

Abstract
Meiotic double-strand breaks (DSBs) are formed by Spo11 in conjunction with at least nine other proteins whose roles are not well understood. We find that two of these proteins, Rec102 and Rec104, interact physically, are mutually dependent for proper subcellular localization, and share a requirement for Spo11 and Ski8 for their recruitment to meiotic chromosomes, suggesting that they work together as a functional unit. Rec102 associated extensively with chromatin loops during leptotene and zygotene and showed preferential binding in the vicinity at least of most DSB sites, consistent with a direct role in DSB formation. However, Rec102 was associated with both DSB-hot and DSB-cold regions, ruling out a simple model in which sites of DSB formation are dictated by where Rec102/104 complexes load. Both proteins persisted on chromatin until pachytene before abruptly disappearing, indicating that they remain on chromosomes well after DSB formation. These studies reveal unexpected behaviors for Rec102 and Rec104, and point to distinct roles and subcomplexes among the DSB proteins.
Keywords: chromosome structure, meiosis, recombination, Rec102, Spo11
Introduction
Meiotic recombination proceeds through the formation and repair of DNA double-strand breaks (DSBs) and is coordinated with changes in higher order chromosome structures. This coordination is essential because reciprocal DNA exchanges are only one part of the physical connections between chromosomes (known as chiasmata) essential for accurate segregation in the first meiotic division: sister chromatid axes must also be locally separated and the axes of recombinant nonsister chromosomes must be reciprocally exchanged (Zickler and Kleckner, 1999; Blat et al, 2002). This ensemble of local DNA exchange and higher order chromosome structures works along with sister chromatid cohesion to ensure that homologous chromosomes remain associated until anaphase I.
Meiotic chromosomes are organized into linear arrays of chromatin loops. The loop bases and associated proteins define a structural axis for each chromatid. Sister chromatid axes are closely joined and, at pachytene, are synapsed with the axes of the homologous sister pair, via the central element of the synaptonemal complex (SC). Cohesins bind preferentially to AT-rich sequences distributed regularly along the chromosome (Blat and Kleckner, 1999; Tanaka et al, 1999) and are components of the chromosome axis (Klein et al, 1999; Eijpe et al, 2000, 2003; Pelttari et al, 2001). These and other considerations lead to the conclusion that cohesin-associated DNA sequences define the bases of the loops. Meiotic DSBs occur preferentially in the sequences between cohesin binding sites, and are thus considered to be in the chromatin loops (Blat et al, 2002), but the recombinosomes that carry out recombination are intimately associated with the axes (Moens et al, 1998). One way to reconcile these facts is to propose that protein–DNA complexes at DSB sites in chromatin loops are recruited to the axis, either as a condition for DNA cleavage or after DSB formation (Zickler and Kleckner, 1999; van Heemst and Heyting, 2000). Thus, the axis/loop organization of meiotic chromosomes defines one level of spatial organization for DSB formation and repair.
A second level of spatial organization is that chiasmata form preferentially in chromosome domains that correspond to GC-rich isochores, or ‘R-bands' (Ashley, 1988; Holmquist, 1992; Eisenbarth et al, 2000; Kong et al, 2002). Much of this positional bias for recombination is dictated during DSB formation in yeast (Baudat and Nicolas, 1997; Gerton et al, 2000; Blat et al, 2002). This bias represents a large-scale position effect, because a recombination reporter placed at different positions takes on the properties of its location: insertions into cold regions give low DSB levels and insertions into hot regions give high DSB levels (Wu and Lichten, 1995; Borde et al, 1999).
DSB formation is catalyzed by Spo11, a relative of archaeal topoisomerases. Spo11 is widely conserved, but it does not act alone: in Saccharomyces cerevisiae at least nine other gene products are also required for DSB formation (reviewed in Keeney, 2001). The list of DSB genes comprises a meiosis-specific set (SPO11 itself plus REC102, REC104, REC114, MEI4, and MER2); a group of genes also involved in DSB repair and other functions in vegetative cells (RAD50, MRE11, and XRS2); and a gene with roles in cytoplasmic RNA metabolism (SKI8/REC103). In addition, DSB formation is reduced in mutants lacking either of two structural components of meiotic chromosomes, Red1 and Hop1 (reviewed in Keeney, 2001). This requirement provides strong evidence that activity of the DSB machinery is coordinated with higher order chromosome structures.
Because of the integration of recombination with other aspects of chromosome structure and dynamics, it is important to understand not only the interactions that connect the DSB proteins to one another, but also the temporal and spatial patterns that govern their association with meiotic chromosomes. This work focuses on Rec102 and Rec104. Neither has a known homolog in species aside from other ascomycetes, or sequence motifs that suggest a biochemical function. Genetic and immunoprecipitation studies connect Rec102 and Rec104 to each other and to Spo11 (Salem et al, 1999; Kee and Keeney, 2002; Jiao et al, 2003), suggesting that Rec102 and Rec104 directly promote DSB formation as part of a multiprotein complex with Spo11. Here we examine interactions between Rec102 and Rec104, and describe the distribution, timing, and genetic requirements for their binding to meiotic chromosomes.
Results
Physical and functional interactions between Rec102 and Rec104
Epitope-tagged versions of Rec102 and Rec104 were expressed under the control of their normal promoters. Rec102 tagged at the C-terminus with multiple copies of the myc or flag epitopes complemented a rec102 null mutant (Kee and Keeney, 2002). Tagging the C-terminus of Rec104 yielded a nonfunctional protein (data not shown), so the protein was tagged instead at its N-terminus; mycREC104 supported normal DSB formation, intragenic recombination, and spore viability (see Materials and methods).
Rec104 protein expression was meiosis-specific, similar to Rec102 (Figure 1A). Steady-state Rec104 levels at 4 h in meiosis were reproducibly lower in rec102, ski8, and mre11 mutants and were elevated 1.5-fold in a mer2 mutant (Figure 1B). For Rec102 as well, ski8 and mre11 mutations gave a noticeable reduction in steady-state levels (Figure 1B). Because ski8 and mre11 mutations cause defects during vegetative growth, it is possible that they affect meiotic gene expression indirectly because of premeiotic defects. This caveat does not apply to the meiosis-specific rec102 mutation, however, so the fact that Rec104 required Rec102 to accumulate to normal levels underscores the close relationship between these proteins.
Figure 1.
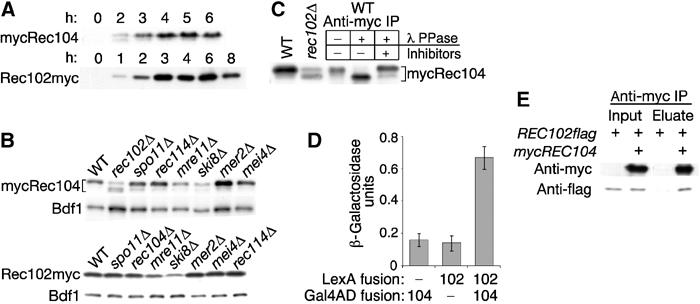
Physical and functional interactions between Rec102 and Rec104. (A) Western blot analysis of a time course of mycRec104 expression. Data for Rec102myc are from Kee and Keeney (2002). (B) Steady-state levels of Rec102myc and mycRec104 in DSB-defective mutants. Whole-cell extracts at 4 h in meiosis were analyzed by Western blot. Blots were stripped and reprobed for Bdf1 as a loading control. (C) Rec104 phosphorylation. Denaturing whole-cell extract was prepared at 4 h in meiosis, then mycRec104 was immunoprecipitated and treated with lambda phosphatase alone or in the presence of inhibitors as indicated. Whole-cell extracts from wild-type (WT) and rec102Δ strains are included for comparison. (D) Yeast two-hybrid analysis of interactions between Rec102 and Rec104. lacZ reporter expression was assayed in strains carrying the indicated fusion constructs. Each value is the mean±s.d. of at least three determinations. (E) Co-immunoprecipitation of Rec102 and Rec104. Nondenaturing whole-cell extracts were prepared from REC102flag strains carrying either REC104 or mycREC104, then immunoprecipitated with anti-myc antibodies. Input extracts and antibody matrix eluates were analyzed by Western blotting with anti-myc and anti-flag antibodies as indicated.
