

Abstract
A sample preparation method for protein C-terminal peptide isolation from cyanogen bromide (CNBr) digests has been developed. In this strategy, the analyte was reduced and carboxyamidomethylated, followed by CNBr cleavage in a one-pot reaction scheme. The digest was then adsorbed on ZipTipC18 pipette tips for conjugation of the homoserine lactone-terminated peptides with 2,2′-dithiobis (ethylamine) dihydrochloride, followed by reductive release of 2-aminoethanethiol from the derivatives. The thiol-functionalized internal and N-terminal peptides were scavenged on activated thiol sepharose, leaving the C-terminal peptide in the flow-through fraction. The use of reversed-phase supports as a venue for peptide derivatization enabled facile optimization of the individual reaction steps for throughput and completeness of reaction. Reagents were replaced directly on the support, allowing the reactions to proceed at minimal sample loss. By this sequence of solid-phase reactions, the C-terminal peptide could be recognized uniquely in mass spectra of unfractionated digests by its unaltered mass signature. The use of the sample preparation method was demonstrated with low-level amounts of a whole, intact model protein. The C-terminal fragments were retrieved selectively and efficiently from the affinity support. The use of covalent chromatography for C-terminal peptide purification enabled recovery of the depleted material for further chemical and/or enzymatic manipulation. The sample preparation method provides for robustness and simplicity of operation and is anticipated to be expanded to gel-separated proteins and in a scaled-up format to high-throughput protein profiling in complex biological mixtures.
Keywords: peptide, ZipTipC18 pipette tips, serial solid-phase derivatization, aminolysis, covalent chromatography, C-terminomics
INTRODUCTION
Positional proteomics technologies, aimed at the isolation of terminal peptides from protein digests, have evolved as valuable tools to characterize protein terminal regions implicated in diverse cellular functions, such as protein degradation, complex formation, and membrane interaction.1 The subsequent structural characterization of the isolates by tandem mass spectrometry (tandem MS) is greatly facilitated by the high sequence-information content at the protein termini, resulting in an increased success rate of protein identification.2
Method developments in this area, also called N/C-terminomics, have focused primarily on the protein N-terminal domain.3 Only a few C-terminal analytical techniques are currently available, including carboxypeptidase ladder sequencing and trypsin-catalyzed 18O-coding.4,5 The efficacy of the enzymatic approach proved dependent on the substrate's amino acid sequence. In the latter strategy, the C-terminal peptide remains unaltered and thus, can be identified by its isotopomer signature. However, the selectivity of 18O-coding is abolished if the C-terminus is occupied by lysine or arginine. An alternative approach proposed to facilitate the mass spectral recognition of the C-terminal cyanogen bromide (CNBr) fragment, carboxylate methyl esterification in methanolic hydrochloric acid (HCl), gave rise to partial deamidation of asparagine and glutamine, as well as oxidative side-reaction complicating data interpretation.6
Methods advocated in the literature for C-terminal peptide isolation from protein digests by negative selection include anhydrotrypsin-based peptide selection and several chemical approaches.7–9. Anhydrotrypsin-mediated C-terminal peptide isolation from tryptic digests relies on affinity depletion of arginine and lysine-terminated internal peptides.7 However, proteins bearing these amino acid residues at their C-termini proved impervious to this approach. Chemical reaction schemes have been proposed that remove internal and N-terminal peptides from lysyl endopeptidase digests on p-phenylenediisothiocyanate (DITC) glass following α-amino group labeling with succinimidyloxycarboxylmethyl tris-(2,4,6-trimethylphenyl) phosphonium bromide (TMPP-Ac-OSu) or alternatively, after chemoselective α-amino group transamination.8,9 However, selective α-amino group labeling by TMPP-Ac-OSu could not be generally demonstrated, and several amino acid residues proved resistant to transamination or ionized poorly in MALDI-TOF MS. Additionally, C-terminal peptides harboring lysine were depleted by the DITC support.
We have recently addressed these shortcomings by the development of a reaction scheme in which protein carboxylates were protected by glycinamidation followed by trypsination.10 The digests was then adsorbed onto C18 reversed-phase supports used as a venue for sequential peptide α-amine acetylation and N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC)-mediated carboxylate condensation with ethylenediamine (EDA) dihydrochloride. The amino group-functionalized N-terminal and internal peptides were then depleted on N-hydroxysuccinimide (NHS)-activated Sepharose, leaving the C-terminal peptide free in solution. This solid-phase sequential reaction format has been exploited recently in conjunction with phosphopeptide derivatization.11
In the conceptually related strategy reported here, whole, intact protein was cleaved by reaction with CNBr. This reagent, in general, hydrolyzes amide bonds C-terminal to methionine residues with exceptionally high efficiency (>80%). In contrast to protein trypsination, this fragmentation method has proved especially useful to prepare hydrophobic proteins for structural characterization by MS.12 The digest was then adsorbed on a C18 reversed-phase support, used as a reaction bed for conjugation of the newly exposed (neo) homoserine lactone-terminated CNBr fragments with 2.2′-dithiobis (ethylamine) dihydrochloride (cystamine dihydrochloride). After reductive release of 2-aminoethanethiol (cysteamine) from the conjugates, the thiol-functionalized internal peptides, along with the N-terminal peptide, were reversibly coupled to activated thiol sepharose, from which the nonbound C-terminal peptide was collected in the flow-through fraction. The intrinsic reactivity of the lactone electrophile toward primary amine- containing compounds has been exploited earlier to attach CNBr fragments in dimethylformamide (DMF) solutions to amino group-functionalized glass for subsequent solid-phase chemical sequencing.13 Only recently, a proteomics application of the chemistry has been reported in which tetrahistidine EDA and biotin EDA were coupled to disulfide-linked CNBr fragments under nonaqueous conditions.14 Cross-linked peptide pairs were then purified from the digests by immobilized metal ion affinity chromatography and on streptavidin agarose.
The chemical incompatibility of the C18 reversed-phase resin with aprotic solvent systems prompted us to optimize the conjugation chemistry under purely aqueous reaction conditions. As shown in the study, conjugation of the homoserine lactone-terminated fragments with the affinity tag proceeded in aqueous reaction media rapidly and at high efficiency. The use of the sample preparation method for selective sampling of C-terminal fragments from CNBr digests was demonstrated with well-characterized model proteins. To our knowledge, this is the first demonstration of a method for selective isolation of C-terminal CNBr from digests derived from whole, intact protein.
We anticipate to exploit the substantial reduction in sample complexity afforded by the chemical approach in future scaled-up applications to sample C-terminal CNBr fragments from digests prepared from subfractions of whole proteomes.
MATERIALS AND METHODS
TFA, EDC, N-hydroxysulfosuccinimide (sulfo-NHS), sulfo-NHS ester of acetic acid (sulfo-NHS acetate), 2-(N-morpholino) ethanesulfonic acid (MES; buffered saline), Bond-Breaker tris (2-carboxyethyl)phosphine (TCEP) solution (0.5 M), Spin Columns-Screw Caps (0.8 mL internal volume), and GelCode Blue Stain Reagent were obtained from Pierce (Rockford, IL, USA). N-Octyl glucoside (OGS) was obtained from Roche Diagnostics (Indianapolis, IN, USA). Ammonium hydrogen carbonate, sodium phosphate dibasic dodecahydrate, and CNBr were purchased from Fluka (Ronkonkoma, NJ, USA). Methanol and acetonitrile were from Burdick & Jackson (Muskegon, MI, USA). 2.2′-Dithiobis (ethylamine) dihydrochloride (cystamine dihydrochloride), EDA dihydrochloride, HCl (11.6 N), iodoacetamide, sodium phosphate monobasic monohydrate, EDTA, acetyl chloride (99%), anhydrous methanol, anhydrous deuterated methanol (CD3OD), aniline-12C6 hydrochloride, aniline-13C6 hydrochloride, 1 M Tris-HCl/0.1 M EDTA buffer (pH 8.0), sodium carbonate, sodium bicarbonate, lysozyme C from chicken egg-white, bovine β-casein, ribonuclease A from bovine pancreas (RNase A), rat serum albumin (RSA), equine alcohol dehydrogenase, bovine β-lactoglobulin, invertase 1 from Saccharomyces cerevisiae, myoglobin from equine heart, bovine carbonic anhydrase, cytochrome c from equine heart, BSA, human angiotensin I acetate salt hydrate [DRVYIHPFHL, mass/charge ratio (m/z) 1296.5], human [Glu1] fibrinopeptide B (EGVNDNEEFFAR, m/z 1570.5) and the somatostatin fragment 3–10 (CKNFFWKT, m/z 1073.2), and bradykinin human acetate salt hydrate (RPGFSPFR, m/z 963.2) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Activated thiol sepharose 4B and urea (ultra-pure grade) were from GE Healthcare (Piscataway, NJ, USA). The α-cyano-4-hyroxycinnamic acid (α-CHCA) preparation was from Agilent Technologies (Palo Alto, CA, USA). Polyacrylamide gels (Criterion Precast Gel, 1 mm, 10%) were from Bio-Rad (Hercules, CA, USA). ZipTipC18 pipette tips (0.6 μL bed volume) and ZipTipμ-C18 pipette tips (0.2 μL bed volume) were purchased from Millipore (Billerica, MA, USA). Microcentrifuge tubes (Protein LoBind/0.5, 1, and 2 mL) were from Eppendorf North America (Hauppauge, NY, USA).
