

Abstract
Myosin VI is involved in a wide variety of intracellular processes such as endocytosis, secretion and cell migration. Unlike almost all other myosins so far studied, it moves towards the minus end of actin filaments and is therefore likely to have unique cellular properties. However, its mechanism of force production and movement is not understood. Under our experimental conditions, both expressed full-length and native myosin VI are monomeric. Electron microscopy using negative staining revealed that the addition of ATP induces a large conformational change in the neck/tail region of the expressed molecule. Using an optical tweezers-based force transducer we found that expressed myosin VI is nonprocessive and produces a large working stroke of 18 nm. Since the neck region of myosin VI is short (it contains only a single IQ motif), it is difficult to reconcile the 18 nm working stroke with the classical ‘lever arm mechanism', unless other structures in the molecule contribute to the effective lever. A possible model to explain the large working stroke of myosin VI is presented.
Keywords: actin, motility, myosin, working stroke
Introduction
In the last 10 years, 18 major classes of myosin motor proteins have been identified (Berg et al, 2001), but apart from the myosins in classes I, II and V, very little is known about their properties or cellular functions. We have focused on class VI myosins that are believed to have roles in vesicle transport and membrane tension maintenance (Buss et al, 1998; Sellers, 2000). Based on sequence comparisons, myosin VI follows the basic myosin organisation (Figure 7) with a highly conserved N-terminal motor domain and a short neck region containing one IQ motif that binds calmodulin. The tail is predicted to consist of a helical region followed by a globular domain (Buss et al, 1998). Myosin VI however has two additional features: firstly, unlike the other myosins, it moves towards the minus end of actin filaments (Wells et al, 1999), and secondly between the motor domain and the IQ motif there is a unique 53-amino-acid insert, which was predicted to be the reverse gear (Wells et al, 1999).
Figure 7.
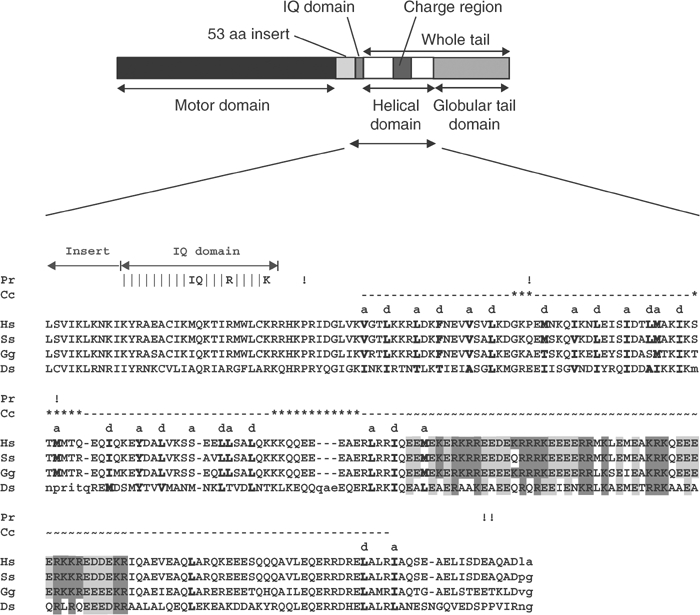
Cartoon showing the different domains of myosin VI together with a sequence alignment and heptad repeat prediction for the myosin VI helical tail domains from different species. The cartoon shows the different domains of myosin VI and below is the sequence alignment of the helical tail domains (amino acids 812–1034) and their analysis using the COILS prediction program (Lupas, 1996). Vertical dashes show the IQ motif with the key residues labelled. The dashed line shows the presence of the putative coiled-coil sequence with hydrophobic a and d residues of the heptad repeat labelled and displayed in bold. Note: The complete sequence alignment includes several species not shown here; hence, hydrophobic residues that are not part of the heptad repeat but are highly conserved in the complete alignment are also shown in bold. Possible breaks in the putative coiled coil are shown by asterisks (***). An exclamation mark pinpoints helix breaking proline residues even if the proline is not conserved in all sequences. ∼∼∼ shows the highly charged region, and within this region, positively and negatively charged residues are shaded in dark and light grey, respectively. Accession numbers for myosin VI sequences are as follows: Hs (Homo sapiens), AAC51654; Ss (Sus scrofa), A54818; Gg (Gallus gallus), CAB96536; Ds (D. melanogaster), Q01989.
Myosin VI is found in higher, multicellular eukaryotic organisms and is ubiquitously expressed (reviewed in Sellers, 2000). It has been implicated in the development and maintenance of the stereocilia in the inner ear in mice (Self et al, 1999) and in various roles in Drosophila melanogaster and Caenorhabditis elegans including oogenesis, cell motility and spermatogenesis (Mermall and Miller, 1995; Kelleher et al, 2000; Geisbrecht and Montell, 2002; Rogat and Miller, 2002). In mammalian cells, myosin VI is involved in endocytosis at the plasma membrane and in the maintenance of Golgi complex morphology and secretion (Buss et al, 1998, 2001; Warner et al, 2003). To understand how myosin VI functions in these diverse cellular events, we need to establish its molecular properties. Since the initial discovery of myosin VI in D. melanogaster (Kellerman and Miller, 1992), the identification of a heptad repeat sequence in the tail domain by the COILS prediction program (Lupas, 1996) has led to the belief that this region forms a stable double helical coiled coil and that myosin VI exists as a dimer. Possibly as a result of this assumption, in vitro motility studies have been carried out on constructs where dimerisation has been enforced by the presence of a C-terminal leucine zipper or myosin II rod domain (De La Cruz et al, 2001; Rock et al, 2001; Nishikawa et al, 2002; Morris et al, 2003). Studies addressing the kinetic properties of myosin VI have been carried out on motor domain constructs, the largest of which includes most of the helical tail region but without the remainder of the ‘cargo-binding' tail (De La Cruz et al, 2001; Morris et al, 2003). The kinetic and in vitro motility studies have demonstrated that these constructs have a high duty ratio (they remain strongly bound to actin for >50% of their ATPase cycle) and move processively along actin filaments with a large step size (∼30 nm). If only the classical ‘lever arm mechanism' (reviewed in Geeves and Holmes, 1999) is involved, the single IQ motif of myosin VI cannot account for the observed large step size of the processively moving dimer, unless additional structures contribute to the effective lever. Different groups (Rock et al, 2001; Nishikawa et al, 2002) have presented alternative models to explain how the large step size could be achieved (see Discussion). Thus, to further explore the properties of myosin VI and see if the presence of the whole tail affected its behaviour, we have expressed and purified the unmodified full-length molecule using the baculovirus system. Here we report that this molecule is a monomeric nonprocessive motor with a large step size that undergoes a large conformational change on the addition of ATP.
Results
Physical characterisation of expressed myosin VI using gel filtration and sucrose density gradients
On a Superdex 200 FPLC column calibrated with standard globular proteins of known Stokes radii, myosin VI eluted as a single peak (Figure 1A) with a Stokes radius (Rs) of 6.0 nm (Figure 1B), indicating that like all the other myosins so far measured, myosin VI is an elongated protein. We compared its elution position with that of HMM (heavy meromyosin) (molecular weight (MW) 350 kDa), a skeletal muscle myosin II motor domain dimer with a short tail (elution volume 7.8 ml), and with that of S1(A2) (subfragment-1) (MW 106 kDa), a single myosin II motor domain with one light chain but no tail (12.0 ml) (Figure 1A). Myosin VI at 10.8 ml eluted close to S1(A2), suggesting that it is monomeric (calculated monomer MW 164 kDa) (Figure 1B).
Figure 1.
