

Abstract
Despite extensive investigation, a comprehensive understanding of the mechanisms whereby stress impacts fertility remains elusive. Since the 1930s, when Hans Selye popularized studying adaptations to stress (Selye, 1937), we have learned that compensatory mechanisms involve a complex interplay of neural and hormonal processes that allow various body functions to adjust to stress, in a coordinated manner. In terms of reproduction, the adjustment to a stressor interferes with integrated functioning at multiple levels of regulation – the hypothalamus, anterior pituitary gland, gonads, and neural centers coordinating behavior. Various mediators are postulated to participate in reproductive suppression. These include catecholamines, cytokines, prostaglandins, endogenous opioid peptides, and hormones of the hypothalamic-pituitary-adrenal axis. This review focuses on one class of mediators, the glucocorticoids, and provides our views on the relevance and mode of action of this inhibitory intermediate within the anterior pituitary gonadotrope, as a potential cellular site whereby glucocorticoids contribute to stress-induced reproductive suppression.
Keywords: glucocorticoids, luteinizing hormone, follicle-stimulating hormone, gonadotropin-releasing hormone, pituitary
1. Introduction to stress
A hallmark of the endocrine response to stress is an increase in glucocorticoid secretion, the final hormonal effector of the hypothalamic-pituitary-adrenal (HPA) axis. The perception of stress by higher brain centers initiates a cascade of hormone synthesis and secretion, which includes corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) from hypothalamic sites into pituitary portal vasculature, and in response, stimulates adrenocorticotrophic hormone (ACTH) release from anterior pituitary corticotrope cells. Glucocorticoids are synthesized in the adrenal cortex in response to activation by ACTH and initiate actions within tissues that express glucocorticoid receptors (GR). It is not surprising that, based on the diverse actions of glucocorticoids, this steroid receptor is found extensively throughout the body to compensate metabolically for the demands imposed by stressors (De Kloet et al., 1998; Sapolsky et al., 2000). Enhanced secretion of glucocorticoids plays a key role in allowing an organism to survive the challenge to homeostasis. Glucose is mobilized from storage sites and diverted to tissues necessary for survival: brain, heart, and muscles. Consequently, stress can result in pathogenic effects on metabolism, growth, tissue repair, immune defenses, and of interest to this review, reproductive physiology and behavior.
The field has clearly established the impact of stressful stimuli on the hypothalamic-pituitary-gonadal axis. Whether the nature of the stressor is physical (e.g., foot-shock, exercise), immunological (e.g., infection, administration of cytokines or endotoxins), or psychological (e.g., isolation, mental performance tasks), each has been shown to decrease circulating levels of gonadotropins (Cates et al., 2004; Dobson and Smith, 2000; Ferin, 1999; Rivier and Rivest, 1991; Saketos et al., 1993; Tilbrook et al., 2002). The observation that stress-induced impairment of reproductive function is typically associated with a concurrent rise in circulating glucocorticoids has led to the hypothesis that enhanced glucocorticoid secretion is relevant to reproductive suppression during stress. This review explores the evidence supporting this hypothesis, places our studies in context with the literature in this area, and identifies avenues for future research regarding the role of glucocorticoids and stress in the management of ovarian cycle disorders and treatment of infertility.
2. Neuroendocrine site of glucocorticoid action during stress
The possibility that elevated glucocorticoids could act at either the hypothalamic or pituitary level to inhibit gonadotropin secretion has received considerable attention over the years. A variety of animals and cell-based models have been employed to discriminate between actions at neuroendocrine sites leading to the conclusion that the relevance of glucocorticoid action likely depends on the species and stress employed.
2.1. The hypothalamus as a site of glucocorticoid action
The hypothalamus is a critical processing center for controlling reproductive function, orchestrating inputs from metabolic, circadian, gonadal systems, and potentially glucocorticoids, upon the GnRH neuron. The effects of elevated glucocorticoids are primarily mediated by GR which are highly expressed in CRH neurons and function in HPA axis regulation (De Kloet et al., 1998), but also in hypothalamic areas critical for GnRH neuron regulation (Dufourny and Skinner, 2002; Takumi et al., 2012), including expression in GnRH neurons in the rat (Ahima and Harlan, 1992). Early seminal studies implicated an action of cortisol at the hypothalamic level to inhibit pulsatile GnRH release by demonstrating in gonadectomized rhesus monkeys and pigs that chronic administration of glucocorticoids suppresses mean LH secretion in the absence of a reduction in pituitary responsiveness to a GnRH challenge (Dubey and Plant, 1985; Estienne et al., 1991). Inhibition at the hypothalamic level is further supported by evidence that glucocorticoids reduce the frequency of LH pulses in ovary-intact female sheep, ovariectomized female rats, and women during the follicular phase of the ovulatory cycle (Breen et al., 2005; Li et al., 2004; Saketos et al., 1993). As LH pulse frequency is generally modulated by the GnRH neurosecretory system, these findings suggest an action of glucocorticoids to suppress the frequency of GnRH pulses. In each of these studies, GnRH secretion was not monitored directly, rather suppression was inferred indirectly, based on LH or on the lack of reduction in pituitary responsiveness to the releasing hormone. More recent work in follicular phase sheep provides the first definitive evidence that glucocorticoids can inhibit GnRH pulses in pituitary portal blood, by modulating the frequency of pulses (Oakley et al., 2009). Whether this inhibition of GnRH by glucocorticoids is relevant to stress-induced suppression of reproduction remains a topic of later discussion (See Section V: Glucocorticoids as a mediator of stress). Indeed, evidence from the Karsch laboratory, using a layered stress paradigm in sheep, demonstrates that the effects of stress on GnRH pulsatility are not reversed by a non-selective glucocorticoid receptor (GR) antagonist (Wagenmaker et al., 2009b), suggesting that stress-induced levels of glucocorticoids may be sufficient to inhibit reproductive neuroendocrine activity, but may not be necessary for this effect. Based on this evidence, we can agree that glucocorticoids can inhibit GnRH and LH; however, we note that the mechanisms underlying suppression may differ between species, animal models, and gonadal steroid status.
Another point of discussion is the cellular mechanism whereby glucocorticoids may inhibit GnRH. In addition to the reduction of secretion noted above, glucocorticoids have been shown to decrease GnRH synthesis in male rat hypothalamus (Gore et al., 2006), illustrating a genomic effect within the GnRH neuron. A direct action within the GnRH neuron itself is supported by evidence that in rats GnRH neurons express GR, and glucocorticoids blunt GnRH synthesis and release from immortalized GnRH neurons, mouse GT1-7 cells (Attardi et al., 1997; DeFranco et al., 1994). However, increasing evidence points to the possibility that glucocorticoids may be acting via a non-GnRH cell target, such as RFamide-related peptide (RFRP) containing neurons. Stress has been shown to stimulate RFRP expression in the dorsomedial nucleus male rat hypothalamus (Kaewwongse et al., 2011), a region in which RFRP neurons are specifically known to coexpress GR (Kirby et al., 2009) and possess projections that extend to the median eminence and preoptic area making putative connections with GnRH neurons (Kriegsfeld et al., 2006). Further support for a mediatory action of the RFRP system in stress-induced suppression of reproductive neuroendocrine activity is demonstrated in a rat model of restraint stress in which adrenalectomy prevents the stress-induced increase in hypothalamic RFRP expression and subsequent suppression of LH (Kirby et al., 2009), highlighting the importance of the RFRP system in suppression of reproductive neuroendocrine activity by glucocorticoids.
