

Abstract
Dendritic cells (DCs) that orchestrate mucosal immunity have been studied in mice. Here we characterize human gut DC populations, and define their relationship to previously studied human and mouse DCs. CD103+Sirpα− DCs were related to human blood CD141+ and to mouse intestinal CD103+CD11b− DCs and expressed markers of cross-presenting DCs. CD103+Sirpα+ DCs aligned with human blood CD1c+ DCs and mouse intestinal CD103+CD11b+ DCs and supported regulatory T cell induction. Both CD103+ DC subsets induced TH17 cells, while CD103−Sirpα+ DCs induced TH1 cells. Comparative transcriptomics revealed conserved transcriptional programs among CD103+ DC subsets and uncovered a selective role for Bcl-6 and Blimp-1 in CD103+Sirpα− and intestinal CD103+CD11b+ DC specification, respectively. These results highlight evolutionarily conserved and divergent programming of intestinal DCs.
The gastro-intestinal tract is challenged by a multitude of antigens, including commensal microflora, food antigens and invasive pathogens. Animal models have begun to define a critical role for dendritic cell (DC) specialization in achieving the balance between tolerogenic and inflammatory immune responses in the intestine. Diversity in intestinal DC phenotype and function has been extensively studied in mice using model systems based on conditional ablation of DCs and engraftment with defined DC precursors1,2. The mouse CD103+CD11b+ (mDP for mouse double positive) and CD103+CD11b− (mSP for mouse single positive) DCs require distinct genetic factors for their development and display unique gene expression profiles3. mSP DCs are closely related to cross-presenting CD8α+ DCs of splenic origin, and both require the transcription factors BATF3, Id2 and IRF8 for development4. mDP DCs are potent inducers of CD4+ T cell responses of both inflammatory and tolerizing nature5-8, and share a requirement for the transcription factors IRF49 and Notch210 with lymphoid resident CD4+CD8α− DC.
Recent studies have identified CD141 as a marker of human cross-presenting cDCs, the putative homologs of mouse CD8α+ DCs, in human blood, spleen, lymph nodes and skin11-13. In addition, CLEC9A+Sirpα−CD103+ DC have been identified in the human ileum9. However the phenotypic features and functional specialization of human intestinal DCs, as well as the transcriptional programs of various DC subsets, remain to be studied.
Our goal here was to identify discrete DC subsets in the human intestinal LP, to relate them to mouse intestinal DC populations by identifying evolutionarily conserved phenotypic features and to start characterizing their functional specialization. We also performed genome-wide expression analysis to define transcriptional fingerprints for each DC subset in the gut, and to correlate gene expression in human and mouse DC subsets from lymphoid and non-lymphoid sites. We identified CD103 (αE) and Sirpα (CD172a) as conserved markers that define three major subpopulations of conventional CD11c+ DCs in the human gut mucosa. Our analyses revealed that human CD103+Sirpα+ (hDP, for human double positive) DCs and mDP DCs have a common set of phenotypic characteristics (i.e. CLEC4A+, CD101+, TLR5+, CCR7hi, CD11b+ and Sirpα+). Human gut CD103+Sirpα− (hSP, for human single positive) DCs share significant transcriptomic similarities with human blood CD141+ and mSP DCs, including expression of CLEC9A, CADM1 and XCR113. We also identified a population of human CD103−Sirpα+ cDCs that had increased frequency in inflamed gut specimens, and expressed transcription factors and gene profiles consistent with monocyte-derived DCs.
Using comparative transcriptomics, we identified transcription factors whose expression was coordinately regulated in these human and mouse intestinal DC subsets. In addition to IRF8, a transcription factor previously implicated in intestinal DC development, our analyses revealed conserved expression of Bcl6 in hSP and mSP DCs, and of Irf4 and Prdm1 (encoding Blimp-1) in hDP and mDP DCs. Selective loss of intestinal mDP DCs was reported in mice with DC-specific IRF4-deficiency9,14. We show here that Bcl-6 and Blimp-1 control the specification of intestinal mSP and mDP DC subsets. Bcl-6 is required for the development of intestinal mSP, as well as for lymphoid tissue CD8α+ DCs, while Blimp-1 deficiency specifically affects the unique mDP subset in the intestine. Hence, in parallel to their counter-regulatory roles in effector T and B cell differentiation, Bcl-6 and Blimp-1 display opposing roles in the specification of mSP and mDP DCs in the intestinal lamina propria.
Results
Sirpα and CD103 define cDC subsets in the human small intestine
To characterize DC subsets in the human small intestine (SI), we prepared cell suspensions from lamina propria (LP) of the jejunum of patients undergoing bariatric surgery. cDCs were defined as CD45+Lin(CD3, CD19, CD14, CD56)−MHCII+CD11c+CD123− cells. We established CD103 and Sirpα as suitable markers to characterize CD11c+ cDC subsets in LP cell preparations. CD103, an integrin implicated in interaction of DCs with epithelial E-cadherin, defines a major migratory intestinal cDC population in the mouse15, distinguishes them from CD103−CD11b+ monocyte-derived LP macrophages and has been described on human intestinal DCs. Sirpα (CD172a), a receptor for CD47, defined discrete subsets of CD103+ and CD103− cDCs (Fig. 1a). CD11b, used to subset mouse gut CD103+ DCs, was expressed preferentially and selectively on the Sirpα+ populations (Fig. 1b), but its staining was consistently weak. CD103 and Sirpα together define four populations of CD11c+ cDCs in the SI LP (Fig. 1a, full gating strategy in Supplementary Fig. 1). Because CD103−Sirpα− DCs were a minor population among LP CD11c+ cDCs, which limited our ability to characterize them, we do not discuss them further.
Fig. 1. Human small intestine lamina propria DC subsets can be distinguished using CD103 and Sirpα.

a) Flow cytometry of lamina propria DCs subpopulations gated on live CD45+ lineage (CD3, CD14, CD16, CD19, and CD56) negative MHCII+ cells. Representative dot plot from fifteen donors is shown here.
b) Flow cytometry of lamina propria DC stained with CD11b, HLA-DR, CD86, CD40, CD80, CD83 and CD64. Representative results of three to five independent experiments are shown.
c) Confocal microscopy (X63) of small intestine villus stained for Sirpα (green), CD11c (red) and CD103 (blue), illustrating intermingling of CD103+Sirpα− (hSP), CD103+Sirpα+ (hDP), and CD103−Sirpα+ CD11c+ cells within the lamina propria. Representative of 4 donors.
d) Relative frequency of intestinal DC subsets among live CD45+lin−HLADR+CD11c+ cells in 15 donors. Open symbols denote two patients with hyperemic epithelium with increased percentage of CD103−Sirpα+ DCs.
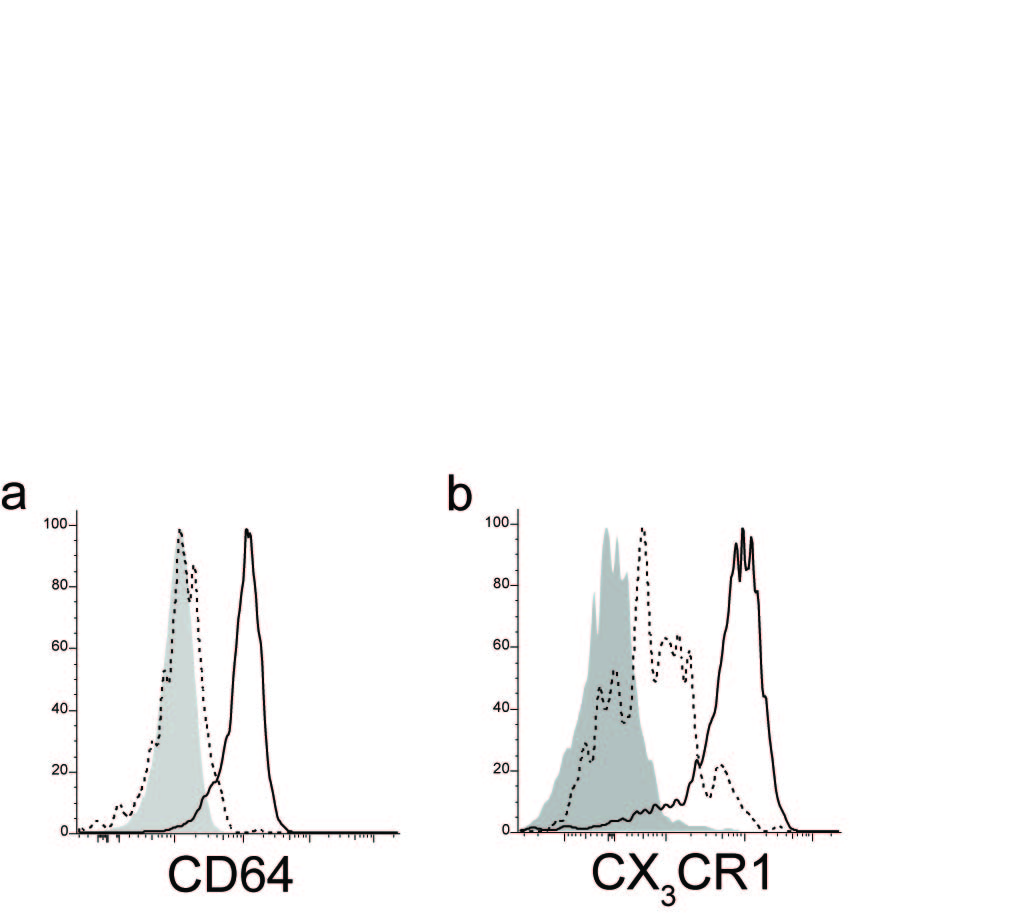
All the DC subsets expressed similar levels of HLA-DR by flow cytometry, but they varied in the expression of other maturation and costimulatory molecules (Fig. 1b). CD103+Sirpα+ (hDP) DCs expressed high amounts of CD86 and CD83, while CD103−Sirpα+ DC had intermediate expression (Fig. 1b). CD103+Sirpα− hSP DCs had low expression of the costimulatory molecule CD86. hDP and (hSP) DCs highly expressed CD40. In contrast to blood DC subsets16, all gut DC subsets express CD40, albeit at varying levels. However, the expression of CD86 and CD80 was similar on both gut (Fig. 1b) and blood DCs12,16. None of the gut DC subsets expressed CD64 (Fig. 1b), a characteristic macrophage marker in both human and mouse (Supplementary Fig. 2a)17
To determine whether the DC subsets occupy discrete microenvironments, we carried out immunofluorescence studies of jejunal/small intestine frozen sections. All the CD103-Sirpα-defined DC subsets were present in the LP within the villus core (Fig. 1c). Histology and cell suspension analysis showed that hDP DCs were the dominant subset in the jejunal LP of most patients (Fig. 1c,d). In two patients whose jejunal tissue presented a hyperemic phenotype indicative of inflammation, CD103−Sirpα+ DCs were dramatically increased in frequency and comprised the majority of CD11c+ DCs (Fig. 1d). The relative representation of DC subsets was different in the colonic LP, where CD103−Sirpα+ and hSP DCs predominated (Supplementary Fig. 3). The increased frequency of hDP DCs in the proximal intestine parallels an increased representation of mDP DCs in the mouse small versus large intestine18. Altogether these data show that the human LP contains three major DC populations that include CD103+Sirpα+ (hDP), CD103+Sirpα− (hSP), and CD103−Sirpα+ DCs.