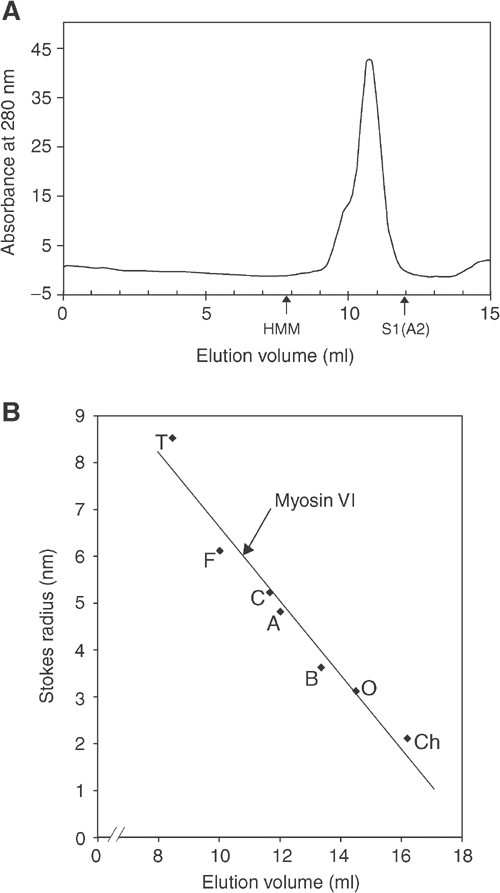
FPLC Superdex 200 gel filtration of myosin VI. (A) Typical elution profile of myosin VI on a Superdex 200 column. The elution positions for myosin II HMM (350 kDa) (a dimer consisting of two motor domains, four IQ domains with four light chains and a short tail) and myosin II S1(A2) (106 kDa) (a single motor domain and one IQ domain with one A2 light chain bound) are shown. (B) Calibration curve of the Superdex 200 gel filtration column using standard proteins of known Stokes radii: (Ch) chymotrypsinogen A, 2.1 nm; (O) ovalbumin, 3.1 nm; (B) BSA, 3.6 nm; (A) aldolase, 4.8 nm; (C) catalase, 5.2 nm; (F) ferritin, 6.1 nm; (T) thyroglobulin, 8.5 nm. The elution position of myosin VI is shown, giving it a Stokes radius of 6 nm.
The sedimentation coefficient of our myosin VI was measured by sucrose density centrifugation together with standard proteins of known sedimentation coefficients and the fractions analysed by SDS–PAGE (Figure 2A). The peak fractions for each protein from eight different runs were determined, the average calculated and used to plot a calibration curve (Figure 2B) from which a sedimentation coefficient (s20,ω) for myosin VI of 6.3 S was determined. Increasing the run temperature from 4 to 25°C, decreasing the NaCl concentration of the buffer from 500 to 25 mM or removal of the N-terminal hexa-his tag with rTEV protease had no effect on the migration position (at protein concentrations up to ∼5 μM). We also phosphorylated myosin VI with myosin I heavy-chain kinase (a kind gift from Dr ED Korn, NIH, Washington, USA) and [32P]-γ-ATP or dephosphorylated it with serine/threonine phosphatase (PP1) (New England Biolabs), but no change in its migration position was seen, indicating that no major conformational changes had occurred.
Figure 2.
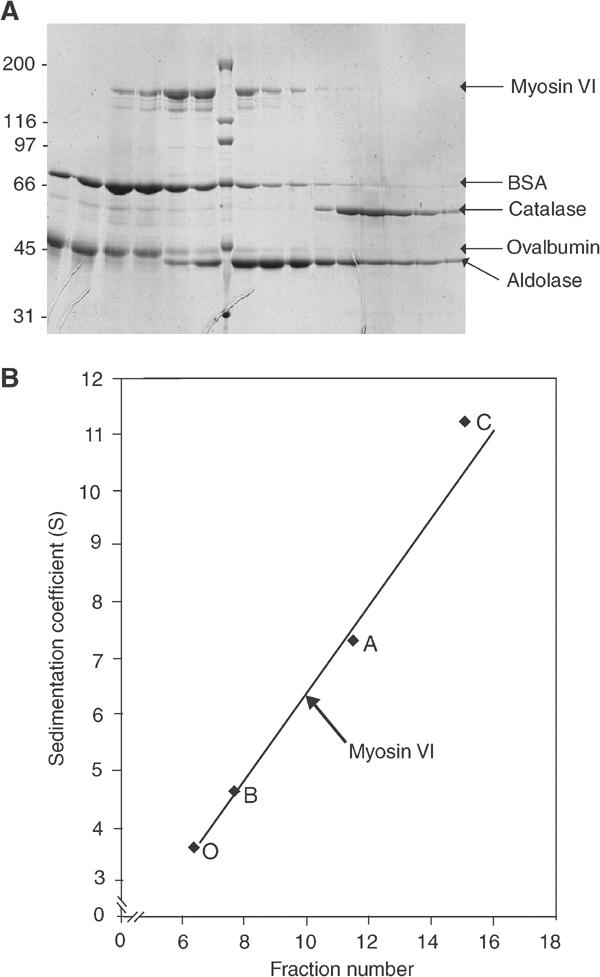
Sucrose density gradient ultracentrifugation of myosin VI. (A) SDS–PAGE gel showing the fractions from a typical sucrose density gradient centrifugation of myosin VI and standard proteins run on a 6–20% sucrose gradient. Note that catalase (232 kDa) and aldolase (158 kDa) consist of tetramers of ∼60 and ∼40 kDa, respectively. Molecular weight markers are indicated on the left and in the middle lane of the gel in kDa. (B) Calibration curve of standard proteins with known sedimentation coefficients from the collected data of eight sucrose density gradient runs similar to that shown in (A). The standard proteins used with their sedimentation coefficients were as follows: (O) ovalbumin, 3.55 S; (B) BSA, 4.6 S; (A) aldolase, 7.3 S; (C) catalase, 11.2 S. The position of myosin VI in the gradient is shown giving it a sedimentation coefficient of 6.3 S.
Cytosolic extracts, prepared from normal rat kidney (NRK) fibroblastic tissue culture cells, were similarly centrifuged on sucrose density gradients (data not shown). Myosin VI was identified in the resulting fractions by Western blotting with specific antibodies. The native myosin VI was found at the same position as the expressed protein on these gradients, demonstrating that under these conditions it too is monomeric.
The sedimentation coefficient (s20,ω) and Stokes radius (Rs) were used to calculate the experimental MW of myosin VI using equation 1 (see Materials and methods). This gave a value of 153 kDa, which compares well with a calculated MW of 164 kDa and clearly demonstrates that both native and expressed myosin VI behave as monomers.
Does the zero-length crosslinker EDC cross link myosin VI?
Expressed myosin VI and myosin II HMM were treated with 5 mM EDC for 30, 60 and 120 min at room temperature (RT) and run on SDS gels (Figure 3A). The dimeric HMM is increasingly crosslinked with time as shown by the increase in the ∼300 kDa dimer band and the decrease in the intensity of the ∼150 kDa monomer band. Myosin VI however remains completely uncross-linked at the protein concentrations tested (up to 3 μM). The EDC-treated myosin VI was also run on sucrose density gradients as described above and found to migrate in the same position as untreated protein.
Figure 3.
