

Abstract
Colonic bacteria, exemplified by Bacteroides thetaiotaomicron, play a key role in maintaining human health by harnessing large families of glycoside hydrolases (GHs) to exploit dietary polysaccharides and host glycans as nutrients. Such GH family expansion is exemplified by the 23 family GH92 glycosidases encoded by the B. thetaiotaomicron genome. Here we show that these are α-mannosidases that act via a single displacement mechanism to utilize host N-glycans. The three-dimensional structure of two GH92 mannosidases defines a family of two-domain proteins in which the catalytic center is located at the domain interface, providing acid (glutamate) and base (aspartate) assistance to hydrolysis in a Ca2+-dependent manner. The three-dimensional structures of the GH92s in complex with inhibitors provide insight into the specificity, mechanism and conformational itinerary of catalysis. Ca2+ plays a key catalytic role in helping distort the mannoside away from its ground-state 4C1 chair conformation toward the transition state.
The human large bowel microbiota, consisting of ~1014 bacteria, contributes to human nutrition and health1. It is estimated that ~10% of caloric intake is derived from plant polysaccharides that are deconstructed exclusively by enzymes produced by the microbiota2. The large bowel microbiota has a generic influence on host-nutrient partitioning, particularly with respect to carbohydrates and fats. An example of this modulation is provided by the observation that the transfer of this bacterial community from a genetically obese mouse to a normal animal induces obesity in its new host3. The colonic microbiota can also influence the host immune system, enhancing the health of the host4,5. Furthermore, butyrate, an end product of colonic fermentation, is thought to have antineoplastic properties and thus plays a role in maintaining a healthy gut6.
Reflecting the critical role the large bowel microbiota plays in maintaining human health, there have been numerous attempts to manipulate the composition of this ecosystem through dietary and probiotic intervention6. Such strategies have been hampered by a paucity of knowledge of the biochemistry of these micro-organisms; particularly with respect to their utilization of complex carbohydrates as nutrients. Researchers have started to unravel the genetic basis for the function of the microbiota. Thus, the genome sequences of selected dominant colonic bacteria are available7–9, and metagenomics is starting to yield more extensive information on the complete microbiota10,11. Commensurate with their nutritional flexibility, the genomes of colonic bacteria, exemplified by B. thetaiotaomicron, have revealed an array of genes encoding glycoside hydrolases (>200). The vast majority of these enzymes are in glycoside hydrolase families (GHs) that contain mainly exo-acting enzymes, which may suggest that the organism is able to use the saccharide decorations appended to the backbone of structural polysaccharides and glycoproteins8. This expansion in exo-GHs is particularly evident in family GH92, which contains 23 members; however, only α-1,2-mannosidase activity has been ascribed to this family12. There are few α-linked mannosyl linkages in nature (Fig. 1), and it is unclear whether the evolutionary drivers for the observed expansion of GH92 in B. thetaiotaomicron reflect substrate or regulatory complexity.
Figure 1. Human N-glycans and the role of α-mannosidases.

(a) Illustration of a Man9(GlcNac)2 N-glycan with the stereochemistry of the linkages indicated. (b) The enzymatic hydrolysis of α-1,2-mannosides, as catalyzed here by many of the GH92 α-mannosidases.
Here we present the analysis of 22 of the 23 GH92 enzymes encoded by the genome of B. thetaiotaomicron. We show that these enzymes are Ca2+-dependent α-mannosidases that perform catalysis with inversion of anomeric configuration. The three-dimensional structures of GH92 α-1,2-mannosidases Bt3990 and Bt2199 reveal bimodular proteins in which an N-terminal β-sheet domain is appended to an (α/α)6 barrel catalytic module in which a shallow surface depression between the two domains comprises the active site. The three-dimensional analysis of inhibitor complexes provides insight into the likely conformational itinerary for mannoside hydrolysis. This study, in addition to providing a biochemical basis for expansion of the GH92 family in a dominant colonic bacterium, informs the use of small-molecule α-mannosidase inhibitors in probing the functional significance of this enzyme family in the survival of B. thetaiotaomicron in the human colon.