Differential expression of chemokine receptors and integrins by LP cDCs
Similar to blood DCs in man (19 and P.W., personal observations), all cDC subsets in the LP exhibited high expression of mucosa associated integrin α4β7 (Fig. 2a). In contrast to mouse DCs, in which CCR9 expression is restricted to pDCs under homeostatic conditions20, CCR9 was expressed by a subset of hDP DCs (Fig. 2a) and was not detected on pDCs (not shown) in the human small intestine. In the mouse LP, CD11c+F4/80hiCD11b+CD103− macrophages have high expression of the chemokine receptor CX3CR1, whereas CD11c+CD11b+CD103− DCs display lower CX3CR1 expression21. Compared to human gut CD14+ macrophages (Supplementary Fig. 2b), which expressed high amounts of CX3CR1, CD103−Sirpα+ DCs showed intermediate expression of CX3CR1 and the marker was absent on the other DC subsets (Fig. 2a).
Fig. 2. Mucosa associated chemokine receptors and integrins expressed by LP DCs.

a) Flow cytometry of lamina propria mononuclear cells stained for the indicated chemokine receptors and integrins. The open (black lines) histograms show the staining profile with indicated antibody while filled (grey) histograms show isotype control staining. Data from one of 3-6 experiments with similar results.
b) Flow cytometry of CD103+Sirpα+ (hDP) DCs from MLN assessing expression of CCR7. Representative result of three independent experiments is shown.
c) Three-color immunofluorescence staining for Sirpα (green), CD103 (blue), and CD11c (red) in cryosections of appendix (X63). Representative result of three independent experiments for (c) and (d) is shown.
d) Same as in c) for MLN. CD103+Sirpα+ (hDP) CD11c+ DCs are present in MLN (arrows) but not appendix.
In the mouse, the chemokine receptor CCR6 controls the localization of CD11c+CD11b+DEC205− DCs in Peyer's patches22. In human LP, CCR6 was highly expressed on both subsets of intestinal LP CD103+ DCs (Fig. 2a), while CXCR3 was selectively expressed on hSP DCs (Fig. 2a). Because intestinal DCs use CCR7 to access lymphatic vessels23, we examined the expression of CCR7 on human lamina propria DC subsets. Similar to migratory mDP DCs, hDP DCs had high expression of CCR7 (Fig. 2a), suggesting that they may migrate via lymphatics to MLNs in vivo. Supporting this idea, hDP DCs were present in the MLN and expressed high amounts of CCR7 (Fig. 2b). Furthermore, immunohistology revealed abundant hDP DCs in the gut-draining MLN, but a complete absence of these DC in the appendix, which lacks afferent lymphatics (Fig. 2c, d). CD103−Sirpα+ and hSP subsets also expressed CCR7 (Fig. 2a), suggesting that they may also be able to enter lymphatics and migrate to the MLN.
The results define conserved and divergent subset expression of chemoattractant and trafficking programs in man vs mouse. Notably, patterns of CX3CR1 expression appear conserved within DC and macrophages in the gut; and CCR7 expression appears to define migratory subsets.
Gene expression and surface receptor profiling of human gut DCs
Next we sorted hDP, hSP and CD103−Sirpα+ subsets from human gut and carried out genome-wide transcriptional profiling. Differentially expressed genes were used to align the three human gut DC subsets with human blood and skin DC and blood monocyte populations as profiled previously12. Hierarchical clustering suggested a close relationship of gut hSP DCs with cross-presenting blood CD141+ DCs, whereas hDP DCs clustered with the major blood CD1c+ DC subset and CD103−Sirpα+ DCs clustered with blood monocytes, consistent with the possibility that they are derived from recruited monocytes (Fig. 3a).
Fig. 3. Transcriptomic and phenotypic comparison of DC subsets in human gut and blood.

a) Hierarchical clustering by correlation of gut, blood and skin DC subsets based on profiles of differentially expressed genes (see online Methods). Significance of clusters of interest is indicated (**p<0.01; *p<0.05).
b) Alignment of blood CD1c+ DCs, CD141+ DCs and CD14+ monocytes with gut DC populations. Data represent pairwise correlation of replicate gut DC samples with mean gene expression profiles of blood CD1c+ DCs, CD141+ DCs or CD14+ monocytes from Haniffa12, using CD1c DC-, CD141 DC- and CD14 monocyte lineage-associated gene sets respectively. Shown with mean and SEM. *=Positive correlation, p<0.01, based on 99% confidence limits.
c) Scatter plot of normalized average gene expression values between gut hSP and blood CD141+ DCs.
d) Comparison of differential gene expression by gut and blood DC subsets. Genes that differ at least two fold in expression between the two gut DCs, hDP and hSP, and also between blood CD1c+ and CD141+ DC are plotted. Colors indicate genes up (red) and down (blue) in skin CD1c+ versus CD141+ DCs. Most genes display coordinate regulation between CD1c+ vs CD141+ DC subsets in all three tissues.
e) Flow cytometry of DC subsets stained for the indicated receptors and transcription factors. CD103+Sirpα+ (hDP) DCs (red), CD103+Sirpα− (hSP) DCs (black), CD103−Sirpα+ DCs (blue), and representative control staining (grey). Data shown are representative of results from 4-5 donors.
f) Relative expression of BTLA, BATF3, TAP1, NLRC5, NLPR3, and IL-22RA2 by gut DC subsets. Arbitrary units (Affymetrix expression) with SEM of the biological replicates shown.
Although each of the clusters highlighted was significant, the association of hDP DCs with CD1c+ DCs by hierarchical clustering was not robust with respect to gene set selection criteria or clustering algorithm. We therefore assessed the pairwise correlation of gene expression in hDP DCs with that of individual blood and skin DC and monocyte populations. Three of the hDP DC samples correlated significantly (p<0.001) with blood CD1c+ DCs, while the other two aligned preferentially with skin CD1c+ (p<0.001), suggesting potential differences in tissue-variant gene expression (Supplementary Fig. 4a). Consistent with the clustering results, hSP and CD103−Sirpα+ DC correlated best with CD141+ blood DC and with monocytes, respectively (Supplementary Fig. 4a). CD103−Sirpα+ intestinal DC also aligned weakly but significantly with CD14+ skin DC, a population related to monocytes as well12.
To assess the relationship of gut DCs with blood DCs and monocytes independently of tissue-specific genes, we took advantage of lineage-associated gene sets developed previously12, from which skin and blood-specific genes were specifically excluded. We additionally excluded gut-specific genes (genes that differed significantly between pooled gut, skin and blood samples) and selected for genes whose expression differed significantly between gut DC subsets (see Supplementary Gene List for Fig. 3 for full gene list used). Using Pearson correlation, we show that hDP DCs are enriched in CD1c lineage genes as evident from alignment of hDP with blood CD1c+ DCs (Fig. 3b). Similarly, hSP align with blood CD141+ DCs in expression of CD141 lineage genes, and CD103−Sirpα+ DCs with blood monocytes in expression of monocyte lineage gene sets (Fig. 3b).