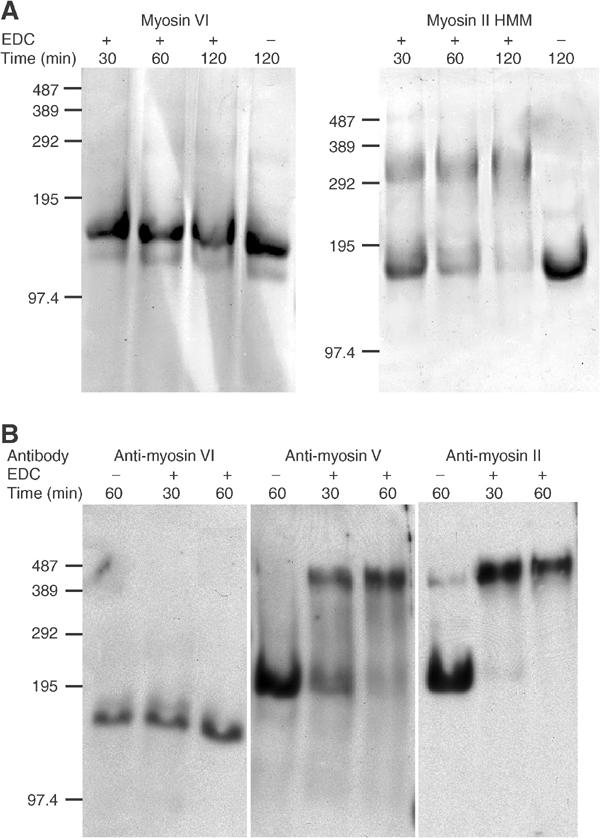
Does EDC crosslink expressed and ‘native' myosin VI? (A) Purified myosin VI and myosin II HMM were treated with 5 mM EDC at RT and samples were taken at 30, 60 and 120 min and run on 3% acrylamide SDS-phosphate PAGE. Controls without EDC were left at RT for 120 min. Molecular weight markers (kDa) are indicated on the left of each gel. (B) NRK cells were grown to confluence and lysed. After centrifugation, the supernatant was incubated at RT with 50 mM EDC for 30 and 60 min or without EDC for 60 min. Samples were run on SDS- phosphate PAGE gels and blotted for myosin VI, myosin V and non-muscle myosin II. Molecular weight markers (kDa) are on the left.
When cytosol prepared from NRK cells was treated in an identical manner but with 50 mM EDC and the resulting samples were run on gels and blotted for myosins VI, V and II, the results show very clearly that cellular myosin VI remains uncrosslinked while myosins II and V become progressively crosslinked (Figure 3B). These results clearly indicate that native myosin VI and expressed myosin VI behave very similarly and are monomeric.
Myosin VI in the electron microscope
A field of negatively stained myosin VI shows that the molecules have a globular and asymmetric appearance (Figure 4A). There is no evidence for dimerisation, and the possibility of two molecules being joined by a connection too thin to be visible at the ∼2 nm resolution of the negative staining method appears unlikely, as dilution experiments showed only increased separation between the molecules and no evidence of pairing (data not shown). Figures 4B and C show selected class averages after single-particle image processing of the molecules stained in the absence of nucleotide (apo) and in the presence of ATP. The averages of the molecules show a large globular region at one end that is 8–9 nm long and this we take to be the motor domain. This region has similarities to the motor domain in image averages of negatively stained myosin II and V (Burgess et al, 1997; Walker et al, 2000). However, the features of the putative motor domain were less distinctive than those of myosin II or V and the image averages were more variable, possibly due to a greater rotational freedom of the molecule about its long axis in attaching to the carbon substrate. At one end of the motor domain is a smaller region that may be the single IQ motif with its light chain and is similar in size to the first light-chain region of myosins II and V outside the motor domain. Beyond this is a second small region similar in size to the first that is presumably the tail of the molecule. The image averages show evidence of a gross conformational change in response to the addition of ATP. The molecules in ATP are relatively straight and have an overall length of about 17 nm. Apo molecules are about 20% shorter and have a strongly bent or hooked appearance in the light chain/tail region, giving the impression that the tail may have folded back across or under the molecule.
Figure 4.
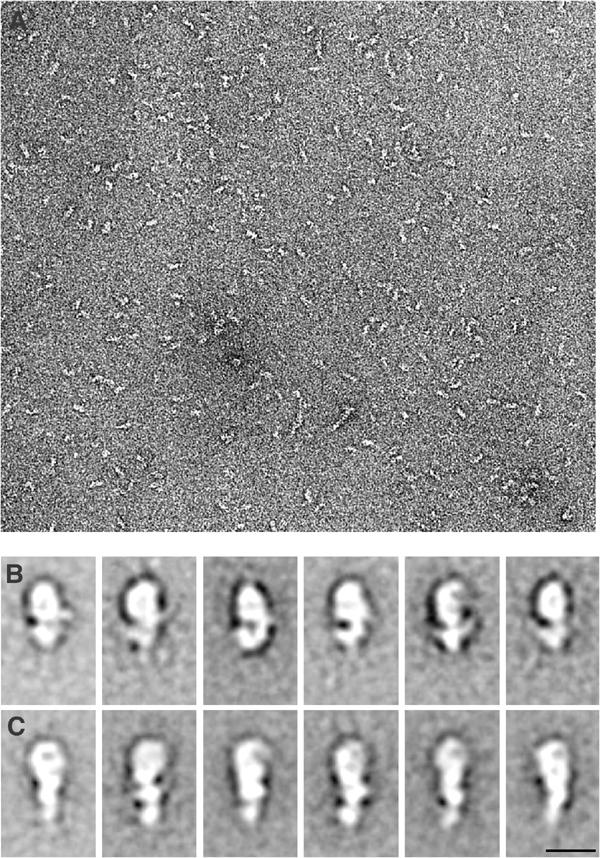
Electron micrographs of negatively stained myosin VI molecules. (A) Field showing molecules stained in the presence of 100 μM ATP; note there is little or no evidence of pairs of molecules that would indicate a dimer. Scale bar, 5 nm. (B, C) Image averages after alignment and classification by single-particle processing of molecules stained without nucleotide (B) and in the presence of 100 μM ATP (C). The large globular region in the upper parts of the panels is tentatively identified as the motor domain. Note the hooked appearance in the apo molecules compared to the straighter molecules in ATP. Scale bar, 10 nm.
What type of motor is full-length myosin VI?
Gliding actin filament assays were performed to determine the velocity of actin translocation by myosin VI. Antibodies against the helical domain and against the globular tail domain were used to anchor myosin VI to the flow cell surface. In an assay buffer containing 5 mM Mg.ATP, myosin VI gave an average velocity of 71±15 nm s−1 with the helical domain antibody. This value was obtained from five different protein preparations and 89 filaments. A very similar velocity of 75±10 nm s−1 was obtained with an antibody against the C-terminal globular tail domain. (Supplementary Figure 1). Our velocities agree well with those previously published for a construct of myosin VI (containing the motor domain, 53 residue insert and IQ motif) of 60–130 nm s−1 (Wells et al, 1999; Morris et al, 2003). Other published data on dimeric constructs of myosin VI give higher velocities of ∼300 nm s−1 (Rock et al, 2001; Morris et al, 2003), which are thought to be the result of dimerisation. Within the field of view, it was common to see 80% of the filaments moving, indicating a low number of nonfunctional myosin VI molecules (dead heads). This was further confirmed using actin sedimentation experiments, where nearly all the myosin VI pelleted with actin and at least 80% was released from the actin pellet with Mg.ATP at high salt. Performing in vitro motility assays at decreasing concentrations of myosin VI resulted in short-lived actin binding. Movement over a single point of attachment characteristic of processive motors like myosin (Howard et al 1989; Mehta et al, 1999) was not observed. Preliminary landing rates (data not shown) suggested a minimum number of two myosin VI molecules required to move an actin filament, consistent with a nonprocessive motor (Howard et al, 1989).
We measured single-molecule mechanical interactions of myosin VI with F-actin using an optical tweezers-based force transducer in three-bead configuration to determine the size of its working stroke (Molloy et al, 1995; Veigel et al, 1998). When myosin VI was attached to beads in the flow cell with either the helical domain or globular tail domain antibody, it produced single, isolated interactions with the actin filament at all ATP concentrations tested (Figure 5A). The working strokes were about 18 nm (Figure 5B) for both antibodies. The histogram of lifetimes of attachment events could be fitted by a single exponential for all ATP concentrations studied (Figure 5C). The detachment rate constants increased with higher ATP concentrations and could be fitted by a saturation growth rate model, kdet=(kmax[ATP])/(Km+[ATP]), yielding a maximum detachment rate kmax of 7.1 s−1 (Figure 5D). The association rate constant (kass) for ATP binding to actomyosin VI obtained for ATP concentrations ⩽0.1 mM was 21 s−1 mM−1. This value is in good agreement with the association rate constant for ATP binding of 18 s−1 mM−1 observed in a solution kinetics study of actomyosin VI (De La Cruz et al, 2001).