RESULTS
Activity and stereochemistry of the GH92 multigene family
We screened the activity of 22 out of 23 B. thetaiotaomicron GH92 enzymes (bt2105 did not express) for mannosidase activity (Supplementary Table 1), and we performed kinetic analysis on 4-nitrophenyl α-D-mannopyranoside (4NP-Man, 1) and the preferred disaccharide (Table 1). All the enzymes had α-mannosidase activity. Of the 16 enzymes that hydrolyzed 4NP-Man, only Bt1032, Bt2948 and Bt3963 did not also cleave D-manno-configured disaccharides. Conversely, while Bt1769, Bt3530 and Bt3773 exhibit disaccharidase activity, they do not tolerate 4NP-Man, which suggests that recognition of a saccharide bound at the +1 (nomenclature in ref. 13) subsite is a critical feature of substrate binding in these enzymes.
Table 1.
Catalytic activity of B. thetaiotaomicron GH92 enzymes
| Bt GH92 | 4-Nitrophenyl-α-D-mannopyranoside
|
Mannobiose
|
Yeast mannana | Ovalbumina | |||||
|---|---|---|---|---|---|---|---|---|---|
| kcat (min−1) | Km (mM) | kcat/Km (min−1 mM−1) | Substrateb | kcat (min−1) | Km (mM) | kcat/Km (min−1 mM−1) | |||
| 1032 | 0.36 ± 0.038 | 1.7 ± 0.36 | 0.21 | NAc | NA | NA | NA | ++ | + |
| 1769 | NA | NA | NA | α1,3-Man2 | 3.8 × 103± 2.1 × 102 | 1.3 ± 0.14 | 2.9 × 103 | +++ | − |
| 1878 | 0.08 ± 0.005 | 0.9 ± 0.13 | 0.089 | α1,3-Man2 | 17 ± 1.3 | 2.0 ± 0.38 | 8.4 | ++ | + |
| 2111 | NA | NA | NA | NA | NA | NA | NA | ++ | ++ |
| 2199 | 6.3 ± 0.29 | 7.5 ± 0.6 | 0.84 | α1,2-Man2 | 2.4 × 103± 1.4 × 102 | 0.68 ± 0.11 | 3.6 × 103 | +++ | +++ |
| 2629 | NA | NA | NA | NA | NA | NA | NA | + | ++ |
| 2948 | ND | ND | 2.56 | NA | NA | NA | NA | ++ | + |
| 3130 | 3.4 × 103± 1.9 × 102 | 0.51 ± 0.097 | 6.7 × 103 | α1,3-Man2 | NDd | ND | 0.2 | +++ | + |
| 3527 | NA | NA | NA | NA | NA | NA | NA | ++ | − |
| 3530 | NA | NA | NA | α1,4-Man2 | 0.06 ± 0.005 | 0.61 ± 0.15 | 0.095 | +++ | ++ |
| 3773 | NA | NA | NA | α1,3-Man2 | 1.1 ± 0.085 | 2.5 ± 0.37 | 0.45 | +++ | ++ |
| 3784 | 4.8 ± 0.39 | 6.9 ± 0.93 | 0.69 | α1,2-Man2 | 27 ± 5.5 | 2.3 ± 0.81 | 11 | +++ | ++ |
| 3858 | 0.025 ± 0.001 | 0.27 ± 0.065 | 0.094 | α1,3-Man2 | 2.4 × 103± 1.5 × 102 | 1.4 ± 0.17 | 1.7 × 103 | +++ | + |
| 3962 | 17 ± 1.6 | 5 ± 0.84 | 3.4 | α1,2-Man2 | 8.2 × 102± 33 | 0.43 ± 0.052 | 1.9 × 103 | +++ | ++ |
| 3963 | 0.027 ± 0.001 | 0.79 ± 0.08 | 0.034 | NA | NA | NA | NA | − | − |
| 3965 | 14 ± 0.4 | 1 ± 0.079 | 14 | α1,4-Man2 | 89 ± 6.8 | 8 ± 1.2 | 11 | +++ | + |
| 3990 | 5.6 ± 0.28 | 4.2 ± 0.42 | 1.3 | α1,2-Man2 | 5.2 × 103± 3 × 102 | 0.76 ± 0.11 | 6.8 × 103 | +++ | +++ |
| 3991 | 0.15 ± 0.021 | 2.7 ± 0.65 | 0.055 | α1,3-Man2 | 2.8 × 102± 18 | 9 ± 1.3 | 31 | +++ | + |
| 3994 | 94 ± 5.6 | 3.7 ± 0.43 | 26 | α1,4-Man2 | ND | ND | 0.005 | +++ | + |
| 4073 | 0.27 ± 0.014 | 1.8 ± 0.21 | 0.14 | α1,4-Man2 | 1.1 × 104± 5.6 × 102 | 0.27 ± 0.049 | 4.1 × 104 | +++ | − |
| 4092 | ND | ND | 0.067 | α1,2-Man2 | 1.1 × 102± 13 | 2.0 ± 0.45 | 54 | +++ | ++ |
| 4093 | 0.67 ± 0.049 | 1.3 ± 0.19 | 0.545 | α1,4-Man2 | 80 ± 3.9 | 35 ± 2.4 | 2.3 | − | + |
Mannose release indicated the enzyme was active against the substrate (+), while no release of the sugar is indicative of no activity (−). The score for each protein is based on the HPLC signal for mannose after 16 h incubations with 1 μM of enzyme: HPLC signals > 500 nanocoulomb (nc), +++; 200–500 nc, ++; <200 nc, +.
