108) were shown to be enriched in the cytoplasm, with Tb11.NT.108 distributed throughout this compartment, whereas Tb3.NT.18 and Tb10.NT.86 appeared somewhat concentrated around the nucleus. Finally, Tb10.NT.90 had a distinct localization in the nucleus, possibly indicative of the nucleolus.
Figure 2.

Localization of the seven small proteins essential in the procyclic life cycle stage. Anti-HA or anti-GFP antibodies were used to detect C-terminal HA- (Tb11.NT.28 and Tb11.NT.29) or GFP- (Tb10.NT.87, Tb10.NT.90, Tb10.NT.86, Tb3.NT.18, and Tb11.NT.108) tagged versions of the proteins by fluorescence microscopy. DNA was stained with Hoechst (blue) and DIC images are shown in the right panels. DIC, differential interference contrast.
Since T. brucei undergoes extensive morphological and metabolic changes during its life cycle alternating between the mammalian (bloodstream) and insect (procyclic) hosts, it was of interest to gauge the essentiality of the seven small proteins in bloodstream forms. Thus, the hairpin constructs were transfected into a bloodstream form cell line competent for RNAi and, following induction, Tb11.NT.29, a potential surface protein, and Tb10.NT.87, a probable mitochondrial protein, were shown to be essential in this stage of the life cycle (Figure 3A and B). Growth curves for the five nonessential proteins can be found in Figure 3C and Additional file 2: Figure S8. Based on the above results, we selected Tb11.NT.29, Tb11.NT.28 and Tb10.NT.87 for further analysis.
Figure 3.

Bloodstream-form growth following RNAi. (A)Tb11.NT.29 RNAi in bloodstream-form cells (BS). (B)Tb10.NT.87 RNAi in BS cells. (C)Tb11.NT.28 RNAi in BS cells. Growth of un-induced (-tet) and induced cells (+ tet) shown in log scale. The data are based on three independent experiments, and average values with standard deviations are presented.
Tb11.NT.29: a putative surface protein
The 796 nucleotide-long Tb11.NT.29 transcript encodes a 62 aa protein, which is highly conserved in kinetoplastids (84% identity between T. brucei and L. major) and has a predicted trans-membrane domain (Figure 4A). Initial immunofluorescence suggested that Tb11.NT.29 might be localized on the surface (Figure 2). To address this possibility, we compared the signal for cells expressing epitope-tagged Tb11.NT.29 that were either permeabilized by detergent prior to antibody exposure or remained non-permeabilized. Under permeabilized conditions we detected both Tb11.NT.29 and the endoplasmic reticulum (ER) protein BiP (Figure 4B). However, when we omitted the permeabilization step, thus limiting access of antibodies to potential surface molecules, no signal was detected for BiP, whereas the signal for Tb11.NT.29 was still visible (Figure 4B). This behavior was similar to procyclin (Figure 4C), a well-characterized T. brucei surface protein specific for the procyclic life-cycle stage [45]. Since the epitope tag was at the C-terminus of the Tb11.NT.29, this result also suggested that this portion of the protein was exposed on the surface. To corroborate this localization, we performed cell fractionation experiments and by Western blot analysis Tb11.NT.29 was enriched in the membrane fraction, similar to procyclin, whereas HSP70 was, as expected, enriched in the cytoplasmic fraction (Figure 4D).
Figure 4.

Characterization of Tb11.NT.29. (A) Sequence conservation of Tb11.NT.29 across kinetoplastids. Amino acid sequences from T. brucei (Tbr), T. cruzi (Tcr) and L. major (Lm) were aligned with ClustalW and conserved residues are shaded with black boxes while similar residues are shaded in grey. The predicted transmembrane domain (TM helix) is indicated. (B) Immunofluorescence analysis on cells expressing a C-terminal HA-tagged version of Tb11.NT.29. Permeabilized cells (left three panels) and non-permeabilized cells (right three panels) were probed for HA (green) and BiP (red) and DNA was stained with Hoechst (blue). (C) Immunofluorescence analysis of procyclin as described in panel (B). (D) Cell fractionation of parasites expressing a GFP-tagged version of Tb11.NT.29. Western blot analysis was performed against GFP-tagged Tb11.NT.29 (top panel), procyclin (second from top), BiP (third panel from top) and HSP 70 (bottom panel) on total (T), cytoplasmic (C), membrane (M), nuclear (N) and cytoskeleton (CSK) fractions.
To begin to probe the potential role of Tb11.NT.29, we monitored the effect of RNAi knockdown on cell cycle progression and cell morphology. In procyclic cells RNAi resulted in a slowdown in growth after two days followed by cell death between day five and six post-induction (Figure 5A), whereas the RNAi effect was more pronounced in bloodstream form cells with cell death occurring between day one and two (Figure 3A). For cell cycle analysis in procyclics, parasites were stained with Hoechst at various time points after induction and the number and position of nuclei and kinetoplasts (mitochondrial kDNA) in each cell were recorded. Cells with one kinetoplast and one nucleus (1K1N) are in G1 of the cell cycle, cells with two kinetoplasts and one nucleus (2K1N) have segregated the kinetoplast and are at the end of S phase, and cells with two kinetoplasts and two nuclei (2K2N) have completed mitosis and are poised for cytokinesis [46]. Any other arrangement is aberrant and might point to defects in cell cycle progression. In wild-type cells, as expected for an asynchronously growing cell population, the majority of parasites will be 1K1N with about 10% of cells having either a 2K1N or 2K2N configuration.
Figure 5.

Growth and cell cycle analysis following RNAi of Tb11.NT.29. (A)Tb11.NT.29 RNAi in procyclics (PC). Growth for un-induced (-tet) and induced cells (+tet) shown in log scale. The data are based on three independent experiments, and average values with standard deviations are presented. (B) Analysis of the K/N configurations after induction of Tb11.NT.29 RNAi in procyclic cells. Cells were scored as having one kinetoplast and one nucleus (1K1N), two kinetoplasts and one nucleus (2K1N), two kinetoplasts and two nuclei (2K2N), one kinetoplast and no nucleus (1K0N), one kinetoplast and two nuclei (1K2N) and more than two nuclei (>2 N). The data are based on three independent experiments and at each time point 1,000 cells were counted. (C) Accumulation of cells with altered morphology after induction of Tb11.NT.29 RNAi in procyclic cells. (D) Example images of a normal cell and cells with altered morphology. Images for DNA content (Hoechst) and DIC are displayed. At each time point 500 cells were counted. DIC, differential interference contrast.
RNAi-induced down-regulation of Tb11.NT.29 in procyclics resulted in the accumulation of cells containing either 1K2N or 1K0N, the latter referred to as zoids (Figure 5B). Zoids and 1K2N cells increased nearly equally in number and after three days of induction they comprised 7.9% and 6.9% of the cell population, respectively. Several morphological changes were observed after the knockdown, including flagellar detachment both as specific areas of separation between the cell membrane and the flagellum, and complete separation with only one visible contact point between the cell body and flagellum (Figures 5C and D). We also noted a change in the shape of the cell body that appeared to be specific to 2K1N or 2K2N cells. After kinetoplast replication and separation, a narrowing of the cell body was evident between the two daughter kinetoplasts (Figure 5D), which might indicate a defect in cytokinesis.