Figure 5.
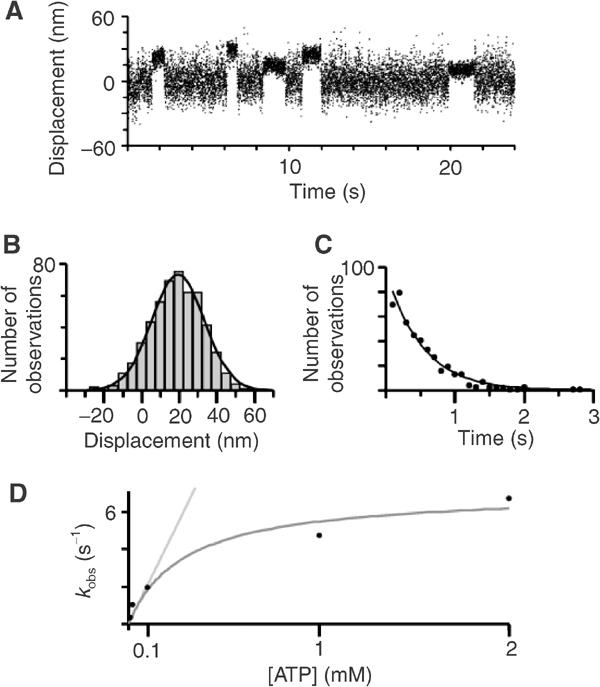
Single-molecule mechanical interactions of myosin VI with actin measured using optical tweezers. (A) Mechanical interactions were measured by monitoring the positions of beads holding the actin filament using two photodetectors (Veigel et al, 1998). The record shows bead movement in parallel with the actin filament axis. Intervals of reduced Brownian noise indicate attachment events of a single myosin VI molecule bound to the actin. [ATP]=0.02 mM. (B) Amplitude of displacements (i.e. working strokes), mean±s.e.m. 19.4±0.8 nm, n=511, [ATP]=0.02 mM. (C) Lifetimes of myosin–actin attachments. The single exponential fit gives a detachment rate kdet of 1.9 s−1, n=441, [ATP]=0.1 mM. (D) Detachment rates of myosin VI from actin at different ATP concentrations. Saturation growth rate fit, kdet=(kmax[ATP])/(Km+[ATP]), gives a maximum detachment rate kmax of 7.1 s−1. Linear fit, kobs=kass[ATP], for ATP concentrations <0.1 mM, gives an association rate kass for ATP binding to actomyosin VI of 21 s−1 mM−1. For conditions, see Materials and methods.
To look more closely at the working stroke of myosin VI, the attachment events were analysed using ensemble-averaging as described by Veigel et al (2002). This suggests that displacements were produced in two distinct phases (Figures 6A and B). An initial large movement of about 17 nm is produced within 15 ms (the time resolution of our experiments) of binding to actin, with the transition therefore too fast to be resolved here. A second small step of about 1 nm occurs after a variable time delay (phase 1) from the onset of binding. The time course of phase 1 could be fitted by a single exponential with an average rate constant k1 of 5.0 s−1, independent of ATP concentration (Figure 6C), suggesting that nucleotide is still bound during this phase or ATP binding was prevented for some reason (Veigel et al, 2002). The value for k1 compares well with the value for ADP release of 6.4 s−1 reported by De La Cruz et al (2001), found by the authors to be the rate-limiting step of the actomyosin VI cycle. The new position was maintained for another variable period (phase 2) terminated by ATP-dependent actomyosin VI dissociation. The lifetimes of phase 2 were also exponentially distributed, with the durations becoming briefer, expressed by an increase in k2, with an increase in ATP concentration (Figure 6C).
Figure 6.
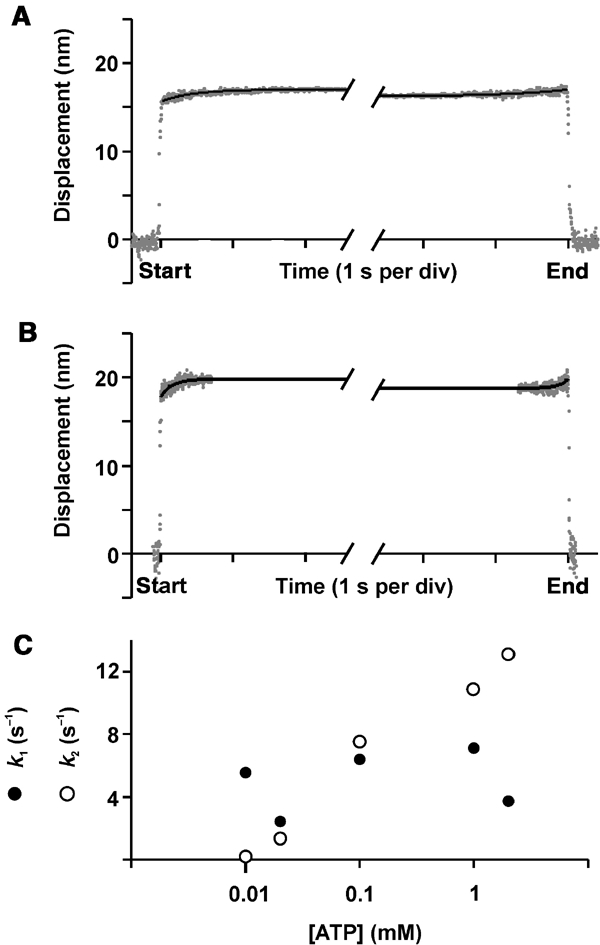
Ensemble-averaged myosin VI–actin attachment events measured at two different ATP concentrations. (A) 0.02 mM and (B) 0.1 mM ATP. Ensemble-averaging the single-molecule myosin VI–actin attachment events (Veigel et al, 2002) showed that the displacements (working strokes) were produced in two distinct phases. The kinetics of the first and second phases were obtained by synchronizing the events with respect to the start and end of each event and by making them of the same duration by extending short events while keeping the level reached at the end and the start of the event, respectively (see Veigel et al, 2002). The time course of the ensemble average was fitted by single exponentials. (A) k1=2.4 s−1, k2=1.3 s−1, n=325, [ATP]=0.02 mM. (B) k1=6.4 s−1, k2=7.5 s−1, n=156, [ATP]=0.1 mM. (C) k1 and k2 at different ATP concentrations. For details and conditions, see Materials and methods.
Discussion
Our hydrodynamic, crosslinking and EM data demonstrate that full-length myosin VI is a monomer under the conditions we have tested. Since its identification in D. melanogaster (Kellerman and Miller, 1992), the first half of the tail sequence of myosin VI has been predicted by programs such as COILS (Lupas, 1996) to form a coiled coil. On this basis, myosin VI has been assumed to be a dimeric molecule, although this has never been experimentally tested. However, a closer analysis of the COILS prediction (Lupas, 1996) for the helical tail domain of myosin VI (Figure 7) shows a number of features that would make the formation of a stable coiled coil questionable. Firstly, there are a number of stutters and stammers within the first part of the predicted coiled-coil region (shown by asterisks above the sequence in Figure 7) where the a and d pattern of hydrophobic repeats breaks down and these may be points where the helix is broken and loops occur. Secondly, the COILS program is well known for its overprediction of E residues in heptad repeats (Lupas, 1996), and this may be the reason why the central ‘charged' region (Figure 7) is predicted to form a coiled coil when it seems highly unlikely that this is possible. Lyu et al (1992) showed that this type of repeating pattern of alternating four positive and four negative charges forms intramolecular salt bridges at the i and i+4 residues (where i is any residue in the helix and i+4 is the four amino-acid residues C-terminal of residue i, that is, at repeating helix positions) rather than intermolecular salt bridges between two helices. Thus, it would seem more likely that this ‘charged' region forms an independent α-helix. If so, one could propose the following speculative model for the monomeric myosin VI tail: initially it could be composed of an intramolecular double helical bundle (possibly in a helix–loop–helix configuration) followed by a flexible salt bridge stabilised helix (‘charge' region), then a region with a predicted weak coiled coil and finally the globular domain that binds membrane receptors and cargo. Clearly, this model needs to be explored experimentally and so efforts are being made to crystallise these tail domains in the presence and absence of receptor-binding peptides.