One-Pot Protein Carboxyamidomethylation and CNBr Cleavage
Model protein (25–200 pmoles), placed into 0.5 mL Protein LoBind microfuge tubes was reduced in 40 μL aqueous 10 mM sodium phosphate /4 M urea solution/0.01% OGS (pH 8.0), supplemented with 2.5 mM TCEP. After 20 min incubation at 55°C, 10 μL 150 mM iodoacetamide solution, prepared in 10 mM sodium phosphate/4 M urea/0.01% OGS, was added to a final concentration of 30 mM, and the incubation continued for 30 min at 37°C in the dark. The CNBr stock solution was prepared by dissolving 10 mg reagent in 0.5 mL 0.5 N aqueous HCl, of which 10 μL was added to the derivatized samples. The cleavage reaction was allowed to proceed overnight for 16 h at ambient temperature with agitation and halted by immobilizing the digests on ZipTipC18 pipette tips, according to the procedure described below. Excess of reagents was removed by passing 100 μL 0.1% TFA and 50 μL water in 10 μL aliquots over the resin. The digests were then processed for MALDI-MS or solid-phase peptide derivatization as described below.
Sample Immobilization
ZipTipC18 pipette tips/ZipTipμ-C18 pipette tips were wetted six times with 10 μL methanol, followed by six washes with 0.1% TFA. Model peptides (10–50 pmoles), prepared in 0.2–0.5% TFA/0.01% OGS solutions, were placed in 10 μL aliquots into 0.5 mL Protein LoBind microfuge tubes and subjected to 10 sample aspiration/dispense cycles. In each cycle, the sample is withdrawn and dispensed once. The immobilized peptides were then washed three times with 10 μL 0.1% TFA. For higher volume sample enrichment (typically 40–60 μl), the CNBr digest solutions and fractions collected from the affinity support were aspirated sequentially in 10 μL aliquots onto ZipTipC18 pipette tips and dispensed into a Protein LoBind 0.5 ml microfuge tube. The partially depleted sample was then transferred back in this step-wise mode to the original microfuge tube. This alternating aspiration /dispense enrichment cycle was repeated five times to maximized peptide binding. The ZipTip pipette tips were then washed 10 times with 10 μL 0.1% TFA. For even more dilute samples, ZipTips were press-fittted onto 200 μL disposable pipette tips and operated with a 200-μL pipettor, as suggested by the manufacturer using 10 load/dispense cycles. Up to 200 μL dilute CNBr digests or test peptide solutions can be processed for binding in this manner (www.millipore.com/publications). To avoid resin de-wetting, the ZipTips tips were processed immediately for on-column derivatization as described below. Controls were loaded with 10 μL 0.1% TFA from 60 μL that had been placed into a 0.5-mL microfuge tube. The ZipTips were left immersed in solvent before elution. In this manner, samples may be stored in 0.1% TFA at −20°C for several days prior to use.
General Procedure for Solid-Phase Peptide Derivatization
Reagents (10 μL) were aspirated three times onto the ZipTipC18 pipette tips or onto ZipTipμ-C18 pipette tips and dispensed to waste. Reagents (10 μL) were then loaded onto the tips from 60 μL that had been placed into 0.5 ml microfuge tubes. During incubation, the tips were left immersed in the reagents. Unless stated otherwise, the samples were desalted by passing 100 μL 0.1% TFA over the resin in 10 μL aliquots. The products were typically eluted from ZipTipC18 pipette tips in 5–10 μL 50% acetonitrile/0.1% TFA/0.01% OGS, of which, 1 μL was used for MALDI-MS analysis. ZipTipμ-C18 pipette tips were eluted in matrix containing 0.1% TFA/0.01% OGS onto the MALDI-TOF plate.
Solid-Phase Homoserine Lactone Conjugation with 2.2′-Dithiobis (Ethylamine) Dihydrochloride (Cystamine Dihydrochloride)
Cystamine dihydrochloride (112.6 mg) was dissolved in 1 mL aqueous 1 M sodium carbonate/2 M urea/0.005% OGS to give a final concentration of 0.5 M (Reagent A; pH 9.5). Additional cystamine dihydrochloride solutions, evaluated during chemistry optimization, were prepared as follows: cystamine dihydrochloride (112.6 mg) was dissolved in 1 mL aqueous 0.0625 M sodium carbonate/2 M urea/0.005% OGS (Reagent B; pH 7.5) or in 1 mL aqueous 0.250 M sodium carbonate/2 M urea/0.005% OGS (Reagent C; pH 8.5). Reagents were prepared fresh for daily use. The CNBr digests were bound to ZipTipC18 pipette tips, as described above. The ZipTips were then flushed three times with 10 μL of the reagents. Reagents (10 μL) were loaded onto the reversed-phase supports, which were then incubated for 30 min at 55°C while immersed in the reagents. After incubation, the ZipTips were washed with 200 μL water, subsequently with 50 μL 0.1% TFA, and finally, with 30 μL water, passed in 10 μL aliquots over the resin. The ZipTips were processed for MALDI-TOF as described above or dried for 5 min in a SpeedVac prior to exposure to gaseous TFA.
Gas-Phase Peptide Lactonization with Consecutive Homoserine Lactone Conjugation
A 2-mL Protein LoBind Eppendorf tube (40 mm×9 mm inner diameter) microfuge tube was used as a reaction vessel. Cores of 10 mm in diameter of Whatman GB005 blotting paper (1.5 mm thick) were used to hold and position the ZipTipC18 pipette tips during incubation (Fig. 1A, inset). The center of these cores was punched out (3 mm) to accommodate the dried ZipTip by insertion to a distance of 10 mm, measured from the top of the tip. Neat TFA (20 μl) was placed into the reactor. The ZipTip/disk assembly was then inserted into the reactor to position the resin bed ∼4 mm above the liquid level. During this operation, care must be taken to avoid resin wetting, which would result in silica leakage from the reversed-phase support. The test tube was capped and incubated in an oven for 30 min at 37°C. Following incubation, the ZipTips were dried for 5 min in a SpeedVac and then flushed three times to waste with 10 μL of the conjugation reagent. The reagent (10 μL) was then aspirated on the support, and the reaction was allowed to proceed for 30 min at 55°C. The ZipTips were desalted with 200 μL water, 50 μL 0.1% TFA, and finally, with 30 μL water, sequentially passed in 10 μL aliquots over the resin. Samples were processed for MALDI-MS, as described above, or incubated under reducing conditions to unmask the thiol groups in the conjugates (see below).
Figure 1.
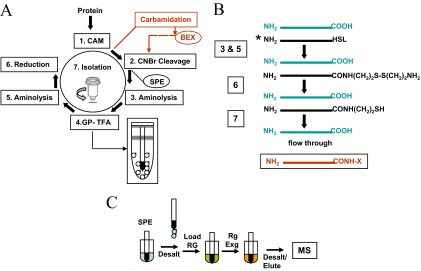
(A) Flow chart of sample preparation for C-terminal CNBr peptide isolation. The reaction steps are numbered and highlighted with boxes. Whole, intact protein is reduced and carboxyamidomethylated (Step 1), followed by CNBr cleavage (Step 2) in a one-pot reaction sequence. The digest is adsorbed on ZipTipC18 reversed-phase supports [solid-phase extraction (SPE)], used as a venue for conjugation of the newly exposed homoserine lactone-terminated N-terminal and internal CNBr fragments with cystamine (Step 3), followed by gas-phase lactonization of residual, homoserine-terminated material (Step 4) and conjugation with cystamine (step 5). After reductive release of 2-aminoethane thiol from the fully formed conjugation products (Step 6), the thiol-functionalized N-terminal and internal peptides are depleted on activate thiol sepharose (Step 7), leaving the C-terminal peptide in the flow-through fraction. CAM denotes carboxyamidomethylation; GP-TFA designates gas-phase reaction step. The reactor is depicted schematically in the inset. The reaction path highlighted in red indicates the incorporation of protein carboxyl group carbamidation in the one-pot reaction scheme (see text for details on the use of the modified reaction scheme). BEX denotes buffer exchange of the carbamidated reaction mixture. (B) Schematic representation of the analyte, the intermediate reaction products, and the C-terminal isolate. HSL denotes homoserine lactone the asterisk denotes conjugation target. The carbamidated C-terminal isolate is highlighted in red. CONH-X denotes the carbamoyl residue. (C) Schematic representation of solid-phase sequential sample handling. The analyte is extracted on the solid-phase support (SPE), followed by a desalting step. The reaction cycle is initiated by loading the reagent onto the support. Reagents are sequentially exchanged directly on the resin bed (Rg Exg), eliminating intermittent sample purification. Note that the C18 reversed-phase tip is left immersed in reagent during incubation. The two-step reaction scheme is concluded by a solvent wash before product elution and MALDI-TOF or electrospray ionization (ESI) tandem MS.
Solid-Phase Disulfide-Bond Reduction
The desalted ZipTips were flushed to waste three times with 10 μL aliquots of 0.1 M sodium phosphate/2 M urea/2 mM EDTA. supplemented with 5 mM TCEP (pH 8.0). The reductant (10 μl) was then aspirated on the support. After 30 min incubation at 45°C, the supports were washed 10 times with 10 μL aliquots of water and then 10 times with 10 μL aliquots 0.1% TFA/2 mM EDTA. The samples were eluted in 50% aqueous acetonitrile/0.1% TFA/0.01% OGS for MALDI-MS analysis or for transfer to activated thiol sepharose (see below).