Tb11.NT.28: a mitochondrial inner membrane protein
The 56-amino acid Tb11.NT.28 protein is 45% identical between T. brucei and L. major and contains a predicted trans-membrane domain (Figure 6A). Initial immunofluorescence staining revealed a similar localization pattern between Tb11.NT.28 and the fluorescent dye MitoTracker Red, a cell-permeable mitochondrion selective probe (Figure 6B). Cell fractionation experiments corroborated the potential mitochondrial localization in that Tb11.NT.28 was enriched in the mitochondrial fraction similar to RNA-editing associated protein (REAP), a known mitochondrial marker [47], whereas the cytoplasmic HSP70 was excluded from this fraction (Figure 6C). To determine in which mitochondrial compartment Tb11.NT.28 might be localized, we exposed whole cells to increasing concentrations of digitonin, a detergent that preferentially solubilizes the plasma membrane and the outer membrane of the mitochondria. As the digitonin concentration is increased, specific mitochondrial compartments have been shown to be solubilized with 0.015% digitonin releasing proteins from the inter-membrane space, 0.025% digitonin solubilizing matrix proteins, 0.04% digitonin resulting in release of outer membrane proteins and 0.1% digitonin solubilizing inner membrane proteins [48,49]. As Tb11.NT.28 was only released upon exposure to 0.1% digitonin, its likely localization is the inner membrane (Figure 6D), since solubilization of trypanosome alternative oxidase (TAO), a known inner membrane protein of T. brucei mitochondria [50], occurred with the same digitonin concentration, whereas a portion of mitochondrial HSP70, a matrix protein, was released with as little as 0.015% digitonin.
Figure 6.

Characterization of Tb11.NT.28. (A) Sequence conservation of Tb11.NT.28 across kinetoplastids. Amino acid sequences from T. brucei (Tbr), T. cruzi (Tcr) and L. major (Lma) were aligned with ClustalW and conserved residues are shaded with black boxes while similar residues are shaded in grey. The predicted transmembrane domain (TM helix) is indicated. (B) Immunofluorescence analysis of cells expressing a C-terminal HA-tagged Tb.11.NT.28. Cells were stained with an anti-HA antibody (green) and MitoTracker (red) and a merge of signal for HA and MitoTracker is shown in yellow. (C) Cell fractionation of cells expressing C-terminal HA-tagged Tb11.NT.28. Western blot analysis was performed against C-terminal HA-tagged Tb11.NT.28 (top panel), RNA-editing associated protein (REAP, middle panel) and cytoplasmic HSP70 (bottom panel) on total (T), cytoplasmic (C), nuclear (N), endoplasmic reticulum (ER) and mitochondrial (Mito) fractions. (D) Digitonin extraction analysis of cells expressing C-terminal HA-tagged Tb11.NT.28. Parasites were exposed to increasing concentrations of the detergent digitonin as indicated above the panels and solubilized proteins were analyzed by Western blot for HA-tagged Tb11.NT.28 (top panel), trypanosome alternative oxidase (TAO, middle panel) and mitochondrial HSP 70 (mtHSP70, bottom panel).
RNAi-induced knockdown of Tb11.NT.28 resulted in cell death in procyclic forms (Figure 7A), but did not affect growth in bloodstream forms (Figure 3C). An analysis of cell cycle progression in the procyclic cells following RNAi did not result in an accumulation of cells containing aberrant DNA amounts. However, a steady increase in the number of 2K1N cells was observed with this cell type constituting one third of all cells four days post-induction (Figure 7B). In this category, 50% of cells had duplicated the kinetoplast, but the daughter kinetoplast remained linked in a dumbbell-shaped body, a larger number than seen in wild-type cells [46]. This indicated that although cells entered S phase it had not been completed.
Figure 7.
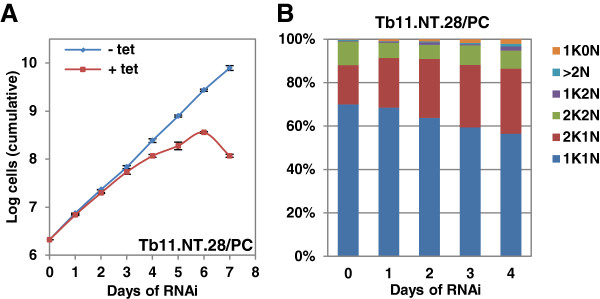
Analysis of growth and cell cycle progression following RNAi of Tb11.NT.28. (A)Tb11.NT.28 RNAi in procyclics (PC). Growth for uninduced (-tet) and induced (+tet) cells shown in log scale. (B) Analysis of the K/N configurations after induction of Tb11.NT.28 RNAi. Cells were scored every 24 hours as having normal DNA (one kinetoplast and one nucleus (1K1N), two kinetoplasts and one nucleus (2K1N) or two kinetoplasts and two nuclei (2K2N)) or abnormal DNA content (one kinetoplast and no nucleus (1K0N), one kinetoplast and two nuclei (1K2N) and more than two nuclei (>2 N). Cell counts were done as described in the legend for Figure 5.
Tb10.NT.87: a mitochondrial matrix protein
Tb10.NT.87, a very basic protein of 64 aa, is highly conserved in kinetoplastids except for the first 10 aa (Figure 8A). Performing fluorescence microscopy on live cells expressing the GFP-tagged RNAi-resistant construct revealed similar localization patterns between Tb10.NT.87 and MitoTracker, indicative of a mitochondrial localization (Figure 8B). Cell fractionation experiments were consistent with the immunofluorescence data (Figure 8D) and digitonin solubilization assays, as described above, suggested that Tb10.NT.87 was a matrix protein, since the solubilization properties were similar to mitochondrial HSP70, an established marker for the matrix (Figure 8E). To further pinpoint its cellular localization, we used immunogold electron microscopy of cells expressing HA-TEV-FLAG tagged Tb10.NT.87. Micrographs of thin sections showed that the protein was in the mitochondrial matrix and its distribution appeared to be uniform (Figure 8C).
Figure 8.

Characterization of Tb10.NT.87. (A) Sequence conservation of Tb10.NT.87 across kinetoplastids. Amino acid sequences from T. brucei (Tbr), T. cruzi (Tcr) and L. major (Lma) were aligned with ClustalW and conserved residues are shaded with black boxes while similar residues are shaded in grey. (B) Live fluorescence microscopy of cells expressing a C-terminal GFP-tagged Tb10.NT.87. GFP-signal (green), MitoTracker (red), GFP and MitoTracker merge (yellow), Hoechst (blue) and DIC images are shown. (C) Immunogold electron microscopy of C-terminal HA-tagged Tb10.NT.87. Mitochondrial tubules indicated with the letter M and kinetoplast (mitochondrial DNA) indicated by an arrow and K. (D) Cellular fractionation of cells expressing C-terminal GFP-tagged Tb10.NT.87. Western blot against C-terminal GFP-tagged Tb10.NT.87 (top panel), editing protein (α-63/P1H3, middle panel) and cytoplasmic HSP70 (bottom panel) on total (T), cytoplasmic (C), nuclear (N), endoplasmic reticulum (ER) and mitochondria (Mito) fractions. (E) Digitonin extraction analysis of cells expressing C-terminal GFP-tagged Tb10.NT.87. Parasites were exposed to increasing concentrations of the detergent digitonin as indicated and solubilized proteins were analyzed by Western blot for GFP-tagged Tb10.NT.87 (top panel), mitochondrial HSP 70 (mtHSP70, middle panel), and trypanosome alternative oxidase (TAO, bottom panel). DIC, differential interference contrast.