2.2. The pituitary gland as a site of glucocorticoid action
Studies in several species suggest that the effects of glucocorticoids can be exerted directly on the pituitary gland to inhibit responsiveness to GnRH. For example, glucocorticoids reduce the amplitude of the LH response to a GnRH challenge in rodents, pigs, cows, and women (Li and Wagner, 1983; Melis et al., 1987; Pearce et al., 1988; Suter et al., 1988). Further, suppression of responsiveness to GnRH in vitro has been observed in rodent, porcine, and bovine pituitary cell cultures, indicating that glucocorticoids can act directly upon the gonadotrope cell to inhibit GnRH-induced LH secretion (Li and Wagner, 1983; Suter and Orosz, 1989; Suter et al., 1988). Consistent with a direct action upon the gonadotrope cell, GR has been identified in mouse (Breen et al., 2012) and rat gonadotrope cells (Kononen et al., 1993) and studies in mouse, rat and pig primary cells suggest that glucocorticoids can modulate signaling mechanisms downstream of the GnRH receptor, including protein kinase C and cyclic AMP (Li, 1994; Suter et al., 1988), which may lead to a reduction in gonadotropin gene expression or hormone release.
Our laboratory and others have focused on the action of steroid hormone regulation, including androgens, progestins, estrogens, and glucocorticoids, on gonadotropin gene expression as a critical regulatory mechanism within the pituitary gland. At the molecular level, LH and FSH are glycoprotein hormones that exist as heterodimers, consisting of a common and abundantly expressed α-subunit complexed with a unique β-subunit that confers biological specificity (Pierce and Parsons, 1981). Synthesis of the β-subunit gene of each hormone is the rate-limiting step in the overall production of LH and FSH (Kaiser et al., 1997; Pierce and Parsons, 1981). As expression of each β-subunit is tightly controlled by endocrine, paracrine and autocrine actions including hypothalamic GnRH, the activin-inhibin-follistatin system, and steroid hormones of gonadal origin (Kaiser et al., 1997; Vale et al., 1977), it is highly plausible that GR regulation of transcriptional activity, at least in part, underlies an inhibitory effect of glucocorticoids, and potentially stress, on the gonadotrope.
3. Mechanisms of glucocorticoid action on gonadotropin genes
3.1. LHβ
We recently initiated two lines of investigation in the mouse to more fully understand the molecular mechanisms whereby elevated glucocorticoids inhibit gonadotrope responsiveness during psychosocial stress. First, we tested the hypothesis that restraint stress, or an elevation in glucocorticoids mimicking the level induced by restraint stress, can disrupt gonadotrope production of LH in female mice (Breen et al., 2012). GnRH-induced LH secretion or LHβ synthesis was monitored in groups of control diestrus mice or mice exposed to 180 min of restraint stress (Fig. 1A & D). In control mice, which were not exposed to restraint stress, corticosterone remained low and exogenous GnRH elicited a robust increase in LHβ expression and circulating LH levels as compared to vehicle-treated animals (Fig. 1B, C & E). Although GnRH significantly increased both LH responses in stressed animals, the increases in synthesis and secretion in response to exogenous GnRH were significantly blunted in restraint-stressed animals compared to each response in control animals (Fig. 1C & E), demonstrating that restraint stress can diminish the ability of the pituitary to respond to GnRH in female mice. Complementing these findings, we also demonstrated that administration of glucocorticoids to ovariectomized female mice, in the absence of stress, acutely reduces LH in circulation, illustrating direct impairment of reproductive neuroendocrine function by glucocorticoids (Breen et al., 2012). Although this restraint stress elicits a lengthening in the diestrus phase of the estrous cyclic in female mice exposed to 180 min of daily restraint for multiple weeks (Breen et al., 2012), it remains to be determined whether this interruption in cyclicity is solely due to a suppression of pituitary responsiveness, considering that glucocorticoids can alter cyclicity in the sheep in part via suppression of the estradiol-induced LH surge (Breen et al., 2005; Wagenmaker et al., 2009a).
FIG. 1.

Acute restraint stress disrupts pituitary responsiveness to GnRH in female mice. A, Schematic depicting events during the 180 min observation period in which animals were maintained in no stress conditions or subjected to restraint stress for measurement of circulating corticosterone and GnRH-induced LH. Time of sacrifice and blood collection are indicated: 0, 30 and 180 min. At 170 min, no stress and stressed animals (group) are divided into two treatments (n=7/group/treatment) receiving either GnRH (200 ng/kg, s.c.) or vehicle. B, Serum corticosterone (ng/ml) was measured in no stress and stress animals. *, indicates significant (p<0.05) effect of stress. C, Serum LH (ng/ml) was measured in no stress and stressed animals that received vehicle or GnRH, respectively, 10 min prior to sacrifice. *, indicates significant (p<0.05) effect of GnRH; #, denotes difference between no stress and stress. D, Schematic depicting events during 180 min observation period in which animals are maintained in no stress conditions or subjected to restraint stress for measurement of GnRH-induced LHβ mRNA. No stress and stressed animals are divided into two groups (n=7/group) receiving either GnRH (200 ng/kg, s.c.) or vehicle at 0 min of observation. Time of sacrifice and blood collection occurred at 180 min. E, Quantitative RT-PCR analysis of LHβ mRNA was performed on individual mouse pituitary glands and the amount of LHβ mRNA compared with the amount of GAPDH mRNA and expressed as relative transcript level. *, indicates significant (p<0.05) effect of GnRH; #, denotes difference between no stress and stress. Reprinted with permission from The Endocrine Society (Breen et al., 2012).
We next conducted a series of studies to examine the molecular mechanisms underlying glucocorticoid regulation of the LHβ promoter utilizing the immortalized mouse Lβ T2 gonadotrope cell line (Alarid et al., 1998; Alarid et al., 1996). This gonadotrope-derived, immortalized cell line is an essential model system, as it provides a pure population of gonadotrope cells in which to study the molecular mechanisms of gonadotropin gene expression. Since gonadotrope cells only comprise 5-15% of the cells within the mouse pituitary gland (Ooi et al., 2004), utilizing primary pituitary culture to investigate gonadotrope-specific mechanisms, poses a limitation for use in molecular studies. The Lβ T2 cell line endogenously expresses many markers of a mature gonadotrope including α GSU, FSHβ, LHβ, and GnRH receptor (Graham et al., 1999; Pernasetti et al., 2001). It also endogenously expresses GR and responds to glucocorticoids (Breen et al., 2012; Thackray et al., 2006), rounding out the numerous desirable characteristics of this gonadotrope cell model.