This relationship is further emphasized by the effect of a rec102 mutation on Rec104 phosphorylation. Rec104 from early meiotic cells (2 h) migrated as a doublet in SDS–PAGE, whereas only the slower-migrating form was observed in extracts from cells at 3 h onward (Figure 1A). The slower-migrating form was converted to the faster one by phosphatase treatment, indicating that Rec104 is a phosphoprotein (Figure 1C). Hypophosphorylated Rec104 remained abundant in a rec102 mutant (Figures 1B and C). Therefore, Rec102 supports normal Rec104 phosphorylation. Because the other DSB genes were not required, Rec104 phosphorylation does not depend on DSB formation.
Rec102 and Rec104 interacted weakly but reproducibly in a yeast two-hybrid system (Figure 1D). Nondenaturing whole-cell extracts from meiotic cells expressing Rec102flag and either mycRec104 or untagged Rec104 were treated with DNase I to eliminate DNA and then immunoprecipitated with anti-myc antibodies. Rec102flag was enriched in the immunoprecipitate from the mycREC104 strain relative to the REC104 control strain (Figure 1E). Similar co-immunoprecipitation results were recently reported with different tagged versions of these proteins (Jiao et al, 2003). Therefore, Rec102 and Rec104 interact directly in vivo.
Dynamics of Rec102 and Rec104 binding to chromosomes
Rec102 is a nuclear protein that associates with chromatin during prophase (Kee and Keeney, 2002). To more precisely define Rec102 association with chromosomes, nuclear spreads were double-stained for Rec102myc and Zip1, a component of the SC central element (Sym et al, 1993). The Zip1 pattern revealed the extent of SC formation and thus the stage in meiosis of any given nucleus (Figure 2). Rec102 first appeared on chromosomes with a punctate or patchy appearance prior to any SC assembly, that is, at or before leptotene (Figure 2A). This result places Rec102 on the chromosomes at the time when DSBs are formed. The staining became brighter through zygotene as the protein accumulated in patchy regions across much of the chromatin (Figure 2B). Staining lasted into early pachytene, indicating that Rec102 persists on the chromatin well past the period when DSBs are made (Figure 2C).
Figure 2.
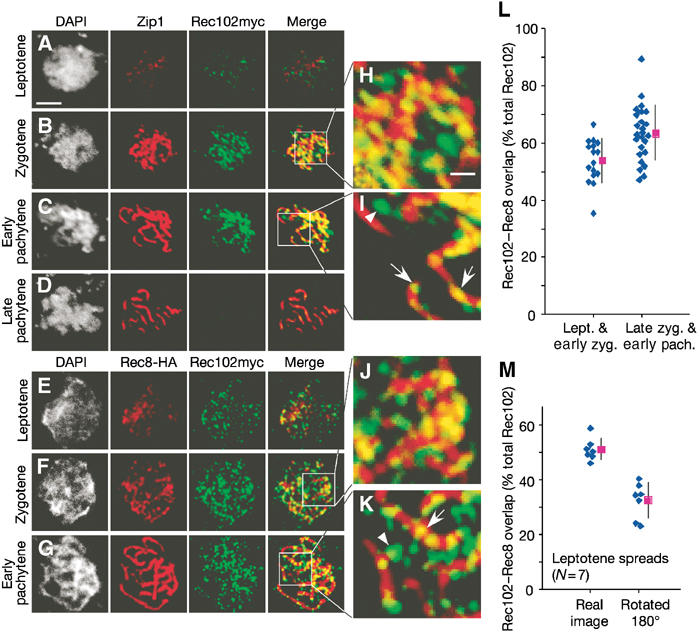
Localization of Rec102 to spread meiotic chromosomes. (A–D) Nuclear spreads from SKY212 (REC102myc) were stained with anti-Zip1 (red) and anti-myc (green) antibodies. (E–G) Spreads from SKY788 (REC102myc REC8HA) were stained with anti-HA (red) and anti-myc (green). Equivalent exposures of representative nuclei are presented, staged according to the extent of SC formation. Scale bars, 3 μm (A–G) or 0.75 μm (H–K). (A) Leptotene or preleptotene. (B) Zygotene (a blow-up of the indicated region is shown in (H)) (C) Early pachytene with extended SCs and significant Rec102 staining (blow-up view in (I)) (D) Late pachytene with more compact SCs and lack of detectable Rec102. (E) Leptotene. (F) Zygotene (blow-up view in (J)). (G) Late zygotene/early pachytene (blow-up view in (K)). Arrows, Rec102 staining overlapping axes; arrowheads, Rec102 staining separated from axes. (L) Overlap of Rec102 immunofluorescence signal with Rec8 in leptotene and early zygotene versus late zygotene and early pachytene spreads. Diamonds, values for individual spreads; squares, mean±s.d. (M) Estimate of random colocalization. The Rec102–Rec8 overlap was measured after 180° rotation of the Rec102 immunofluorescence channel.
Under these spreading conditions, pachytene nuclei could be divided into two classes: those with longer, overlapping SCs (Figure 2C) and those with more compact, distinct SCs (Figure 2D). A total of 28 pachytene nuclei were examined in detail. All (14/14) of the nuclei with extended SCs retained at least some Rec102 staining, whereas all (14/14) of the nuclei with more compact SCs were negative for Rec102 staining (compare Figures 2C and D). Because nuclei of both classes often lay side by side, it is unlikely that the differences between them are an artifact of spreading or uneven staining. We interpret the two classes to represent distinct stages of pachytene, with the more compact class later. Rec102 thus dissociates from the chromosomes during a specific transition period between the two pachytene stages.
Rec104 immunostaining patterns were very similar to those of Rec102 (Figure 3). The protein first showed faint, patchy staining at leptotene, accumulated to maximal levels at zygotene, and disappeared during pachytene.
Figure 3.