Tb10.NT.87 was shown to be essential in both procyclic and bloodstream forms (Figures 9A and 3B) and a significant increase in cells with more than two nuclei and grossly enlarged cell bodies was noted after knockdown of Tb10.NT.87 in procyclics (Figures 9B and C). After four days of induction, 12.5% of cells had between three and eight nuclei. Most of these cells contained a single kinetoplast, indicating that although mitosis was occurring there was not a corresponding replication and division of mitochondrial DNA. The enlarged cell body remained as one unit with a single flagellum as expected in a cell with one kinetoplast. A substantial number of zoids (13.7%) and 1K2N cells (16%) also accumulated by this time point. In accordance with the presence of a single kinetoplast in many cells containing multiple nuclei, a potential defect in kDNA replication was further manifested by the appearance of cells with small kDNA or no detectable kDNA (Figures 9D and E). For example, after three days of Tb10.NT.87 RNAi, 32% of cells had normal-sized kDNA, whereas 65% of the cells were scored as having a small kDNA.
Figure 9.

Analysis of growth and cell cycle progression following RNAi of Tb10.NT.87. (A)Tb10.NT.87 RNAi in procyclics (PC). Growth for uninduced (-tet) and induced (+tet) cells shown in log scale. (B) Cell cycle progression in procyclic cells after RNAi of Tb10.NT.87. Cells were scored every 24 hours as having normal DNA (one kinetoplast and one nucleus (1K1N), two kinetoplasts and one nucleus (2K1N) or two kinetoplasts and two nuclei (2K2N)) or abnormal DNA content (one kinetoplast and no nucleus (1K0N), one kinetoplast and two nuclei (1K2N) and more than two nuclei (>2 N)). (C) Example images of abnormal morphology noted after RNAi of Tb10.NT.87. Hoechst staining and DIC are shown. (D) Examination of kDNA size after Tb10.NT.87 RNAi in procyclic cells. Cells were scored as having normal kDNA size, small kDNA, absent kDNA or asymmetrical kDNA, which is one cell with two daughter kDNA of different sizes, by visually comparing the kDNA size to that in the uninduced control. (E) Examples of cells with normal kDNA, small kDNA (both in G1 and dividing) and absent kDNA. Hoechst and DIC images are displayed. Cell counts were done as described in the legend for Figure 5. DIC, differential interference contrast.
Discussion
Although genome sequencing projects have provided a wealth of information about genome structure and organization, they also encountered a challenge to catalogue all protein-coding genes. Since gene annotation programs do not perform well in predicting small proteins, that is, less than 100 aa, it has been common practice to set an artificial length cutoff of 100 aa to avoid flawed predictions. However, recent computational and functional studies highlighted the existence and importance of small proteins in numerous organisms [16-20,51]. Our approach to gauge the extent of the small proteome in T. brucei integrated experimental data (transcriptome and MS data) and evolutionary conservation. Starting with 1,117 transcribed sequences not mapping to an annotated CDS [28], 987 have the potential to code for one or more proteins of at least 25 amino acids. This data set was then examined for conserved proteins in related kinetoplastids and representative eukaryotes. A similar strategy was applied in yeast, where more than 60% of the 299 small proteins identified had significant similarities with annotated proteins in other eukaryotes, including humans [19]. Likewise, an analysis of the M. musculus small proteome revealed that two-thirds of the potential sORFs identified were conserved in rat and 50% were conserved in human [17]. In contrast, only 1.3% (13) of the predicted ORFs in T. brucei had potential homologues in representative eukaryotes, with half of them coding for ribosomal proteins. One possible explanation for this stark difference in evolutionary conservation might be the early divergence of T. brucei from other eukaryotes. We observed a comparable lack of conservation when searching for potential homologues in related kinetoplastid species: 16.3% (161) of the 987 transcripts have predicted ORFs with a significant homology to annotated proteins or a six-frame translation of the available genomes. This result was somewhat unexpected, since more than half of the small proteins identified in Arabidopsis had homologues in four closely related plants [16] and more than 3,000 sORFs were found to be conserved between D. melanogaster and a closely related species, D. pseudoobscura[15]. On the other hand, since it has been argued that conservation in closely related species is evidence for translation, the 173 small proteins identified by evolutionary conservation might represent a substantial proportion of the small proteome. The currently available MS data support this view, since the peptide hits were largely confined to the set of 173 proteins. The only exception was the identification of five T. brucei-specific proteins. Thus, to fully expose the catalogue of small proteins, future approaches will have to concentrate on the identification of T. brucei-specific ORFs through MS analysis or ribosome profiling [52,53].
Examining the functional importance of small proteins in yeast, revealed that 22 (15.5%) of 140 sORF knockout cell lines had an essential phenotype in specific growth conditions [19] while in Arabidopsis, overexpression of 10% of a handpicked set of almost 500 small proteins resulted in an abnormal phenotype [20]. Our results showed that 16.7% of the ORFs tested were essential in procyclic cells, while an additional 12% altered normal growth patterns. For example, Tb11.NT.28 was essential in procyclics but not in bloodstream-form parasites. We further provided evidence that this small protein of 56 amino acids is likely localized to the inner membrane of the mitochondria. In T. brucei, both the size and activity of the mitochondria vary dramatically between life cycle stages. In the bloodstream form, mitochondrial respiratory activity is repressed and a single tubule of the organelle is maintained. On the other hand, in procyclic cells the mitochondrion forms an extensive, branching network and has active respiration. Although further experiments will be required to elucidate the function of Tb11.NT.28, examination of cell cycle progression following RNAi induction revealed a decreased ability of the cells to divide replicated kinetoplasts. In contrast, Tb10.NT.87, a mitochondrial matrix protein of 64 amino acids, is essential in both procyclic- and bloodstream-form parasites. Ablation of this protein in procyclics resulted in the accumulation of cells containing multiple nuclei and a single kinetoplast, as well as cells with small or no kinetoplast, suggesting that kinetoplast replication is impaired. A similar scenario was observed in bloodstream-form cells upon RNAi of Tb10.NT.87 (data not shown). The basic nature of Tb10.NT.87 (pI of 11.5) is intriguing and will need further investigation. Nevertheless, our current data are consistent with Tb10.NT.87 playing a role in kinetoplast replication, which is an essential process in both life cycle stages [54]. Finally, Tb11.NT.29, indispensable in both procyclic and bloodstream stages, is most likely localized on the cell surface. Two days post-induction cells appeared with an asymmetrical hourglass shape and a kinetoplast sequestered in the smaller half of the cell, while the remaining portion contains 1K1N or 1K2N. The proportional accumulation of zoids and 1K2N cells that began after these cells arose suggested that an aberrant cytokinesis of these asymmetrical hourglass cells occurred.