Seminal work by early investigators clearly identified inhibitory effects of glucocorticoids within the reproductive neuroendocrine axis, and postulated that glucocorticoids may alter pituitary gene expression. Our findings expand upon these early in vivo studies by demonstrating that glucocorticoids can act directly upon the anterior pituitary gonadotrope cell to suppress GnRH induction of LHβ gene expression (Breen et al., 2012). Rather than by directly binding a hormone responsive element, GR is recruited to the 5′ region of the rat LHβ gene in Lβ T2 cells and blunts GnRH induction of LHβ by interfering with the genomic effects of early growth response factor 1 (Egr1) on the proximal rat LHβ promoter (Fig. 2A). The rat LHβ promoter contains two sets of Egr1 and Steroidogenic Factor 1 (SF1) response elements, which are highly conserved among a variety of species, including the cow and horse (reviewed in (Jorgensen et al., 2004)), as well as the human (Fortin et al., 2009). Indeed, within the proximal 150 bases, the rat LHβ sequence shows great homology across mammals, whereas the distal region of the promoter diverges, highlighting the evolutionary importance of this GnRH responsive region. Both the Egr1 and SF1 elements, as well as the factors themselves, are necessary for GnRH to induce LHβ gene activity (Jorgensen et al., 2004). We find that GR disrupts GnRH-induced activation of rat LHβ via interfering with Egr1-mediated induction at the DNA level. Direct binding of GR to DNA is unnecessary, whereas binding by Egr1 to the rat LHβ promoter is required for glucocorticoid repression, suggesting that the physical interaction between GR and Egr1 is responsible for recruitment of GR to the rat LHβ promoter (Breen et al., 2012).
FIG. 2.
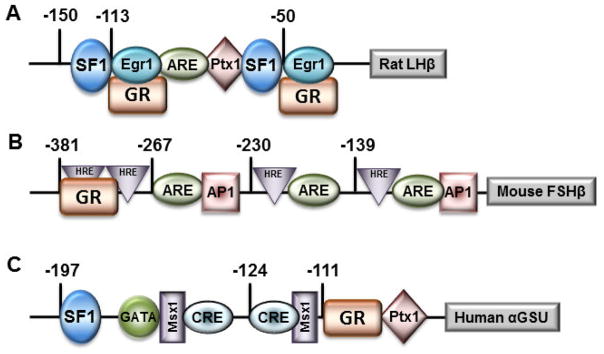
Transcription factors, including GR, and associated binding sites involved in expression of the rat LHβ, murine FSHβ, and human α GSU promoters. Sites are labeled as hormone response elements (HRE) if they play a role in regulation by 3-keto steroid hormones or activin responsive elements (ARE) if they bind SMAD proteins or other activin-induced transcription factors.
3.2. FSHβ
In contrast to the inhibitory effects on LHβ, glucocorticoids have been found to induce transcription of FSHβ in the pituitary of male rats (McAndrews et al., 1994; Ringstrom et al., 1991), and increase basal FSHβ expression in rat pituitary cultures (Bohnsack et al., 2000; Kilen et al., 1996; Leal et al., 2003). Those studies postulated that the effect of glucocorticoids on FSHβ was likely at the level of transcription, as no evidence for impairment of FSHβ mRNA half-life was found (Kilen et al., 1996). We expanded upon these initial observations by identifying the molecular mechanism whereby glucocorticoids, as well as androgens and progestins, increase FSHβ transcription. Each of these 3-keto steroids possesses a similar chemical structure and mechanism of action, binding DNA directly at highly conserved hormone response elements containing the half-site TGTTCT (Ham et al., 1988; Mangelsdorf et al., 1995). Although the LHβ promoter lacks a binding site that resembles a hormone response element, the murine FSHβ promoter contains numerous fairly conserved hormone response elements (Fig. 2B) that are also present in numerous other species (Thackray et al., 2006). One element located at −381 from the start site of mouse FSHβ transcription contains two half sites, and exhibits homology in one of the half sites (5 of 6 base pair match) to the FSHβ promoter region in both human and sheep. This element binds steroid receptors for progesterone, androgen and glucocorticoids, and is critical for mediating glucocorticoid responsiveness of the murine FSHβ promoter (Thackray et al., 2006). For glucocorticoid induction, GR is recruited to the proximal promoter and requires receptor dimerization and direct DNA binding.
3.3. α GSU
The glycoprotein hormone α-subunit (αGSU) gene is expressed in pituitary gonadotropes and thyrotropes in all mammalian species. This subunit heterodimerizes with unique β subunits for the pituitary glycoprotein hormones, including LHβ, FSHβ, and TSHβ to generate the biologically active, heterodimeric hormones (Gharib et al., 1990). In addition to LHβ and FSHβ, transcriptional effects of glucocorticoids within the pituitary have been demonstrated on the α GSU promoter. Schwartz and colleagues provided some of the first evidence that glucocorticoids could modulate the gonadotropin genes, showing that α-subunit mRNA was either increased or decreased depending on the duration of glucocorticoid treatment in rat primary cultures (Kilen et al., 1996; Ringstrom et al., 1991). Our laboratory has examined the responsiveness of the human αGSU promoter to glucocorticoids at the level of the gonadotrope and characterized the mechanism of action in the LβT2 gonadotrope-derived immortalized cell line (Sasson et al., 2008). We have identified two elements within the proximal promoter that mediate induction by glucocorticoids, a cAMP-response element located at −124 relative to the transcription start site and a glucocorticoid-response element at −111 (Fig. 2C). GR binds directly to this glucocorticoid-response element and repression is dependent on cofactor binding to the neighboring cAMP-response element, suggesting transcription factor stabilization via AP1 or CREB.
3.4. GnRH receptor gene
In addition to the gonadotropin subunit genes, strong evidence suggests that regulation of the GnRH receptor is another target gene underlying the anti-gonadal effect of stress. The GnRH receptor is a 7-transmembrane G protein-coupled receptor present on the gonadotrope cell. GnRH binding to the GnRH receptor elicits the synthesis of the gonadotropin subunits, as well as the stimulus-secretion coupling that results in mature LH and FSH release from secretory granules residing within the gonadotrope cell. Glucocorticoids have been shown to regulate transcription of the GnRH receptor gene (Adams et al., 1999; Maya-Nunez and Conn, 2003). Reporter and chromatin immunoprecipitation assays show that dexamethasone treatment can increase transcription of the GnRHR by interaction of GR with steroid receptor coactiator-1 protein recruitment to the activating protein-1 (AP1) region of the murine GnRHR promoter (Kotitschke et al., 2009).