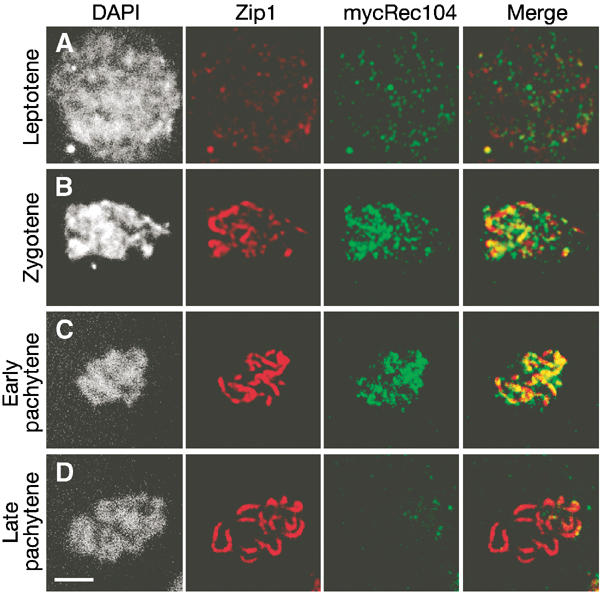
Localization of Rec104 to spread meiotic chromosomes. Nuclear spreads from SKY791 (mycREC104) were analyzed by indirect immunofluorescence with anti-Zip1 (red) and anti-myc (green) antibodies. Scale bar, 3 μm. (A) Leptotene or preleptotene. (B) Zygotene. (C) Early pachytene. (D) Late pachytene.
Spatial organization of chromosomal Rec102
Rec102 signals showed only partial overlap with Zip1 in zygotene cells, suggesting that at least a fraction of the Rec102 protein was located in the chromatin loops rather than on the axes, at least in the regions where SC had formed (Figures 2B and H). To further examine Rec102 localization, we double-stained nuclei for Rec102myc and Rec8-HA, a meiosis-specific cohesin that is axis-associated prior to SC formation (Klein et al, 1999; Blat et al, 2002; Eijpe et al, 2003). Representative images are shown in Figures 2E–G. Again, the early Rec102 signal overlapped only partially with the chromosome axes (Figures 2E, F and J).
The staining patterns of both Rec8 and Rec102 are often irregular rather than being limited to discrete foci. We therefore evaluated overlap between these proteins on a pixel-by-pixel basis using the fluorescence intensities for each protein as recorded by the CCD camera. Based on visual inspection of the images, thresholds were set to define individual pixels as positive or negative for Rec8, and separately as positive or negative for Rec102. Then, the total fluorescence signal over background for Rec102 was measured and divided into two categories: the portion falling within Rec8-positive pixels (i.e. overlapping with axial staining) and the portion falling within Rec8-negative pixels (i.e. non-overlapping). In control experiments, Zip1 showed ∼90% overlap with Rec8 by this method (data not shown).
When this analysis was applied to cells in leptotene through early zygotene, 53.7±7.7% of the total Rec102 signal overlapped with Rec8 staining (mean±s.d., N=16; Figure 2L). Put another way, almost half of the total Rec102 is clearly not colocalized with Rec8. Thus, Rec102 is broadly distributed across large chromosome regions early in prophase, and a substantial fraction of it is in the chromatin loops. Because of limits on resolution of the fluorescence signal, colocalization tends to be overestimated when two proteins lie nearby but are not truly colocalized. Therefore, our data are likely a conservative estimate of the size of the non-axial Rec102 population.
The molecular nature of the Rec102 subpopulation that overlaps with Rec8 staining is less clear. One trivial possibility is that fortuitous overlap results from confinement of two unrelated protein distributions within the area of a nuclear spread (Gasior et al, 1998). To test this possibility, we measured overlap in a set of symmetrically spread leptotene nuclei after rotating the Rec102 image 180°. The rotated spreads showed overlap of 32.7±6.5%, decreased from 51.1±4.0% for the true images of these nuclei (Figure 2M; statistically significant at P<0.0001, Student's t-test). Thus, although fortuitous overlap may account for a fraction of the Rec102–Rec8 colocalization, it cannot account for all of it. It is possible that a subpopulation of Rec102 is intimately associated with axes. Alternatively, Rec102 and Rec8 may be components of distinct structures (e.g. chromatin loops versus axes) that only appear to colocalize because they are spatially coordinated with one another by virtue of being parts of a larger entity. The small size of yeast chromosomes and limits on the resolution of light microscopy preclude distinguishing between these possibilities.
For cells later in meiotic prophase (late zygotene through early pachytene), a substantial fraction of Rec102 was still spatially separated from axes (arrowheads in Figures 2I and K), but there was an increase in the overlapping fraction to 63.2±9.6% of total Rec102 (N=25; Figure 2L; arrows in Figures 2I and K). The difference between earlier and later cells is statistically significant at P<0.002. Analogous changes in spatial distribution have been reported for Spo11 and Ski8 in Sordaria (Storlazzi et al, 2003; Tessé et al, 2003). This difference may reflect a change in the steady-state distribution such that more Rec102 is axis-associated later in meiosis. However, we note that the chromatin tends to be more compact at these later stages (see DAPI images in Figures 2C and G), so apparent overlap of distinct but spatially correlated populations would be expected to increase under these circumstances.
Genetic requirements for recruitment to meiotic chromatin
We next tested whether any of the other DSB proteins were required for the association of Rec102 and Rec104 with chromatin, using a subcellular fractionation assay outlined schematically in Figure 4A. In wild-type cells, ∼70% of Rec102 was in the first pellet, consistent with nuclear localization (Figure 4A). Most of the protein remained insoluble after detergent extraction, but was solubilized by DNase I digestion (fraction S3), indicating that the majority of Rec102 is chromatin-associated. In spo11 and rec104 strains in contrast, ∼90% of the Rec102 protein was recovered in the first soluble extract and little if any was recovered in the chromatin fraction (S3) (Figure 4B). The remaining ∼10% of the protein was recovered in the final pellet, and presumably represents nonspecific aggregation and/or unlysed cells. A similar pattern was observed in a ski8 mutant, except that a small portion (10–15% of total Rec102) was consistently recovered in the chromatin fraction. Thus, Rec102 chromatin association requires Rec104, Spo11, and, to a lesser extent, Ski8. A DSB-defective spo11 missense mutant (spo11-Y135F) supported nearly normal Rec102 fractionation behavior (Figure 4B), indicating that Spo11 protein is required, but not the ability to make DSBs. None of the other DSB proteins was required.
Figure 4.
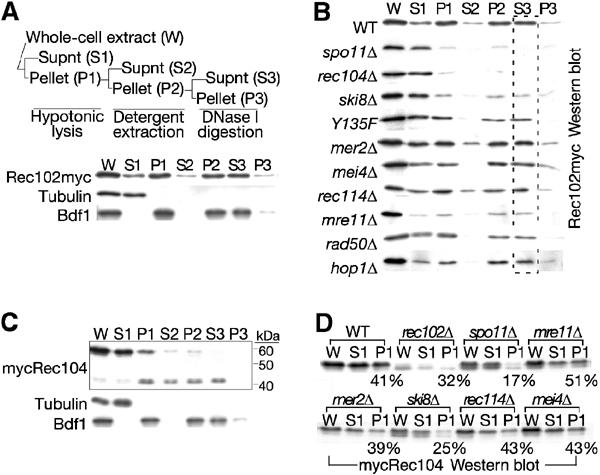
Genetic requirements for chromatin association of Rec102 and Rec104. (A) The subcellular fractionation assay is outlined schematically at the top. The distribution of Rec102myc at 4 h in meiosis in a wild-type strain was followed by Western blotting with anti-myc antibodies. Lanes contain equal cell equivalents of each fraction. Tubulin illustrates the behavior of a soluble protein in this assay; the transcription factor Bdf1 demonstrates the behavior of a known chromatin-associated protein. (B) Effect of DSB-defective mutations on Rec102myc chromatin association. The indicated mutants were analyzed at 4 h in meiosis. (C) Behavior of mycRec104 in the subcellular fractionation assay in a wild-type background at 4 h in meiosis. Endogenous protease activities degrade mycRec104 to yield an ∼44 kDa species. (D) Effect of DSB-defective mutations on mycRec104 chromatin association. The first lysis step of the subcellular fractionation assay was carried out in mycREC104 strains carrying the indicated mutations. The yield of mycRec104 in the pellet fraction for each strain (as percent of total mycRec104) is given.