Conclusions
Our study provides evidence for the existence and importance of small proteins in the human pathogen T. brucei. At the same time, it is somewhat puzzling that an unexpectedly low number of transcripts not matching annotated proteins or having conservation with closely related species were identified as containing functional ORFs. Even though there may be small proteins expressed at low levels not yet detectable by MS and others that might be expressed at specific stages of the life cycle, it is tempting to speculate that the T. brucei transcriptome includes a substantial number of non-coding RNAs. As all the small proteins identified as essential are unique to kinetoplastids, they may become new targets to block the survival of trypanosomes in the insect vector and/or the mammalian host. The next important question to be tackled is the mechanism of action of these small proteins.
Methods
Standard methods
Western blots [55], transfection of procyclic cells [56] and RNA isolation [56] were performed using previously published protocols. Oligonucleotides used to prepare clones and probes for Northern blots are listed in Additional file 2: Figure S9.
Bioinformatics
The 987 transcripts were translated using the getorf program of the European Molecular Biology Open Software Suite setting a lower limit of 25 aa and including only ORFs that contained a start and stop codon [57].
We used the NCBI BLAST suite (BLAST 2.2.28, [35]) for our protein searches. The predicted 4,699 T. brucei ORFs were used as queries for blastp to search all non-redundant kinetoplastid protein sequences (taxid: 5653), using an e-value cutoff of 0.1. T. brucei (taxid: 5691). T. b. gambiense (taxid: 31285) and T. b. brucei (taxid: 5702) sequences were excluded from the search. Similarly, tblastn was used to search the kinetoplastid translated nucleotide database with an e-value cutoff of 0.1. The annotated proteins of S. cerevisiae (taxid: 4932), C. elegans (taxid: 6239), A. thaliana (taxid: 3702), D. melanogaster (taxid: 7227), M. musculus (taxid: 10090) and H. sapiens (taxid: 9606) were queried with the same strategy. All alignments were manually inspected and verified to exclude false positives due to the relaxed threshold.
The predicted T. brucei proteins were scanned for domains using the NCBI CD-Search Tool (cdsearch/cdd v3.10; [36]). Transmembrane helices in proteins were predicted using the TMHMM Server v. 2.0 [38,58] and the presence and location of signal peptide cleavage sites were scanned at the SignalP 4.1 Server [37,59]; both servers are at the Technical University of Denmark.
Mass spectrometry analysis
Procyclic-form trypanosomes (MiTat 1.4) were lysed in 50 mM Tris (pH 7.3) with 4% SDS. To decrease sample complexity, the samples were filtered directly using different (3 kDa, 10 kDa, 30 kDa and 50 kDa molecular weight cut off (MWCO)) Amicon Ultracel centrifugal filter units (Millipore, Billerica, MA, USA) and under strong denaturing conditions with 8 M urea. The filtrate was precipitated using 4 volumes of ethanol and subsequently resuspended in 20 μl 8 M urea. The samples were reduced with 1 mM dithiothreitol (DTT) and alkylated using 5 mM iodoacetamide, prior to digestion with 0.2 μg Lys-C (Wako, Richmond, VA, USA) for three hours followed by digestion with 0.2 μg trypsin (Promega, Madison, WI, USA) overnight. The peptides were desalted using a StageTip [60]. For MS analysis, peptides were separated by a nanoflow liquid chromatography EASY-nLC system on a capillary packed with Reprosil-C18 (Dr. Maisch) with an acetonitrile gradient from 2% to 60% at a flow rate of 250 nl/minute for 230 minutes. The Orbitrap XL mass spectrometer was operated in a data-dependent acquisition mode performing Top10 MS/MS per full cycle. Data analysis was done with MaxQuant version 1.2.0.18 [26] using a concatenated database of TREU 927 v.2.3 (10,533 entries, tritrypDB.org) and the hits generated by the RNA-Seq analysis (987 entries, Additional file 1). Enzyme search specificity was set for tryptic peptides. Up to two miscleavages for each peptide were allowed. Carbamidomethylation on cysteines was set as fixed modification, while methionine oxidation and protein N-acetylation were considered as variable modifications. The search was performed with an initial mass tolerance of 6 ppm mass accuracy for the precursor ion and 0.5 Da. False discovery rate was fixed at one percent on peptide and protein level. For the second data set [22], we reanalyzed previously generated MS data using MaxQuant standard settings with the above described concatenated database. The MS proteomics data have been deposited to the ProteomeXchange Consortium [61] via the PRIDE partner repository [62] with the dataset identifier PXD000711 [22] and PXD000712 (this study).
Cell culture
T. brucei 29.13.6 Lister 427 procyclic cells [41] were cultured at 28°C with 5% CO2 in Cunningham’s media supplemented with 10% Tet-system approved heat inactivated fetal bovine serum (FBS, Clontech, Mountain View, CA, USA), 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamicin, 15 μg/mL G418 and 50 μg/ml hygromycin B. A total of 1 × 107 29.13.6 cells were used for each procyclic-form transfection. Cells were spun down, washed in Cytomix (20 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4, 25 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (Hepes), 2 mM ethylenediaminetetraacetic acid (EDTA) and 5 mM MgCl2, pH7.6), then resuspended in 500 μl Cytomix. Then, 25 μg of linearized plasmid DNA was added to the solution and cells were pulsed twice at 1,600 V with a time constant of 0.6 ms on a GenePulser Xcell (BioRad, Hercules, CA, USA). Cells were allowed to rescue for 24 hours before selective drug was added. Phleomycin and blasticidin were added to a final concentration of 2.5 μg/ml and 10 μg/ml, respectively. Transfected cells were cloned at least 24 hours after transfection. Serial dilutions of the cells were then made in media with 20% serum and the presence of 3 × 107 29.13.6 cells that had not been transfected. A total of 200 μl of transfected cells were added to 1.8 ml of the cloning media and further diluted in six, five-fold dilutions. Each dilution was plated in a 96-well plate and clones were selected from dilutions where fewer than 30% of wells had growth. T. brucei SM Lister 427 bloodstream-form cells [41] were maintained at 37 °C with 5% CO2 in HMI-9 media supplemented with 10% Tet-system approved heat inactivated FBS (Clontech), 100 units/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamicin, and 2.5 μg/ml G418. For bloodstream form transfections, 3 × 107 cells were centrifuged, washed quickly with Tb-BSF buffer [63], resuspended in 100 μl Tb-BSF buffer containing 10 μg of plasmid DNA and transfected with protocol X-01 in an AMAXA Nucleofector®. Transfected cells were placed in 30 ml pre-warmed HMI-9 media and two 10-fold serial dilutions were plated in a 24-well plate. Six hours after transfection, pre-warmed medium supplemented with appropriate selectable drugs was added. The final concentration of selectable markers was 2.5 μg/ml phleomycin and 5 μg/ml blasticidin. For induction of hairpin or RNAi-resistant construct expression, 10 μg/ml and 1 μg/ml of doxycycline was added to procyclic- and bloodstream-form cells, respectively.
Plasmid constructions
Constructs encoding hairpin RNAs
We followed the Gateway®-adapted cloning scheme developed by Margaret Phillips and colleagues [40]. Briefly, a 300 to 400 base pair region was PCR amplified and TA-cloned (deoxythymidine, T, deoxyadenosine, A) into plasmid pCR/8GW/TOPO (Invitrogen, Grand Island, NY, USA) to generate an entry clone. The entry clone was then recombined with the destination vector pTrypRNAiGate. Final constructs were verified by restriction enzyme digestions and DNA sequencing.