Blood CD141+ DCs are efficient antigen cross-presenting cells, and share phenotypic and functional properties with mouse cross-presenting CD8α+ lymphoid DCs12. Using a scatter plot of normalized gene expression we found a strong correlation between gut hSP and blood CD141+ DCs (Fig. 3c). The two populations shared expression of CD141, as well as of core cross-presenting DC signature genes, including the chemokine receptor XCR1 (Fig. 3c) (supports migration of DCs to XCL1 expressed by CD8+ T cells), the lectin receptor CLEC9A (Fig. 3c, e) (mediates uptake of apoptotic cells), and the adhesion receptor CADM1 (Fig. 3c), (may promote induction of CTL responses24). Notably, hSP DCs expressed higher levels of IDO1, CLEC1A and BTLA, molecules associated with dampening of the immune response (Fig. 3c, f), and NLRC5 (Fig. 3c, f), a key regulator of MHC class I expression for controlling immune responses against intracellular pathogens25. Flow cytometry confirmed that hSP DCs expressed high amounts of CD141 and CLEC9A (Fig. 3e) but lacked CLEC4A (DCIR, the 33D1 antigen), CD101, CD206 (Fig. 3e) and CD11b (Fig. 1b). Collectively, these results indicate a close relationship between intestinal hSP DCs and CD141+ cross-presenting DCs in blood and other tissues.
hDP DCs express a number of receptors, e.g. CD101, CLEC4A, CD206 and CD209 (DC-SIGN) that have been described as markers of mDP DCs (Fig. 3c, e; and comparative transcriptomics analysis below). The immunoregulatory lectin receptors CLEC4A (Fig. 3e) and CLEC10A (data not shown) and the immunoregulatory Ig family member CD10126 were highly expressed by hDP, but absent or poorly expressed by hSP DCs (Fig. 3e). Both CD101 and CLEC4A were also expressed at intermediate levels on CD103−Sirpα+ DCs (Fig. 3e). TLR5 was expressed by the two Sirpα+ DC subsets, but not by hSP DCs (Fig. 3e). Expression of the endocytic receptors CD206, CD209 and CD207 (Langerin) was restricted to hDP DCs (Fig. 3e). The transcription factor IRF4 was also highly expressed in hDP, but absent in hSP DCs (Fig. 3e). Thus, hDP gut DCs share many phenotypic features with their mDP intestinal DC counterparts.
A number of genes associated with tolerance induction were preferentially expressed by hDP DCs, such as VSIG4, a member of the B7 superfamily and a negative regulator of T cell activation (Fig. 3d), IRAK3, a key inhibitor of the IRAK-NF-κB pathway in chronic inflammation, TGFBR2, a receptor involved in TGF-β signaling and the inhibitory receptor PILRA, which also serves as an entry receptor for herpes simplex (data not shown). hDP DCs also specifically expressed transcripts for molecules involved in regulating IL-2227, a cytokine that can promote pathological inflammatory response in the intestine28. These included the cytosolic sensor NLRP3, caspase-1, IL-18 and the soluble IL-22 cytokine receptor IL-22RA2 (Fig. 3f).
We assessed the genes co-regulated in gut hDP DC and blood CD1c+ subsets by comparing each of them to CD141+ DCs derived from the same tissue. Plotting the fold change in gene expression between CD1c+ and CD141+ DC in blood versus that between hDP DCs and CD141+ hSP DCs in the gut revealed that the majority of differentially expressed genes were coordinately regulated in the gut and blood DC subsets (Fig. 3d). Most of these genes were also coordinately regulated between skin CD1c+ and CD141+ DCs (Fig. 3d). However, a number of genes expressed by gut hDP and blood CD1c+ DCs, including CLEC10A, CLEC4A, ALOX5 and GIMAP4, were downregulated in skin CD1c+ DCs. Genes preferentially expressed by CD1c+ DCs in all sites (ie, by blood CD1c vs blood CD141+ DC, skin CD1c+ vs skin CD141+ DC, and gut hDP vs hSP DCs), included CD1c, CD1e, Sirpα, IgSF6, IRF4 and MS4A6A (Fig. 3d).
Gut CD103−Sirpα+ DC gene expression showed a significant correlation with that of blood monocytes (Supplementary Fig. 4), especially CD14+ monocytes. Importantly, CD103−Sirpα+ DCs fulfilled current criteria that distinguish DCs from macrophages. First, they expressed CD11c, but lacked the canonical macrophage antigens CD14 and CD64 (Fig. 1b). Second, intestinal CD103−Sirpα+ DCs were as efficient as hDP DCs, and better than hSP DCs, at promoting T cell expansion in allogenic co-culture, a feature that distinguishes DCs from macrophages (Supplementary Fig. 5a). Finally, the PU.1/Mafb ratio, which determines whether a myeloid precursor acquires a macrophage or DC fate29 was similar in CD103−Sirpα+ DCs and other gut DCs, and much lower than in macrophages (Supplementary Fig. 5b).
Together the results establish a close relationship between intestinal hSP DCs and CD141+ cross-presenting DCs in blood and other tissues, and between gut hDP and the blood CD1c+ DC lineage. They suggest that gut CD103−Sirpα+ comprise a true DC population, likely derived from inflammatory recruitment of circulating monocytes. The latter hypothesis was consistent with the dramatic increase in the frequency of CD103−Sirpα+ DCs in the lamina propria isolates of two specimens with hyperemic intestinal mucosa (Fig. 1d).
Human intestinal DC subsets in CD4+ T cell education
Intestinal DCs moderate gut immune responses in part by inducing regulatory CD4+ T cells (Treg)18. In humans, Treg cells are phenotypically identifiable as FOXP3+CD127loCD25hiCD4+ T cells30. We primed human allogeneic naïve peripheral blood CD4+ T cells with DC subsets sorted from lamina propria and assessed the expression of FOXP3, CD25 and CD127 on responding CD4+ T cells after 9 days. While both hDP and CD103−Sirpα+ DCs supported robust T cell proliferation, hDP DCs were the most efficient inducers of FOXP3+CD127lo cells (Fig. 4a, b). Co-culture with hSP DCs induced comparatively less T cell proliferation (Supplementary Fig. 5a) and fewer T cells upregulated FOXP3 (Fig. 4a).
Fig. 4. Human intestinal DC subsets in CD4 T cell education.

a) Flow cytometry of CD4+ T cells activated by incubation for 9 days with allogeneic sorted gut DC subsets. CD4+ T cells were stained for CD25, CD127, CD4 and FOXP3. Shown are plots of FOXP3 versus CD127 for CD25hi CD4 cells (black dots) and for CD25lo CD4 cells (grey density contours). Gates for FOXP3 were set based on isotype control. Numbers indicate CD25hi FOXP3+CD127lo Treg cells as percentage of total CD4+ T cells. Representative for four experiments.
b) Composite data of four experiments expressed as FOXP3+CD127lo cells as percentage of total CD4+ T cells, * p<0.05 Mann Whitney, mean ± SEM.
c) RALDH activity in the three gut DC subsets assessed by ALDEFLUOR assay. Fold change in mean fluorescence intensity (MFI) of ALDEFLUOR staining in the presence or absence of the RALDH inhibitor DEAB. Representative of six independent experiments.
d) Flow cytometry of CD4+ T cells activated as in a). FACS plots illustrate representative CCR9 and α4β7 induction. Composite data of three experiments are presented in the bar graphs. Middle: percent of CD4 cells staining for α4β7, with SEM. Right: percent of CD4 cells staining for CCR9 (mean±SEM).
e,f) Staining of IL-17 and IFNγ in CD4+ T cells differentiated in vitro by co-culturing naïve CD4+CD45RA+CD25− T cells with sorted lamina propria DC. Numbers (left panel) denote percent positive, and are from one of three independent replicates included in the graph at right (symbols indicate different replicates). Data in graph are shown with mean ± SEM. * p<0.05, paired t-test.
Induction of Treg cells by mouse gut DCs is mediated in part by DC expression of aldehyde dehydrogenase (ALDH) activity, which produces retinoic acid for presentation to responding T cells7. To assess whether the differences in induction of FOXP3+CD127lowCD4+ T cells correlated with ALDH activity, human DC subsets were incubated with the fluorogenic substrate ALDEFLUOR, in the presence or absence of the ALDH inhibitor DEAB, and fluorescence was assessed by flow cytometry. Both Sirpα+ DC subsets exhibited high ALDH activity compared with Sirpα− DCs (Fig. 4c). Similarly, mDP DCs display the highest RALDH activity and efficiency at inducing Foxp3+ CD4+ Treg cells18. In the context of intestinal immune responses, DC-driven production of retinoic acid also imprints responding T cells with trafficking receptors for homing to the small intestine31. Consistent with the pattern of ALDH activity in the DC subsets, hDP DCs were strong inducers of CCR9 on CD4+ T cells, but all subsets of gut DCs could induce the receptor (Fig. 4d). In humans and mice, gut microbiota and their metabolites act via resident DCs at steady-state to promote IL-17 production in T cells in the SI lamina propria32,33. We examined the capacity of human gut DC subsets sorted from the LP to stimulate and polarize naïve CD4+ T cells in an allogeneic mixed leukocyte reaction. After 9 days of coculture, we restimulated the responding allogeneic CD4+ T cells with PMA+ionomycin and stained for intracellular cytokines. Lamina propria hDP and hSP DCs induced the highest frequency of TH17 cells (Fig. 4e), whereas CD103−Sirpα+ DCs were relatively inefficient. This result is consistent with the TH17-skewing capacity of mDP DCs from the small intestine9,14,18. In contrast, the CD103−Sirpα+ DC subset was most efficient at skewing responding CD4+ T cells towards IFN-γ production (Fig. 4f), similar to monocyte-derived mouse intestinal inflammatory DCs (CD103− CX3CR1int DCs) which are also efficient inducers of TH1 cells34.