Solid-Phase Acetylation with Consecutive Benzoylation or Amination
Aniline hydrochloride was dissolved in aqueous 0.1 M MES to a final concentration of 0.2 M (pH 4.7). EDC and sulfo-NHS were dissolved together in aqueous 0.1 M MES to a final concentration of 0.2 M and 10 mM, respectively. EDA dihydrochloride was dissolved in aqueous 0.1 M MES to a final concentration of 0.8 M (pH 4.7). The nucleophile solutions were combined with the condensation reagent in a ratio of 1:1. A 20-mM sodium phosphate buffer, pH 8.0, was supplemented with 20 mM sulfo-NHS acetate. ZipTipC18 pipette tips were loaded with 10 μL of the reagent, incubated for 20 min at 55°C and dispensed to waste. The ZipTipC18 pipette tips were flushed three times with the benzoylation or amination reaction medium, of which 10 μL was then loaded on the reversed-phase supports. The carbamidation and the amination reaction were allowed to proceed at 37°C for 3.5 h and 1.5 h, respectively. After incubation, the ZipTips were washed with 50 μL water and subsequently with 100 μL 0.1% TFA, passed in 10 μL aliquots over the resin. Sample processing for MALDI-TOF was as described above. Reagents were prepared fresh for daily use.
Covalent Chromatography
Activated thiol sepharose (1 g) was allowed to swell for 10 min in 10 ml water. The gel was washed by vacuum filtration with a total of 150 ml water added in 15 ml aliquots, suspended in 10 ml 10% aqueous ethanol, and stored refrigerated until use. A gel slurry (100 μL of 75%) was placed into the spin column that had been washed with 200 μL acetonitrile by centrifugation at 1500 rpm for 1 min before use, a centrifugation speed and time setting that were used throughout the protocol. The affinity medium was centrifuged, resuspended in 200 μL 50 mM sodium phosphate/2 mM EDTA, pH 8.0, or in 50 mM Tris-HCl/5 mM EDTA, pH 8.0, followed by centrifugation; the flow-through was discarded. The spin column was then sealed with the plastic plugs supplied by the manufacturer. The CNBr digest that had been thiol-functionalized, as described above, was eluted from the ZipTipC18 pipette tip with 10 μL 50% acteonilrile/0.1% TFA/0.01% OGS directly into 40 μL of the thiol-coupling buffer composed of 50 mM sodium phosphate/2 mM EDTA//0.01% OGS (pH 8.0) or of 50 mM Tris-HCl/2 mM EDTA/0.01% OGS (pH 8.0). The mixture was transferred to the spin column, which was then agitated gently to resuspend the medium pellet. The spin column was capped, inserted into 1.5 mL microfuge tubes, secured with Parafilm, and incubated end-over-end for 1 h at room temperature (RT) using a rotary mixer and subsequently centrifuged to recover unbound material, i.e., the C-terminal CNBr fragment. The affinity medium was then washed consecutively with 50 μL of the thiol coupling buffer, 50 μL of 60% aqueous acetonitrile/0.1% TFA, and 50 μL of 80% aqueous acetonitrile/0.1% TFA. The combined flow-through fractions were reduced in volume by SpeedVac evaporation to 35 μL and acidified with 5 μL TFA to a final concentration of 1%. The isolates were immobilized on ZipTipC18 pipette tips, which were then desalted with 100 μL of 0.1% TFA, passed over the resin in 10 μL aliquots. Isolates were eluted from the support with 10 μL of 50% acetonitrile/0.1% TFA/0.01% OGS before MALDI-TOF analysis. Low-level amounts of peptide were enriched on micro ZipTips, washed 10 times with 10 μL aliquots of 0.1% TFA, and deposited in matrix containing 0.1% TFA/0.01% OGS directly onto the MALDI target. The disulfide-linked peptides, i.e., the internal and N-terminal fragments, can be optionally recovered from the support. To this purpose, the affinity medium was washed with 50 μL thiol-coupling buffer to neutralize residual TFA; the flow-through was discarded. The coupling buffer (50 μL), containing 5 mM TCEP, was added to the sealed spin column, which was then incubated for 30 min at RT with continuous end-over-end mixing. The reductively released peptides were collected from the affinity support by centrifugation, which was then washed with 50 μL reductant and subsequently with the organic solvents, as described above. The combined flow-through fractions were reduced in volume to 35 μL and acidified with 5 μL of 10% TFA to a final concentration of 1%. The material was bound to ZipTipC18 pipette tips, washed with 100 μL of 0.1% TFA, and passed over the resin in 10 μL aliquots. Prior to MALDI-MS, peptides were eluted from the support with 10 μL of 50% acetonitrile/0.1% TFA/0.01% OGS. Peptides immobilized on Micro ZipTips were deposited in matrix containing 0.1% TFA/0.01% OGS directly onto the MALDI target.
ZipTipC18 Pipette Tip Alkali Compatibility Evaluation
The efficiency of relative peptide recovery of peptide from the silica-based reversed-phase support before and after the alkali treatment was assessed using the isotope dilution procedure, as described previously.11 Efficiency and reproducibility of relative peptide recovery from the support were assessed as follows: aliquots of angiotensin I solutions in 0.2% TFA/0.01% OGS containing 50 pmoles peptide were bound to ZipTipC18 pipette tips, which were then washed three times with 10 μL of 0.1% TFA to remove unbound material, and eluted twice with 10 μL of 50% acetonitrile/0.1% TFA/0.01% OGS. Eluates were dried in a SpeedVac, dissolved in 25 μL of the isotopically labeled methanolic HCl, incubated for 90 min at 12°C, and subsequently dried. The methyl-d3-esterified peptides were then dissolved in 10 μL of 50% acetonitrile/0.1% TFA, and 5 μL of the peptide solutions were mixed with 5 μL of the methyl-d0-esterified reference peptide, followed by addition of 10 μL MALDI matrix. The mixture (1 μL) was applied to the MALDI target for relative quantitation of the eluted peptides.
To assess the potential impact of silica alkali exposure on peptide recovery, 50 pmoles peptide was immobilized in replicates, as described above. The ZipTipC18 pipette tips were rinsed three times with 10 μL of 0.1% TFA, then loaded with 10 μL 0.5 M cystamine dihydrochloride in aqueous 1 M sodium carbonate/2 M urea/0.005% OGS (pH 9.5), and incubated for 30 min at 55°C. After a solvent wash, another 10 μL portion of the conjugation reagent was loaded onto the support, and the incubation continued for 30 min at 55°C. The samples were desalted and loaded with 10 μL 0.1 M sodium phosphate /2 M urea/2 mM EDTA/5 mM TCEP (pH 8.0). After 30 min incubation at 45°C, the ZipTipC18 pipette tips were desalted and eluted twice with 10 μL of 50% acetonitrile/0.1% TFA/0.01% OGS. The eluates were dried and methyl-d3-esterified under the conditions described above. The methyl-d3-esterified peptides were then dissolved in 10 μL of 50% acetonitrile/0.1% TFA, and 5 μL of the peptide solutions was mixed with 5 μL of the methyl-d0-esterified reference peptide, followed by addition of 10 μL MALDI matrix. The mixture (1 μL) was applied to the MALDI target for relative quantitation of the eluted peptides. Potential alkali-induced sample loss from the reversed-phase support was determined by measuring the ratio of the relative abundance of the methyl-d3 and methyl-d0-esterified peptides in the mass spectra.
MS
A Voyager-DE STR (Applied Biosystems, Foster City, CA, USA) was used and operated in the reflector mode at an accelerating voltage of 20 kV, 67% grid voltage, and 250 ns extraction delay time. Laser intensity was typically set at 1400–1600, and spectra were acquired using 100 laser shots/spectrum. When operated in the linear mode, the grid voltage was typically set at 95% and the extraction delay time at 150 ns. The data shown are based on three accumulated acquisitions. Some spectra were obtained using 80 laser shots/spectrum, and spectra acquired in the linear mode from eight different positions were averaged. The analyte was prepared on target in the dried droplet mode using α-CHCA as matrix. ZipTipμ-C18 pipette tips were eluted in matrix containing 0.1% TFA/0.01% OGS onto the target.
RESULTS AND DISCUSSION
Reaction Scheme
As schematically illustrated in Fig. 1A, the sample preparation scheme is initiated by protein disulfide bond reduction, followed by carboxyamidomethylation of the liberated thiols (Step 1). The protein is then subjected to CNBr cleavage (Step 2). The one-pot reaction mixture is adsorbed onto ZipTipC18 pipette tips (SPE) for conjugation of the newly formed homoserine lactone-terminated fragments, i.e., the internal and N-terminal species, with cystamine dihydrochloride (Step 3). The subsequent reaction steps involve exposure of the reversed-phase support to gaseous TFA (Step 4) and conversion of the resultant lactonized material to its cystamine-conjugated counterpart (Step 5). The reaction scheme is concluded by a reduction step to release 2-aminoethanethiol (cysteamine) from the conjugates (Step 6), followed by sample elution and transfer to activated thiol sepharose. The thiol group-functionalized N-terminal and internal peptides are captured on affinity support, leaving the C-terminal peptide in the flow-through fraction (Step 7). The intermediate and final reaction products formed during the serial solid-phase reaction cycle are depicted schematically in Fig. 1B.