4. Hormonal Interactions on LH and FSH
4.1. Interactions with GnRH
It is not surprising, with the broad range of regulatory processes involving glucocorticoid actions, that its mechanism of transcriptional modulation within the gonadotrope could be diverse as well. Similar to other steroid hormone receptors, GR mediates transcriptional regulation of target genes via a host of direct and indirect mechanisms and our findings in gonadotrope cells suggest that GnRH may contribute to the mechanism of GR action. For example, genes encoding FSHβ, αGSU and GnRHR are each induced by glucocorticoids alone (Kotitschke et al., 2009; McGillivray et al., 2007; Sasson et al., 2008), and induction of either the murine FSHβ or human α GSU gene occurs via direct GR binding to DNA at conserved glucocorticoid response elements (Sasson et al., 2008; Thackray et al., 2006). In contrast, using cell models either devoid of or expressing GR (CV-1 vs. Lβ T2 cells, respectively), we determined that neither an intact DNA-binding domain nor dimerization domain is necessary for GR repression of the rat LHβ gene, implicating a genomic action that occurs via indirect GR binding (Breen et al., 2012). Rather, in the case of LHβ, GR physically interacts with Egr1 and is recruited to the LHβ promoter chromatin by this GnRH-induced factor (Fig. 2A). In contrast to GR induction of FSHβ and α GSU, which require intact DNA-binding and dimerization domains, GR repression of the LHβ promoter occurs via tethering of this receptor to at least one other promoter-bound transcription factor (e.g. Egr1), illustrating the differential effect of glucocorticoids on gene transcription within the gonadotrope cell.
4.2. Interactions with Activin
In addition to the modulatory role of GnRH, activin is potent regulator of gonadotropin synthesis and plays a critical role in enhancing the actions of steroids on FSH during the ovulatory cycle. Activin is produced within the anterior pituitary, as well as the gonads, and acts in an endocrine, paracrine, and autocrine manner (Corrigan et al., 1991). An interaction between activin and glucocorticoids in the regulation of FSHβ has been demonstrated in primary rat pituitary cultures (Leal et al., 2003), and our laboratory has identified a mechanism of direct crosstalk between activities of GR and the activin-activated SMAD transcription factors (McGillivray et al., 2007; Thackray et al., 2010). The synergistic interaction between activin and glucocorticoids on gonadotropin gene expression is interesting on multiple levels. First, activin and glucocorticoids synergistically activate transcription of FSHβ via an interaction that occurs at composite response elements for GR and activin-activated SMAD proteins that are as many as 100 base pairs apart (Fig. 2B). One such element is an activin response element (ARE) located at −267 in the murine FSHβ promoter that has been shown to bind SMAD proteins (Bernard, 2004; Gregory et al., 2005) and the other is a hormone response element at −381 that has been shown to bind GR (Thackray et al., 2006). Blockade of transcription factor binding or inhibition of physical interaction prevents this synergy. Collectively, GnRH and activin play important roles in regulating gonadotropin gene expression and interaction between either of these hormones and GR may be another layer of transcriptional control.
5. Glucocorticoids as a mediator of stress
From an experimental perspective, this commentary has highlighted overwhelming evidence that stress can suppress the activity of the reproductive neuroendocrine axis and, in some species, elevations in glucocorticoids can mimic the effects of stress. For example, we recently demonstrated direct impairment of reproductive function by glucocorticoids, as evident by blunted LHβ synthesis and LH secretion in ovariectomized female mice, in response to a stress level of corticosterone (Breen et al., 2012). By utilizing the female sheep model, we have come to understand that cortisol, by itself, is sufficient to account for stress-induced suppression of reproductive neuroendocrine activity at the pituitary gland, by reducing the LH response to GnRH, but cortisol is not sufficient to suppress GnRH (Breen and Karsch, 2004). These findings underscore the importance in conducting studies that not only address whether glucocorticoids are sufficient to mediate reproductive dysfunction, but whether glucocorticoids are necessary for these effects. In contrast to sufficiency, necessity is more difficult to establish because it requires preventing glucocorticoid synthesis or action during stress. The remainder of this commentary summarizes research utilizing such models to answer the question: Do glucocorticoids mediate reproductive neuroendocrine deficits induced by stress?
Psychosocial stress is pervasive in today’s society and considered a major factor in functional hypothalamic amenorrhea (Berga, 1996), a common anovulatory menstrual cycle disorder described over 50 years ago (Reifenstein, 1946). This condition is associated with enhanced glucocorticoid (Brundu et al., 2006) and reduced pulsatile LH secretion (Berga et al., 1989). In non-human primates, psychosocial stress combined with other types of stress interferes with reproductive hormone secretion and disrupts the ovulatory cycle (Williams et al., 2007; Xiao et al., 2002). As a member of the Fred Karsch laboratory, we devised a paradigm of acute psychosocial stress for investigation using sheep, a species unsurpassed as a model for monitoring GnRH secretion and discriminating hypothalamic from pituitary effects. This layered stress paradigm, which involves sequential hourly layering of isolation, restraint, blindfold, and predator cues (barking dog sound), robustly stimulates the secretion of cortisol, the natural glucocorticoid in sheep, and reduces the amplitude of LH pulses, but it has little or no influence on LH pulse frequency in sheep that are either ovariectomized (Breen and Karsch, 2004) or in the follicular phase of the estrous cycle (Wagenmaker et al., 2010). We found that there are two causes of the blunted LH pulses, a hypothalamic effect reducing the amplitude of GnRH pulses in pituitary portal blood (Wagenmaker et al., 2009b), and a pituitary effect inhibiting responsiveness to GnRH (Breen and Karsch, 2004). Using RU486 to antagonize GR, we found that blockade of cortisol action eliminated the inhibitory effect of psychosocial stress on pituitary responsiveness to GnRH (Breen et al., 2004). Intriguingly, RU486 did not prevent this stress from lowering GnRH pulse amplitude (Wagenmaker et al., 2009b). This observation is keenly interesting because it suggests that the rise in cortisol during this psychosocial stress is necessary for suppression of pituitary responsiveness to GnRH, but glucocorticoids are not required for the hypothalamic effect on GnRH pulses.