In our single-molecule mechanical experiments, myosin VI produced single, isolated interactions with the actin filament (Figure 5A). The same observations have been made for nonprocessive myosins, both monomeric and dimeric (Molloy et al, 1995; Veigel et al, 1999; Warshaw et al, 2000; Ruff et al, 2001), and for monomeric constructs of processive myosins (Moore et al, 2001; Purcell et al, 2002; Veigel et al, 2002). As we have not observed processive interactions (neither in the motility assays nor in the optical tweezer studies), a monomeric myosin VI would be sufficient to explain our mechanical results. We observed a large working stroke for myosin VI of about 18 nm (Figure 5B). Ensemble-averaging of the attachment events showed that myosin VI, like some other myosins (Veigel et al, 1999, 2002), produces its working stroke in two steps (Figures 6A and B). The phase following the initial displacement of 17 nm appears to be a nucleotide-bound state, possibly actomyosin-ADP, as its lifetime is independent of ATP concentration. The second step of 1 nm seems to lead to a nucleotide-free rigor state, as the lifetime of the subsequent phase is ATP dependent. Despite its small size we believe that this second step is a feature of myosin VI, as it is consistently present, the time courses for both phases are in general agreement with the myosin VI biochemical data (De La Cruz et al, 2001) and electron microscopy of the rigor state showed movement of the putative lever upon addition of ADP (Wells et al, 1999).
While there is no universal agreement (see Kitamura et al, 1999; Tanaka et al, 2002), a general consensus has emerged that the size of the working stroke of a given myosin depends in a linear fashion on the length of the light-chain binding domain (lever arm) (Molloy et al, 1995; Veigel et al, 1999; Warshaw et al, 2000; Moore et al, 2001; Ruff et al, 2001; Purcell et al, 2002). Thus, the stepping behaviour of most of the myosins so far studied can be explained by the tilting lever model of force production (reviewed in Geeves and Holmes, 1999). Electron microscopy of myosin VI suggests a movement of the putative lever as a function of the nucleotide state (ATP and rigor in this study, and ADP and rigor in Wells et al, 1999) hinting at a lever arm mechanism for myosin VI. Can the working stroke observed here be explained with a lever arm mechanism? Kohler et al (2003) observed that myosin I isoforms with similar lever arm lengths can produce working strokes of different sizes, and suggested that this could be explained by different degrees of rotation of their lever arms. In the crystal structures of a number of class II myosins (Rayment et al, 1993; Dominguez et al, 1998; Houdusse et al, 1999, 2000), the orientation of the lever arm is thought to be either near its prepower stroke or near its rigor position, and the rotation between these positions varies significantly when structures of different myosin isoforms are compared (Houdusse et al, 1999, 2000). Visualising different myosins bound to actin using cryo-electron microscopy and image processing showed that the lever arm rotation associated with ADP release also varies between different myosins (Jontes et al, 1995; Whittaker et al, 1995; Wells et al, 1999). In myosin VI, it was found that the ADP orientation of the lever arm points about 65° towards the minus end of the actin filament, with the rigor one pointing even further, at about 85°, towards the same end (Wells et al, 1999). The authors point out that despite the relatively large rotation of about 20° associated with ADP release, not much movement can be expected parallel to the actin filament. Thus, the orientation of the lever arm is relevant for determining the size of the myosin working stroke.
Therefore, the size of the working stroke of a given myosin depends not only on the length of the lever arm but also on its orientation with regard to the actin filament and degree of rotation. These findings are reflected in a simple geometrical model in Figure 8 where the size of the working stroke is determined by a combination of lever arm length, orientation and rotation. According to this model, a displacement parallel to the actin filament towards its minus end can be expressed as follows: displacement=lever arm length(sin(final orientation)−sin(starting orientation)). Is the model sufficient to account for the myosin VI working strokes observed by us? The single IQ motif of myosin VI is expected to contribute about 4 nm to its effective lever arm (Rayment et al, 1993). It is conceivable that in myosin VI, the single IQ motif, the unique 53-amino-acid insert and possibly the first part of the helical tail region could form an extended lever arm of about 10 nm. A lever arm of this length is also consistent with the putative levers visualised for myosin VI using electron microscopy in this study (Figure 4) and by Wells et al (1999). In our model, such a lever of only 10 nm can account for the two substeps of 17 and 1 nm measured here and is also consistent with the ADP (65°) and rigor (85°) orientations reported previously (Wells et al, 1999). In order for a 10 nm lever to produce the first substep of 17 nm, finishing in the ADP orientation (65°), a prepower stroke orientation of −53° is calculated. In the model, the movement of a 10 nm lever from its 65° ADP to its 85° rigor orientation accounts for a substep of 0.9 nm, very similar to the 1 nm substep observed here and consistent with the small movement parallel to the actin filament expected by Wells et al (1999) for that substep. In total then, the swing of the myosin VI lever arm would be 138°. The strongly bent or hooked appearance of the lever arm/tail region in rigor (at the end of the working stroke) (Figure 4B) is consistent with the model.
Figure 8.
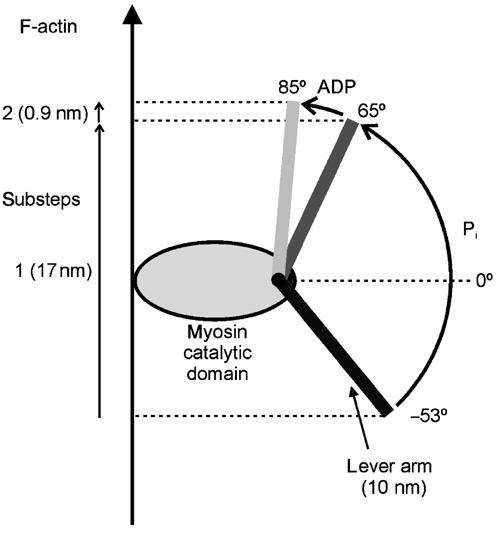
A possible model to explain the working stroke of our myosin VI. The cartoon shows how we assume that the working stroke taken by myosin VI results from lever arm length, orientation and rotation. A displacement parallel to the actin filament towards its minus end can be expressed as follows: displacement (working stroke)=lever arm length(sin(final orientation)−sin(starting orientation)). An assumed lever arm of 10 nm can account for the two substeps of 17 and 1 nm measured here and is also consistent with the ADP (65°) and rigor (85°) orientations reported previously by Wells et al (1999). In order for the lever to produce the first substep of 17 nm, finishing in the ADP orientation (65°), a prepower stroke orientation of −53° is calculated. The movement of the lever from its 65° ADP to its 85° rigor orientation accounts for a substep of 0.9 nm. At present, we stress that it is still an assumption that other regions of the molecule act as the additional effective rigid lever and that it rotates by 118° before ADP release.