Sample Handling
As noted in earlier reports,10,11,15 and demonstrated in the present study, the solid-phase derivatization format using ZipTipC18 pipette tips as highly miniaturized reaction beds (0.2–0.6 μL) offers some notable advantages over the classical in-solution-based methods, the predominant sample preparation technique in current proteomic studies. It affords easy sample clean-up before and after the derivatization. This simple desalting step replaced repeated vacuum concentration, commonly used in contemporary protocols to ensure complete removal of reagent and solvent from CNBr digests.14 This sample manipulation can cause adsorptive peptide loss, ranging up to 50% or more of the starting solution after a single evaporation step, especially as seen with low-level analyte.16 The process of sample adsorption concentrates the analyte on the support, allowing the reaction to proceed in situ, in general, at higher efficiency and faster kinetics than in solution. Dilute digests, in which chemical reactions inherently proceed at a slow reaction rate, are enriched preferentially on ZipTipμ-C18 pipette tips, from which products can be transferred to the MALDI target nearly undiluted. In this manner, femtomole-level mass detection can be readily attained. Finally, the solid-phase procedure should lend itself to automation on ZipTipC18 pipette tip-compatible liquid-handling stations. As the introduction of this sample handling format to phosphoprotein characterization,17 its apparent benefits have been exploited to prepare samples for de novo sequence analysis.18,19 We note here that this reaction format has evolved over the past two decades as the predominant sample preparation technique for trace detection of bioorganic compounds in toxicology, environmental, and pharmaceutical studies.20,21
Liquid-phase serial-reaction schemes, commonly practiced in proteomics studies, are considered as problematic because of the high potential of cumulative adsorptive sample loss incurred during intermittent sample purification. In contrast, during solid-phase serial sample preparation, reagents are replaced directly on the support, obviating the need for sample transfer between the reaction steps (see Fig. 1C).10,11 Owing to this minimal sample handling, optimal product formation is maintained throughout the derivatization cycle, allowing the final reaction products to be recovered at minimal sample loss. In the current application of this reaction format, the presence of the chaotropic agent in the reagent solutions is perceived to enhance the solubility of the analyte-facilitating processing of large peptides (>2 kDa), commonly seen in a CNBr digest (see Fig. 5).
Figure 5.

Application of a method to whole, intact protein. A CNBr digest generated from carboxyamidomethylated RSA (25 pmoles) was conditioned for affinity purification, as described in Fig. 4. MALDI-MS spectra of (A) alkylated digest and (B) alkylated digest after solid-phase conjugation and disulfide bond reduction. (A and B) Arrows indicate the characteristic net mass addition of 77 Da imparted on the homoserine lactone-terminated peptides. (A) Asterisk designates a nonspecific truncation product (see text for details). (B) Filled arrowhead indicates C-terminal peptide recognized by its unaltered mass signature. The ion at m/z 1876.0 mass matched to the homoserine lactone-terminated fragment QRFGERAFKAWAVARM, spanning residues 228–243. The ion at m/z 1892.0 and the ion at m/z 1858.9 correspond to the peptide's mono-oxidized analog and its pyroglutamate counterpart, respectively. The adjoining fragment at m/z 5209.5 is annotated with the starting and ending amino acid residue. The thiol group-functionalized digest was submitted to covalent chromatography on activated thiol sepaharose. The flow-through fraction was bound to a ZipTipC18 pipette tip, eluted, and analyzed by MALDI-MS (C). Asterisk denotes low-level, unidentified coisolate. The isolate at m/z 4003.2 mass matched within the known protein sequence to the carboxyamidomethylated C-terminal peptide GDFAQFVDKCCKAADKDNCFATEGPNLVARSKEALA, spanning residues 573–608. One-tenth of the eluates was applied to the target. (Inset) Experiment at 5 pmole sample load. The isolate was eluted from a ZipTipμ-C18 pipette tip in matrix onto the target.
As schematically depicted in Fig. 1C, sample handling during sequential solid-phase derivatization is exceedingly simple, involving peptide adsorption on ZipTipC18 reversed-phase pipette tips, followed by a solvent wash to remove unwanted matrix components. The tips are then loaded with reagent and left immersed in reagent during incubation. The subsequent chemical reaction is initiated by a reagent exchange in situ. The reaction cycle is concluded by a clean-up step, followed by product elution prior to MALDI-MS or sample transfer to activated thiol sepharose.
The use of Poros R1-coated micropipette tips as alternate solid-phase reaction support for sequential peptide derivatization deserves consideration. Poros R1 is a polystyrene-divinylbenzene copolymer resin designed to facilitate reversed-phase fractionation of hydrophobic proteins and polypeptides. Accordingly, integration of this commercially available micro-extraction device into the workflow for C-terminal peptide purification holds the promise to promote the recovery of large hydrophobic C-terminal isolates. In addition, the less-retentive ZipTipC4 pipette tips may provide another avenue to facilitate elution of this class of peptides. These alternate chromatographic supports are expected to prove especially useful to process CNBr digests prepared from integral membrane proteins.
To minimize adsorptive sample loss during subsequent peptide isolation by affinity chromatography, a miniaturized spin column format was adopted to accommodate a relatively small resin bed (<100 μL), and buffers used in this reaction vessel were supplemented with trace amounts of OGS (0.01%). This MALDI-compatible, nonionic detergent has been shown minimize nonspecific protein/peptide adsorption to plastics and to enhance peptide solubility.22 To exploit this benefit, reagent solutions used for protein derivatization were also supplemented with OGS. Inclusion of the additive in the α-CHCA matrix has been shown to promote peptide ionization in MALDI-MS, especially as seen for large, hydrophobic peptides in the mass range between 4 and 11 kDa.23 To take advantage of this effect, peptides were eluted from the ZipTipC18 pipette tips in the presence of the detergent. Furthermore, the additive proved fully compatible with liquid chromatography (LC)-ESI MS, owing to its strong retention on reversed-phase columns.24
Sample Preparation at the Protein Level
As illustrated in Fig. 1A, derivatization at the protein level involved disulfide reduction, followed by alkylation of the liberated thiols. We have evaluated the efficiency of these sequential reactions in an earlier report and found that the thiol-blocking step proceeded to completion.11 In agreement with these results, the test proteins, examined in the current report, reacted to completion, as discussed later in the text. The one-pot reaction scheme was concluded by CNBr cleavage of the modified proteins. In standard literature protocols, samples were taken to dryness before and after the fragmentation in concentrated trifluoroacetic or formic acid solutions. These sample manipulations may cause substantial cumulative adsorptive loss, especially as seen with low-level peptide solutions.14,16 We have avoided this complication by allowing the CNBr cleavage to take place under dilute, moderately acidic condition. The digests could then be adsorbed directly on ZipTipC18 pipette tips from this aqueous reactant mixture. This aqueous solvent condition has been found advantageous previously to render highly hydrophobic proteins susceptible to CNBr cleavage and to minimize hydrolytic protein degradation.12 As assessed by gel-electrophoretic analysis of digests prepared from various test proteins, the CNBr cleavage proceeded under the described reaction conditions at high efficiency (>80%), yielding fragments that were observed in the gel images within the predicted MW range of 5–30 kDa (results not shown). The C-terminal fragments of the proteins were observed by MALDI-MS in the mass range between 2 and 5 kDa, as shown later in the report.
We are currently adapting the solution-phase CNBr cleavage protocol to the in-gel reaction format. The gel-based method is expected to provide for viable tools to prepare low-level amounts of proteins for C-terminal peptide isolation.
Sample Preparation at the Peptide Level
Sample preparation at the peptide level relies on the intrinsic reactivity of the homoserine lactone electrophile toward exogenous primary amine-containing compounds, resulting in amide bond formation between the reactants. Earlier applications of this chemistry used anhydrous DMF or DMSO as reaction media.13,14 The silica-based reversed-phase support proved incompatible with the use of these anhydrous solvent systems and therefore, prompted us to optimize the chemistry under pure aqueous conditions. EDA dihydrochloride, which we used initially as a reagent for solid-phase conjugation, proved highly suitable to incorporate an affinity tag into homoserine lactone-terminated fragments. However, subsequent depletion of the amino group-functionalized peptides on NHS-activated sepharose consumed ∼16 h, limiting the overall throughput of the method. To remedy this situation, we devised a strategy by which a masked thiol group was incorporated into the internal/N-terminal peptides using 2.2′-dithiobis (ethylamine) dihydrochloride (cystamine dihydrochloride) as a primary amine-containing compound. The disulfide-linked 2-aminoethanethiol moiety was then released reductively from the conjugates. The internal/N-terminal peptides, thiol-functionalized in this manner, were then reversibly captured on activated thiol sepharose, leaving the nonretained C-terminal CNBr fragment free in solution. This affinity-purification procedure was completed in <90 min.
CNBr digests prepared from β-casein were used as a model system to optimize the chemistry for throughput and efficiency of conjugation. In these experiments, the digests were immobilized on ZipTipC18 pipette tips and incubated in 0.5 M cystamine dihydrochloride solutions that had been adjusted with sodium carbonate to pH 7.5, 8.5, and 9.5. After incubation for 30 min at 55°C, the ZipTipC18 pipette tips were desalted and eluted. The recovered digest along with the untreated control were subjected to MALDI-MS analysis. Comparison of the MALDI-MS spectra produced from these samples revealed that the various homoserine lactone fragments, recognized by the derivatization mass shift of 152 Da, reacted poorly with the diamine at the pH near neutrality (Fig. 2A and B). The conjugation efficiency noticeably improved under the moderately basic reaction conditions, albeit owing to the competing ester bond hydrolysis; a considerable portion of analyte remained in its unreactive, homoserine form (Fig. 2C). When the reaction was performed at pH 9.5, the conversion of the homoserine lactone peptides, observed at m/z 1281.9 and m/z 1333.6 to their conjugates at m/z 1433.9 and m/z 1485.9, respectively, reached near completion (Fig. 2D). A minor fraction (<30%) of the homoserine lactone peptide at m/z 3188.2 remained converted to the homoserine-terminated form (Fig. 2D, arrow). Taken together, the data underscore the requirement of the more stringent alkaline reaction conditions to ensure for efficient conjugation product formation. Lowering the concentration of the reagent while maintaining the pH of the reaction media at 9.5 resulted, as expected, in noticeably diminished reaction yields. We recognize the need for expanded data to fully establish the general use of the method. However, this is, thus far, the first demonstration of a method enabling efficient aminolysis of homoserine lactone-bearing peptides with amines under pure aqueous conditions.
Figure 2.