Analysis of Rec104 proved more complicated because the protein was degraded during the assay (Figure 4C). Roughly half of the protein remained in the first pellet, consistent with nuclear localization of at least a fraction of the protein, and a prominent proteolytic product of 44 000 Mr could be solubilized by DNase I treatment, consistent with chromatin association. The protein was relatively stable until exposed to nonionic detergent (data not shown), perhaps reflecting activation of proteases. We therefore carried out only the first (detergent-free) lysis step to determine the effects of various mutants. Whereas ∼40–50% of total Rec104 protein partitioned to the pellet in wild-type cells and in mer2, mre11, rec114, and mei4 mutants under these conditions, yields in the pellet were reduced approximately two-fold in spo11 and ski8 mutants (Figure 4D). The pellet fraction was also reduced in a rec102 mutant, on top of the reduction in steady-state protein levels noted above.
Whole-cell immunostaining extended these observations. In wild-type cells, Rec102 was predominantly nuclear prior to the first meiotic division as previously described (Kee and Keeney, 2002), but was dispersed throughout the nucleus and cytoplasm during prophase I in a rec104 mutant (Figure 5A). Nuclear localization was also impaired in spo11 and ski8 mutants, but to a different extent: 60–70% of cells showed diffuse staining throughout the cell but 30–40% showed discernible enrichment in the nucleus in addition to diffuse cytoplasmic staining (Figure 5A). Likewise, Rec104 protein was nuclear in wild type, dispersed throughout the cell in a rec102 mutant, and showed a mixture of staining classes for spo11 and ski8 mutants (Figure 5B). Thus, Rec102 and Rec104 are fully interdependent for nuclear localization and chromatin association and share at least a partial requirement for Spo11 and Ski8. These results further define functional subgroups among the DSB proteins, specifically, Rec102+Rec104 and Spo11+Ski8.
Figure 5.
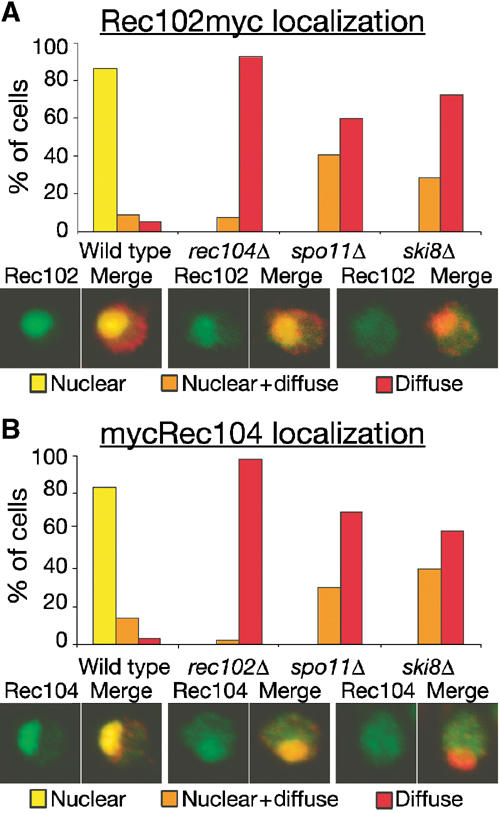
Nuclear localization of Rec102 and Rec104. Cells from wild type and the indicated mutant strains carrying REC102myc (A) or mycREC104 (B) were fixed at 4 h in meiosis and stained with anti-myc antibodies (green) and with DAPI (shown in red). Representative cells are shown for the three staining classes observed: exclusively nuclear localization, diffuse staining of the entire cell, or diffuse cytoplasmic staining along with discernible enrichment in the nucleus. For each strain, ⩾50 cells were analyzed.
Preferred binding sites for Rec102 are not restricted to recombinationally hot domains of chromosome III
To examine Rec102 localization at higher resolution, we performed in vivo crosslinking and chromatin immunoprecipitation (ChIP). Immunoprecipitated DNA was 32P-labeled and hybridized to an array of 133 DNA fragments (average length ∼3 kb) spaced along chromosome III. This approach was previously used by others to study the localization of proteins important for DSB formation and repair and/or for higher order chromosome structure (Blat and Kleckner, 1999; Blat et al, 2002).
To control for timing variations, cultures were staged relative to the first meiotic division (see Materials and methods). For cultures in which few cells had reached zygotene, Rec102 ChIP signals were only slightly over background (data not shown), so these samples were not analyzed further. Cultures with mostly leptotene/zygotene cells or later gave substantial Rec102 ChIP signals. One set of cultures analyzed (Figure 6A) contained cells half way through prophase I. Based on the timing of divisions in these cultures and the immunofluorescence patterns described above for independent cultures, we infer that the Rec102 ChIP signal was derived primarily from leptotene and zygotene cells. However, we cannot rule out the possibility that these cultures contain a significant subpopulation of early pachytene cells, which would also contribute to the observed patterns. In the second set of cultures (Figure 6B), half the cells had reached the first division. Because Rec102 disappears from chromosome spreads during pachytene, we infer that the ChIP signal in these later cultures was derived from the slower cells in the population that were still in late zygotene or early pachytene.
Figure 6.
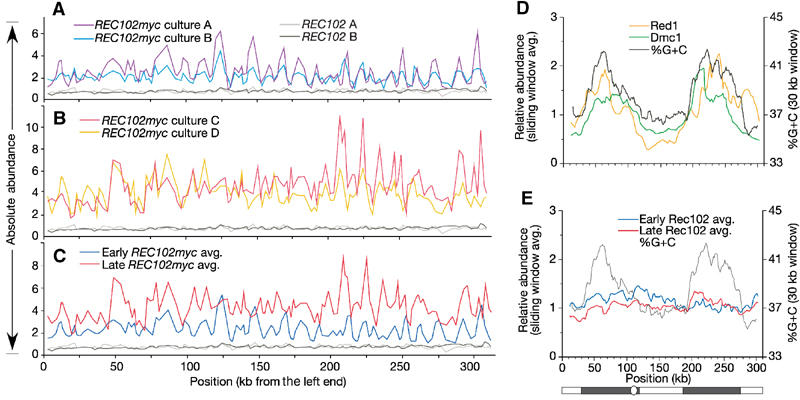
ChIP analysis of the distribution of Rec102 on chromosome III. (A) Absolute signals for two cultures of a myc-tagged strain relative to cultures of an isogenic untagged strain. Each hybridization signal is plotted at the position of the probe fragment midpoint. The REC102myc cultures represent mostly leptotene and zygotene cells. (B) ChIP analysis of two independent REC102myc cultures representing mostly late zygotene and early pachytene cells. (C) Average of the two early REC102myc cultures (from panel A) compared to the average of the two later REC102myc cultures (from panel B). (D, E) Low-resolution views. In the cartoon of chromosome III at the bottom of the panel, the dark gray regions indicate the DSB-hot domains, the white regions are the DSB-cold domains, and the white circle indicates the centromere. (D) Relative abundances for Dmc1 and Red1 ChIP (from Blat et al, 2002) averaged in a nine-fragment sliding window (equivalent to ∼30 kb) and compared to sequence composition (%G+C content averaged over a 30 kb sliding window). (E) Similar sliding window analysis of the early Rec102 average (blue line) and the late Rec102 average (red line), compared to sequence composition.