Differences in CD4+ T cell priming could reflect differences in steady-state cytokine expression by the DCs. However, genes for key cytokines involved in T cell differentiation, namely IL-6, IL-23, IL-12 and IL-10 were poorly expressed if at all (mean raw value <150) in all DCs subsets at steady-state. It will be interesting to analyze cytokine expression by activated DC subsets in vitro, and by DC in inflamed settings in this regard.
Comparison of mouse and human DC subsets
We next sought to compare the gene expression profiles of human and mouse gut DC subsets in order to further assess their relationships, and to define evolutionarily conserved DC subset signatures. We identified sets of human genes or their mouse homologs that were differentially expressed among the three human gut DC subsets profiled here; among mouse gut mSP, mDP DCs and CD11b+ macrophages; and among mouse spleen CD4+ and CD8α+ DCs and red pulp macrophages (data from Immgen.org; see Methods). Normalized expression of genes common to the three gene sets above was used to cluster human and mouse DC and monocyte-macrophage cell types (Fig. 5a, see Supplementary Gene List for Fig. 5a for full gene list). Hierarchical clustering using a Euclidean distance metric indicated a significant similarity between hSP DCs, intestinal mSP and cross-presenting splenic CD8α+ DCs (cluster 1 DCs hereafter). Intestinal hDP DCs clustered with mDP and spleen CD4+ DCs (cluster 2 DCs; Fig. 5a). Human intestinal CD103−Sirpα+ DCs clustered with mouse blood Ly6c+ monocytes (Fig. 5a), supporting their relationship with human blood monocytes (Fig. 3a). Mouse gut CD11b+ macrophages and spleen red pulp macrophages, both thought to represent resident macrophages, formed a separate cluster. Transcriptomic data on mouse inflammatory CD103−CD11b+CX3CR1int DCs from lamina propria was not available and thus we did not compare their gene expression profile with human CD103−Sirpα+CX3CR1int DCs.
Fig. 5. Transcriptomic relationship between human intestinal DC and mouse DC subsets.

a) Hierarchical clustering illustrates transcriptional alignment of human intestinal DC with mouse DCs from gut and lymphoid organs. Euclidean distances were computed using genes selected as differentially expressed (top 15% most diverse in comparisons of cell types isolated from each organ); mean expression >120 for at least one mouse and one human population; and differentially expressed between human gut DC subsets (P<0.05). Significant clusters (P<0.01) of interest are highlighted. GEO accession code for human intestinal microarray data: GSE50380; for others see materials and method section.
b) Heat map illustrating expression of transcripts that are most differentially expressed (ANOVA P<0.01, at least 4 fold difference) by the three groups of DCs or DCs and monocytes defined by the highlighted clusters in the dendrogram in 5a.
c) Scatterplot of the ratios of the normalized averaged values between SP and DP DCs of mice against men. Depicted genes are at least 2-fold differentially expressed between SP and DP in both species. Mouse gene expression data were obtained from the NCBI GEO website (GSE15907) and originate from the Immgen Consortium38. All probes were selected for >120 expression value in all replicates in at least one sample, and for an uncorrected p-value of <0.05 between the two subsets in each species.
We identified genes whose expression differed at least 4 fold between the clusters for heatmap visualization (Fig. 5b). DC cluster 1 was enriched in transcripts belonging to a wide-range of functional groups, including solute carriers, inhibitory and activating ligands, adhesion and signaling molecules. In particular, XCR1, CLEC9A, TLR3 and CADM1 appeared as highly conserved markers of cluster 1 (hSP, mSP, CD8α+ cross-presenting-like) DCs in mouse and human (Fig. 5b). Transcription factors IRF8 and BATF3, which are critical for the differentiation of mSP and lymphoid CD8α+ DCs, were also expressed by gut hSP and by human blood CD141+ DCs (Fig. 5b and Fig. 3c), suggesting conservation of genes regulating cluster 1 differentiation. Genes preferentially expressed by cluster 2 DCs included the canonical CD4+ DC marker Clec4a (33D1 antigen), CD101 (Igsf2) and the transcription factor IRF4, implicated in CD4+ and mDP DC development in the mouse (Fig. 5b). Cluster 2 DCs also shared preferential expression of ABCB1 (an ATP dependent xenobiotic efflux transporter, variants of which are associated with inflammatory bowel disease), and were enriched in genes that enhance leukocyte motility, including the Rac GTPase activating proteins Vav2 and Dock4. A number of genes were shared by human and mouse monocytes and by CD103−Sirpα+ DCs, such as the IFN induced anti-viral protein IFITM2 and the C5A receptor, and distinguished them from DCs derived from committed DC precursors. Identification of core tissue and species-independent signature genes for CD103−Sirpα+ DCs must await gene profiling of mouse intestinal inflammatory (monocyte-derived) DCs.
Interestingly, a number of genes shared by intestinal hDP DC and cluster 3 (CD103−Sirpα+ DCs/monocyte group) were less well conserved in expression by spleen and MLN CD4+ DCs. These genes, which include Fc receptors, EMILIN2, ALOX5, THBS1, C3AR1, PRDM1, CLEC10A and VWA5A, may be induced in cluster 2 and 3 DC in response to the gut inflammatory environment. Thus the results support the conservation of core lineage-specific gene signatures, but also reveal conserved responses to local tissue environments.
Conserved signatures of SP and DP intestinal DCs in mouse and man
We next looked more closely at conserved transcriptional profiles of SP vs DP DCs. Among ∼700 genes that differed at least 2 fold between human or mouse SP versus DP DCs, only 84 showed coordinate up- or downregulation in both species: 18 genes were elevated in both mouse and human SP versus DP DCs; and 66 genes were more highly expressed in the DP DC populations (Fig. 5c and Supplementary Table 1). Interestingly, differential expression of 14 of these genes was exclusively seen in the gut: these genes were either not differentially (<2 fold) expressed or were discordantly regulated between cluster 1 and cluster 2 DC subsets in lymphoid organs or skin; and all but one (GYPC) of these genes were expressed more highly in DP than SP DCs. These 14 genes (Supplementary Table 1, bold) include genes encoding chemokine receptor CCR1, activation-associated migration regulator CD69, inhibitory receptor PILRA, IL13RA1, as well as transcription factor Blimp-1 discussed below. Together, the results show that genes conserved in their differential expression by SP vs DP intestinal DC comprise both core signature genes distinguishing cluster 1 from cluster 2 DC, as well as genes whose differential expression is gut-selective. They also suggest that DP and other cluster 2 lineage DCs may be more responsive to local (tissue-specific) environmental imprinting than cluster 1 lineage DC.
Differential expression of Bcl-6 and Prdm1 in DC subsets
We hypothesized that transcription factors (TF) that were differentially expressed between intestinal DP versus SP DCs in both mouse and man might contribute to conserved gut DC specialization. Only seven TF showed conserved differential expression: IRF8 and Bcl-6 were preferentially expressed in SP DCs compared to DP DCs, while IRF4, Prdm1, ZEB2, HIVEP2 and FoxN3 were more highly expressed in DP DCs (Fig. 6a). Id2 and BATF3, also implicated in intestinal DC development, were elevated in human and mouse SP versus DP DCs, but did not meet the criteria of 2-fold differential expression in both species (data not shown). The roles of ZEB2 or FoxN3 in hematolymphoid cell development remain unclear, while Bcl-6 and Blimp-1 (encoded by Prdm1) are transcriptional repressors with counter-regulatory roles in effector B and T cell fate determination35,36. Bcl-6 is downregulated by Blimp-1 in both lymphoid lineages37. Bcl-6 expression was highest in SP LP DC, and was preferentially expressed by cross-presenting (SP or CD8α+) DCs versus CD11b+ DCs across all tissues investigated (Fig. 6b). Prdm1 had an overall low expression in CD11b+ splenic DCs, and high expression in DP intestinal DCs (Fig. 6b).
Fig. 6. Differential expression of Bcl-6 and Blimp-1 in DC subsets.