Chemistry optimization at the peptide level: peptide conjugation. β-Casein (25 pmoles) was reduced, carboxyamidomethylated, and submitted to CNBr cleavage in the one-pot reaction sequence. The digest was adsorbed on ZipTipC18 pipette tips and reacted with 0.5 M cystamine dihydrochloride solutions that had been adjusted to pH 7.5, 8.5, and 9.5. MALDI-MS spectra of (A) unmodified digest; (B) after conjugation at pH 7.5; (C) after conjugation at pH 8.5; and (D) after conjugation at pH 9.5. (A and B) Arrows denote homoserine lactone peptides and reaction products differing in mass, as indicated by 152 Dalton. The homoserine lactone peptides are annotated in A with starting and ending amino acids. (C) Asterisk denotes thiol-group functionalized species. (D) Arrow denotes residual homoserine-terminated species. One-tenth of the eluates was applied to the target.
To address the minor signal dilution of the conjugate at m/z 3339.8, the immobilized sample was exposed for 20 min at 37°C to gaseous TFA in a capped microfuge tube, followed by incubation with cystamine dihydrochloride under the conditions described above (Fig. 1A, Steps 4 and 5). MALDI-MS of the samples revealed that the conversion of homoserine to homoserine lactone proceeded to completion (Fig. 3B). Subsequent aminolysis with cystamine dihydrochloride gave rise to the fully formed conjugate at m/z 3340.0 (Fig. 3C). Furthermore, the gas-phase lactonization procedure had no impact on the integrity of the sample. In our experience, exposure of protein digests to neat TFA, as practiced in the traditional conversion procedure, resulted, in some cases, in significant peptide degradation. This solution-phase reaction is halted by SpeedVac sample concentration, a handling step that may cause adsorptive sample loss.
Figure 3.

Chemistry optimization at the peptide level: peptide lactonization. Carboxyamidomethylated β-casein (25 pmoles) was submitted to CNBr fragmentation. The digest was bound to ZipTipC18 pipette tips and reacted with cystamine under the conditions described in Fig. 2D. The sample was then exposed for 20 min at 37°C to gaseous TFA in a capped microfuge tube, followed by incubation with cystamine under conditions described in Fig. 2D. MALDI-MS spectra of (A) digest before gas-phase lactonization; (B) after gas-phase lactonization; and (C) after subsequent reaction with cystamine. (A) Arrow denotes residual homoserine terminated species. (A and B) Arrow indicates low-abundance, homoserine-terminated peptide at m/z 3206.6 and its conversion product at m/z 3188.9, respectively. Note characteristic mass signature of 18 Da arising from homoserine lactone regeneration. Note resultant, essentially quantitative formation of the conjugate at m/z 3340.0. Asterisk indicates preformed thiol group-functionalized species. One-tenth of the eluates was applied to the target.
In replicate experiments, the test protein was carried through the series of chemical reactions at the protein and peptide levels, as described above. MALDI-MS analysis of this preparation showed that the protein was cleaved and then derivatized at the peptide level in a highly reproducible manner (Fig. 4A). The preparation, although immobilized on ZipTipC18 pipette tips, was exposed subsequently to a sodium PBS containing 5 mM TCEP (pH 8.0). After 30 min incubation at 45°C, the reversed-phase supports were desalted. As revealed by MALDI-MS of the eluates, this treatment resulted in the reductive release of cysteamine from the conjugates and the contaminant formation of the thiol-functionalized derivatives (Fig. 4B). The reaction was accompanied by the mass shift of 75 Da imparted on all peptides, with exception of the peptide at m/z 2663.4 that could thus be recognized as the C-fragment by its unaltered mass signature. Comparable results were obtained with RSA, the additional test protein examined. Representative data from this experiment are illustrated in Fig. 5, displaying the MALDI mass maps produced from the unmodified CNBr digest (Fig. 5A) and the thiol-functionalized preparation (Fig. 5B). The homoserine lactone fragments were recognized by net mass addition of 77 Da and hence, were distinguishable from the C-terminal peptide at m/z 4003.4 that was unaffected by the chemical reaction. The peptide at m/z 1876.2 and at m/z 1892.2 mass matched to the sequence QRFGERAFKAWAVARM, spanning residues 228–243 and its singly oxidized form, respectively, indicating that the peptide's tryptophan residues have been targeted by this modification. In addition, the peptide was partially converted to pyroglutamate, as indicated by the loss of 17 Da. The peptide at m/z 1392.5 had no counterpart among the in silico-generated CNBr fragments. The peptide was assigned tentatively by FindPept computational analysis to the nonspecific cleavage product AAVRQRMKCSSM bearing methionine sulfoxide, spanning residues 216–227. The peptide's sequence aligned with the C-terminal section of the CNBr fragment, spanning residues 148–227. Such C-terminal truncation events have been noted to occur during CNBr cleavage of the additional proteins examined, as discussed later in the text. As demonstrated by the experiments described below, C-terminal truncation products were, as expected, subject to affinity depletion by covalent chromatography and thus, had no impact on the selectivity of the C-terminal peptide-affinity purification.
Figure 4.

Application of a method to whole, intact protein. A CNBr digest, generated from caroboxyamidomethylated β-casein (25 pmoles), was adsorbed on ZipTipC18 pipette tips, reacted with cystamine dihydrochloride, as described in Fig. 2D, and then carried through the sequential reaction scheme, as described in Fig. 3. The preparation was then exposed to TCEP in a sodium PBS (pH 8.0) and analyzed by MALDI-MS. MALDI-MS spectra of (A) digest before disulfide bond reduction and (B) digest after disulfide bond reduction. (A) Arrow indicates preformed thiol derivative. (A and B) Arrows indicate the 75-Da mass shift corresponding to the loss of cysteamine from the thiol group-functionalized counterparts. (B) Filled arrowhead denotes the C-terminal peptide at m/z 2663.7, identified by its unaltered mass signature. The thiol group-functionalized digest was submitted to covalent chromatography on activated thiol sepaharose. The flow-through fraction was concentrated on a ZipTipC18 pipette tip, eluted, and analyzed by MALDI-MS. Note that the C-terminal peptide at m/z 2663.8 was recovered selectively from the resin (C). The peptide was aligned by database to PIQAFLLYQEPVLGPVRGPFPIIV, spanning residues 201–224. One-tenth of the eluates was applied to the target. (Inset) Experiment at 5 pmole sample load. The isolate was eluted from a ZipTipμ-C18 pipette tip in matrix onto the target.
C-terminal CNBr fragments, present in mixtures, such as derived by fragmentation of high MW proteins or protein complexes, are expected, owing to the signal suppression effect, to be identified with difficulty by differential peptide mapping. In this situation, C-terminal peptide identification relies on affinity-based depletion of the redundant material, as demonstrated by the experiments described below.
Covalent Chromatography
The method relies on capture of proteins/peptides from a mixture on the activated thiol sepharose by thiol-disulfide interchange, removal of the nonbound material, followed by reductive release of the disulfide-linked material. This approach in its original implementation exploits the reactivity of protein/peptide cysteinyl groups toward the 2, 2′-dipyridyl ligand of affinity support.25 In the application of this technique to the isolation of C-terminal CNBr fragments, efficient depletion of the thiol-functionalized peptides was considered to be a prerequisite to preclude co-isolation of redundant material in the flow-through fraction. In addition, nonspecific peptide adsorption during this process is of concern, as it would reduce the recovery of the species of interest. We reasoned that micro adaptation of the method would minimize this effect expected to become especially noticeable with low-level analyte. To this purpose, a miniaturized spin-column format was adopted to accommodate a relatively small reaction bed (<100 μL). In addition, specialty microfuge tubes were used as collection vessels to minimize sample-to-surface binding.
To test the reaction system's use to capture a thiol-containing peptide, mimicking a thiol-functionalized CNBr fragment, 5 pmole of the cysteinyl peptide somatostatin (CKNFFWKT, m/z 1073.2) immobilized on a ZipTipC18 pipette tip and eluted in 10 μL of 50% acetonitrile/0.1% TFA/0.01% OGS into 40 μL 50 mM sodium phosphate/2 mM EDTA/0.01% OGS. A slurry of 100 μL activated thiol sepharose, placed into the micro spin column, was centrifuged briefly and resuspended with the peptide solution. The reaction vessel was end-over-end incubated for 1 h at RT and then centrifuged to collect nonbound material. After an organic solvent wash of the resin, the combined fractions were concentrated on ZipTipC18 pipette tips along with controls, and ∼1 pmole peptide was deposited onto the MALDI target. A total of 640 laser shots, sampled from eight different spot positions, was summed for each spectrum. The MALDI-TOF spectra revealed that an estimated 5% of the starting material was typically found in the flow-through fraction, indicating that optimal coupling efficiency could be achieved (data not illustrated). The bound peptide was then released reductively during 30 min incubation in sodium phosphate buffer supplemented with 5 mM TCEP. After 30 min end-over-end incubation, the material was collected by centrifugation. The flow-through fraction was combined with the subsequent organic solvent wash, concentrated, and submitted to ZiptipC18 pipette tip purification. MALDI-MS analysis of the eluates showed that the signal from the starting material and from the recovered peptide was observed at similar abundance, indicating that the test peptide was retrieved effectively from the affinity resin. Hence, after isolation of the C-terminal CNBr fragment, the disulfide-bonded internal fragments can be optionally released from the affinity support.