At this stage however it should be stressed that one should not exclude alternatives to the tilting lever mechanism for the myosin VI stepping mechanism. Rock et al (2001) have speculated that the large step size of their myosin VI dimer was the result of a small working stroke in the ‘lead' head attached to actin coupled to a large conformational change, which allows the free ‘trailing' head to extend to the next available binding site on the actin filament. They suggest that unfolding the 53-residue insert between the converter domain and light-chain binding domain, along with adjacent structures, causes the large conformational change. Nishikawa et al (2002) however proposed that the lever arm length of their myosin VI dimer was not critical for its processivity or its large step size. Based on electron microscopy, they postulated that binding of the first head causes conformational changes in the actin filament that allows the second head to bind 36 nm along the filament. While the structure of the neck and tail domains remains to be determined, it is difficult to establish whether or not they or parts of them contribute towards the effective lever arm and whether they would be stiff enough to generate displacement under load. Further EM studies on the conformational changes induced in myosin VI on addition of nucleotide especially when bound to actin should also help to test our model.
Previous in vitro motility and single-molecule mechanical experiments have shown that dimeric myosin VI constructs, where dimerisation was ensured by the inclusion of either a leucine zipper (Rock et al, 2001) or a short segment of myosin II tail (Nishikawa et al, 2002), move processively with large steps of 30–36 nm. Our expressed full-length myosin VI however is monomeric and produces a large 18 nm working stroke, and does not show processive behaviour. Myosin V produces a power stroke of 25 nm while stepping 36 nm when moving processively, with the 11 nm disparity thought to be bridged by diffusive movement of the free head (Veigel et al, 2002). For myosin VI, interestingly, the difference between the processive step size of 30 nm observed by (Rock et al, 2001) and the 18 nm power stroke observed here is similar, and a diffusive movement of 12 nm by the free head of a dimeric myosin VI would be sufficient to reach its observed binding position on actin.
The in vitro results presented here and the previous studies (Rock et al, 2001; Nishikawa et al, 2002) raise the intriguing possibility that myosin VI may be able to function as a monomer and as a dimer in vivo. Our cellular localisation and functional studies suggest that myosin VI has multiple roles in cells, and certain functions such as those involving maintenance of the Golgi complex structure (Warner et al, 2003) might require a nonprocessive monomer, while other functions such as transport of endocytic vesicles (Buss et al, 2001) might benefit more from a processive dimer. In these cases, it is conceivable that myosin VI binding to one of its binding partners, or a post-translational modification of myosin VI such as phosphorylation, might cause dimerisation at the site of action such as on a lipid membrane. The ability to regulate whether myosin VI is a monomer or dimer would be an important way in which multiple cellular functions might be controlled, and experiments are underway to explore these intriguing possibilities.
Materials and methods
Protein expression and purification
Myosin VI from chicken intestinal brush border cells containing the large insert (residues 1–1276) (Buss et al, 1998) was cloned into the Bac-to-Bac (Invitrogen, UK) vector FasBacHT such that the expressed protein contained a hexa-his tag at the N-terminus. The Bac-to-Bac protocol as described in the Invitrogen instruction manual was used to produce recombinant baculovirus. Sf9 cells were simultaneously infected with myosin VI virus and calmodulin virus (a gift from Dr J Sellers, NIH, Washington, USA).
Sf9 cell pellets expressing myosin VI were frozen in liquid nitrogen and thawed in 500 mM NaCl, 50 mM Tris–HCl (pH 8.0), 0.7% Nonidet P-40, 10% glycerol, 1–2 complete EDTA-free protease inhibitor tablets per 50 ml (Roche Diagnostics), 2 mM ATP, 5 mM MgCl2 and 50 mM glucose and lysed by sonication. The lysate was spun at 85 000 g for 30 min, and 0.2 mg ml−1 hexokinase (Sigma) and 0.2 mg ml−1 F-actin were added to the supernatant and incubated for 1 h at 4°C with constant agitation. The supernatant was then centrifuged again at 85 000 g for 30 min and the actomyosin VI pellet was resuspended by homogenisation in 500 mM NaCl, 10 mM imidazole (pH 7.4), 10 mM Na2PO42− (pH 7.2), one protease inhibitor tablet, 5 mM ATP, 5 mM MgCl2, 0.1 mM EGTA and 5 mM β-mercaptoethanol and incubated with 0.5 ml Ni-NTA agarose (Qiagen) on a rotary mixer at 4°C for 30 min. After washing on a column with the above solution (5 × 5 ml), myosin VI was eluted from the resin with elution buffer A (100 mM NaCl, 150 mM imidazole (pH 7.4), 10 mM Na2PO42− (pH 7.2), 1 mM MgCl2, 0.1 mM EGTA and 2.5 mM β-mercaptoethanol). The main fractions containing myosin VI were pooled and dialysed against 30 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 20 mM MOPS (pH 7.5) and 1 mM DTT. The N-terminal His tag could be removed using rTEV protease (Invitrogen, UK) specific for a cleavage site located between the His tag and the myosin VI reading frame. Myosin VI tail constructs were expressed using bacterial expression systems and purified as described previously (Buss et al, 1998).
F-actin was prepared from chicken skeletal muscle acetone powder (Kendrick-Jones et al, 1970; Pardee and Spudich, 1982), and myosin II, myosin II HMM, myosin II S1(A1) and myosin II S1(A2) were from chicken skeletal muscle (Margossian and Lowey, 1982).
Antibody preparation and affinity purification
Polyclonal rabbit antibodies were raised against the whole tail (residues 846–1276) and predicted globular tail domain (residues 1062–1273) of myosin VI (Buss et al, 1998), the tail of myosin V (Lionne et al, 2001) and against non-muscle myosin II (Scholey et al, 1982). Antibodies against myosin VI and V were affinity-purified before use (Buss et al, 1998). SDS–PAGE and Western blotting using different expressed constructs of the myosin VI tail were used to determine the specificity of the whole tail antibody for the helical domain and the globular tail antibody for the globular tail domain.
Chemical crosslinking of purified protein and cellular extracts
In all, 100 μl of purified myosin VI at 0.2–0.5 mg ml−1 or skeletal muscle myosin II HMM at 0.2 mg ml−1, both in elution buffer A (see above for composition), was mixed with a final concentration of 5 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, Sigma, UK) at RT. At 30, 60, 90 and 120 min, samples were taken and added to phosphate gel sample buffer (28 mM NaH2PO4, 72 mM Na2HPO4, 0.2% SDS, 6 M urea, 0.9 M β-mercaptoethanol, 0.01% bromophenol blue) and immediately frozen on dry ice. SDS phosphate gels (3% acrylamide, 28 mM NaH2PO4, 72 mM Na2HPO4, 0.2% SDS) were prepared as described in the Sigma technical bulletin MWS-877X. Crosslinked phosphorylase-b molecular weight marker (Sigma, UK) was used to calibrate the gels.
NRK and A431 (human epithelial) cells were grown in Dulbecco's modified Eagle's medium with Glutamax-1, sodium pyruvate, 4500 mg l−1 glucose and pyridoxine (Invitrogen) supplemented with 10% FCS (Fetaclone 1, Hyclone), penicillin and streptomycin (100 μg ml−1, Invitrogen) at 37°C in 5% CO2. Cells were grown in 90 mm dishes to confluence, lysed in 500 mM NaCl, 50 mM Tris–HCl (pH 8.0), 0.7% Nonidet P-40, one protease inhibitor tablet per 50 ml (Roche Diagnostics), 2 mM ATP and 5 mM MgCl2, sonicated and centrifuged for 30 min at 195 000 g. To the supernatant, termed NRK or A431 cytosol, EDC was added to a final concentration of 50 mM and a time course reaction was carried out and samples were run on SDS phosphate gels as described above. Western blots using anti-myosin VI whole tail, anti-myosin V tail and anti-myosin II antibodies were carried out to detect these myosins. Aliquots of the NRK cytosol were also run on sucrose density gradients.