a) VENN diagrams of all transcription factors at least 2-fold differentially expressed by human intestinal SP vs DP DCs or by mouse SP vs DP DCs. Genes within the intersections showed conserved SP or DP-selective expression in mice and humans.
b) Microarray data for pdrm1 (Blimp1-encoding) and Bcl-6 genes in small intestinal SP versus DP DCs. Bar graphs show the mean and SEM of raw expression values.
c) BCL-6 staining on human intestinal DCs. hSP DC gates and data are shown in orange, hDP in blue, non-stained control in gray. Bar graph shows the MFI of BCL-6 staining in each subset (n=3, p<0.05 paired t test.
d) Expression of a YFP-Bcl-6 and a Prdm1-YFP reporter by gut associated mouse cDCs. Contour plots show gating strategies. Pre-gated on live, lineage negative (CD3, CD19, NK1.1), CD11chi, MHCIIhi cells. Histograms depict YFP-Bcl-6 expression in the indicated DC subsets (green: CD103−CD11b+, blue: mDP, orange: mSP) and a wildtype control (gray). Data are representative of two independent experiments (n=3).
e) MFIs for YFP-Bcl-6 and YFP-Prdm1 expressed in the indicated DC subsets in the mLNs and SI LP. Mean of three mice per group with SEM. *p=0.05, ***p<0.0009, ****p<0.0001; unpaired t-test.
f) For pLNs and spleen, histograms show expression of YFP-Bcl-6 and YFP-Prdm1 within CD8α+ cDCs (purple) and CD11b+ cDCs (light blue). Representative of 2 technical replicates, n=3
In line with gene expression data, flow cytometry showed BCL-6 protein expression was higher in hSP DCs compared to hDP DCs (Fig. 6c). We also analyzed Bcl-6 expression using YFP-Bcl-6 reporter mice. In the small intestine LP, mSP DCs expressed YFP-Bcl-6, whereas the mDP DCs showed no Bcl-6-reporter expression. However, mLN-resident mDP DCs, as well as mSP DCs were both YFP-positive (Fig. 6d, e). Moreover, all DCs in the spleen and peripheral lymph nodes (pLN) expressed Bcl-6-YFP (Fig. 6f), albeit lower than SI LP mSP DCs or mLN mSP DCs. CD8α+ DCs expressed more Bcl-6-YFP than CD8α− (CD11b+) DCs, but this difference was more prominent in spleen than pLN DCs. Taken together, high Bcl-6 expression marked developmentally and functionally related mSP and CD8α+ DCs.
Using reporter mice expressing YFP under control of the Prdm1 promoter, we detected high expression of Prdm1-YFP in mDP DCs in intestinal lamina propria DCs, while mSP DCs were YFP-negative (Fig. 6d, e). The difference in expression was less pronounced in mLN DCs, where mLN mSP DCs expressed Prdm1-YFP, although less than mDP DCs. CD11b+CD103− DC in the mLN (which represent a DC population unrelated to LP CD11b+CD103− macrophages38) did not express Prdm1-YFP. We did not detect Prdm1-YFP expression in steady-state spleen or pLNs (Fig. 6f), consistent with the low constitutive Prdm1 expression in spleen DCs (Immgen.org). This suggests that Bcl-6 could contribute to DC specification across all organs tested, whereas Blimp-1 might have a selective role in the specification of the mDP intestinal DC subset at steady-state.
Blimp-1 and Bcl-6 have opposing roles in DC subset specification
To investigate the role of Bcl-6 in DC fate determination we assessed DC subsets in Bcl-6-deficient, heterozygous and wild-type littermate mice. Phenotypically healthy Bcl-6-deficient mice and wild-type mice had similar frequencies of plasmacytoid DCs versus total cDCs among CD11c+ MHC class II+ DCs within the mLNs (Fig. 7a) and other lymphoid tissues (data not shown), but Bcl-6-deficient mice displayed a specific loss of antigen cross-presenting DC subsets including the mSP DCs in mLNs, small intestinal LP and colon (Fig. 7b), and CD8α+ DCs in the spleen and pLN (Fig. 7c). To determine whether Bcl-6 acts cell-intrinsically to control DC development, heterozygous CD45.1/CD45.2 hosts were reconstituted with a mixture of CD45.2+ Bcl-6-deficient and CD45.1+ wild-type bone marrow (Supplementary Figure 6). In these chimeric mice, mSP cDCs were derived almost exclusively from wild-type bone marrow, whereas Bcl-6-deficient and wild-type progenitors contributed equally to mDP DCs (Fig. 7d, f). Similarly, CD8α+ cDCs in pLNs and spleen were derived primarily or exclusively from wild-type donors (Fig. 4e), whereas both Bcl-6-deficient and wild-type donors contributed to CD8α− lymphoid organ DCs. Taken together, this suggest that Bcl-6 has a critical and cell intrinsic role in the specification or survival of major classes of cross-presenting DCs.
Fig. 7. Bcl-6-deficient mice lack splenic CD8α+and intestinal mSP DCs.

a) Plots show live-gated lineage-negative (CD3, CD19, NK1.1) mLN cells. Relative frequency of cDC (CD11c+, B220neg) versus pDC (CD11c+, B220pos) in wild type, Bcl-6 heterozygous, and Bcl-6-deficient mice. Representative of 4 biological replicates.
b) Plots show the relative abundance of CD11bsp, CD103+CD11b+ (mDP), and CD103+CD11b− (mSP) DC in mLNs, SI LP, and colon (gated on live, CD3, CD19, and NK1.1 negative, CD11c+, MHCIIhi cells). Representative of at least ten biological replicates.
c) Relative abundance of subsets defined by CD8α and CD11b in wild type versus Bcl-6 knockout mice in pLN and spleen. Gated on live, CD3, CD19, and NK1.1 negative, CD11c positive, MHC class II high cells. Representative of at least ten biological replicates.
d) Subset composition of cDC derived from wild type versus Bcl-6 knockout donor BM in mixed bone marrow chimeras. Left column plots show wild type donor derived DC, right column plots show Bcl-6 knockout donor-derived DC. Representative of three replicates (n=3).
e) Wild type and Bcl-6 knockout BM-derived cDC populations in pLN and spleen.
f) SI LP DC subset contribution of wild type and Bcl-6 knockout derived donor cells to mDP and mSP DC. Cells were gated on live, lineage negative (CD3, CD19, NK1.1), CD11c+, and MHC class II+. Bars show the mean contribution in percent to the defined subsets (mDP, mSP), wild type-derived cells in white, Bcl-6-derived cells in black. **p=0.002, unpaired t-test.
To assess the role of Blimp-1 in intestinal DCs, we generated DC-specific Blimp-1-deficient mice by crossing CD11c-driven Cre-recombinase-expressing mice with mice carrying a floxed stop cassette in front of the prdm1 gene (called DC-BlimpKO from here on)39. DC-BlimpKO mice had a higher percentage of mSP DCs in the mLNs, with a corresponding decrease in mDP DCs (Fig. 8a). The relative loss of mDP DCs was more pronounced in the intestinal LP, while the percentages of mSP DCs among total CD11c+ cells in the LP were similar in DC-BlimpKO and littermate control mice. The ratio of mDP to mSP DCs was reproducibly decreased in DC-BlimpKO vs. WT mouse SI and mLN (Fig. 8c). Consistent with its pattern of expression, DC-specific Blimp-1 deficiency had no significant effect on the relative representation of DC subsets in spleen and pLNs (Fig. 6 and Fig. 8b). Similarly, Blimp-1 deficiency had no effect on DC subsets in the colonic LP, where mDP DCs are a minor population (data not shown). Thus, Blimp-1 and Bcl-6 reciprocally and cell-intrinsically control SP and DP DCs in the small intestine.
Fig. 8. Blimp-1 deficiency specifically impairs intestinal mDP frequencies.

a) Subset composition of gut-associated cDCs derived from Blimp-1fl/fl littermate controls (wt) versus Blimp-1fl/fl crossed to CD11c-cre mice (DCBlimp-1KO). Cells were gated on the live, lineage negative (CD3, B220, NK1.1), CD11chi, MHCIIhi population. Representative for two individual stainings of three pairs each and three individual stainings of three pairs each.
b) Subset composition of cDCs populations defined by CD8α and CD11b expression from the same mice as in c) in spleens and peripheral LNs.
c) Ratios of mDP to mSP DCs within CD11chiMHCIIhi, lineage negative (CD3, B220, NK1.1) live cells of mLNs and SI LP of DCBlimpKO (black bars) and littermate controls (grey bars); three mice each, mLN: **p = 0.005, SI LP: *p = 0.03
Discussion
This study defines three major cDC subsets in the human small intestine, relates them to mouse DC subsets to elucidate evolutionarily conserved and divergent phenotypic and functional properties, and identifies novel roles for the transcriptional regulators Bcl-6 and Blimp-1 in DC development and specialization. CD103+Sirpα− (hSP) DCs align with the human and mouse cross-presenting DC lineage, and share preferential expression of Bcl6 which we show contributes to differentiation of this lineage. CD103+Sirpα+ (hDP) DCs appear related to the human CD1c+ DC lineage, and display functional, phenotypic and transcriptomic similarities with mouse CD103+CD11b+ (mDP) DCs including expression of Blimp-1, which we show controls intestinal mDP specification. Finally, we identify CD103−Sirpα+ DCs as a distinct subset related to blood monocytes and with characteristics similar to those of mouse monocyte-derived CD103− CX3CR1int inflammatory DCs.
Both transcriptomic and phenotypic analyses confirm the close relation of hSP intestinal DCs to antigen cross-presenting DC lineages, including CD141+ DCs in man and CD8α+ lymphoid tissue and CD103+CD11b− (mSP) intestinal cDCs in the mouse. hSP express CADM1, XCR1 and CLEC9A, molecules with important roles in cross-priming CD8+ T cells: CLEC9A is involved in sensing and endocytosis of dead cells40, XCR1 mediates cross-talk with XCL1-secreting CD8+ T cells41 and CADM1 engages with CRTAM on CD8+ T cells42 and promotes IL-22 production43. hSP DCs also preferentially (compared to hDP DC) express transcripts for BATF3 and IRF8, genetic factors that drive development of the CD8α+ DC lineage in the mouse.