Immune inflammatory stress also disrupts the menstrual cycle of primates (Xiao et al., 1998). This stress may be modeled by administering endotoxin, a component of bacteria that induces flu-like symptoms. In sheep, endotoxin profoundly stimulates cortisol secretion, interrupts the follicular phase of the cycle and interferes with female sexual behavior (Battaglia et al., 2000). It acts by suppressing both pulsatile GnRH secretion (Battaglia et al., 1997) and pituitary responsiveness to GnRH independent of changes in GnRH secretion (Williams et al., 2001). A different picture emerged for immune inflammatory stress. In this case, we used metyrapone to block cortisol synthesis and found that cortisol is not necessary for endotoxin to inhibit either GnRH secretion or pituitary responsiveness to GnRH (Debus et al., 2002). Instead, pro-inflammatory molecules such as cytokines and prostaglandins mediate reproductive neuroendocrine suppression at both the hypothalamic and pituitary levels in response to endotoxin (Harris et al., 2000). Thus, immune inflammatory and psychosocial stress share two distinct disruptive effects at both hypothalamic and pituitary levels.
6. Closing perspective
This review has highlighted the field, including some of our studies, addressing mechanisms by which glucocorticoids inhibit reproductive neuroendocrine activity and the relevance of this hormone in mediating stress-induced suppression of gonadotropin secretion. As stress is not a specific stimulus, but a disruption of, or threat to, homeostasis, it is reasonable to expect that the relevant mediator(s) depend on the nature of the disruptor. Indeed, the above observations highlight the qualification that different categories of stressors (e.g. psychosocial, metabolic, immune, etc.) are perceived in distinct ways and likely stimulate different pathways that lead to reproductive suppression (Herman et al., 2003). In this regard, a variety of mediators (including CRH, AVP, glucocorticoids, catecholamines, endogenous opioid peptides, cytokines, prostaglandins, etc.) have been implicated in stress-induced suppression of gonadotropin secretion and disruption of ovarian cyclicity (Ferin, 1999; Rivier and Rivest, 1991; Tilbrook et al., 2000). The relative importance of each, however, appears to depend not only on the nature of the stressor employed, but also the species under investigation (Bethea et al., 2008; Li et al., 2010).
It is of utmost importance to not only understand the relevance of inhibitory intermediates to the disruption of gonadotropin secretion and ovarian cyclicity resulting from different types of stress, but to tease apart the cellular and molecular mechanism whereby these intermediates lead to reproductive suppression. Collectively, the present observations create a theoretical framework whereby stress elicits enhanced systemic glucocorticoids that have the capacity to blunt LH production in the gonadotrope, reducing the amplitude of LH pulses in circulation and interfering with ovarian cyclicity (Breen et al., 2005; Breen and Karsch, 2004; Breen et al., 2012). Thus, the blockade of high amplitude LH pulses would be quite costly from a reproduction standpoint. As for reconciling the relevance of enhanced FSHβ, GnRH receptor and α GSU gene expression in response to elevated glucocorticoids, we can only speculate, as it remains an intriguing research avenue to pursue. As it is well established that gonadal steroids differentially modulate LH and FSH synthesis, we demonstrate that glucocorticoids are not inhibitory to these gonadotrope genes, rather we observe that glucocorticoids readily contribute to maintenance of high levels of FSHβ, GnRH receptor and α GSU gene expression. We speculate that enhancement of these gonadotropin genes contributes to elevated FSH and preservation of reproductive function following stress. Evidence in support of the resilience of the ovarian cycle, in the face of stress-induced perturbations in LH secretion in the female sheep provides support for this hypothesis (Wagenmaker et al., 2010). As developed in this review, studies from mice to sheep support the conclusion that glucocorticoids appear to be particularly important for suppression of gonadotrope responsiveness to GnRH during psychosocial stress; however, it remains an ongoing challenge for future studies to utilize well-defined in vivo and in vitro models to determine which intermediates are relevant to specific stress types and define the neuroendocrine site and cellular mechanism whereby reproductive suppression occurs.
Highlights.
Glucocorticoids can inhibit reproductive activity
Gonadotropin gene expression is altered by glucocorticoids
Relevance of glucocorticoids to changes in LH and FSH depends on the stress type
Acknowledgments
The authors are grateful to Dr. Varykina G. Thackray, Dr. Djurdjica Coss, Dr. Amy Oakley, Ms. Elizabeth Wagenmaker, and Dr. Fred Karsch for their excellent assistance in conducting and interpreting the studies highlighted in this review.
This work was supported by NIH grants R01 HD072754 and R01 DK044838 (to P.L.M.) and by the Eunice Kennedy Shriver NICHD/NIH through cooperative agreement (U54 HD012303) as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (P.L.M.). P.L.M. was also partially supported by P30 CA023100, P30 DK063491, and P42 ES010337. K.M.B. was supported by NIH grant K99/R00 HD060947.
Footnotes
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams TE, Sakurai H, Adams BM. Effect of stress-like concentrations of cortisol on estradiol-dependent expression of gonadotropin-releasing hormone receptor in orchidectomized sheep. Biol Reprod. 1999;60:164–168. doi: 10.1095/biolreprod60.1.164. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Harlan RE. Glucocorticoid receptors in LHRH neurons. Neuroendocrinology. 1992;56:845–850. doi: 10.1159/000126315. [DOI] [PubMed] [Google Scholar]
- Alarid ET, Holley S, Hayakawa M, Mellon PL. Discrete stages of anterior pituitary differentiation recapitulated in immortalized cell lines. Mol Cell Endocrinol. 1998;140:25–30. doi: 10.1016/s0303-7207(98)00025-2. [DOI] [PubMed] [Google Scholar]
- Alarid ET, Windle JJ, Whyte DB, Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development. 1996;122:3319–3329. doi: 10.1242/dev.122.10.3319. [DOI] [PubMed] [Google Scholar]