Substrate: the substrate used was the most active for the enzyme determined from the screen reported in Supplementary Table 1.
NA: no activity was detected.
ND: the individual kinetic values could not be determined as Km was above the maximum substrate concentration deployed.
There are three enzymes that are highly active α-1,2-mannosidases (Bt2199, Bt3962 and Bt3990) (Table 1), while Bt1769, Bt3858 and Bt3991 are α-1,3-mannosidases that display particularly high activity against α-1,3-mannobiose (2). Bt3965, Bt4073 and Bt4093 exhibit specificity for α-1,4-mannobiose (3) (a substructure unknown in human glycans). The GH92 proteins Bt2111, Bt2629 and Bt3527 display no activity against small mannose-configured substrates, but release mannose from eukaryotic glycans, suggesting that they may contain an array of reducing end “positive”13 subsites that are poorly mimicked by disaccharide substrates. Bt3130 displays high activity against 4NP-Man and trace activity against α-1,3-mannobiose, but it is active against both eukaryotic glycans assessed. It is possible that the natural substrate recognized by Bt3130 is a complex glycan or a currently undefined glycoconjugate.
The capacity of selected GH92s to hydrolyze high-mannose N-glycans was assessed. Bt3990 and Bt2199 hydrolyze Man9GlcNAc2 to Man5GlcNAc2 (Supplementary Fig. 1), and thus cleave all the α-1,2-mannoside linkages in high-mannose glycans. The α-1,3-mannosidase Bt3991, while unable to hydrolyze Man9GlcNAc2, converts Man5GlcNAc2 (generated from Man9GlcNAc2 by Bt3990) into Man3GlcNAc2, indicating that the two α-1,3-mannosidic linkages in N-glycans are available to the enzyme once the terminal α-1,2-linked mannosyl residues have been removed. The capacity of Bt3994 to convert Man5GlcNAc2 to Man4GlcNAc2, and Man3GlcNAc2 to Man2GlcNAc2, indicates that the enzyme hydrolyzes the distal α-1,6-mannosidic linkage in high-mannose N-glycans. As Bt3994 displays trace activity against disaccharides, the active site likely requires longer substrates. The two other α-1,3-mannosidases, Bt3858 and Bt1769, convert Man4GlcNAc2 (generated by the α-1,2-mannosidase Bt3990 and the α-1,6-mannosidase Bt3994) to Man3GlcNAc2, but did not attack Man5GlcNAc2, indicating that these enzymes cannot tolerate α-1,6-Man side chains.
1H NMR analysis of reaction products revealed that the GH92 enzymes Bt3990, Bt3858 and Bt4073 cleave α-mannosidic bonds with inversion of anomeric configuration (Supplementary Fig. 2), and thus the catalytic apparatus of the three enzymes is predicted14 to comprise a catalytic acid to aid leaving group departure and a catalytic base to activate the nucleophilic water for a single-displacement nucleophilic substitution. All the GH92 enzymes analyzed were inactivated by EDTA, and activity was fully recovered by the addition of calcium (although partial reactivation was achieved with other divalent metals; Supplementary Table 2). These data indicate that GH92 is a calcium-dependent family, consistent with the conservation of the amino acids that coordinate the metal ion within the active site of these enzymes (see below). The pH optimum of a representative GH92 enzyme, Bt3991, was 6.5 (Supplementary Fig. 3).