Comparative transcriptional analyses suggest a relationship between human gut DP DC and CD1c+ DC in human blood and skin. These DC populations also share expression of lineage-associated surface antigens including CD1c itself. However, in contrast with the clear correlation of gene expression between hSP DC and their blood CD141+ DC counterparts, the transcriptional alignment of hDP and CD1c+ DC was robust only when tissue-specific genes were eliminated from analysis. These observations suggest that, CD1c+ lineage DCs may be more sensitive than CD141+/hSP cross-presenting DCs to local environmental programming44.
hDP also align phenotypically and functionally with mouse DP intestinal DCs and, more distantly, with mouse lymphoid tissue CD4+ DC. hDP and mDP intestinal DC similarly induce TH17 and Treg, and both express CLEC4A, CD101, and TLR5. These conserved molecules regulate cytokine production and autoimmunity26,45, and likely mediate evolutionarily critical roles in intestinal immune homeostasis. Our comparative genomics analysis also revealed conserved mouse and human DP DC expression of transcriptional regulators IRF4, recently implicated in CD4+ DC and mDP development9, and Prdm1, shown here to selectively control intestinal mDP specification.
Our findings also identify CD103−Sirpα+ intestinal DCs. In two patients with hyperemic jejunal mucosa, there was an increase in this CD103−Sirpα+ DC population consistent with their recruitment or expansion in inflammation. Transcriptomic similarities between CD103−Sirpα+ DCs and human and mouse blood monocytes lead us to propose that they may develop from monocytes recruited in response to gut inflammation, similar to a recently described mouse CX3CR1int cDC34. CD103−Sirpα+ DCs and mouse CX3CR1int cDCs both induce efficient CD4+ T cell proliferation and promote T cell IFNγ production. Given the diversity in CX3CR1+ populations in the mouse, we cannot of course exclude heterogeneity within the CD103−Sirpa+ CX3CR1int or indeed the other populations studied here. Further investigation of CD103−Sirpα+ DCs in colitis and other intestinal inflammatory conditions is warranted. Developmental relationships of CD103−Sirpα+ DCs with CD14negCD11cneg anergic macrophages46 need further investigation.
Interestingly, our analyses of DC in gut associated lymphoid tissues suggest that both hSP and hDP subsets can access the draining mesenteric LN. hDP also display the highest amounts of CCR7 and express genes associated with enhanced cell motility: thus, as proposed for mouse mDP, they may be a principal migratory subset that can transport gut antigenic and environmental signals to lymph nodes. hSP DCs express less CCR7 than hDP DCs: this may suggest a difference in relative efficiency of exit, but chemokine receptors can be rapidly modulated. Significant if low expression of CCR7 by Sirpα+CD103− DCs suggests that they may also have the capacity to migrate to draining LN.
We show that Bcl-6 is more highly expressed in human and mouse SP DCs than in DP DCs, and in lymphoid tissue cross-presenting DCs as well; and that it plays an important role in development of these cross-presenting DC populations. Previous studies suggested an overall reduction in mature cDCs in the absence of Bcl-647, a finding we could not confirm in healthy Bcl-6 deficient mice. In contrast to Bcl-6, Prdm1 is most highly expressed by small intestinal DP DCs, and this expression correlates with the preferential effect of Blimp-1-deficiency on small intestinal DC development in our study. Surprisingly, expression is significantly higher in LP mDP DCs than in mDP DCs in the mLN, even though mLN mDP DCs are thought to represent recent migrants from the LP48. This raises the possibility that Prdm1 is actively and potentially reversibly regulated by the local environment in the small intestines, contributing to its predominant effect on SI mDP DCs. Prdm1 expression is even lower, and Blimp-1 reporter expression undetectable, among CD8α−CD11b+ DCs in spleen and CD103−CD11b+ DCs in the mLN, correlating with a lack of significant effect of Blimp-1 deficiency on these populations in our study. These results suggest a selective role for Blimp-1 in the specification and fate of mDP DCs within the small intestines. Indeed, even colon mDP DCs expressed little Blimp-1-YFP and were apparently unaffected by Blimp-1 deficiency. Blimp-1 has also been implicated in DC-intrinsic tolerance maintenance39, and it may therefore contribute to intestinal immune homeostasis by modulating DP DC responses as well as their numbers. Although not studied here, Prdm1 is also highly expressed in subsets of skin DCs and in CD11b+CD103− mononuclear phagocytes in the SI LP (data not shown): a role for Blimp-1 in these cells should be investigated. Our results suggest that Bcl-6 and Blimp-1 play key roles in an evolutionarily conserved transcription factor network that has evolved to control terminal fate decisions within each of the major lineages involved in the adaptive immune response.
In conclusion, we have characterized the dendritic cell network in the human small intestine, and have defined developmental parallels with the mouse DC system. The findings lay a framework for investigating the function of gut DC subsets in intestinal immunity and in inflammatory conditions such as food allergies, IBD, and gastritis.
Online Material and Methods
Preparation of LPMCs
LPMCs were isolated from resected jejunal/colon specimens using modification of previously described technique49. Briefly, the mucosa was dissected free from the muscularis in the tissue plane of sub-mucosa using fine scissors. The dissected mucosa was incubated in calcium and magnesium-free HBSS (Cellgro) containing 5% heat-inactivated fetal bovine serum and 1mM DTT (Sigma-Aldrich) to remove mucus. The epithelial layer was removed by incubating the mucosa three times for 15min in HBSS containing 5mM EDTA at 37°C with stirring. Tissue was collected and treated with Liberase TL (25μg/ml, Roche) containing HBSS for 30 minutes at 37°C. The remaining tissue was mashed and cells were collected by centrifugation (10min, 4°C, 400g). The mononuclear cells were subjected to centrifugation in a 70%/44%/20% percoll density gradient to separate lymphocytes from contaminating epithelial and stromal cells. All human tissue samples were de-identified and approved by IRB.
DC Enrichment
cDCs were enriched from LPMCs by negative selection with biotin conjugated mouse anti-human lineage marker mAbs against CD3, CD14, CD16, CD19, CD56 and glycophorin, followed by their removal using biotin negative selection cocktail (19153, Stemcell Technologies). Enriched DCs were labeled with antibodies and sorted using BD FACS Aria III (100μm nozzle, BD Biosciences). For microarray studies, the DCs were collected directly into RLT buffer (Qiagen).
Antibodies and Flow Cytometry
Anti-HLA-DR (L243), anti-CD103 (Bly7), anti-CD123 (6H6), anti-CD3 (OKT3), anti-CD14 (61D3), anti-CD16 (3G8), anti-CD83 (HB15e), anti-CD101 (BB27), anti-IRF4 (3E4), and anti-CD40 (5C3) were purchased from eBioscience. Anti-CLEC4A (I3-612), and anti-CD11b (ICFR44) were obtained from BD Biosciences. Anti-CD141 (M80), anti-CD172a (SE5A5), anti-CD11c (Bu15), anti-CD56 (MEM-188), anti-CLEC9A (8F9), anti-CX3CR1 (2A9-1), anti-CXCR3 (TG1) antibodies were purchased from Biolegend. Anti-CD207 (DCGM4) antibody was obtained from Immunotech. Anti-CCR7 (150503) antibody from R&D Systems. Anti-α4β7(Act-1) and anti-CCR9 (3E3) were produced in our lab from hybridomas. BCL-6 was stained with APC-conjugated αBCL-6 (clone K112-91) from BD Biosciences using the Foxp3 Staining Buffer Set from eBioscience. The cells were stained in with combinations of above listed fluorochrome-conjugated antibodies. Lamina propria leukocytes were incubated at 37° C for two hours prior to staining when indicated to allow cells to reexpress internalized chemokine receptors: this procedure was important for detection of CCR9. Staining was analyzed using BD LSRFortessa and FlowJo software (Tree Star Inc). ALDH activity was determined using the ALDEFLOUR staining kit (STEMCELL Technologies Inc.) according to the manufacturer's protocol as modified50.
Microscopy
Appendix and jejunum samples were placed in Tissue-Tek (Sakura) and frozen at −80°C. Cryosections of acetone-fixed tissue were incubated sequentially with rabbit anti-human CD11c (Epitomics), Dylight 549 donkey-anti-rabbit IgG (Jackson Immunoresearch), mouse-anti-human Sirpα (SE5A5), Dylight 488 donkey-anti-mouse IgG (Jackson Immunoresearch), mouse serum, and Alexa 647 conjugated mouse-anti-human CD103 (eBioscience). Staining was imaged on an LSM 710 confocal microscope (Carl Zeiss).
In vitro Treg and CD4+ T helper differentiation
Naïve CD4+ T cells (5×104 cells/well) were cultured with small intestine DC (1 × 104 cells/well) subsets in Yssel's medium with 2% human serum. After 9 days, cultured cells were harvested and analyzed for intracellular Foxp3 expression by flow cytometry. Cocultured cells were re-stimulated for 6 hours with PMA (50ng/ml; Sigma) and ionomycin (500ng/ml; Sigma) in the presence of GolgiStop (BD Pharmingen). CD4+ T cells producing IFNγ and IL-17 were analyzed by flow cytometry.