- Attardi B, Tsujii T, Friedman R, Zeng Z, Roberts JL, Dellovade T, Pfaff DW, Chandran UR, Sullivan MW, DeFranco DB. Glucocorticoid repression of gonadotropin-releasing hormone gene expression and secretion in morphologically distinct subpopulations of GT1-7 cells. Mol Cell Endocrinol. 1997;131:241–255. doi: 10.1016/s0303-7207(97)00102-0. [DOI] [PubMed] [Google Scholar]
- Battaglia DF, Bowen JM, Krasa HB, Thrun LA, Viguie C, Karsch FJ. Endotoxin inhibits the reproductive neuroendocrine axis while stimulating adrenal steroids: a simultaneous view from hypophyseal portal and peripheral blood. Endocrinology. 1997;138:4273–4281. doi: 10.1210/endo.138.10.5449. [DOI] [PubMed] [Google Scholar]
- Battaglia DF, Krasa HB, Padmanabhan V, Viguie C, Karsch FJ. Endocrine alterations that underlie endotoxin-induced disruption of the follicular phase in ewes. Biol Reprod. 2000;62:45–53. doi: 10.1095/biolreprod62.1.45. [DOI] [PubMed] [Google Scholar]
- Berga SL. Functional hypothalamic amenorrhea. In: Adashi EY, JAR, Rosenwaks Z, editors. Reproductive endocrinology, surgery, and technology. Lipincott-Raven; Philadelphia: 1996. pp. 1061–1072. [Google Scholar]
- Berga SL, Mortola JF, Girton L, Suh B, Laughlin G, Pham P, Yen SS. Neuroendocrine aberrations in women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab. 1989;68:301–308. doi: 10.1210/jcem-68-2-301. [DOI] [PubMed] [Google Scholar]
- Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone beta subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. doi: 10.1210/me.2003-0264. [DOI] [PubMed] [Google Scholar]
- Bethea CL, Centeno ML, Cameron JL. Neurobiology of stress-induced reproductive dysfunction in female macaques. Mol Neurobiol. 2008;38:199–230. doi: 10.1007/s12035-008-8042-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack BL, Szabo M, Kilen SM, Tam DH, Schwartz NB. Follistatin suppresses steroid-enhanced follicle-stimulating hormone release in vitro in rats. Biol Reprod. 2000;62:636–641. doi: 10.1095/biolreprod62.3.636. [DOI] [PubMed] [Google Scholar]
- Breen KM, Billings HJ, Wagenmaker ER, Wessinger EW, Karsch FJ. Endocrine basis for disruptive effects of cortisol on preovulatory events. Endocrinology. 2005;146:2107–2115. doi: 10.1210/en.2004-1457. [DOI] [PubMed] [Google Scholar]
- Breen KM, Karsch FJ. Does cortisol inhibit pulsatile luteinizing hormone secretion at the hypothalamic or pituitary level? Endocrinology. 2004;145:692–698. doi: 10.1210/en.2003-1114. [DOI] [PubMed] [Google Scholar]
- Breen KM, Stackpole CA, Clarke IJ, Pytiak AV, Tilbrook AJ, Wagenmaker ER, Young EA, Karsch FJ. Does the type II glucocorticoid receptor mediate cortisol-induced suppression in pituitary responsiveness to gonadotropin-releasing hormone? Endocrinology. 2004;145:2739–2746. doi: 10.1210/en.2004-0123. [DOI] [PubMed] [Google Scholar]
- Breen KM, Thackray VG, Hsu T, Mak-McCully RA, Coss D, Mellon PL. Stress levels of glucocorticoids inhibit LHbeta-subunit gene expression in gonadotrope cells. Mol Endocrinol. 2012;26:1716–1731. doi: 10.1210/me.2011-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundu B, Loucks TL, Adler LJ, Cameron JL, Berga SL. Increased cortisol in the cerebrospinal fluid of women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab. 2006;91:1561–1565. doi: 10.1210/jc.2005-2422. [DOI] [PubMed] [Google Scholar]
- Cates PS, Li XF, O’Byrne KT. The influence of 17beta-oestradiol on corticotrophin-releasing hormone induced suppression of luteinising hormone pulses and the role of CRH in hypoglycaemic stress-induced suppression of pulsatile LH secretion in the female rat. Stress. 2004;7:113–118. doi: 10.1080/1025389042000218988. [DOI] [PubMed] [Google Scholar]
- Corrigan AZ, Bilezikjian LM, Carroll RS, Bald LN, Schmelzer CH, Fendly BM, Mason AJ, Chin WW, Schwall RH, Vale W. Evidence for an autocrine role of activin B within rat anterior pituitary cultures. Endocrinology. 1991;128:1682–1684. doi: 10.1210/endo-128-3-1682. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- Debus N, Breen KM, Barrell GK, Billings HJ, Brown M, Young EA, Karsch FJ. Does cortisol mediate endotoxin-induced inhibition of pulsatile luteinizing hormone and gonadotropin-releasing hormone secretion? Endocrinology. 2002;143:3748–3758. doi: 10.1210/en.2002-220291. [DOI] [PubMed] [Google Scholar]
- DeFranco DB, Attardi B, Chandran UR. Glucocorticoid receptor-mediated repression of GnRH gene expression in a hypothalamic GnRH-secreting neuronal cell line. Ann NY Acad Sci. 1994;746:473–475. doi: 10.1111/j.1749-6632.1994.tb39289.x. [DOI] [PubMed] [Google Scholar]
- Dobson H, Smith RF. What is stress, and how does it affect reproduction? Anim Reprod Sci. 2000;60–61:743–752. doi: 10.1016/s0378-4320(00)00080-4. [DOI] [PubMed] [Google Scholar]
- Dubey AK, Plant TM. A suppression of gonadotropin secretion by cortisol in castrated male rhesus monkeys (Macaca mulatta) mediated by the interruption of hypothalamic gonadotropin-releasing hormone release. Biol Reprod. 1985;33:423–431. doi: 10.1095/biolreprod33.2.423. [DOI] [PubMed] [Google Scholar]
- Dufourny L, Skinner DC. Progesterone receptor, estrogen receptor alpha, and the type II glucocorticoid receptor are coexpressed in the same neurons of the ovine preoptic area and arcuate nucleus: a triple immunolabeling study. Biol Reprod. 2002;67:1605–1612. doi: 10.1095/biolreprod.102.005066. [DOI] [PubMed] [Google Scholar]
- Estienne MJ, Barb CR, Kesner JS, Kraeling RR, Rampacek GB. Luteinizing hormone secretion in hypophysial stalk-transected gilts given hydrocortisone acetate and pulsatile gonadotropin-releasing hormone. Domest Anim Endocrinol. 1991;8:407–414. doi: 10.1016/0739-7240(91)90008-8. [DOI] [PubMed] [Google Scholar]
- Ferin M. Clinical review 105: Stress and the reproductive cycle. J Clin Endocrinol Metab. 1999;84:1768–1774. doi: 10.1210/jcem.84.6.5367. [DOI] [PubMed] [Google Scholar]
- Fortin J, Lamba P, Wang Y, Bernard DJ. Conservation of mechanisms mediating gonadotrophin-releasing hormone 1 stimulation of human luteinizing hormone beta subunit transcription. Mol Hum Reprod. 2009;15:77–87. doi: 10.1093/molehr/gan079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharib SD, Wierman ME, Shupnik MA, Chin WW. Molecular biology of the pituitary gonadotropins. Endocrine Rev. 1990;11:177–199. doi: 10.1210/edrv-11-1-177. [DOI] [PubMed] [Google Scholar]