Isolation of C-Terminal Fragments by Covalent Chromatography
The covalent purification protocol was applied to the β-casein and RSA digests that had been conditioned for C-terminal peptide isolation by the serial reactions described above. MALDI-MS of the flow-through fraction collected from the β-casein digest revealed the prominent ion at m/z 2663.8, and there was no background contamination (Fig. 4C). The peptide mass matched within the known protein sequence to the C-terminal peptide PIQAFLLYQEPVLGPVRGPFPIIV, spanning residues 201–224. The RSA digest yielded the isolate at m/z 4003.2 that aligned with the protein's C-terminal CNBr fragment GDFAQFVDKCCKAADKDNCFATEGPNLVARSKEALA, spanning residues 573–608 (Fig. 5C). We note that ions corresponding to incompletely carboxyamidomethylated analogs of this peptide were not observed in the spectrum of the starting material (Fig. 5A). The data show that the thiol-blocking step at the protein level proceeded to completion, thereby precluding diminished recovery of the isolate. The presence of the minor co-isolate at m/z 2003.1 in the flow-through fraction bears evidence of an unspecific cleavage event that must have occurred during sample preparation, as discussed later in the text (Fig. 5C, asterisk).
In the following experiment, the affinity support was treated with sodium phosphate-buffered TCEP (pH 8.0). By this procedure, the internal CNBr fragments were released from the resin, providing a means to collect such material for further manipulation, e.g., phosphorylation-site mapping (data not illustrated).15 Taken together, the data validate the use of miniaturized a sample processing/covalent chromatographic system to isolate C-terminal peptides from whole, intact protein at high selectivity and with minimal sample loss.
To assess the overall sensitivity of the C-terminal isolation strategy, 5 pmoles β-casein and RSA was carried through the sample preparation scheme using ZipTipμ-C18 pipette tips for peptide derivatization and submitted to covalent chromatography. The flow-through fractions were concentrated on a ZipTipμ-C18 pipette tip and deposited in matrix onto to the target. The high quality of spectra suggests that lower quantities of protein should be amenable to the purification strategy (Figs. 4C, inset, and 5C, inset).
Expanded Evaluation of CNBr Cleavage under Dilute Acidic Conditions
To explore the general usefulness of the CNBr fragmentation conditions, a panel of eight proteins, ranging MW from 11.6 to 66.3 Da, was probed with the protocol. The MALDI-MS spectra prepared from these samples are shown in Fig. 6. On the basis of the known sequences of these proteins, the identity of the C-terminal fragments could be ascertained readily. C-terminal fragments harboring cysteinyl residues were recovered in their fully carboxyamidomethylated form as ions that would reveal that incomplete alkylation was not observed in the spectra (Fig. 6B, C, and E). We note here that the C-terminal fragments were observed in MALDI-MS at relatively high abundance, suggesting that the peptides were desorbed efficiently in the acidic aqueous organic solvent. Only trace amounts of peptides were recovered when the samples were subjected to a second elution step (data not shown).
Figure 6.
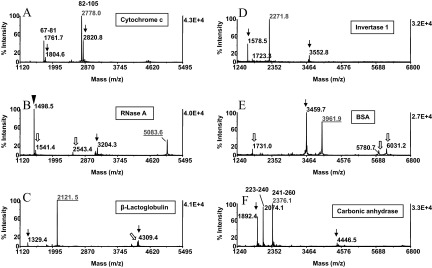
CNBr cleavage of model proteins. Proteins (50 pmoles) were reduced, carboxyamidomethylated, and subjected to CNBr cleavage in the one-pot reaction scheme. The reaction mixture was absorbed onto ZipTipC18 pipette tips, which were desalted and eluted. The eluates were analyzed by MALDI-MS. The MALDI-MS spectra were annotated with the individual proteins examined. Masses highlighted in red designate the C-terminal CNBr fragments identified by database search. Fragments recognized as proximal to each other in the protein sequences are annotated with the starting and ending amino acids. Arrows and open arrows denote homoserine lactone-terminated peptides, recognized as truncation products and internal, unspecific degradation products, respectively (see text for details). Filled arrowhead indicates the protein's N-terminal CNBr fragment. One-tenth of the eluates was applied to the target. Note that C-terminal fragments, harboring cysteinyl residues, were recovered in their fully carboxyamidomethylated form (B, C, and E). The sequences of the CNBr C-terminal fragments ascertained by database search are listed in Table 1. One-tenth of the eluates was applied to the target.
The spectra displayed ions that were identified as homoserine lactone peptides by the presence of the low-abundance homoserine satellite peaks. However, the majority of these fragments could not be matched to their in silico predicted counterparts, indicating that they must have been generated by C-terminal truncation of the corresponding larger fragments (arrows). Such truncation products are inherently targeted for affinity depletion by covalent chromatography. We reasoned that the presence of these peptides in the digests reflects acid-induced degradation events occurring during protein CNBr cleavage. Likewise, the formation of the low-abundance peptides that appeared as singlets in the spectra and hence, represent carboxyl group-terminated fragments, was considered to be attributable to this side-reaction (Fig. 6B, C, and E, open arrows). This subpopulation of peptides must have been generated by cleavage within the peptide chains and as such, would not be captured on the affinity support. As shown in Fig. 5C, such low-level co-isolates would not compromise the selectivity of the affinity purification. However, in a complex sample setting, these co-isolates may complicate data interpretation. A reaction scheme devised to eliminate this background contamination is discussed later in the report.
The sequences of the CNBr C-terminal fragments ascertained by database search are listed in Table 1. The data set shows that these fragments cover a mass range between 1678.9 and 5083.6 Da. Hence, the majority of these fragments should be amenable to structural characterization by collision-induced dissociation using MALDI-TOF/TOF or electrospray tandem MS. Owing to the high specificity of the amino acid sequence in the C-terminal protein domain, sequence tags, comprising as few a four to five contiguous amino acid residues, have been shown to narrow the search space drastically for protein identification by database search.2 Of course, a Fourier transform ion cyclotron resonance (FTICR) mass spectrometer would be the most suitable platform to characterize high mass CNBr C-terminal fragments (>3 kDa), such as derived from BSA, RSA, and RNase A, expected in general to occur in protein digests.26 In addition, this top-down analytical approach lends itself on a chromatographic time-scale to high-throughput operation.
TABLE 1.
MALDI-MS Masses and Sequences of CNBr C-Terminal Protein Fragments
| Mra | Proteinb | [MH]+ Calc | [MH]+ Obs | Peptide amino acid sequencec |
|---|---|---|---|---|
| 36.6 | Alcohol dehydroginase | 1678.9 | 1679.5 | EKGQIVGRYVVDTSK (334–348) |
| 18.2 | β-Lactoglobulind | 2122.0 | 2121.5 | HIRLSFNPTQLEEQCHI (162–178) |
| 58.5 | Invertase | 2272.2 | 2271.8 | TTGVDNLFYIDKFQVREVK (514–532) |
| 16.9 | Myoglobin | 2512.3 | 2512.4 | TKALELFRNDIAAKYKELGFQG (113–154) |
| 28.9 | Carbonic anhydrase | 2376.3 | 2376.1 | LANWRPAQPLKNRQVRGFPK (241–260) |
| 23.6 | β-Caseind | 2663.7 | 2663.8 | PIQAFLLYQEPVLGPVRGPFPIIV (201–224) |
| 11.6 | Cytochrome c | 2779.5 | 2778.0 | IFAGIKKKTEREDLIATLKKATNE (82–105) |
| 14.3 | Lysozyme C | 2973.5 | 2972.7 | NAWVAWRNRCKGTDVQAWIRGCR (124–147) |
| 66.3 | BSA | 3960.8 | 3961.9 | ENFVAFVDKCCAADDKEACFAVEGPKLVVSTQTALA (572–607) |
| 65.9 | RSAd | 4003.8 | 4003.2 | GDFAQFVDKCCKAADKDNCFATEGPNLVARSKEALA (573–608) |
| 13.6 | RNase A | 5084.4 | 5083.6 | SITDCRETGSSKYPNCAYKTTQANKHIIVACEGNPYVPVHFDASV (106–150) |
Mr, kDa;
carboxyamidomethylated
conditioned for C-terminal fragment isolation by covalent chromatography
sequences annotated with starting and ending amino acid residues. [MH]+ Calc, Calculated; Obs, Observed.
ZipTipC18 Pipette Tips Alkali Compatibility
Long-term C18 reversed-phase column testing (>1 week) with mobile phases at pH 9–12.3 has been shown to result in minor leakage (∼2%) of the silane-bonded stationary phase, attributable to mechanical attrition around the covalently attached silanes, eventually causing splitting off of the bonded organic phase.27 To examine whether this effect could potentially diminish analyte recovery from the silica-based ZipTip support, we assessed the relative recovery of the model peptide angiotensin I (DRVYIHPFHL, m/z 1296.6) from ZipTips, before and after the alkali treatment, using an earlier reported, stable isotope-dilution technique.11 To assess the recovery of this hydrophobic peptide from the untreated, solid-phase support, 50 pmoles peptide was immobilized in replicates, washed to remove unbound material, eluted, methyl-d3-esterified, and mixed with the methyl-d3-esterified internal reference peptide. Portions of the mixtures were then analyzed by MALDI-MS. As estimated from the spectral abundance ratio calculated for the replicates, an average of 77.3% of peptide, loaded onto the support, was found in the eluted fractions, indicating that the hydrophobic peptide was desorbed at notably high efficiency (Fig. 7B). The reproducibility and effectiveness of peptide recovery from the untreated resin noted here agree with an earlier evaluation of ZipTipc18 pipette tips using 14C-accelerator MS for peptide quantitation.28
Figure 7.