Statistical analysis
The statistical significance of differences between the two sets of data was assessed by Student's t-test unless stated otherwise. Analytic methods for significance of differential gene expression are indicated in the text. Significance of clusters was determined by modified bootstrap resampling51.
Microarray Analysis
All intestinal DC samples generated in this study are deposited in GEO, accession code GSE50380. RNA was isolated from the sorted DC subsets using Qiagen RNeasy Plus Micro kit. 5-20ng of total RNA from each sample was used for amplification, labeling, and hybridization by Expression Analysis, Inc (Durham, NC). Hybridization was performed on Affymetrix Genechip® Human Gene 1.0 ST Array. GeneSpring GX 11.5 and Partek Genomic Suite (6.6) were used for processing and analyzing the data. Genespring preprocessing and default normalization (RMA-16) was applied to each dataset [i.e., separately for Illumina data from Haniffa et al12 (NCBI GSE35458); for our human Affymetrix Human Gene 1.0 ST Array data (to be deposited); and for mouse Affymetrix data from Immgen.org (NCBI GSE15907)] for the populations included in the specific analyses. Translation of mouse gene lists to their human homologs was performed in Genespring. For analyses involving data from different platforms, normalized gene expression values were exported from Genespring and combined for analysis in Partek after removal of gene duplicates in Excel.
For clustering and correlation analyses, genes that discriminate the studied DC or monocytes subsets within each tissue type were selected. For human subset comparisons, genes with the 15% most variable expression between subsets isolated from blood or skin, and between the gut DC subsets, with expression above threshold values (>120 for Affymetrix data; >160 for Illumina data) by at least one subset in each tissue, and differing significantly in expression between the three gut DC types (p<0.01 ANOVA) were used. The 535 genes so selected are listed in the Supplementary Gene list document as Gene List for Fig. 3a and Supplementary Figure 4.
To assess the relationship of gut DC to blood DC and monocytes independently of tissue-specific genes, blood vs skin-specific gene-depleted CD1c+ DC-, CD141+ DC-, and CD14+ monocyte- lineage gene sets from Haniffa12 were filtered as follows to remove gut-specific genes: Samples isolated from each tissue type (skin, blood or gut) were pooled, and genes that discriminated gut, blood or skin pools (P<0.05 ANOVA) were excluded. Samples included for these analyses were those presented in the cluster dendrograms. Remaining genes were filtered for differential expression by the gut DC subsets (P<0.05), yielding common (tissue-specific-gene depleted) lineage-associated gene sets that are presented as lineage associated genes for Fig. 3b in Supplementary Gene lists for Figure 3. CD1c+ DC lineage-associated genes were used to compare CD1c+ blood DC with gut DC subsets; and the CD141+ DC- and CD14+ monocyte- lineage gene sets to compare our samples with CD141+ DC and CD14+ monocytes, respectively (Fig. 3b).
For display of the correlation of genes discriminating blood CD1c+ vs CD141+ DC with those discriminating human gut CD103+ DC subsets (Fig. 3d) genes whose mean expression differed at least two fold between gut CD103+Sirpα+ vs CD103+Sirpα− DC, and also between CD1c+ vs CD141+ blood DC were selected (246 genes, Fig. 3d, Supplemental Gene List for Fig. 3).
For mouse/human comparisons, human genes or their mouse homologs differentially expressed (top 15% most diverse) a) among the three human gut DC subsets, b) among mouse gut CD103+CD11b−, CD103+CD11b+ DCs and CD11b+ macrophages, and c) among mouse spleen CD4+ and CD8α+ DCs and red pulp macrophages were selected. Genes common to the three gene sets above were further selected for threshold expression (>120 by at least one mouse and one of the human DC subsets clustered) and for significance in comparisons of human gut DC subsets (p<0.01, Benjamini-Hochberg corrected), and were then used to cluster human gut DC (replicate samples) with mean expression profiles of human and mouse DC and monocyte/macrophage cell types (Fig. 5a). The 455 genes so selected are listed in Supplementary Gene List for Fig. 5a. Genes that differed significantly (p<0.01, ANOVA) and by at least 4 fold between the three DC or DC/monocyte clusters defined in Fig. 5a were used for the heatmap in Fig. 5b.
For generation of gene-expression datasets comparing human and mouse intestinal DP versus SP DCs (Fig. 5c and transcription factor expression, Fig. 6a), mouse gene expression data from the Immgen Consortium38 were obtained from the NCBI GEO website (GSE15907). Gene lists were created in Genespring as follows: Genes were filtered by expression value (at least one replicate in either subset had to show a raw value above 120), followed by statistical analysis to exclude samples with excessive variability (unpaired, two-tailed t-test between both DC subsets >0.05, uncorrected). We then filtered on probes that were at least 2-fold differentially expressed between the mSP and mDP DC subsets. The same filtering criteria were applied to our human datasets to identify genes differing at least 2 fold between hDP and hSP DCs. The mouse gene list was then translated into the human experiment in Genespring. The intersection of both lists was used for the ratio-depicting scatter plot, generated in Partek Genomic Suite. For transcription factor identification, we used GO Term 0003700: sequence-specific DNA binding transcription factor activity.
Mice
Bcl-6-deficient (Bcl-6-KO) mice were provided by A. Dent (Indiana University) and maintained and bred in specific pathogen-free conditions in the animal facility of Veterans Affairs Palo Alto Health Care Systems (VAPAHCS). YFP-Bcl-6 mice52 were provided by Dr. T. Okada (RIKEN, Japan), and Prdm-1-YFP mice53 were provided by Eric Meffre (Yale University). Both strains were bred and maintained at UCSF. DCBlimpKO mice and control mice were bred in the animal facility of The Feinstein Institution for Medical Research (FIMR) under specific pathogen-free conditions and repeats were performed with mice bred at VAPAHCS. All animal work was approved by the IACUC committee at the VAPAHCS, or by relevant animal care committees at the collaborating institutions.
Mouse lymphocyte isolation
Spleens, peripheral (inguinal, axillary and brachial), and mesenteric LNs were isolated and digested with RPMI media containing 5% FCS, 0.5 mg/ml of collagenase IV (Sigma Aldrich) and 1 unit/ml of DNaseI (Sigma) for 30 minutes at 37°C and made into single cell suspension. For the isolation of lamina propria residing dendritic cells from small intestine and colon, full-length SI (with Peyer's Patches removed) and colon were cut open longitudinally and rinsed twice in HBSS (without Ca2+ and Mg2+) with 2% FCS and cut into small pieces. After 1mM EDTA treatment for 40 minutes total to release the epithelium, tissues were digested in 10 ml RPMI containing 5% FCS and 0.5 μg/ml of collagenase IV (Sigma C2139) at 37°C for 15 minutes, 3 times. Supernatant containing LP cells was collected. Mononucleated cells from both colon and SI LP supernatant were isolated by gradient separation with 40% and 70% Percoll solutions (GE, 17-0891-01) at 2500 rpm for 15 minutes at room temperature. Cells of interest were located at the interface.
Flow-cytometry on mouse cells and analysis
Cells were first blocked with FACS buffer (PBS with 0.5% BSA) containing 1μg/ml αCD16/CD32 (clone 93, eBioscience). The following antibodies were purchased from either eBiosciene, Biolegend or BD Biosciences and used for staining: αCD3-PECy7/AF780 (145-2C11), αCD19-PECy7/AF780 (ID3), αNK1.1-PECy7/AF780 (PK136), αB220-PECy7/AF700/AF780 (RA3-6B2), αMHCII-AF700/AF780 (M5/114.15.2), αCD11c-PB/PE-Cy7 (N418), αCD103-PE/Fitc/APC (M290), αCD11b-PerCP-Cy5.5 (M1/70), αCD8α-APC/V500 (53-6.7), αCD45.1-APC/A780 (A20), αCD45.2-FITC (RA3-6B2), αCLEC4A-PE/APC (33D1), αCD101-PE (Moushi101), αTLR3-APC (11F8). Dead cells were excluded by propidium iodide staining (Sigma). Flow cytometry data was acquired on either a LSRII or a Fortessa (BD), using Diva software (BD). Further analysis was performed using FlowJo from Treestar. Statistics of all flow cytometry-based data were calculated with the unpaired Student's t-test using GraphPad Prism.