- Gore AC, Attardi B, DeFranco DB. Glucocorticoid repression of the reproductive axis: effects on GnRH and gonadotropin subunit mRNA levels. Mol Cell Endocrinol. 2006;256:40–48. doi: 10.1016/j.mce.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Graham KE, Nusser KD, Low MJ. Lβ T2 gonadotroph cells secrete follicle stimulating hormone (FSH) in response to activin A. J Endocrinol. 1999;162:R1–R5. doi: 10.1677/joe.0.162r001. [DOI] [PubMed] [Google Scholar]
- Gregory SJ, Lacza CT, Detz AA, Xu S, Petrillo LA, Kaiser UB. Synergy between activin A and gonadotropin-releasing hormone in transcriptional activation of the rat follicle-stimulating hormone-beta gene. Mol Endocrinol. 2005;19:237–254. doi: 10.1210/me.2003-0473. [DOI] [PubMed] [Google Scholar]
- Ham J, Thomson A, Needham M, Webb P, Parker M. Characterization of response elements for androgens, glucocorticoids and progestins in mouse mammary tumour virus. Nucleic Acids Res. 1988;16:5263–5276. doi: 10.1093/nar/16.12.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TG, Battaglia DF, Brown ME, Brown MB, Carlson NE, Viguie C, Williams CY, Karsch FJ. Prostaglandins mediate the endotoxin-induced suppression of pulsatile gonadotropin-releasing hormone and luteinizing hormone secretion in the ewe. Endocrinology. 2000;141:1050–1058. doi: 10.1210/endo.141.3.7393. [DOI] [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Jorgensen JS, Quirk CC, Nilson JH. Multiple and overlapping combinatorial codes orchestrate hormonal responsiveness and dictate cell-specific expression of the genes encoding luteinizing hormone. Endocr Rev. 2004;25:521–542. doi: 10.1210/er.2003-0029. [DOI] [PubMed] [Google Scholar]
- Kaewwongse M, Takayanagi Y, Onaka T. Effects of RFamide-related peptide (RFRP)-1 and RFRP-3 on oxytocin release and anxiety-related behaviour in rats. J Neuroendocrinol. 2011;23:20–27. doi: 10.1111/j.1365-2826.2010.02077.x. [DOI] [PubMed] [Google Scholar]
- Kaiser UB, Conn PM, Chin WW. Studies of gonadotropin-releasing hormone (GnRH) action using GnRH receptor-expressing pituitary cell lines. Endocr Rev. 1997;18:46–70. doi: 10.1210/edrv.18.1.0289. [DOI] [PubMed] [Google Scholar]
- Kilen SM, Szabo M, Strasser GA, McAndrews JM, Ringstrom SJ, Schwartz NB. Corticosterone selectively increases follicle-stimulating hormone beta-subunit messenger ribonucleic acid in primary anterior pituitary cell culture without affecting its half-life. Endocrinology. 1996;137:3802–3807. doi: 10.1210/endo.137.9.8756550. [DOI] [PubMed] [Google Scholar]
- Kirby ED, Geraghty AC, Ubuka T, Bentley GE, Kaufer D. Stress increases putative gonadotropin inhibitory hormone and decreases luteinizing hormone in male rats. Proc Natl Acad Sci USA. 2009;106:11324–11329. doi: 10.1073/pnas.0901176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononen J, Honkaniemi J, Gustafsson JA, Pelto-Huikko M. Glucocorticoid receptor colocalization with pituitary hormones in the rat pituitary gland. Mol Cell Endocrinol. 1993;93:97–103. doi: 10.1016/0303-7207(93)90144-9. [DOI] [PubMed] [Google Scholar]
- Kotitschke A, Sadie-Van Gijsen H, Avenant C, Fernandes S, Hapgood JP. Genomic and nongenomic cross talk between the gonadotropin-releasing hormone receptor and glucocorticoid receptor signaling pathways. Mol Endocrinol. 2009;23:1726–1745. doi: 10.1210/me.2008-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegsfeld LJ, Mei DF, Bentley GE, Ubuka T, Mason AO, Inoue K, Ukena K, Tsutsui K, Silver R. Identification and characterization of a gonadotropin-inhibitory system in the brains of mammals. Proc Natl Acad Sci USA. 2006;103:2410–2415. doi: 10.1073/pnas.0511003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal AM, Blount AL, Donaldson CJ, Bilezikjian LM, Vale WW. Regulation of follicle-stimulating hormone secretion by the interactions of activin-A, dexamethasone and testosterone in anterior pituitary cell cultures of male rats. Neuroendocrinology. 2003;77:298–304. doi: 10.1159/000070896. [DOI] [PubMed] [Google Scholar]
- Li PS. Modulation by cortisol of luteinizing hormone secretion from cultured porcine anterior pituitary cells: effects on secretion induced by phospholipase C, phorbol ester and cAMP. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:107–112. doi: 10.1007/BF00178214. [DOI] [PubMed] [Google Scholar]
- Li PS, Wagner WC. In vivo and in vitro studies on the effect of adrenocorticotropic hormone or cortisol on the pituitary response to gonadotropin releasing hormone. Biol Reprod. 1983;29:25–37. doi: 10.1095/biolreprod29.1.25. [DOI] [PubMed] [Google Scholar]
- Li XF, Edward J, Mitchell JC, Shao B, Bowes JE, Coen CW, Lightman SL, O’Byrne KT. Differential effects of repeated restraint stress on pulsatile lutenizing hormone secretion in female Fischer, Lewis and Wistar rats. J Neuroendocrinol. 2004;16:620–627. doi: 10.1111/j.1365-2826.2004.01209.x. [DOI] [PubMed] [Google Scholar]
- Li XF, Knox AM, O’Byrne KT. Corticotrophin-releasing factor and stress-induced inhibition of the gonadotrophin-releasing hormone pulse generator in the female. Brain Res. 2010;1364:153–163. doi: 10.1016/j.brainres.2010.08.036. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Nunez G, Conn PM. Transcriptional regulation of the GnRH receptor gene by glucocorticoids. Mol Cell Endocrinol. 2003;200:89–98. doi: 10.1016/s0303-7207(02)00419-7. [DOI] [PubMed] [Google Scholar]
- McAndrews JM, Ringstrom SJ, Dahl KD, Schwartz NB. Corticosterone in vivo increases pituitary follicle-stimulating hormone (FSH)-beta messenger ribonucleic acid content and serum FSH bioactivity selectively in female rats. Endocrinology. 1994;134:158–163. doi: 10.1210/endo.134.1.8275929. [DOI] [PubMed] [Google Scholar]
- McGillivray SM, Thackray VG, Coss D, Mellon PL. Activin and glucocorticoids synergistically activate follicle-stimulating hormone β-subunit gene expression in the immortalized LβT2 gonadotrope cell line. Endocrinology. 2007;148:762–773. doi: 10.1210/en.2006-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis GB, Mais V, Gambacciani M, Paoletti AM, Antinori D, Fioretti P. Dexamethasone reduces the postcastration gonadotropin rise in women. J Clin Endocrinol Metab. 1987;65:237–241. doi: 10.1210/jcem-65-2-237. [DOI] [PubMed] [Google Scholar]