ZipTipC18 pipette tip alkali-compatibility evaluation. Angiotensin I, DRVYIHPFHL at m/z 1296.5 (50 pmoles), was immobilized on ZipTipC18 pipette tips, which were washed briefly to remove unbound material. The tips were then exposed to the akali reaction conditions, as described in Figs. 2D and 4B, or left untreated. Peptides with and without incubation were desorbed, methyl-d3-esterified, and mixed in equimolar amounts with the methyl-d0-esterified reference peptide. Experiments were in triplicate. MALDI-MS spectra were acquired using 400 laser shots/spectrum, accumulated from five different spot positions. MALDI-MS spectra of (A) methyl-d3-esterified peptide from untreated sample mixed with methyl-d0-esterfied internal reference peptide and (B) methyl-d3-esterified peptide from alkali-exposed sample mixed with methyl-d0-esterified internal reference peptide. (B and C) Labels designate the relative peptide abundance ratios. The average alkali-induced sample loss is highlighted in the box. Approximately 0.5 pmoles peptides was applied to the target.
To assess the peptide recovery after alkali exposure, ZipTipC18 pipette tips were loaded with 50 pmoles peptide and washed briefly. Samples were then carried through the conjugation reaction (pH 9.5) at 55°C for a total of 1 h and then treated with the reductant for 30 min at 45°C (pH 8.0). The ZipTipC18 pipette tips were desalted and eluted. The eluates were methyl-d3-esterified, reconstituted, mixed with the methyl-d0-esterified peptide internal reference, and analyzed by MALDI-MS. As estimated from the spectral abundance ratio calculated for the replicates, an average of 69.2% of peptide could be reproducibly recovered from the chemically treated supports (Fig. 7C). The comparison data show that only a minor fraction of peptide (average 8.1%) was lost during sample clean-up. Accordingly, the integrity of the silica-bonded stationary phase was impacted little by the chemical treatment. In a separate experiment, the immobilized peptide was exposed to gaseous TFA for 30 min at 37°C. In parallel, the peptide was adsorbed on the reversed-phase support from the moderately acidic CNBr reaction medium. The eluates were methyl-d3-esterified, redissolved, mixed with the methyl-d0-esterified peptide internal reference, and analyzed by MALDI-MS. The spectra showed that the peptide's elution characteristics were unaffected by the chemical treatment, rendering the reversed-phase support highly suitable for these applications (results not illustrated).
Chemistry Side-Reactions
Side-reactions known to occur during CNBr cleavage include cysteinyl group partial conversion to cysteic acid and deamidation of asparagine and glutamine side-chains.12 The oxidative side-reaction was prevented by the sulfhydryl group alkylation, used as an initial reaction step in the sample preparation method. We note here that partial tryptophan oxidation, identified in our study as potential CNBr fragmentation side-reaction, has thus far not been reported in the literature.
Exposure of peptides and proteins at 50°C to 2.74 N HCl for up to 16 h has been shown to induce in model peptides limited, successive C-terminal truncation and partial internal peptide bond cleavage at the C-terminal side of asparagine, at the N-terminal sides of serine and threonine, and at the C- and N-terminal sides of glycine, as well as deamidation of asparagine and glutamine side-chains.29 When the reaction was performed in a 0.1-N HCl solution, only deamidation was observed. Unexpectedly, peptide degradation occurred under comparable but the less-stringent reaction condition used in our study (i.e., 0.1 N HCl/16 h/RT). As noted above, this side-reaction resulted primarily in C-terminal truncation of internal CNBr peptides. As a result, these hydrolysis products became subject to depletion by covalent chromatography. In contrast, other hydrolytic fragments, since destined for co-isolation by covalent chromatography, were generated by cleavage within the peptides chains (see Figs. 5 and 6). With the proteins so far examined, the side-reaction appeared to occur at low efficiency. A reaction scheme devised for depletion of these co-isolates from complex samples is discussed below.
Current and Future Developments
Large-scale protein profiling
We have begun to explore the use of commercially available, high-capacity, reversed-phase spin columns for chemistry scale-up, needed to adapt the sample preparation method to large-scale C-terminal peptide profiling in digests of complex biological mixtures. In the case of S. cerevisiae, frequently used as a biological model system, ∼44,000 CNBr peptides are predicted to be generated based on the 6300-gen Saccharomyces Genome Database, assuming that eight fragments will be generated from each protein. As selective isolation of C-terminal peptides results in substantial sample complexity reduction (by one order of magnitude), comprehensive proteome coverage of small proteomes at the subset level of C-terminal fragments should thus be achievable using state-of-the art LC/tandem MS instrumentation. Importantly, C-terminal peptide sampling, as advanced in our report, reconciles the need for high-throughput peptide analysis while maximizing efficient use of LC-tandem MS-based peptide identification We note here that orthogonal prefractionation at the protein level is undoubtedly required to render mammalian proteomes predicted to yield at least 30,000 C-terminal CNBr fragments amenable to comprehensive positional analysis. In these efforts, FTICR-MS, providing for high-resolution/high-accuracy mass measurement, is considered as an ideal analytical platform to characterize this subpopulation of C-terminal CNBr fragments.
Differential stable isotope labeling
We have used glycinamidation previously as a means to protect protein carboxyl groups to enable negative selection of C-terminal tryptic fragments.10 In this procedure, carboxyamidomethylated protein preparations were reacted with 0.4 M glycineamide in a MES-buffered solution containing 0.1 M EDC, 5 mM sulfo-NHS, and 4 M urea for 1.5 h at 37°C. In recent experiments, we found that these conditions proved effective to label the carboxylates of whole, intact proteins using aniline 12C6 hydrochloride and in separate experiments, aniline 13C6 hydrochloride, the stable isotope-coded counterpart, as nucleophiles in the EDC-mediated condensation reaction.30 Derivatization of the test peptide [Glu1] fibrinopeptide B (EGVNDNEEFFAR) with the “light” or “heavy” version of the reagent revealed in MALDI-MS the expected mass difference of 30 Da between the peptide isotopomers.
As illustrated in Fig. 1A, the sample preparation workflow can be readily modified to accommodate the carbamidation step as a means to introduce a differential isotopic signature into whole intact proteins. In the practical implementation of the method that we begun to evaluate in small-scale experiments with protein model systems, the carboxylate labeling is halted by gel-filtration on Zeba Desalt Spin columns (7 kDa MW cutoff), as described previously,10 except that the reaction mixture was buffer-exchanged into 10 mM phosphate /4 M urea/0.01% OGS (pH 8.0). The recovered protein preparations (100μL) are then supplemented with 20 μL of the CNBr stock solution prepared in 0.6 N HCL (20 mg CNBr/mL). The CNBr digests, prepared from the differentially isotopic-labeled preparations, are mixed, adsorbed on the solid-phase support, and carried through the serial chemical reactions required to condition the internal N-terminal fragments for depletion by covalent chromatography. The unbound isolates are recovered from the flow-through fraction for subsequent tandem mass spectrometric analysis to determine the proteins' relative abundance. Carboxylate labeling at the protein level is considered advantageous, as peptides derived from the original protein C-terminus may be validated upon tandem MS by the presence of y-type product ions in the tandem MS spectrum containing the label's characteristic mass signature, thereby adding an another level of selectivity to the C-terminal peptide purification approach.
The method in its current sample-handling configuration permits processing ∼1 nmole digest, confining its use to small-scale applications, such as to quantify changes in protein abundance in protein complexes. Future work will focus on evaluation of high-capacity sample-handling components to adapt the sample preparation method to proteome-wide applications.
Quantitative assessment of C-terminal-progressive truncation, resulting from carboxypeptidase proteolysis, known a ragging, is of considerable interest, because of potential involvement of this protein-processing event in the modulation of cellular functions. The differential isotope coding strategy described above should provide an avenue to monitor and quantitate this protein-processing event. Owing to the substoichiometric nature of this post-translational event, the co-enriched C-terminal truncation products are expected to be generated in low-level amounts. Hence, in complex analytical settings, removal the aforementioned (acid-induced) cleavage products, generated during sample preparation, would ensure that the C-terminal truncation products are recognized unambiguously by tandem MS as part of the proteolytic signature of the sample (see Figs. 5 and 6). In addition, fragments generated by internal proteolytic processing would escape depletion by covalent chromatography and thus, would contribute to the undesired background level. The principles of the expanded sample processing/affinity-purification procedure that would afford depletion of the various co-isolates are depicted schematically in Fig. 8A. In this strategy, the flow-through fraction, collected from the activated thiol sepharose resin, containing the differentially isotope-coded C-terminal peptide (and its truncation products) and the co-isolates bearing free carboxyl groups, is concentrated on the reversed-phase support. Subsequent reactions on the solid-phase involve amino group acetylation, followed by carboxyl group amination using EDA as a nucleophile in the EDC-catalyzed condensation reaction. This sequence of reactions, schematically depicted in Fig. 8B, allows selective conversion of the co-isolates to amino group-functionalized species, which are then captured on NHS-sepharose. The C-terminal fragment (and its truncation products), protected by acetylation from coupling to the affinity resin, is then recovered from the flow-through fraction. We note here that this depletion procedure has been used successfully in an analogous manner for the selective isolation of C-terminal tryptic peptides from digest prepared from glycinamidated proteins.10 Chemistry scale-up will be required to exploit the benefits of the expanded sample processing/affinity-purification strategy for quantitative monitoring of C-terminal processing events in subfractions derived from whole proteomes.
Figure 8.