Generation of mixed bone marrow chimeras
Donor bone marrow cells were collected from femurs and tibias and bone marrow was flushed out using a 27G needle. After red blood cell lysis, cells were counted and 5 × 106 cells total were injected into lethally irradiated recipients (1000 Rad). To ensure equal engraftment by different donor cells, following groups were set up: Bcl-6 knockout-derived on CD45.2 allogeneic background with wild type-derived donor cells on a CD45.1 background as experimental group and wild type-derived donor cells on both backgrounds as a control group. For the latter, we used littermates from Bcl-6 knockout mice as CD45.2 wild type mice to ensure for proper engraftment by this strain. At least six different mixed bone marrow groups were set up with successful engraftment from both donor cell populations in all groups tested.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank J. Sweere and Dr. H. Kiefel for critical reading of the manuscript, L. Rott for assistance with flow cytometry and cell sorting, E. Resurreccion for assistance with immunohistochemistry, Dr. K. Bhat for writing python scripts, all Butcher lab members for fruitful discussions, and the Immgen consortium and the Merad lab (Mount Sinai, School of Medicine) for generating the publicly available microarray data from sorted mouse intestinal DC. Supported in part by NIH grants R01 AI093981, R01 DK084647, R37 AI047822 and an award from the Department of Veterans Affairs to ECB and by the FACS Core Facility under the Stanford Digestive Diseases grant P30 DK056339. KL was supported by fellowships from the German Research Foundation (DFG), the Crohn's and Colitis Foundation of America, and the ITI Young Investigator Award from Stanford. M. Lee was supported by NIH 5T32AI007290-25 Postdoctoral Training in Molecular and Cellular Immunobiology and is a recipient of Research Fellows Award from Crohn's & Colitis Foundation of America. RZ was supported by the Agency for Science, Technology And Research, Singapore. D.B. was supported by the Swiss National Science Foundation (PBBEP3-133516) and the Swiss Foundation for Grants in Biology and Medicine (PASMP3-142725). S.J.K. was supported by NIH NIAMS K01 AR59378. K.M.A. is a Leukemia & Lymphoma Society Scholar. H.H. was supported by a grant from the California Institute for Regenerative Medicine (CIRM) and by an Investigator Career Award from the Arthritis Foundation and was a fellow under NIH Training Grant AI07290.
Footnotes
Author contributions: PW designed and performed human DC experiments including microarrays, performed gene expression analysis, and wrote the manuscript; KL designed and performed all mouse experiments, carried out gene expression analysis and wrote the manuscript; ML performed RNA preparation and quality control for microarray studies; JM provided human tissue; SJK designed and performed DCBlimpKO experiments; DB provided YFP-Bcl-6 and Blimp-1-YFP mice and helped to design and to perform the experiments; RZ helped to perform YFP-Bcl-6 experiments; KMA provided feedback and YFP-Bcl-6 and Prdm-1-YFP mice; AD provided Bcl-6-/- mice and feedback; BD provided DCBlimpKO mice; HH drove initial interest and provided advice; and ECB performed gene expression analyses, guided the study and wrote the manuscript.
Database Accession Numbers: All human intestinal microarray data is deposited at GEO and has the accession code GSE50380. Other human datasets were derived from Haniffa et al12 (NCBI GSE35458). Mouse gene expression data were obtained from the NCBI GEO website (GSE15907) and originate from the Immgen Consortium38
References
- 1.Varol C, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31:502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 2.Bogunovic M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller JC, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol. 2012;13:888–899. doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helft J, Ginhoux F, Bogunovic M, Merad M. Origin and functional heterogeneity of non-lymphoid tissue dendritic cells in mice. Immunol Rev. 2010;234:55–75. doi: 10.1111/j.0105-2896.2009.00885.x. [DOI] [PubMed] [Google Scholar]
- 5.Uematsu S, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol. 2008;9:769–776. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 6.Kinnebrew MA, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunol Rev. 2011;241:206–227. doi: 10.1111/j.1600-065X.2011.01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Persson EK, et al. IRF4 Transcription-Factor-Dependent CD103CD11b Dendritic Cells Drive Mucosal T Helper 17 Cell Differentiation. Immunity. 2013;38:958–969. doi: 10.1016/j.immuni.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Lewis KL, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity. 2011;35:780–791. doi: 10.1016/j.immuni.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poulin LF, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood. 2012;119:6052–6062. doi: 10.1182/blood-2012-01-406967. [DOI] [PubMed] [Google Scholar]
- 12.Haniffa M, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. 2012;37:60–73. doi: 10.1016/j.immuni.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villadangos JA, Shortman K. Found in translation: the human equivalent of mouse CD8+ dendritic cells. J Exp Med. 2010;207:1131–1134. doi: 10.1084/jem.20100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlitzer A, et al. IRF4 Transcription Factor-Dependent CD11b Dendritic Cells in Human and Mouse Control Mucosal IL-17 Cytokine Responses. Immunity. 2013;38:970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaensson E, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med. 2008;205:2139–2149. doi: 10.1084/jem.20080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mittag D, et al. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J Immunol. 2011;186:6207–6217. doi: 10.4049/jimmunol.1002632. [DOI] [PubMed] [Google Scholar]
- 17.Tamoutounour S, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol. 2012;42:3150–3166. doi: 10.1002/eji.201242847. [DOI] [PubMed] [Google Scholar]
- 18.Denning TL, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol. 2011;187:733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumgart DC, et al. Patients with active inflammatory bowel disease lack immature peripheral blood plasmacytoid and myeloid dendritic cells. Gut. 2005;54:228–236. doi: 10.1136/gut.2004.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hadeiba H, et al. CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nat Immunol. 2008;9:1253–1260. doi: 10.1038/ni.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerovic V, et al. Intestinal CD103(-) dendritic cells migrate in lymph and prime effector T cells. Mucosal Immunol. 2013;6:104–113. doi: 10.1038/mi.2012.53. [DOI] [PubMed] [Google Scholar]
- 22.Ito T, Carson WFt, Cavassani KA, Connett JM, Kunkel SL. CCR6 as a mediator of immunity in the lung and gut. Exp Cell Res. 2011;317:613–619. doi: 10.1016/j.yexcr.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang MH, et al. CCR7 Is Critically Important for Migration of Dendritic Cells in Intestinal Lamina Propria to Mesenteric Lymph Nodes. The Journal of Immunology. 2006;176:803–810. doi: 10.4049/jimmunol.176.2.803. [DOI] [PubMed] [Google Scholar]
- 24.Takeuchi A, et al. CRTAM confers late-stage activation of CD8+ T cells to regulate retention within lymph node. J Immunol. 2009;183:4220–4228. doi: 10.4049/jimmunol.0901248. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. 2012;12:813–820. doi: 10.1038/nri3339. [DOI] [PubMed] [Google Scholar]
- 26.Bouloc A, Bagot M, Delaire S, Bensussan A, Boumsell L. Triggering CD101 molecule on human cutaneous dendritic cells inhibits T cell proliferation via IL-10 production. Eur J Immunol. 2000;30:3132–3139. doi: 10.1002/1521-4141(200011)30:11<3132::AID-IMMU3132>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 27.Huber S, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 29.Bakri Y, et al. Balance of MafB and PU.1 specifies alternative macrophage or dendritic cell fate. Blood. 2005;105:2707–2716. doi: 10.1182/blood-2004-04-1448. [DOI] [PubMed] [Google Scholar]
- 30.Liu W, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mora JR. Homing imprinting and immunomodulation in the gut: role of dendritic cells and retinoids. Inflamm Bowel Dis. 2008;14:275–289. doi: 10.1002/ibd.20280. [DOI] [PubMed] [Google Scholar]
- 32.Ivanov II, et al. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atarashi K, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 34.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 36.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–120. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller JC, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol. 2012 doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SJ, Zou YR, Goldstein J, Reizis B, Diamond B. Tolerogenic function of Blimp-1 in dendritic cells. J Exp Med. 2011;208:2193–2199. doi: 10.1084/jem.20110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sancho D, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dorner BG, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity. 2009;31:823–833. doi: 10.1016/j.immuni.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 42.Galibert L, et al. Nectin-like protein 2 defines a subset of T-cell zone dendritic cells and is a ligand for class-I-restricted T-cell-associated molecule. J Biol Chem. 2005;280:21955–21964. doi: 10.1074/jbc.M502095200. [DOI] [PubMed] [Google Scholar]
- 43.Yeh JH, Sidhu SS, Chan AC. Regulation of a late phase of T cell polarity and effector functions by Crtam. Cell. 2008;132:846–859. doi: 10.1016/j.cell.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 44.Sigmundsdottir H, Butcher EC. Environmental cues, dendritic cells and the programming of tissue-selective lymphocyte trafficking. Nat Immunol. 2008;9:981–987. doi: 10.1038/ni.f.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujikado N, et al. Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nat Med. 2008;14:176–180. doi: 10.1038/nm1697. [DOI] [PubMed] [Google Scholar]
- 46.Smythies LE, et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. 2005;115:66–75. doi: 10.1172/JCI19229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohtsuka H, et al. Bcl6 is required for the development of mouse CD4+ and CD8alpha+ dendritic cells. J Immunol. 2011;186:255–263. doi: 10.4049/jimmunol.0903714. [DOI] [PubMed] [Google Scholar]
- 48.Milling S, Yrlid U, Cerovic V, MacPherson G. Subsets of migrating intestinal dendritic cells. Immunol Rev. 2010;234:259–267. doi: 10.1111/j.0105-2896.2009.00866.x. [DOI] [PubMed] [Google Scholar]
- 49.Bull DM, Bookman MA. Isolation and functional characterization of human intestinal mucosal lymphoid cells. J Clin Invest. 1977;59:966–974. doi: 10.1172/JCI108719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeng R, et al. Retinoic acid regulates the development of a gut-homing precursor for intestinal dendritic cells. Mucosal Immunol. 2013;6:847–856. doi: 10.1038/mi.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 52.Kitano M, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. 2011;34:961–972. doi: 10.1016/j.immuni.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 53.Rutishauser RL, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.