Gel filtration
A measure of 150–250 μl samples of purified protein at 0.3–0.8 mg ml−1 were applied to a Superdex 200 (30 × 1.5 cm) analytical column (Amersham Pharmacia Biotech) equilibrated in 25 mM NaCl, 10 mM Tris (pH 7.5), 10 mM imidazole (pH 7.4), 2 mM MgCl2 and 1 mM EGTA and controlled using an AKTA purifier (Amersham Pharmacia Biotech). The column was calibrated with chymotrypsinogen A, ovalbumin, BSA, aldolase, catalase, ferritin and thyroglobulin standards. The same protocol was carried out using a Sephacryl S300 (Amersham Pharmacia Biotech) (90 × 1.5 cm) column under gravity flow and similar results were obtained.
Sucrose density gradients
The 6–20% sucrose gradients contained 500 mM NaCl, 150 mM imidazole (pH 7.4), 10 mM Na2PO42− (pH 7.2), 5 mM MgCl2 and 1 mM EGTA. Samples of ∼1.5 mg ml−1 of purified myosin VI or 300 μl of NRK cell cytosol, together with protein standards of 0.5 mg ovalbumin, 0.25 mg BSA, 0.5 mg aldolase and 0.5 mg catalase, were layered on top of the gradient in a volume of 400 μl. The gradients were centrifuged for 18 h at 38 000 rpm in an SW Ti 60 Beckman rotor. Following centrifugation, 200 μl fractions were taken starting from the top and aliquots run on 6%–20% acrylamide gradient SDS–PAGE gels. In the case of tissue culture cell cytosol, myosin VI was detected by Western blotting using anti-myosin VI whole tail affinity-purified antibody.
Calculation of native molecular weight
The native molecular weight of myosin VI was calculated using its Stokes radius measured by gel filtration and its sedimentation coefficient determined by sucrose density gradient centrifugation using the following equation as described in Post et al (2002):
where Avogadro's number N=6.02 × 1023, viscosity coefficient n=1 × 10−2 g s−1 cm−1, solution density ρ=1 g cm−3 and partial specific volume υ=0.72 cm3 g−1 (assumed to be that of an average soluble protein).
Motility assays
The procedure was adapted from that described by Kron et al (1991) and Sellers et al (1993). The assay buffer was 25 mM NaCl, 20 mM Tris–HCl (pH 8.0), 20 mM imidazole (pH 7.4), 5 mM MgCl2, 1 mM EGTA and 10 mM DTT. Myosin VI was bound to the nitrocellulose-coated surface of the flow cell (volume 20 μl) by affinity-purified polyclonal myosin VI antibodies (0.1 μg ml−1) against the helical domain or globular tail domain. All the assays were carried out at 28°C. Images were taken at set time intervals and viewed in Adobe Photoshop. To obtain the velocity of any actin filament, the x and y coordinates of one end of the filament were obtained for 10–15 images. The distance c between pairs of coordinates in consecutive frames could then be calculated using the equation c=√((a2)+(b2)), where a=x2–x1 and b=y2–y1. The distance c in pixels was converted to μm and divided by the time elapsed between each image to give the velocity of the filament between two images. The average velocity of the actin filament was obtained by dividing the sum of all these velocities by n−1, where n is the number of images.
Optical tweezers
The optical trapping procedure and conditions using rhodamine–phalloidin-labelled rabbit F-actin and N-ethyl maleimide (NEM)-modified rabbit myosin were as described previously (Veigel et al, 1998, 1999). In the flow cell, stationary beads coated with nitrocellulose were first treated with affinity-purified polyclonal antibodies (0.1 μg ml−1) against the helical domain or globular tail region of myosin VI and then blocked with 0.5 mg ml−1 BSA (Sigma). Myosin VI was then applied at a sufficiently low density of 0.02 μg ml−1 for single acto-myosin molecular interactions to occur (binding events were separated by long intervening periods of free bead diffusion and ∼70% of the stationary beads tested showed no interactions with the actin filament). Mechanical interactions were measured by monitoring the positions of the microspheres holding the actin filament using two photodetectors (Veigel et al, 1998). Myosin VI produced single, isolated interactions with the actin filament, which were identified by the change in variance of the Brownian noise of the signal, corresponding to sudden changes in system stiffness as myosin bound to actin (Molloy et al, 1995). The lifetime and amplitude of each attachment were measured, the latter relative to the mean rest position for the bead before and after attachment, and corrected for series elastic components in the system. Individual displacements were biased in one direction, determined by the orientation of the actin filament. The working stroke was calculated by analysing the amplitudes of a minimum of 100 displacement events observed for an individual actin filament and by combining the results of at least three filaments. Attachment events were also closely inspected for the presence of a substep by ensemble-averaging the data as described previously (Veigel et al, 2002). ATP concentrations between 0.01 and 2 mM were used.
Electron microscopy of myosin VI using negative stain
The negative staining procedure was similar to that described previously (Walker et al, 1985; Burgess et al, 1997). Myosin VI samples diluted 5–10 times with 25 mM KCl, 10 mM MOPS, 1 mM EGTA, 1 mM MgCl2 and 2 mM K2PO4 (pH 7.0) (final concentration ∼80 nM) were applied to grids coated with a thin (∼10 nm) continuous film of carbon rendered hydrophilic by irradiation under a UV lamp for about 40 min. The grids were then stained with three drops of 1% uranyl acetate, wicked dry and examined in a Jeol 1200EX electron microscope operating at 80 kV. Images were recorded at a magnification of × 40 000. Image averages were aligned and classified by single-particle processing of the stained molecules as described (Walker et al, 2000).
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank RT Tregear for technical support and discussion, SA Burgess for advice on image processing, D Woolfson and A Lupas for help with the coiled-coil prediction programs, and the MRC, The Royal Society, BBSRC, NIH and Wellcome Trust for financial support.
References
- Berg JS, Powell BC, Cheney RE (2001) A millennial myosin census. Mol Biol Cell 12: 780–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SA, Walker ML, White HD, Trinick J (1997) Flexibility within myosin heads revealed by negative stain and single-particle analysis. J Cell Biol 139: 675–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F, Arden SD, Lindsay M, Luzio JP, Kendrick-Jones J (2001) Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J 20: 3676–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F, Kendrick-Jones J, Lionne C, Knight AE, Cote GP, Paul Luzio J (1998) The localization of myosin VI at the golgi complex and leading edge of fibroblasts and its phosphorylation and recruitment into membrane ruffles of A431 cells after growth factor stimulation. J Cell Biol 143: 1535–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz EM, Ostap EM, Sweeney HL (2001) Kinetic mechanism and regulation of myosin VI. J Biol Chem 276: 32373–32381 [DOI] [PubMed] [Google Scholar]
- Dominguez R, Freyzon Y, Trybus KM, Cohen C (1998) Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell 94: 559–571 [DOI] [PubMed] [Google Scholar]
- Geeves MA, Holmes KC (1999) Structural mechanism of muscle contraction. Annu Rev Biochem 68: 687–728 [DOI] [PubMed] [Google Scholar]
- Geisbrecht ER, Montell DJ (2002) Myosin VI is required for E-cadherin-mediated border cell migration. Nat Cell Biol 4: 616–620 [DOI] [PubMed] [Google Scholar]