RNAi-resistant constructs
To rescue the lethal RNAi phenotype, RNAi-resistant versions of the CDS were synthesized by GeneWiz, Inc, Cambridge, MA, USA. A silent mutation was introduced every twelfth nucleotide [44], a C-terminal HA-TEV-FLAG tag was added and the CDS was flanked at the 5’ and 3’ end by a Hind III and Bam HI restriction site, respectively. The RNAi-resistant construct was cloned into the inducible pLew100v5 BSR plasmid flanked by the GPEET procyclin 5’ UTR and an aldolase 3’UTR. In addition, inducible GFP-tagged RNAi-resistant constructs were generated in pLew100v5 BSR. The plasmids containing the RNAi-resistant constructs were digested with Not I to allow for recombination into a T. brucei ribosomal RNA spacer region following transfection. For each knock-down, ten clones were tested for a phenotype.
Northern blotting
Total RNA was separated on 1.5% agarose gels in the presence of 6.3% formaldehyde in 40 mM 3-morpholinopropane-1-sulfonic acid (MOPS) and 2 mM EDTA. RNA ladder 0.5 to 10 kb (Invitrogen) was used as marker. The RNA was transferred overnight to a Hybond-N nylon membrane (GE Healthcare, Little Chalfont, United Kingdom) by capillary transfer with 10× SSC (0.15 M sodium citrate, 1.5 M sodium chloride), UV cross-linked to the membrane and stained with methylene blue. The membrane was pre-hybridized for one hour in 5× SET (0.75 M sodium chloride, 5 mm EDTA, 0.15 M Tris-HCl, pH 7.4), 10× Denhardt’s solution, 1% SDS and 100 μg/ml yeast RNA, and then hybridized overnight in the same solution. DNA probes were internally labeled by synthesis with specific dsDNA templates, sense and antisense primers and Pfx DNA polymerase (Invitrogen) in the presence of [α-32P]dCTP. The membrane was washed two to three times (10 minutes each) with 2× SSC, 0.1% SDS and hybridization signals were detected by PhosphorImager.
Semi-quantitative reverse transcriptase (RT)-PCR
For each sample, 5 μg of total RNA was treated with 2 units of DNase RQ (Promega), phenol extracted and ethanol precipitated. DNase-treated RNA was reverse transcribed using random primers (Promega) and Superscript II enzyme (Invitrogen) according to the manufacturer’s protocol. Twenty-two cycles of PCR were then performed using Platinum Pfx (Invitrogen), according to the manufacturer’s instructions, for each knockdown with histone 4 used as a control. An annealing temperature of 50°C was used for all oligonucleotides with an extension time of 30 seconds.
Immunofluorescence
A total of 5 to 8 × 106 cells were spun down and washed twice with cell wash (20 mM Tris–HCl (pH 7.5), 100 mM NaCl and 3 mM MgCl2) before being placed on slides coated with poly-L-lysine and settling for five minutes. Then, 4% paraformaldehyde (PFA) was added and the cells were fixed for 30 minutes at 4°C. Cells were washed twice and then exposed to 0.1% NP-40 detergent in a solution of 2% goat serum. Slides were washed again and blocked with 10% goat serum for 10 minutes. Upon removal of blocking solution, primary antibody, diluted in 2% goat serum, was administered for one hour. Anti-GFP (Roche, Basel, Switzerland), anti-HA (Covance, Princeton, NJ, USA), and α-GPEET procyclin (Cedarlane labs, Burlington, Ontario, Canada) antibodies were obtained commercially. The antibodies to BiP were generously provided by Jay Bangs. Cells were washed five times, before the addition of secondary antibody and 5 μg/ml Hoechst (Cell Signaling Technology, Inc, Beverly, MA, USA). Cells were exposed to secondary antibody, diluted in 2% goat serum, for one hour. All secondary antibodies (Alexa Fluor 488-conjugated goat anti-mouse, 594-conjugated goat anti-mouse, 488-conjugated goat anti-rabbit and 594-conjugated goat anti-rabbit (Invitrogen)) were used at a 1:1,000 dilution. Samples were washed five times for 10 minutes total. Wash solution was removed and FluorSave (Calbiochem, Darmstadt, Germany) was added. Next, the coverslip was placed on the slide, and FluorSave reagent dried for two hours to overnight before cells were imaged on a Zeiss (Jena, Germany) Axioplan 2 fluorescence microscope. For kDNA size estimation, cells were fixed and stained with Hoechst, following the protocol outlined above. Cells were imaged and kDNA size was compared visually between un-induced cells and induced cells.
MitoTracker
A total of 1 × 107 cells was spun down, washed once in cell wash (20 mM Tris–HCl (pH 7.5), 100 mM NaCl and 3 mM MgCl2) and resuspended in Cunningham’s media with no added serum. MitoTracker Red CM-H2xRos (Invitrogen) was added to a final concentration of 1 μM. Cells were incubated at 28°C and 5% CO2 for 10 to 15 minutes, centrifuged, washed and placed in fresh media without serum. Cells were rescued in media without MitoTracker for 25 minutes, then Hoechst (5 μg/ml) was added; cells were spun down and finally resuspended in 20 to 50 μl of PBSG.
Live cell imaging
A total of 5 to 8 × 106 cells was collected, Hoechst (5 μg/ml) was added and the cells were incubated in the dark for two minutes. Cells were centrifuged, washed once in phosphate-buffered saline with glucose (PBSG), resuspended in 20 to 50 μl of PBSG, and imaged on a Zeiss Axioplan 2 fluorescence microscope.
Cell fractionations
Cell compartment qproteome kit (Qiagen)
Each fractionation used 1 × 109 cells expressing GFP-tagged Tb11.NT.29 and the manufacturer’s (Qiagen, Venlo, Limburg, The Netherlands) instructions were followed. The anti-HSP70 and anti-BiP antibodies were generously provided by Jay Bangs.
Mitochondrial isolation qproteome kit (Qiagen)
Each fractionation used 1 × 109 cells expressing GFP-tagged Tb10.NT.87 or Tb11.NT.28 and the manufacturer’s (Qiagen) instructions were followed. The antibodies to REAP and TbMP63 were generously provided by Steve Hajduk and Ken Stuart, respectively.
Digitonin solubilization assay
Cells (1 × 108) expressing GFP-tagged Tb10.NT.87 or HA-tagged Tb11.NT.28 were spun down for each assay, washed twice with 20 mM sodium phosphate (pH 7.9), 20 mM glucose and 0.15 M NaCl, and then resuspended in 500 μl SoTE buffer (20 mM Tris–HCl (pH 7.5), 0.6 M sorbitol, 2 mM EDTA). Next, 500 μl of SoTE buffer with varying digitonin amounts was added to each sample to a final concentration of detergent of 0.015%, 0.025%, 0.04%, 0.05% or 0.1% [48,49]. Samples were incubated for five minutes at 4°C followed by centrifugation for three minutes at 5,000 g at 4°C. SDS-sample buffer was added to the supernatants and samples were analyzed by SDS-PAGE and Western blotting. The antibodies to TAO and mtHSP70 were generously provided by Minu Chaudhuri and Jay Bangs, respectively.