- Oakley AE, Breen KM, Clarke IJ, Karsch FJ, Wagenmaker ER, Tilbrook AJ. Cortisol reduces gonadotropin-releasing hormone pulse frequency in follicular phase ewes: influence of ovarian steroids. Endocrinology. 2009;150:341–349. doi: 10.1210/en.2008-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi GT, Tawadros N, Escalona RM. Pituitary cell lines and their endocrine applications. Mol Cell Endocrinol. 2004;228:1–21. doi: 10.1016/j.mce.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Pearce GP, Paterson AM, Hughes PE. Effect of short-term elevations in plasma cortisol concentration on LH secretion in prepubertal gilts. J Reprod Fertil. 1988;83:413–418. doi: 10.1530/jrf.0.0830413. [DOI] [PubMed] [Google Scholar]
- Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, Mellon PL. Cell-specific transcriptional regulation of FSH by activin and GnRH in the L T2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Ann Rev Biochem. 1981;50:465–495. doi: 10.1146/annurev.bi.50.070181.002341. [DOI] [PubMed] [Google Scholar]
- Reifenstein EC., Jr Psychogenic or hypothalamic amenorrhea. Med Clin North Am. 1946;30:1103–1114. doi: 10.1016/s0025-7125(16)35908-9. [DOI] [PubMed] [Google Scholar]
- Ringstrom SJ, McAndrews JM, Rahal JO, Schwartz NB. Cortisol in vivo increases FSH beta mRNA selectively in pituitaries of male rats. Endocrinology. 1991;129:2793–2795. doi: 10.1210/endo-129-5-2793. [DOI] [PubMed] [Google Scholar]
- Rivier C, Rivest S. Effect of stress on the activity of the hypothalamic-pituitary-gonadal axis: peripheral and central mechanisms. Biol Reprod. 1991;45:523–532. doi: 10.1095/biolreprod45.4.523. [DOI] [PubMed] [Google Scholar]
- Saketos M, Sharma N, Santoro NF. Suppression of the hypothalamic-pituitary-ovarian axis in normal women by glucocorticoids. Biol Reprod. 1993;49:1270–1276. doi: 10.1095/biolreprod49.6.1270. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Sasson R, Luu SH, Thackray VG, Mellon PL. Glucocorticoids induce human glycoprotein hormone alpha-subunit gene expression in the gonadotrope. Endocrinology. 2008;149:3643–3655. doi: 10.1210/en.2007-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selye H. The Significance of the Adrenals for Adaptation. Science. 1937;85:247–248. doi: 10.1126/science.85.2201.247. [DOI] [PubMed] [Google Scholar]
- Suter DE, Orosz G. Effect of treatment with cortisol in vivo on secretion of gonadotropins in vitro. Biol Reprod. 1989;41:1091–1096. doi: 10.1095/biolreprod41.6.1091. [DOI] [PubMed] [Google Scholar]
- Suter DE, Schwartz NB, Ringstrom SJ. Dual role of glucocorticoids in regulation of pituitary content and secretion of gonadotropins. Am J Physiol. 1988;254:E595–600. doi: 10.1152/ajpendo.1988.254.5.E595. [DOI] [PubMed] [Google Scholar]
- Takumi K, Iijima N, Higo S, Ozawa H. Immunohistochemical analysis of the colocalization of corticotropin-releasing hormone receptor and glucocorticoid receptor in kisspeptin neurons in the hypothalamus of female rats. Neurosci Lett. 2012;531:40–45. doi: 10.1016/j.neulet.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Thackray VG, McGillivray SM, Mellon PL. Androgens, progestins and glucocorticoids induce follicle-stimulating hormone β-subunit gene expression at the level of the gonadotrope. Mol Endocrinol. 2006;20:2062–2079. doi: 10.1210/me.2005-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray VG, Mellon PL, Coss D. Hormones in synergy: Regulation of the pituitary gonadotropin genes. Mol Cell Endocrinol. 2010;314:192–203. doi: 10.1016/j.mce.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilbrook AJ, Turner AI, Clarke IJ. Effects of stress on reproduction in non-rodent mammals: the role of glucocorticoids and sex differences. Rev Reprod. 2000;5:105–113. doi: 10.1530/ror.0.0050105. [DOI] [PubMed] [Google Scholar]
- Tilbrook AJ, Turner AI, Clarke IJ. Stress and reproduction: central mechanisms and sex differences in non-rodent species. Stress. 2002;5:83–100. doi: 10.1080/10253890290027912. [DOI] [PubMed] [Google Scholar]
- Vale W, Rivier C, Brown M. Regulatory peptides of the hypothalamus. Ann Rev Physiol. 1977;39:473–527. doi: 10.1146/annurev.ph.39.030177.002353. [DOI] [PubMed] [Google Scholar]
- Wagenmaker ER, Breen KM, Oakley AE, Pierce BN, Tilbrook AJ, Turner AI, Karsch FJ. Cortisol interferes with the estradiol-induced surge of luteinizing hormone in the ewe. Biol Reprod. 2009a;80:458–463. doi: 10.1095/biolreprod.108.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmaker ER, Breen KM, Oakley AE, Tilbrook AJ, Karsch FJ. Psychosocial stress inhibits amplitude of gonadotropin-releasing hormone pulses independent of cortisol action on the type II glucocorticoid receptor. Endocrinology. 2009b;150:762–769. doi: 10.1210/en.2008-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmaker ER, Breen KM, Oakley AE, Tilbrook AJ, Karsch FJ. The estrous cycle of the ewe is resistant to disruption by repeated, acute psychosocial stress. Biol Reprod. 2010;82:1206–1215. doi: 10.1095/biolreprod.109.078774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CY, Harris TG, Battaglia DF, Viguie C, Karsch FJ. Endotoxin inhibits pituitary responsiveness to gonadotropin-releasing hormone. Endocrinology. 2001;142:1915–1922. doi: 10.1210/endo.142.5.8120. [DOI] [PubMed] [Google Scholar]
- Williams NI, Berga SL, Cameron JL. Synergism between psychosocial and metabolic stressors: impact on reproductive function in cynomolgus monkeys. Am J Physiol Endocrinol Metab. 2007;293:E270–276. doi: 10.1152/ajpendo.00108.2007. [DOI] [PubMed] [Google Scholar]
- Xiao E, Xia-Zhang L, Barth A, Zhu J, Ferin M. Stress and the menstrual cycle: relevance of cycle quality in the short- and long-term response to a 5-day endotoxin challenge during the follicular phase in the rhesus monkey. J Clin Endocrinol Metab. 1998;83:2454–2460. doi: 10.1210/jcem.83.7.4926. [DOI] [PubMed] [Google Scholar]
- Xiao E, Xia-Zhang L, Ferin M. Inadequate luteal function is the initial clinical cyclic defect in a 12-day stress model that includes a psychogenic component in the Rhesus monkey. J Clin Endocrinol Metab. 2002;87:2232–2237. doi: 10.1210/jcem.87.5.8500. [DOI] [PubMed] [Google Scholar]