- Houdusse A, Kalabokis VN, Himmel D, Szent-Gyorgyi AG, Cohen C (1999) Atomic structure of scallop myosin subfragment S1 complexed with MgADP: a novel conformation of the myosin head. Cell 97: 459–470 [DOI] [PubMed] [Google Scholar]
- Houdusse A, Szent-Gyorgyi AG, Cohen C (2000) Three conformational states of scallop myosin S1. Proc Natl Acad Sci USA 97: 11238–11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ, Vale RD (1989) Movement of microtubules on single molecules. Nature 342: 154–158 [DOI] [PubMed] [Google Scholar]
- Jontes JD, Wilson-Kubalek EM, Milligan RA (1995) A 32 degree tail swing in brush border myosin I on ADP release. Nature 378: 751–753 [DOI] [PubMed] [Google Scholar]
- Kelleher JF, Mandell MA, Moulder G, Hill KL, L'Hernault SW, Barstead R, Titus MA (2000) Myosin VI is required for asymmetric segregation of cellular components during C. elegans spermatogenesis. Curr Biol 10: 1489–1496 [DOI] [PubMed] [Google Scholar]
- Kellerman KA, Miller KG (1992) An unconventional myosin heavy chain gene from Drosophila melanogaster. J Cell Biol 119: 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick-Jones J, Lehman W, Szent-Gyorgyi AG (1970) Regulation in molluscan muscles. J Mol Biol 54: 313–326 [DOI] [PubMed] [Google Scholar]
- Kitamura K, Tokunaga M, Iwane AH, Yanagida T (1999) A single myosin head moves along an actin filament with regular steps of 5.3 nanometres. Nature 397: 129–134 [DOI] [PubMed] [Google Scholar]
- Kohler D, Ruff C, Meyhofer E, Bahler M (2003) Different degrees of lever arm rotation control myosin step size. J Cell Biol 161: 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron SJ, Toyoshima YY, Uyeda TQ, Spudich JA (1991) Assays for actin sliding movement over myosin-coated surfaces. Methods Enzymol 196: 399–416 [DOI] [PubMed] [Google Scholar]
- Lionne C, Buss F, Hodge T, Ihrke G, Kendrick-Jones J (2001) Localization of myosin Va is dependent on the cytoskeletal organization in the cell. Biochem Cell Biol 79: 93–106 [PubMed] [Google Scholar]
- Lupas A (1996) Prediction and analysis of coiled-coil structures. Methods Enzymol 266: 513–525 [DOI] [PubMed] [Google Scholar]
- Lyu PC, Gans PJ, Kallenbach NR (1992) Energetic contribution of solvent-exposed ion pairs to alpha-helix structure. J Mol Biol 223: 343–350 [DOI] [PubMed] [Google Scholar]
- Margossian SS, Lowey S (1982) Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol 85 (Part B): 55–71 [DOI] [PubMed] [Google Scholar]
- Mehta AD, Rock RS, Rief M, Spudich JA, Mooseker MS, Cheney RE (1999) Myosin-V is a processive actin-based motor. Nature 400: 590–593 [DOI] [PubMed] [Google Scholar]
- Mermall V, Miller KG (1995) The 95F unconventional myosin is required for proper organisation of the Drosophila syncytial blastoderm. J Cell Biol 129: 1575–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy JE, Burns JE, Kendrick-Jones J, Tregear RT, White DC (1995) Movement and force produced by a single myosin head. Nature 378: 209–212 [DOI] [PubMed] [Google Scholar]
- Moore JR, Krementsova EB, Trybus KM, Warshaw DM (2001) Myosin V exhibits a high duty cycle and large unitary displacement. J Cell Biol 155: 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CA, Wells AL, Yang Z, Chen LQ, Baldacchino CV, Sweeney HL (2003) Calcium functionally uncouples the heads of myosin VI. J Biol Chem [DOI] [PubMed] [Google Scholar]
- Nishikawa S, Homma K, Komori Y, Iwaki M, Wazawa T, Hikikoshi Iwone A, Saito J, Ikebe R, Katayama E, Yanagida T, Ikebe M (2002) Class VI myosin moves processively along actin filaments backward with large steps. Biochem Biophys Res Commun 290: 311–317 [DOI] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA (1982) Purification of muscle actin. Methods Enzymol 85 (Part B): 164–181 [DOI] [PubMed] [Google Scholar]
- Post PL, Tyska MJ, O'Connell CB, Johung K, Hayward A, Mooseker MS (2002) Myosin-IXb is a single-headed and processive motor. J Biol Chem 277: 11679–11683 [DOI] [PubMed] [Google Scholar]
- Purcell TJ, Morris C, Spudich JA, Sweeney HL (2002) Role of the lever arm in the processive stepping of myosin V. Proc Natl Acad Sci USA 99: 14159–14164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM (1993) Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261: 50–58 [DOI] [PubMed] [Google Scholar]
- Rock RS, Rice SE, Wells AL, Purcell TJ, Spudich JA, Sweeney HL (2001) Myosin VI is a processive motor with a large step size. Proc Natl Acad Sci USA 13: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogat AD, Miller KG (2002) A role for myosin VI in actin dynamics at sites of membrane remodeling during Drosophila spermatogenesis. J Cell Sci 115: 4855–4865 [DOI] [PubMed] [Google Scholar]
- Ruff C, Furch M, Brenner B, Manstein DJ, Meyhofer E (2001) Single-molecule tracking of myosins with genetically engineered amplifier domains. Nat Struct Biol 8: 226–229 [DOI] [PubMed] [Google Scholar]
- Scholey JM, Smith RC, Drenckhahn D, Groschel-Stewart U, Kendrick-Jones J (1982) Thymus myosin. Isolation and characterization of myosin from calf thymus and thymic lymphocytes, and studies on the effect of phosphorylation of its Mr=20,000 light chain. J Biol Chem 257: 7737–7745 [PubMed] [Google Scholar]
- Self T, Sobe T, Copeland NG, Jenkins NA, Avraham KB, Steel KP (1999) Role of myosin VI in the differentiation of cochlear hair cells. Dev Biol 214: 331–341 [DOI] [PubMed] [Google Scholar]
- Sellers JR (2000) Myosins: a diverse superfamily. Biochim Biophys Acta 1496: 3–22 [DOI] [PubMed] [Google Scholar]
- Sellers JR, Cuda G, Wang F, Homsher E (1993) Myosin-specific adaptations of the motility assay. Methods Cell Biol 39: 23–49 [DOI] [PubMed] [Google Scholar]
- Tanaka H, Homma K, Iwane AH, Katayama E, Ikebe R, Saito J, Yanagida T, Ikebe M (2002) The motor domain determines the large step of myosin-V. Nature 415: 192–195 [DOI] [PubMed] [Google Scholar]
- Veigel C, Bartoo ML, White DC, Sparrow JC, Molloy JE (1998) The stiffness of rabbit skeletal actomyosin cross-bridges determined with an optical tweezers transducer. Biophys J 75: 1424–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veigel C, Coluccio LM, Jontes JD, Sparrow JC, Milligan RA, Molloy JE (1999) The motor protein myosin I produces its working stroke in 2 steps. Nature 398: 530–533 [DOI] [PubMed] [Google Scholar]
- Veigel C, Wang F, Bartoo ML, Sellers JR, Molloy JE (2002) The gated gait of the processive molecular motor, myosin V. Nat Cell Biol 4: 59–65 [DOI] [PubMed] [Google Scholar]
- Walker M, Knight P, Trinick J (1985) Negative staining of myosin molecules. J Mol Biol 184: 535–542 [DOI] [PubMed] [Google Scholar]
- Walker ML, Burgess SA, Sellers JR, Wang F, Hammer JA III, Trinick J, Knight PJ (2000) Two-headed binding of a processive myosin to F-actin. Nature 405: 804–807 [DOI] [PubMed] [Google Scholar]
- Warner CL, Stewart A, Luzio JP, Steel KP, Libby RT, Kendrick-Jones J, Buss F (2003) Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell's waltzer mice. EMBO J 22: 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshaw DM, Guilford WH, Freyzon Y, Krementsova E, Palmiter KA, Tyska MJ, Baker JE, Trybus KM (2000) The light chain binding domain of expressed smooth muscle heavy meromyosin acts as a mechanical lever. J Biol Chem 275: 37167–37172 [DOI] [PubMed] [Google Scholar]
- Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, Carragher BO, Milligan RA, Sweeney HL (1999) Myosin VI is an actin-based motor that moves backwards. Nature 401: 505–508 [DOI] [PubMed] [Google Scholar]
- Whittaker M, Wilson-Kubalek EM, Smith JE, Faust L, Milligan RA, Sweeney HL (1995) A 35-Å movement of smooth muscle myosin on ADP release. Nature 378: 748–751 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1