Electron microscopy
Samples were fixed in 4% PFA/0.1% gluteraldehyde in PBS for 30 minutes followed by further fixation in 4% PFA for one hour, rinsed in PBS, scraped and re-suspended in 10% gelatin. Chilled blocks were trimmed, placed in 2.3 M sucrose overnight on a rotor at 4°C, transferred to aluminum pins and frozen rapidly in liquid nitrogen. The frozen blocks were cut on a Leica Cryo-EMUC6 UltraCut and 65 nm thick sections were collected using the Tokoyasu method [64] and placed on carbon/formvar coated grids and floated in a dish of PBS for immunolabeling. Grids were placed section side down on drops of 0.1 M ammonium chloride to quench untreated aldehyde groups, then blocked for nonspecific binding on 1% fish skin gelatin in PBS. Single labeled grids were incubated on a primary antibody mouse anti-HA (Covance) 1:50 dilution, which required a rabbit anti-mouse bridge (JacksonImmuno, West Grove, PA, USA). The secondary antibody was 10 nm Protein A gold (Utrecht Medical Center). All grids were rinsed in PBS, fixed using 1% gluteraldehyde for five minutes, rinsed again and transferred to a UA/methylcellulose drop before being collected and dried. Samples were viewed using a FEI Tencai Biotwin TEM at 80 Kv. Images were taken using Morada CCD and iTEM (Olympus) software.
Abbreviations
aa: amino acids; BLAST: Basic Local Alignment Search Tool; CDS: coding sequences; dsRNA: double-stranded RNA; EDTA: ethylenediaminetetraacetic acid; ER: endoplastic reticulum; GFP: green fluorescent protein; MS: mass spectrometry; PBS: phosphate-buffered saline; PFA: paraformaldehyde; REAP: RNA-editing associated protein; RNA-Seq: RNA sequencing; RRM: RNA recognition motif; RT-PCR: reverse transcriptase-polymerase chain reaction; snoRNA: small nucleolar RNA; snRNA: small nuclear RNA; sORF: short open reading frame; TAO: trypanosome alternative oxidase; uORF: upstream open reading frame; UTR: untranslated region.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ME, MAJ, FB, MM, EU and CT participated in the study design. ME, MAJ and FB performed the experiments. CT and FB performed the bioinformatics analyses. MM contributed novel instrumentation. ME, MAJ, FB, EU and CT contributed to the data analysis. ME and CT wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Coding potential of the 987 transcripts described in Figure 1. Setting a lower limit of 25 aa, all the potential ORFs are listed. For example Tb1.NT.1_1 refers to the novel transcript Tb1.NT.1 according to the nomenclature by Kolev et al. [28] and _1 indicates ORF #1. The numbers in parenthesis specify the CDS.
Length distribution of the predicted 4,699 ORFs. Figure S2. Growth analysis of the five small proteins with faster or slower growth in procyclics following induction of RNAi. Figure S3. Growth analysis of the seven small proteins essential in procyclics following induction of RNAi with and without expression of an RNAi-resistant construct. The data are based on three independent experiments. Figure S4. Northern blot analysis of essential ORFs. Northern blots for six (Tb11.NT.108, Tb10.NT.90, Tb10.NT.86, Tb10.NT.87, Tb11.NT.28 and Tb3.NT.18) of the seven essential transcripts. Marker sizes in kb are indicated on the left. The predicted transcript size is indicated below each panel. Northern blot analysis for Tb11.NT.29 can be found in Kolev NG, Franklin JB, Carmi S, Shi H, Michaeli S, Tschudi C: The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog 2010, 6:e1001090 [28]. Figure S5. Semi-quantitative RT-PCR analysis of RNAi knockdown. RT-PCR was performed on the transcripts encoding each essential small protein from procyclic cells un-induced (-) or induced (+) for RNAi. Histone 4 (H4) was used as a control. The small protein transcript and H4 are indicated to the right of each panel. Figure S6. Listing of the seven essential proteins in procyclics with a C-terminal GFP or HA tag. All constructs rescued the lethal RNAi phenotype. Figure S7. Western blot analysis of GFP-tagged proteins encoded by an RNAi-resistant construct. Size markers (kDa) are shown at the left. Figure S8. Growth analysis of bloodstream-form cells following RNAi against four small proteins. Growth for un-induced (tet-) and induced (tet+) cells are shown in log scale. The data are based on three independent experiments. Figure S9. Listing of oligonucleotides used in this study.
Listing of the 178 transcripts (sheet 178 ORFs) and 42 transcripts (sheet 42 ORFs) encoding potential ORFs with matching peptides and hits in Kinetoplastids. Panigrahi et al.: A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics 2009, 9:434–450 [21]. Butter et al.: Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite's host adaptation machinery. Mol Cell Proteomics 2013, 12:172–179 [22]. viv, T. vivax; cru, T. cruzi; Leish, Leishmania; con, T. congolense; evansi, T. evansi. PTM Abbreviations: P, phosphorylation; S, sumoylation; PM, palmitoylation; G, glycosylation. Phosphorylation prediction: NetPhos http://www.cbs.dtu.dk/services/NetPhos/; Sumoylation prediction: http://sumosp.biocuckoo.org/; Palmitoylation Prediction: http://csspalm.biocuckoo.org/; Glycosylation Prediction: http://www.cbs.dtu.dk/services/YinOYang/.
T. brucei predicted ORFs conserved in representative eukaryotes. BLAST bit scores of T. brucei predicted ORFs with potential homologs in representative eukaryotes. Bit scores are shown after the accession number and they represent a normalized version of the raw BLAST alignment score.
Survey of conserved domains in 173 T. brucei predicted ORFs.
Contributor Information
Megan Ericson, Email: megan.ericson@yale.edu.
Michael A Janes, Email: michaelannjanes@gmail.com.
Falk Butter, Email: F.Butter@imb-mainz.de.
Matthias Mann, Email: mmann@biochem.mpg.de.
Elisabetta Ullu, Email: elisabetta.ullu@yale.edu.
Christian Tschudi, Email: christian.tschudi@yale.edu.
Acknowledgements
This work was supported by Public Health Service grants AI28798 and AI56333 to EU and AI043594 to CT. ME was supported by a grant from the National Institutes of Health (T32 AI007404 to CT). Work in the Mann laboratory was supported by the Max Planck Society. We thank J. Bangs for providing BiP and Hsp70 antibodies, M. Chaudhuri for the antibodies against alternative oxidase, S. Hajduk for REAP antibodies and K. Stuart for antibodies against TbMP63. We especially thank M. Phillips for providing the Gateway® compatible vector and for advice and guidance. The authors acknowledge the Center for Cellular and Molecular Imaging at Yale contribution, P. De Camilli for the use of the AMAXA Nucleofector®, and advice and comments from N. Kolev.