Principles of sample preparation for the quantitative analysis of C-terminal truncation events. Proteins derived from different cell states are carboxyamido-methylated and subsequently, carbamidated using the light and heavy version of the nucleophile (i.e., aniline hydrochloride) in the EDC-catalyzed condensation reaction. The differentially labeled preparations are desalted and subjected CNBr cleavage. The digests are mixed and adsorbed on a C18 reversed-phase support .and carried through the sequence of chemical reaction to condition the internal/N-terminal peptides for depletion by covalent chromatography on activated thiol sepharose (see Fig. 1A). (A) Depletion of hydrolytic cleavage products. The carbamidated C-terminal peptides (and the respective truncation products), along with the subpopulation of nonspecific cleavage products bearing free carboxyl groups, are concentrated from the flow-through fraction on a C18 reversed-phase support (SPE). The immobilized material is subjected to α-amine acetylation (Step 1) and subsequently to carboxyl group amination using EDA as a nucleophile in the EDC-catalyzed condensation reaction (Step 2). By this sequence of reactions, an amino group-functionalized affinity tag is introduced selectively into the co-isolates for subsequent depletion on NHS-sepharose (Step 3). The purified C-terminal peptides (and the respective truncation products) are then analyzed by LC-tandem MS to determine their relative abundance. (B) Schematic illustration of the analyte and reaction products. AC, Acetyl group; CONH-X, carbamoyl moiety. Asterisk denotes depletion target. The amino group-functionalized affinity tag is highlighted in red.
CONCLUSIONS
A sample preparation method for protein C-terminal peptide isolation from protein cyanogen digests has been developed. The approach uses a series of chemical reactions at the protein and peptide levels. Whole, intact proteins were carboxyamidomethylated and subsequently subjected to CNBr cleavage in a one-pot reaction scheme. Subsequent sample processing by this strategy involved conjugation of the homoserine lactone-terminated fragments with cystamine, followed by reductive release of the aminothiol moiety from amide-bonded derivatives. The use of reversed-phase supports as miniaturized reaction beds promoted the efficiency of the sequential chemical reactions, enabling sample processing with minimal sample loss. By this sequence of solid-phase reactions, the C-terminal peptide could be recognized uniquely in mass spectra of unfractionated digests. The thiol group-functionalized N-terminal and internal peptides were reversibly captured on activated thiol sepharose, leaving the C-terminal peptide in the flow-through fraction. The use of the liquid/solid-phase sample preparation method was demonstrated with low-level amounts of model protein. The C-terminal peptides were retrieved selectively from the affinity resin. The sample preparation method combines robustness with simplicity of operation using standard equipment, readily available in most biological laboratories, and is expected to be readily expanded to gel-separated proteins and in a scaled-up format, to sampling of C-terminal CNBr fragments from digests prepared from complex biological samples.
ACKNOWLEDGMENTS
This work has been funded by U.S. National Institutes of Health Grants R33CA101150 and HHSN266200400054C to R.H.A. The authors thank the Albert Einstein College of Medicine for generous support, Mr. Edward Nieves and Dr. Richard Stanley for helpful discussions, and Ms. Junko Hihara for editorial assistance in preparing this manuscript.
DISCLOSURE
The authors declare no conflict of interest associated with financial support.
REFERENCES
- 1. Chung JJ, Shikano S, Hanyu Y, Li M.Functional diversity of protein C-termini: more than zipcoding. Trends Cell Biol 2002;12:146–150 [DOI] [PubMed] [Google Scholar]
- 2. Wilkins MR, Gasteiner E, Tonella L, et al. Protein identification with N and C-terminal sequence tags in proteome projects. J Mol Biol 1998;278:549–608 [DOI] [PubMed] [Google Scholar]
- 3. Nakazowa T, Yamaguchi M, Okamura T, Ando E, Nishimura O, Tsunasawa S. Terminal proteomics: N- and C-terminal analyses for high-fidelity identification of proteins using MS. Proteomics 2008;8:673–685 [DOI] [PubMed] [Google Scholar]
- 4. Samyn B, Sergeant K, Castanheira P, Faro C, van Beeumen J. A new method for C-terminal sequence analysis in the proteomic era. Nat Methods 2005;2:193–200 [DOI] [PubMed] [Google Scholar]
- 5. Julka S, Dielman D, Young SA. Detection of C-terminal peptides of proteins using isotope coded strategies. J Chromatogr 2008;874:101–110 [DOI] [PubMed] [Google Scholar]
- 6. Murphy CM, Fenselaue C.Recognition of the caroboxy-treminal peptides in cyanogen bromide digests of proteins. Anal Chem 1995;67:1644–1645 [Google Scholar]
- 7. Sechi S, Chait BT. A method to define the carboxyl terminus of proteins. Anal Chem 2000;72:2374–3378 [DOI] [PubMed] [Google Scholar]
- 8. Kuyama H, Shima K, Sonomura K, et al. A simple and highly successful C-terminal sequence analysis of proteins by mass spectrometry. Proteomics 2008;8:1539–1550 [DOI] [PubMed] [Google Scholar]
- 9. Sonomura K, Kuyama H, Matsuo E, Tsunasawa S, Nishimura O.The specific isolation of C-terminal peptides of proteins through a transamination reaction and its advantage for introducing functional groups into the peptide. Rapid Commun Mass Spectrom 2009;23:611–618 [DOI] [PubMed] [Google Scholar]
- 10. Nika H, Nieves E, Hawke DH, Angeletti RH. C-terminal protein characterization by mass spectrometry using micro scale liquid and solid-phase derivatization. J Biomol Tech 2013;24:17–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nika H, Nieves E, Hawke DH, Angeletti RH. Optimization of the β-elimination/Michael addition chemistry on reversed-phase supports for mass spectrometry analysis of O-linked protein modifications. J Biomol Tech 2013;24:132–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodriguez JC, Jennings PA. The solvent in CNBr cleavage reactions determines the fragmentation efficiency of ketosteroid isomerase fusion proteins used in the production of recombinant peptides. Protein Expr Purif 2003;28:224–231 [DOI] [PubMed] [Google Scholar]
- 13. Horn JM, Laursen RA. Solid-phase Edman degradation: attachment of carboxyl-terminal homoserine peptides to an insoluble resin. FEBS Lett 1973;36:285–288 [DOI] [PubMed] [Google Scholar]
- 14. Shi T, Weerasekera R, Yan C, et al. Method for affinity purification of covalently linked peptides following cyanogen bromide cleavage of proteins. Anal Chem 2009;81:9885–9895 [DOI] [PubMed] [Google Scholar]
- 15. Nika H, Lee JH, Willis IM, Angeletti RH, Hawke DH. Phosphopeptide characterization by mass spectrometry using reversed-phase supports for solid-phase β-elimination/Michael addition. J Biomol Tech 2012;23:2012–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Speicher DT, Kolbas O, Harper D, Speicher DW. Systematic analysis of peptide recoveries from in-gel digestions for protein identifications in proteome studies. J Biomol Tech 2000;11:74–86 [PMC free article] [PubMed] [Google Scholar]
- 17. Nika H, Hawke DH, Kobayashi R. Derivatization on reversed-phase supports for enhanced detection of phosphorylated peptides. 52nd ASMS Conference on Mass Spectrometry and Allied Topics, Nashville, TN, USA, 2004, Abstr ThPN 292 [Google Scholar]
- 18. Conrotto P, Hellman U.Sulfonation chemistry as a powerful tool for MALDI TOF/TOF de novo sequencing and posttranslational modification analysis. J Biomol Tech 2005;16:441–452 [PMC free article] [PubMed] [Google Scholar]
- 19. Cindric M, Cepo T, Skrlin A, Vuletic M, Bindila L.Accelerated on-column lysine derivatization and cysteine methylation by imidazole reaction in a deuterated environment for enhanced product ion analysis. Rapid Commun Mass Spectrom 2006;20:694–702 [DOI] [PubMed] [Google Scholar]
- 20. Rosefeld JM. Solid-phase analytical derivatization: enhancement of sensitivity and selectivity of analysis. J Chromatogr A 1999;843:19–27 [DOI] [PubMed] [Google Scholar]
- 21. Johnson ME, Carpenter TS. The use of solid-phase supports for derivatization in chromatography and spectroscopy. Appl Spectrosc Rev 2005;40:391–412 [Google Scholar]
- 22. Katayama H, Nagasu T, Oda Y.Improvement of in-gel digestion protocol for peptide mass fingerprinting by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2001;15:1416–1421 [DOI] [PubMed] [Google Scholar]
- 23. Cohen SL, Chait BT. Influence of matrix solution conditions on the MALDI-MS analysis of peptides and proteins. Anal Chem 1996;68:31–37 [DOI] [PubMed] [Google Scholar]
- 24. Katayama H, Tabata T, Ishihama Y, Sato T, Oda Y, Nagasu T.Efficient in-gel digestion procedure using 5-cyclohexyl-1-pentyl-β-D-maltoside as an additive for gel-based membrane proteomics. Rapid Commun Mass Spectrom 2004;18:2388–2394 [DOI] [PubMed] [Google Scholar]
- 25. Brocklehurst K, Carlsson J, Marek P, Kierstan J, Crook EM. Covalent chromatography. Preparation of fully active papain from dried papaya latex. Biochem J 1973;133:573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelleher NL, Lin HY, Valaskovic GA, et al. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J Am Chem Soc 1999;121:806–912 [Google Scholar]
- 27. Kirkland JJ, van Straten MA, Claessens HA. High pH mobile phase effects on silica-based reversed-phase high-performance liquid chromatography. J Chromatogr A 1995;691:3–19 [DOI] [PubMed] [Google Scholar]
- 28. Palmblad M, Vogel JS. Quantitation of binding, recovery and desalting effiency of peptides and proteins in solid phase extraction micropipette tips. J Chromatogr B 2005;814:309–313 [DOI] [PubMed] [Google Scholar]
- 29. Takayama N, Matsui T, Sakai T, Tsugita A.A method for peptide successive C-terminal degradation using dilute hydrochloric acid. J Biolmol Tech 1999;10:194–198 [PMC free article] [PubMed] [Google Scholar]
- 30. Panchaud A, Hansson J, Affolter M, et al. ANIBAL, stable isotope-based quantitative proteomics by aniline and benzoic acid labeling of amino and carboxylic groups. Mol Cell Proteomics 2008;7:800–812 [DOI] [PubMed] [Google Scholar]