The Rec102 binding pattern showed a series of peaks and valleys across the length of chromosome III, well above background levels in control cultures lacking myc-tagged protein (Figures 6A and B). Peak positions were reproducible when cultures at the same stage were compared. However, early cultures differed significantly from late cultures, with only 35–45% peak overlap compared to 70–80% overlap for cultures of similar timing (Figure 6C; discussed further below). These results indicate that Rec102 binds preferentially to specific sites or regions in the genome, but that the array of preferred sites varies during the course of meiosis. The later cultures also showed higher overall Rec102 signals (Figure 6C).
Early and late Rec102 binding patterns differed from previously established meiotic features of chromosome III, and did not meet the simplest expectation if the only role of Rec102 were to act locally to promote DSB formation. Previous studies defined the binding patterns for mitotic cohesins Smc1 and Mcd1/Scc1, for Red1 (a structural component of meiotic chromosomes), and for Dmc1 (a strand exchange protein required to repair meiotic DSBs) (Blat and Kleckner, 1999; Blat et al, 2002). At low resolution, assessed by averaging ChIP signals in a sliding window of nine probe fragments (∼30 kb), Red1 and Dmc1 bound preferentially within two large domains, one in each of the chromosome arms (Figure 6D). These recombinationally hot regions correspond to relatively GC-rich isochores (Figure 6D) (Baudat and Nicolas, 1997; Blat and Kleckner, 1999; Blat et al, 2002). In contrast, Rec102 was distributed more uniformly, with no obvious preference for DSB-hot domains (Figure 6E). These results show that Rec102 is not targeted solely to recombinationally active domains.
Rec102 binds preferentially to chromatin loops
In early cultures, 29 reproducible peaks of Rec102 binding were observed (Figures 6 and 7). These peaks had a remarkably regular, even spacing (mean±s.d. 10.5±3.4 kb; range 4.1–18.2 kb). Peak heights were relatively constant across the length of the chromosome, and binding to the sequences in between the peaks was often close to background (Figure 6A).
Figure 7.
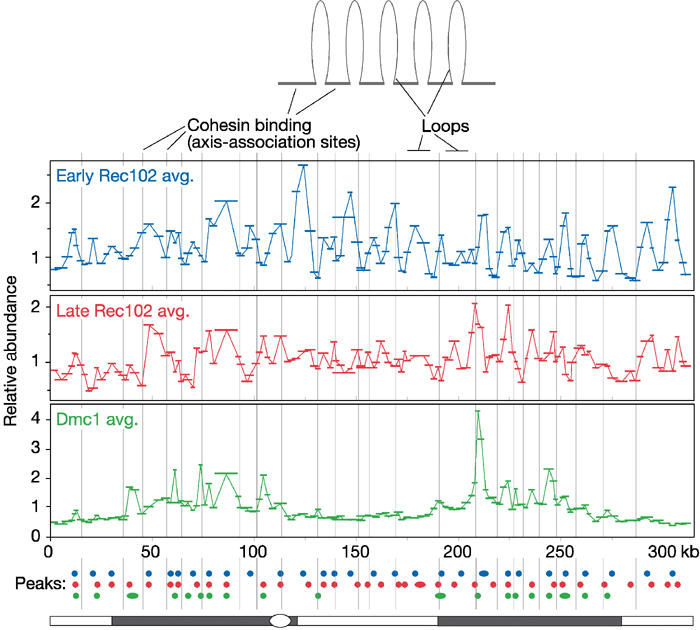
Comparisons of early Rec102, late Rec102, Dmc1, and cohesin distributions. Average relative abundance values for the early Rec102 cultures (blue line, top panel), the two late Rec102 cultures (red line, middle panel), and four Dmc1 samples (green line, bottom panel) are plotted as a function of position on the chromosome. The lengths of individual probe fragments are indicated by the horizontal bars. Peak binding positions are summarized by the filled circles below, color-coded to match the graphs. Vertical gray lines indicate the positions of cohesin binding sites (assigned in Blat and Kleckner, 1999; Blat et al, 2002).
The preferred sites of Rec102 binding in the early cultures were precisely complementary to sites of cohesin binding (Figure 7, top panel). Of the 29 Rec102 peaks, only three (10%) overlapped with previously identified cohesin binding sites (indicated by the gray vertical lines in Figure 7). As discussed in Introduction, the local maxima for cohesin binding are inferred to be the sequences preferentially associated with the chromosome axis, with the sequences in between constituting the chromatin loops (Blat and Kleckner, 1999). Thus, these results strongly suggest that Rec102 binds preferentially to sequences in the chromatin loops. Consistent with this idea, the periodicity of Rec102 peaks is in the range of the genome-wide average of 20 kb for yeast loop sizes estimated from measurements in electron micrographs (Moens and Pearlman, 1988). The ChIP data are consistent with the observation that a significant fraction of Rec102 is spatially separated from the axes in chromosome spreads.
To test the relationship of Rec102 binding to DSB sites, the early Rec102 signal was also compared with the Dmc1 pattern, which was previously shown to represent the DSB pattern well (Blat et al, 2002). Dmc1 (and the DSBs to which it is bound) occurs preferentially in the chromatin loops within GC-rich isochores (Figure 7, lower panel) (Baudat and Nicolas, 1997; Blat et al, 2002). Interestingly, even though both Dmc1 and Rec102 occurred preferentially between the cohesin binding sites, only 37% (7/19) of the Dmc1 peaks matched precisely with Rec102 peaks. Thus, although preferential binding of Rec102 occurs in the vicinity of most if not all DSB sites, maximal Rec102 accumulation may occur up to a few kilobase pairs away. Rec102 also differed from Dmc1 in that Rec102 binding extended equally well over the DSB-hot and DSB-cold regions, with the same periodicity and with no obvious distinction in peak height or width.
Changes in the Rec102 distribution as cells progress through meiosis
Rec102 binding patterns differed in later cultures; this later pattern is inferred to reflect an average of binding in late zygotene and early pachytene cells. These cultures showed more broadly distributed binding, less even spacing of the peaks, more variation in peak height, and less distinction between peaks and valleys in many parts of the chromosome (Figure 6C). Notably, binding in the valleys was significantly higher than background, unlike the case for the early cultures. More of the late Rec102 peaks overlapped with cohesin binding sites (23% of the late Rec102 peaks versus 10% of the early ones), although this still left much of the Rec102 signal on the loops (Figure 7, middle panel). These observations appear to correlate with the increased overall signal and increased overlap with axes in later cells by immunocytology (see above).