References
- Jacquier A. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet. 2009;12:833–844. doi: 10.1038/nrg2683. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Kondo T, Hashimoto Y. Coding vs non-coding: translatability of short ORFs found in putative non-coding transcripts. Biochimie. 1981–1986;12:93. doi: 10.1016/j.biochi.2011.06.024. [DOI] [PubMed] [Google Scholar]
- Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;12:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. Life with 6000 genes. Science. 1996;12:547–563. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H. et al. The transcriptional landscape of the mammalian genome. Science. 2005;12:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Jung HW, Tschaplinski TJ, Wang L, Glazebrook J, Greenberg JT. Priming in systemic plant immunity. Science. 2009;12:89–91. doi: 10.1126/science.1170025. [DOI] [PubMed] [Google Scholar]
- Anginot A, Espeli M, Chasson L, Mancini SJ, Schiff C. Galectin 1 modulates plasma cell homeostasis and regulates the humoral immune response. J Immunol. 2013;12:5526–5533. doi: 10.4049/jimmunol.1201885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;12:1137–1146. doi: 10.1016/S0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- Chilley PM, Casson SA, Tarkowski P, Hawkins N, Wang KL, Hussey PJ, Beale M, Ecker JR, Sandberg GK, Lindsey K. The POLARIS peptide of Arabidopsis regulates auxin transport and root growth via effects on ethylene signaling. Plant Cell. 2006;12:3058–3072. doi: 10.1105/tpc.106.040790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi H, Huang YC, Chen IC, Lin CD, Yeh HF, Pai LM. Identification of 11-amino acid peptides that disrupt Notch-mediated processes in Drosophila. J Biomed Sci. 2011;12:42. doi: 10.1186/1423-0127-18-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pueyo JI, Couso JP. The 11-aminoacid long Tarsal-less peptides trigger a cell signal in Drosophila leg development. Dev Biol. 2008;12:192–201. doi: 10.1016/j.ydbio.2008.08.025. [DOI] [PubMed] [Google Scholar]
- Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;12:336–339. doi: 10.1126/science.1188158. [DOI] [PubMed] [Google Scholar]
- Magny EG, Pueyo JI, Pearl FM, Cespedes MA, Niven JE, Bishop SA, Couso JP. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;12:1116–1120. doi: 10.1126/science.1238802. [DOI] [PubMed] [Google Scholar]
- Ladoukakis E, Pereira V, Magny EG, Eyre-Walker A, Couso JP. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 2011;12:R118. doi: 10.1186/gb-2011-12-11-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Zhang X, Borevitz JO, Li WH, Shiu SH. A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res. 2007;12:632–640. doi: 10.1101/gr.5836207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Forrest AR, Nourbakhsh E, Pang KC, Kai C, Kawai J, Carninci P, Hayashizaki Y, Bailey TL, Grimmond SM. The abundance of short proteins in the mammalian proteome. PLoS Genet. 2006;12:e52. doi: 10.1371/journal.pgen.0020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;12:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenmayer JP, Ni L, Chu A, Kitchen LE, Au WC, Yang H, Carter CD, Wheeler D, Davis RW, Boeke JD, Snyder MA, Basrai MA. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 2006;12:365–373. doi: 10.1101/gr.4355406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Higuchi-Takeuchi M, Okamoto M, Yoshizumi T, Shimizu M, Nakaminami K, Nishi R, Ohashi C, Iida K, Tanaka M, Horii Y, Kawashima M, Matsui K, Toyoda T, Shinozaki K, Seki M, Matsui M. Small open reading frames associated with morphogenesis are hidden in plant genomes. Proc Natl Acad Sci U S A. 2013;12:2395–2400. doi: 10.1073/pnas.1213958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ogata Y, Zikova A, Anupama A, Dalley RA, Acestor N, Myler PJ, Stuart KD. A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics. 2009;12:434–450. doi: 10.1002/pmic.200800477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butter F, Bucerius F, Michel M, Cicova Z, Mann M, Janzen CJ. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite's host adaptation machinery. Mol Cell Proteomics. 2013;12:172–179. doi: 10.1074/mcp.M112.019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. Decoding human cytomegalovirus. Science. 2012;12:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitton DA, Wood V, Scutt PJ, Grallert A, Yates T, Smith DL, Hagan IM, Miller CJ. Augmented annotation of the Schizosaccharomyces pombe genome reveals additional genes required for growth and viability. Genetics. 2011;12:1207–1217. doi: 10.1534/genetics.110.123497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitton DA, Smith DL, Connolly Y, Scutt PJ, Miller CJ. An integrated mass-spectrometry pipeline identifies novel protein coding-regions in the human genome. PLoS One. 2010;12:e8949. doi: 10.1371/journal.pone.0008949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;12:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, Böhme U, Hannick L, Aslett MA, Shallom J, Marcello L, Hou L, Wickstead B, Alsmark UC, Arrowsmith C, Atkin RJ, Barron AJ, Bringaud F, Brooks K, Carrington M, Cherevach I, Chillingworth TJ, Churcher C, Clark LN, Corton CH, Cronin A. et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;12:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Kolev NG, Franklin JB, Carmi S, Shi H, Michaeli S, Tschudi C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010;12:e1001090. doi: 10.1371/journal.ppat.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson D, Gunasekera K, Mani J, Osteras M, Farinelli L, Baerlocher L, Roditi I, Ochsenreiter T. Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. PLoS Pathog. 2010;12:e1001037. doi: 10.1371/journal.ppat.1001037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel TN, Hekstra DR, Wang X, Dewell S, Cross GA. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 2010;12:4946–4957. doi: 10.1093/nar/gkq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitch NJ, Johnson PC, Trivedi U, Terry S, Wildridge D, MacLeod A. Digital gene expression analysis of two life cycle stages of the human-infective parasite, Trypanosoma brucei gambiense reveals differentially expressed clusters of co-regulated genes. BMC Genomics. 2010;12:124. doi: 10.1186/1471-2164-11-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information. Sequence Read Archive. http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi.
- The EuPathDB Project Team. TriTrypDB: The Kinetoplastid Genomics Resource. http://www.tritrypdb.org.
- Logan-Klumpler FJ, De Silva N, Boehme U, Rogers MB, Velarde G, McQuillan JA, Carver T, Aslett M, Olsen C, Subramanian S, Phan I, Farris C, Mitra S, Ramasamy G, Wang H, Tivey A, Jackson A, Houston R, Parkhill J, Holden M, Harb OS, Brunk BP, Myler PJ, Roos D, Carrington M, Smith DF, Hertz-Fowler C, Berriman M. GeneDB–an annotation database for pathogens. Nucleic Acids Res. 2012;12:D98–D108. doi: 10.1093/nar/gkr1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;12:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;12:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;12:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;12:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kolev NG, Tschudi C, Ullu E. RNA interference in protozoan parasites: achievements and challenges. Eukaryot Cell. 2011;12:1156–1163. doi: 10.1128/EC.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalidas S, Li Q, Phillips MA. A Gateway(R) compatible vector for gene silencing in bloodstream form Trypanosoma brucei. Mol Biochem Parasitol. 2011;12:51–55. doi: 10.1016/j.molbiopara.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;12:89–101. doi: 10.1016/S0166-6851(99)00002-X. [DOI] [PubMed] [Google Scholar]
- Vilela C, McCarthy JE. Regulation of fungal gene expression via short open reading frames in the mRNA 5'untranslated region. Mol Microbiol. 2003;12:859–867. doi: 10.1046/j.1365-2958.2003.03622.x. [DOI] [PubMed] [Google Scholar]
- Oyama M, Itagaki C, Hata H, Suzuki Y, Izumi T, Natsume T, Isobe T, Sugano S. Analysis of small human proteins reveals the translation of upstream open reading frames of mRNAs. Genome Res. 2048–2052;12:14. doi: 10.1101/gr.2384604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringpis GE, Lathrop RH, Aphasizhev R. iCODA: RNAi-based inducible knock-in system in Trypanosoma brucei. Methods Mol Biol. 2011;12:23–37. doi: 10.1007/978-1-61779-018-8_2. [DOI] [PubMed] [Google Scholar]
- Richardson JP, Beecroft RP, Tolson DL, Liu MK, Pearson TW. Procyclin: an unusual immunodominant glycoprotein surface antigen from the procyclic stage of African trypanosomes. Mol Biochem Parasitol. 1988;12:203–216. doi: 10.1016/0166-6851(88)90150-8. [DOI] [PubMed] [Google Scholar]
- Woodward R, Gull K. Timing of nuclear and kinetoplast DNA replication and early morphological events in the cell cycle of Trypanosoma brucei. J Cell Sci. 1990;12:49–57. doi: 10.1242/jcs.95.1.49. [DOI] [PubMed] [Google Scholar]
- Madison-Antenucci S, Sabatini RS, Pollard VW, Hajduk SL. Kinetoplastid RNA-editing-associated protein 1 (REAP-1): a novel editing complex protein with repetitive domains. EMBO J. 1998;12:6368–6376. doi: 10.1093/emboj/17.21.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Bouzaidi-Tiali N, Chanez AL, Bulliard L. ATP production in isolated mitochondria of procyclic Trypanosoma brucei. Methods Mol Biol. 2007;12:379–387. doi: 10.1007/978-1-59745-365-3_27. [DOI] [PubMed] [Google Scholar]
- Povelones ML, Tiengwe C, Gluenz E, Gull K, Englund PT, Jensen RE. Mitochondrial shape and function in trypanosomes requires the outer membrane protein, TbLOK1. Mol Microbiol. 2013;12:713–729. doi: 10.1111/mmi.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri M, Ajayi W, Temple S, Hill GC. Identification and partial purification of a stage-specific 33 kDa mitochondrial protein as the alternative oxidase of the Trypanosoma brucei brucei bloodstream trypomastigotes. J Eukaryot Microbiol. 1995;12:467–472. doi: 10.1111/j.1550-7408.1995.tb05892.x. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kondo T, Kageyama Y. Lilliputians get into the limelight: novel class of small peptide genes in morphogenesis. Dev Growth Differ. 2008;12:S269–S276. doi: 10.1111/j.1440-169X.2008.00994.x. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;12:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez JJ, Hon CC, Vanselow JT, Schlosser A, Siegel TN. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Nucleic Acids Res. in press. [DOI] [PMC free article] [PubMed]
- Dean S, Gould MK, Dewar CE, Schnaufer AC. Single point mutations in ATP synthase compensate for mitochondrial genome loss in trypanosomes. Proc Natl Acad Sci U S A. 2013;12:14741–14746. doi: 10.1073/pnas.1305404110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Djikeng A, Tschudi C, Ullu E. Argonaute protein in the early divergent eukaryote Trypanosoma brucei: control of small interfering RNA accumulation and retroposon transcript abundance. Mol Cell Biol. 2004;12:420–427. doi: 10.1128/MCB.24.1.420-427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo H, Tschudi C, Gull K, Ullu E. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc Natl Acad Sci U S A. 1998;12:14687–14692. doi: 10.1073/pnas.95.25.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;12:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- TMHMM Server v. 2.0, Prediction of transmembrane helices in proteins. http://www.cbs.dtu.dk/services/TMHMM/
- SignalP 4.1 Server. http://www.cbs.dtu.dk/services/SignalP/
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 1896–1906;12:2. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- The ProteomeXchange consortium. http://www.proteomexchange.org.
- Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O’Kelly G, Schoenegger A, Ovelleiro D, Pérez-Riverol Y, Reisinger F, Ríos D, Wang R, Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;12:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann Burkard G, Jutzi P, Roditi I. Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol Biochem Parasitol. 2011;12:91–94. doi: 10.1016/j.molbiopara.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Tokuyasu KT. A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol. 1973;12:551–565. doi: 10.1083/jcb.57.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coding potential of the 987 transcripts described in Figure 1. Setting a lower limit of 25 aa, all the potential ORFs are listed. For example Tb1.NT.1_1 refers to the novel transcript Tb1.NT.1 according to the nomenclature by Kolev et al. [28] and _1 indicates ORF #1. The numbers in parenthesis specify the CDS.
Length distribution of the predicted 4,699 ORFs. Figure S2. Growth analysis of the five small proteins with faster or slower growth in procyclics following induction of RNAi. Figure S3. Growth analysis of the seven small proteins essential in procyclics following induction of RNAi with and without expression of an RNAi-resistant construct. The data are based on three independent experiments. Figure S4. Northern blot analysis of essential ORFs. Northern blots for six (Tb11.NT.108, Tb10.NT.90, Tb10.NT.86, Tb10.NT.87, Tb11.NT.28 and Tb3.NT.18) of the seven essential transcripts. Marker sizes in kb are indicated on the left. The predicted transcript size is indicated below each panel. Northern blot analysis for Tb11.NT.29 can be found in Kolev NG, Franklin JB, Carmi S, Shi H, Michaeli S, Tschudi C: The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog 2010, 6:e1001090 [28]. Figure S5. Semi-quantitative RT-PCR analysis of RNAi knockdown. RT-PCR was performed on the transcripts encoding each essential small protein from procyclic cells un-induced (-) or induced (+) for RNAi. Histone 4 (H4) was used as a control. The small protein transcript and H4 are indicated to the right of each panel. Figure S6. Listing of the seven essential proteins in procyclics with a C-terminal GFP or HA tag. All constructs rescued the lethal RNAi phenotype. Figure S7. Western blot analysis of GFP-tagged proteins encoded by an RNAi-resistant construct. Size markers (kDa) are shown at the left. Figure S8. Growth analysis of bloodstream-form cells following RNAi against four small proteins. Growth for un-induced (tet-) and induced (tet+) cells are shown in log scale. The data are based on three independent experiments. Figure S9. Listing of oligonucleotides used in this study.
Listing of the 178 transcripts (sheet 178 ORFs) and 42 transcripts (sheet 42 ORFs) encoding potential ORFs with matching peptides and hits in Kinetoplastids. Panigrahi et al.: A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics 2009, 9:434–450 [21]. Butter et al.: Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite's host adaptation machinery. Mol Cell Proteomics 2013, 12:172–179 [22]. viv, T. vivax; cru, T. cruzi; Leish, Leishmania; con, T. congolense; evansi, T. evansi. PTM Abbreviations: P, phosphorylation; S, sumoylation; PM, palmitoylation; G, glycosylation. Phosphorylation prediction: NetPhos http://www.cbs.dtu.dk/services/NetPhos/; Sumoylation prediction: http://sumosp.biocuckoo.org/; Palmitoylation Prediction: http://csspalm.biocuckoo.org/; Glycosylation Prediction: http://www.cbs.dtu.dk/services/YinOYang/.
T. brucei predicted ORFs conserved in representative eukaryotes. BLAST bit scores of T. brucei predicted ORFs with potential homologs in representative eukaryotes. Bit scores are shown after the accession number and they represent a normalized version of the raw BLAST alignment score.
Survey of conserved domains in 173 T. brucei predicted ORFs.