Discussion
At least nine proteins in addition to Spo11 are required to make the DSBs that initiate recombination, but the functions of most of them are poorly understood. We report here detailed characterization of the behaviors and chromosomal association of two of these proteins. Rec102 and Rec104 interact physically, are mutually dependent for proper subcellular localization, and share a requirement for Spo11 and Ski8 for their recruitment to meiotic chromosomes. Moreover, Rec102 is required for Rec104 to accumulate to normal steady-state levels and to be properly phosphorylated. These results, combined with multicopy suppression analyses of temperature-sensitive alleles (Salem et al, 1999; Jiao et al, 2003), establish Rec102 and Rec104 as a functional unit during meiosis and define multiple distinct subgroups among the DSB proteins. As discussed below, our findings are consistent with Rec102 and Rec104 playing a direct role in DSB formation, but their spatial and temporal distributions are not as expected for proteins localized only to DSB sites.
The early Rec102/Rec104 distribution reflects the basic axis/loop organization of meiotic chromosomes
Immunocytology revealed significant Rec102 association with chromatin domains away from the axes in early prophase, and ChIP analysis revealed a remarkable ∼10 kb periodicity of Rec102 binding sites precisely complementary to the array of cohesin binding sites thought to define the DNA component of the chromosome axis. Importantly, the single-cell immunocytology analysis agreed well with the population-average ChIP analysis. We interpret these findings to indicate that Rec102 localizes preferentially to sites within chromatin loops in leptotene and zygotene cells, at the time when DSBs are being formed and before they are processed into further recombination intermediates. Where analyzed, Rec104 behaved similarly, so it is likely that this conclusion applies to Rec104 as well. Crossreactivity of anti-flag antibodies with endogenous nuclear protein(s) in spreads has prevented us from testing directly the expected colocalization of Rec102 and Rec104 (unpublished results).
Because ChIP yields a population average measurement, we cannot assess whether Rec102 binds every loop or only a subset of loops in any given cell. Moreover, limits on the spatial resolution of the ChIP analysis make it difficult to distinguish whether the Rec102 peaks represent binding to highly localized sites or binding that is spread across several kilo bases. The immunofluorescence pattern suggests a broader binding pattern in that Rec102 (and Rec104) appeared as diffuse, irregular patches rather than the discrete foci seen for proteins with more localized binding sites (e.g. Dmc1). Higher resolution ChIP analysis will be required to resolve this question.
In addition to the loop-associated Rec102, a substantial portion of the protein overlapped with axes of spread chromosomes, although there was little such overlap apparent in ChIP analysis of early prophase cells. These findings may indicate that there are separate axis-associated and loop-associated Rec102 populations that show different tendencies to be recovered in ChIP experiments. Alternatively, Rec102 may localize exclusively to chromatin loops, with overlap of Rec102 and Rec8 immunofluorescence signals attributable to the proximity of loop arrays to the axes on chromosome spreads combined with limitations on the resolution of light microscopy. A third, intermediate possibility is that Rec102 bound to sequences in a chromatin loop is recruited to the axis along with the loop, as part of a ‘tethered loop–axis complex'. As discussed in Introduction, such tethered complexes can account for the observation that DSBs occur in loop sequences, whereas recombinosomes are intimately associated with axes. The current studies do not allow us to distinguish between these possibilities.
Rec102 distribution does not reflect the DSB map
The broad distribution of Rec102 seen by immunofluorescence and ChIP was not as expected for a protein localized only to DSB sites. Moreover, maximal Rec102 binding by ChIP was often offset several kilobases from peaks of DSBs (as indicated by Dmc1 binding). These findings indicate that Rec102 loading cannot be the determining factor for specification of local DSB hotspots, nor of larger hot domains. One possibility is that DSB positions are governed by a combinatorial process involving overlap of distinct protein distributions. In this model, it is conceivable that no one protein is restricted solely to DSB hotspots, and that DSB patterns emerge only from the intersection of long-range (large domains), medium-range (loop/axis), and short-range (localized targeting of catalytic machinery) spatial organizations. Alternatively, the distribution of DSB sites could be controlled by restrictions on the spatial distribution of just a few DSB-promoting factors. Notably, ChIP analysis of Mre11 shows a pattern that correlates well with DSB hotspots (Borde et al, 2004; K Ohta, personal communication).
Recruiting Rec102/104 to chromosomes requires Spo11/Ski8
Spo11 and Ski8 form a complex critical for DSB formation (Arora et al, 2004). The studies here show that normal accumulation of Rec102 and Rec104 in the nucleus and their assembly on chromosomes requires this Spo11–Ski8 complex. Rec102 and Rec104 sizes are below the diffusion limit for nuclear pores, and both were found in the cytoplasm as well as the nucleus in spo11 and ski8 mutants by whole-cell staining, yet showed little or no stable association with chromatin in the fractionation assay. Thus, it is likely that Rec102 and Rec104 move freely in and out of the nucleus but are most stably sequestered there only when they can form a complex on chromosomes. However, it is also possible that interactions with Spo11/Ski8 affect the kinetics of nuclear import or export. Our results are consistent with the observation that fusing Spo11 to a sequence-specific DNA binding domain can target DSBs to new sites but still dependent on Rec102 and Rec104, suggesting that Spo11 can recruit these proteins in order to promote DSB formation (Peciña et al, 2002).
However, the findings appear to raise a paradox because Spo11 must bind to every DSB site, yet the Rec102 distribution does not reflect the distribution of DSB sites. Perhaps, Spo11/Ski8 complexes are themselves distributed more widely than just DSB sites. Ski8 immunolocalization patterns in S. cerevisiae and Spo11 and Ski8 localization patterns in Sordaria macrospora lend support to this possibility (Storlazzi et al, 2003; Tessé et al, 2003; Arora et al, 2004). Alternatively, initial Rec102/104 recruitment to chromosomes may occur in a Spo11-dependent fashion at DSB sites, and then spread locally across broader regions. Such spreading might account for the changes in the ChIP pattern in later cells.
Possible roles for Rec102 and Rec104 in DSB formation
With any cytological or ChIP analysis, it is difficult to prove that one is observing the functional population of the target molecule. However, several observations suggest that the early localization patterns described here reflect Rec102 function in DSB formation: appearance on chromosomes at or prior to the DSB stage; dependence on Spo11/Ski8 but not on DSB formation, indicating that Rec102/104 chromosomal association is tied to Spo11 function but is not a downstream consequence of break formation; and preference for chromatin loops.
What roles might Rec102 and Rec104 play in DSB formation consistent with the observed localization patterns? The association of Rec102 with chromatin loops, along with its prominent binding near but not always at major DSB sites, could indicate that Rec102/104 complexes promote alterations of local chromatin structure within loops that stabilize Spo11-containing complexes at hotspots, or that activate hotspot-associated Spo11 complexes to cleave DNA. It has been suggested that changes in chromatin structure (or the resulting stress on the chromosome) could provide a physical force that irreversibly commits Spo11 to DSB formation through disruption of the Spo11 dimer interface (Keeney, 2001). Another possibility, suggested by the possible colocalization of a fraction of Rec102/104 with axes, is that they participate in loop–axis interactions that promote DSB formation or repair in spatial proximity to the axis. Interestingly, Rec104 overexpression can suppress DSB defects caused by the absence of the axis-associated protein Hop1 (Friedman et al, 1994).
A transition point during pachytene
Rec102 and Rec104 disappeared from chromosomes during pachytene. It does not appear that Rec102 is targeted for efficient degradation at this point because Rec102 levels remained high in whole-cell extracts to at least 8 h (when 80–90% of the cells had completed the first division) (Figure 1A and data not shown). This behavior is reminiscent of that of certain DSB proteins in other organisms. For example, the Drosophila Mei-P22 protein is associated with chromosomes until mid-pachytene (Liu et al, 2002), and in Sordaria Spo11 and Ski8 appear to dissociate from chromosomes during pachytene (Storlazzi et al, 2003; Tessé et al, 2003). However, the same does not appear to be true for all DSB proteins: Ski8 in yeast appears to persist on chromosomes into late pachytene, although perhaps at reduced levels (Arora et al, 2004 and unpublished results).
Disappearance of Rec102 and Rec104 correlated with a change in the appearance of spread chromosomes. Earlier electron and light microscopy studies defined an apparently similar transition between distinct pachytene stages, correlated with changes in chromatin compaction (R Padmore, PhD thesis; N Kleckner, personal communication). The functional significance of this transition is not known, but it may correspond to transitions described in other organisms. For example, transit through mid-pachytene in mouse spermatocytes is accompanied by morphological changes in the SC and functional changes in the properties of the chromosomes, including synaptic adjustment and relaxation of barriers against the use of sister chromatids for DSB repair (Moses et al, 1984; Mahadevaiah et al, 2001). Rec102/104 dissociation will be a useful cytological marker for studies of this transition in yeast.
The persistence of Rec102 and Rec104 on chromosomes into pachytene is intriguing. It is possible that this persistence has no functional significance. However, their dissociation from chromosomes at a defined stage suggests that their persistence is regulated, raising in turn the possibility that these proteins play continued roles after DSB formation. If so, the fact that no late roles have been revealed by genetic analyses may indicate that such roles lie downstream of DSB formation, perhaps including functions in processing recombination intermediates, controlling the distribution of crossovers, and/or integrating recombination with higher order chromosome structures.
Materials and methods
Strains and culture methods
Meiotic cultures were prepared as described (Kee and Keeney, 2002). Yeast strains (SK1 background) are listed in Supplementary Table I. Two-hybrid assays were as described (Arora et al, 2004). REC8-HA3 (Klein et al, 1999) was provided by F Klein (University of Vienna). REC102myc9∷URA3 and REC102flag3∷TRP1 were described earlier (Kee and Keeney, 2002). REC104 was tagged using a tag-URA3-tag strategy as described in Supplementary data. The mycREC104 allele supported normal meiotic recombination initiation in vivo: intragenic recombination frequencies at arg4 in return-to-growth assays were 0.051±0.016 compared to 0.053±0.014 for untagged (prototrophs per viable cell, mean±s.d., N⩾3); maximal steady-state DSB levels at his4LEU2 site I in RAD50 cells were 3.5% of total DNA versus 3.3% in untagged; and spore viability was 95 versus 93% for untagged (⩾20 tetrads for each).
Cytology
Chromosome spreads were prepared and stained using variations on published methods (Loidl et al, 1991; Gasior et al, 1998). Details are provided in Supplementary data. Fluorescence images were captured using a Cooke Sensicam cooled CCD camera and processed using the Slidebook program (Intelligent Imaging Innovations). Fluorescence channels were aligned by imaging 0.2-μm-diameter beads that emit fluorescence in all channels (Tetraspek beads, Molecular Probes). Specificity of staining for myc-tagged proteins was demonstrated previously (Kee and Keeney, 2002). Colocalization of Rec102myc and Rec8-HA was quantified as described in the text and Supplementary data.
Protein analyses
For subcellular fractionation, ∼109 cells were digested for 10 min at 30°C with 80 μg/ml zymolyase 100T, then spheroplasts were pelleted and placed on ice. Subsequent steps were at 0–4°C. The pellet was resuspended in five volumes of hypotonic buffer (HB: 100 mM MES–NaOH, pH 6.4, 1 mM EDTA, 0.5 mM MgCl2) plus protease inhibitors. After 5 min, 120 μl of this whole-cell lysate (W) was layered on a 120 μl 30% sucrose cushion in HB and centrifuged for 10 min at 16 000 g. The supernatant was saved (S1), the pellet was resuspended in 100 μl EBX (50 mM HEPES–NaOH, pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 0.05% Triton X-100, plus protease inhibitors), and an aliquot was reserved (P1). A 90 μl portion of the P1 suspension was centrifuged at 16 000 g for 10 min. The supernatant was collected (S2), the pellet was resuspended in 75 μl EBX, and 4 μl of 1 mg/ml DNase I and 2 μl of 1 M MgCl2 were added. The suspension was incubated at room temperature for 10 min, an aliquot was reserved (P2), and the remaining 60 μl was centrifuged for 10 min at 16 000 g. The chromatin supernatant (S3) was collected and the pellet was resuspended in 50 μl EBX (P3). Fractions were analyzed by Western blotting with chemiluminescent detection and quantified by densitometry scans of the film.
Nondenaturing extracts and immunoprecipitations were as described (Kee and Keeney, 2002). Antibodies for Western blotting were mouse monoclonal anti-myc (1:1000, Covance), mouse monoclonal anti-flag (1:1000, Sigma), rabbit polyclonal anti-Bdf1 (1:1000, GS Roeder), and peroxidase-conjugated secondary antibodies (1:10 000, Jackson ImmunoResearch). Phosphatase treatment was performed on anti-myc immunoprecipitates as described in Supplementary data.
Chromatin immunoprecipitation
Rec102 ChIP was performed as described (Blat and Kleckner, 1999; Blat et al, 2002, http://www.mcb.harvard.edu/kle ckner/ChIP.html) in SKY212 (REC102-myc9∷URA3). Control experiments were performed in SKY165 (REC102). For each ChIP sample, 600 ml of synchronized meiotic culture (2 × 107 cells/ml) was processed. At various times (see below), cells were fixed with formaldehyde and chromatin was purified and immunoprecipitated with anti-myc antibodies (Covance) at 4°C overnight followed by protein G–Sepharose 4 Fast Flow (Amersham Biosciences) at 4°C for 6 h. DNA was purified, radiolabeled, and hybridized to a membrane containing an array of fragments spanning chromosome III.
Data were analyzed as described (Blat et al, 2002). ‘Absolute abundance' values control for hybridization efficiency of different DNA fragments, and were obtained by dividing the counts in each ChIP filter spot by the corresponding counts on a filter probed with a sample of the input chromatin. ‘Relative abundance' further normalizes for differences in total signal strength and thus facilitates comparison of ChIP data for different proteins. Relative abundance was calculated by first dividing the counts in each ChIP filter spot by the sum of all the counts on the filter. The same analysis was performed for each input filter spot. The fractional value for each ChIP filter spot was then divided by the corresponding fractional value for each input filter spot.
The kinetics of meiosis can vary substantially from culture to culture even in the same strain. To control for these variations, an aliquot of each culture was allowed to continue meiosis after cells were collected for formaldehyde treatment. Meiotic divisions were scored and the time of formaldehyde crosslinking was compared to the time when 50% of the unfixed cells completed the first division (MI50%). Details of the cultures in Figure 6 are as follows. Untagged control culture A was crosslinked at 5 h (MI50%=10.5 h) and control culture B was crosslinked at 7 h (MI50%=9.5 h). REC102myc culture A was crosslinked at 4 h (MI50%=7 h), REC102myc culture B was crosslinked at 6 h (MI50%=9 h), REC102myc culture C was crosslinked at 6 h (MI50%=5.5 h), and REC102myc culture D was crosslinked at 7 h (MI50%=6.5 h). Notably, REC102myc cultures B and C (i.e. one early and one late) were processed in parallel in the same experiment, so differences between early and late cultures are not an artifact of day-to-day variations in experimental conditions.
Supplementary Material
Supplemental material
Acknowledgments
We thank Michael Lichten and Nancy Kleckner for critical comments on the manuscript and Shirleen Roeder for antibodies. This work was supported in part by NIH grant R01 GM58673 (to SK). RUP was supported by NSF Minority Postdoctoral Fellowship DBI-0001979 and by NIH grant R01 GM44794 (to N Kleckner).
References
- Arora C, Kee K, Maleki S, Keeney S (2004) Antiviral protein Ski8 is a direct partner of Spo11 in meiotic DNA break formation, independent of its cytoplasmic role in RNA metabolism. Mol Cell 13: 549–559 [DOI] [PubMed] [Google Scholar]
- Ashley T (1988) G-band position effects on meiotic synapsis and crossing over. Genetics 118: 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Nicolas A (1997) Clustering of meiotic double-strand breaks on yeast chromosome III. Proc Natl Acad Sci USA 94: 5213–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blat Y, Kleckner N (1999) Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell 98: 249–259 [DOI] [PubMed] [Google Scholar]
- Blat Y, Protacio RU, Hunter N, Kleckner N (2002) Physical and functional interactions among basic chromosome organizational features govern early steps of meiotic chiasma formation. Cell 111: 791–802 [DOI] [PubMed] [Google Scholar]
- Borde V, Wu T-C, Lichten M (1999) Use of a recombination reporter insert to define meiotic recombination domains on chromosome III of Saccharomyces cerevisiae. Mol Cell Biol 19: 4832–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borde V, Lin W, Novikov E, Petrini JH, Lichten M, Nicolas A (2004) Association of Mre11p with double-strand break sites during yeast meiosis. Mol Cell 13: 389–401 [DOI] [PubMed] [Google Scholar]
- Eijpe M, Heyting C, Gross B, Jessberger R (2000) Association of mammalian SMC1 and SMC3 proteins with meiotic chromosomes and synaptonemal complexes. J Cell Sci 113: 673–682 [DOI] [PubMed] [Google Scholar]
- Eijpe M, Offenberg H, Jessberger R, Revenkova E, Heyting C (2003) Meiotic cohesin REC8 marks the axial elements of rat synaptonemal complexes before cohesins SMC1beta and SMC3. J Cell Biol 160: 657–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth I, Vogel G, Krone W, Vogel W, Assum G (2000) An isochore transition in the NF1 gene region coincides with a switch in the extent of linkage disequilibrium. Am J Hum Genet 67: 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB, Hollingsworth NM, Byers B (1994) Insertional mutations in the yeast HOP1 gene: evidence for multimeric assembly in meiosis. Genetics 136: 449–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior SL, Wong AK, Kora Y, Shinohara A, Bishop DK (1998) Rad52 associates with RPA and functions with Rad55 and Rad57 to assemble meiotic recombination complexes. Genes Dev 12: 2208–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton JL, DeRisi J, Shroff R, Lichten M, Brown PO, Petes TD (2000) Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA 97: 11383–11390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist GP (1992) Chromosome bands, their chromatin flavors, and their functional features. Am J Hum Genet 51: 17–37 [PMC free article] [PubMed] [Google Scholar]
- Jiao K, Salem L, Malone R (2003) Support for a meiotic recombination initiation complex: interactions among Rec102p, Rec104p, and Spo11p. Mol Cell Biol 23: 5928–5938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee K, Keeney S (2002) Functional interactions between SPO11 and REC102 during initiation of meiotic recombination in Saccharomyces cerevisiae. Genetics 160: 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S (2001) Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol 52: 1–53 [DOI] [PubMed] [Google Scholar]
- Klein F, Mahr P, Galova M, Buonomo SB, Michaelis C, Nairz K, Nasmyth K (1999) A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell 98: 91–103 [DOI] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31: 241–247 [DOI] [PubMed] [Google Scholar]
- Liu H, Jang JK, Kato N, McKim KS (2002) mei-P22 encodes a chromosome-associated protein required for the initiation of meiotic recombination in Drosophila melanogaster. Genetics 162: 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loidl J, Nairz K, Klein F (1991) Meiotic chromosome synapsis in a haploid yeast. Chromosoma 100: 221–228 [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JMA, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS (2001) Recombinational DNA double strand breaks in mice precede synapsis. Nat Genet 27: 271–276 [DOI] [PubMed] [Google Scholar]
- Moens PB, Pearlman RE (1988) Chromatin organization at meiosis. BioEssays 9: 151–153 [DOI] [PubMed] [Google Scholar]
- Moens PB, Pearlman RE, Heng HH, Traut W (1998) Chromosome cores and chromatin at meiotic prophase. Curr Top Dev Biol 37: 241–262 [DOI] [PubMed] [Google Scholar]
- Moses MJ, Dresser ME, Poorman PA (1984) Composition and role of the synaptonemal complex. Symp Soc Exp Biol 38: 245–270 [PubMed] [Google Scholar]
- Peciña A, Smith KN, Mezard C, Murakami H, Ohta K, Nicolas A (2002) Targeted stimulation of meiotic recombination. Cell 111: 173–184 [DOI] [PubMed] [Google Scholar]
- Pelttari J, Hoja MR, Yuan L, Liu JG, Brundell E, Moens P, Santucci-Darmanin S, Jessberger R, Barbero JL, Heyting C, Hoog C (2001) A meiotic chromosomal core consisting of cohesin complex proteins recruits DNA recombination proteins and promotes synapsis in the absence of an axial element in mammalian meiotic cells. Mol Cell Biol 21: 5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem L, Walter N, Malone R (1999) Suppressor analysis of the Saccharomyces cerevisiae gene REC104 reveals a genetic interaction with REC102. Genetics 151: 1261–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storlazzi A, Tessé S, Gargano S, James F, Kleckner N, Zickler D (2003) Meiotic double-strand breaks at the interface of chromosome movement, chromosome remodeling and reductional division. Genes Dev 17: 2675–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sym M, Engebrecht JA, Roeder GS (1993) ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72: 365–378 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Cosma MP, Wirth K, Nasmyth K (1999) Identification of cohesin association sites at centromeres and along chromosome arms. Cell 98: 847–858 [DOI] [PubMed] [Google Scholar]
- Tessé S, Storlazzi A, Kleckner N, Gargano S, Zickler D (2003) Localization and roles of Ski8p in Sordaria macrospora meiosis and delineation of three mechanistically distinct steps of meiotic homolog juxtaposition. Proc Natl Acad Sci USA 100: 12865–12870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heemst D, Heyting C (2000) Sister chromatid cohesion and recombination in meiosis. Chromosoma 109: 10–26 [DOI] [PubMed] [Google Scholar]
- Wu T-C, Lichten M (1995) Factors that affect the location and frequency of meiosis-induced double-strand breaks in Saccharomyces cerevisiae. Genetics 140: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zickler D, Kleckner N (1999) Meiotic chromosomes: integrating structure and function. Annu Rev Genet 33: 603–754 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material