Three-dimensional structures of Bt3990 and Bt2199
The crystal structure of the B. thetaiotaomicron GH92 α-1,2-mannosidase Bt3990 was solved at a resolution of 1.9 Å (details in Methods and Supplementary Table 3). The structure was subsequently determined in complex with a nonhydrolyzable substrate mimic, thio α-1,2-mannobioside (thiomannobioside, 4), and the inhibitors swainsonine (5), mannoimidazole (6) and kifunensine (7) at resolutions from 2.4 to 1.9 Å. The structure of a related GH92 α-1,2-mannosidase, Bt2199, has also been solved and is discussed later in light of experiments on substrate specificity. Bt3990 adopts a two-domain structure (Fig. 2a), with the catalytic center derived from elements of both structural domains (Fig. 2b). The N-terminal domain (residues 20–288) displays a β-sandwich that exhibits similarity to the accessory domains of other glycosidases, including GH38 α-mannosidases (top scoring result from SSM15 was the bovine lysosomal α-mannosidase16, with r.m.s.d. 3 Å over 158 matched Cα positions showing just 4% sequence identity). The N-terminal module terminates with an extended α-helix, residues 271–288, with residues 289–293 and 296–309 contributing the first β-strand and α-helix, respectively, of the larger C-terminal domain.
Figure 2. Three-dimensional structure of GH92 α-1,2-mannosidase Bt3990.

(a) Protein cartoon of the Bt3990 structure, in divergent (“wall-eyed”) stereo with the N-terminal domain in red and the C-terminal domain in blue, and with the thiomannobioside (4) substrate mimic shown in ball-and-stick. (b) The solvent-accessible surface of Bt3990 (colored as in a), highlighting how the thiomannobioside substrate mimic (with gray surface) nestles at the interface. (c–e) The maximum-likelihood/σA weighted 2Fobs − Fcalc densities (contoured at 1σ) for mannoimidazole (c), swainsonine (3) (d) and kifunensine (5) (e). The critical Ca2+ ion is shown as a green sphere.
The C-terminal module (residues 289–755) is an adorned (α/α)6 barrel that shows similarity, at least in the core barrel (calculated using SSM15), to the (α/α)6 of some other glycosidases, most notably GH37 trehalases17 (r.m.s.d. 3.3 Å over 276 matched Cα positions showing 7% sequence identity). The GH92 barrel contains an unusual feature: a six-stranded β-sheet of unknown function adorning the C terminus of the structure, with the first strand derived from the crossover of the N-terminal domain and the remaining five strands provided from residues 676–735. The extended loop regions of the core (α/α)6 barrel (at the N-terminal end of helices 1, 5 and 6) and the protrusions from the β-barrel N-terminal domain (residues 32–41 and 84–90) contribute to the topography and substrate specificity of the active center.
Active center structure and catalysis
The complex of Bt3990 with the inhibitors swainsonine, mannoimidazole and kifunensine (Ki values of 5, 140 and 96 μM for Bt3990, and 14, 93 and 233 μM for Bt2199, respectively) revealed the −1 (ref. 13) subsite interactions (Fig. 2c–e). Exo-specificity is conferred through an active site nestled in a shallow pocket in which Asp348 provides the recognition of O4 and O3 of the mannoimidazole, with O6 hydrogen bonding with the side chain of Ser86 (Fig. 3a). The hydrophobic element ‘below’ the sugar is provided primarily by Met394. Of particular importance is the coordination of both O2 and O3 by a calcium ion, consistent with the kinetic data described earlier. This Ca2+ is octa-coordinate with three water ligands, O2 and O3 from the −1 mannoside, the side chain carbonyls of both Asn601 and Gln602, and one of the carboxylate oxygens from Asp644. The role of calcium in the catalytic mechanism of GH92 enzymes is supported by the conservation of Asn601 (asparagine or aspartate), Gln602 (glutamine or glutamate) and Asp644 (invariant) (Supplementary Fig. 4). The disposition of carboxylate side chains suggests the Brønsted acid and potential base contributions to catalysis. Glu533 lies on the “α-” face of the ligand, in a position where it can interact with the glycosidic oxygen of an α-linked mannoside substrate (confirmed in the analysis of the thio-linked substrate mimic, below), and thus provides protonic assistance for leaving group departure. Lying above the “C1” of the ligand is a solvent molecule in the appropriate position to function as the catalytic water. This water is coordinated by the Ca2+, but also by Asp644 and Asp642, both of which lie in the canonical14 position one would expect for a general base in the inverting mechanism (Fig. 3b). Twin carboxylates in the base “position” have been observed in other enzyme families18. It is not formally possible to identify which aspartate acts as the catalytic base, but sequence alignments show Asp644 is invariant, whereas Asp642 may also be asparagine in the GH92 family (Supplementary Fig. 4), implying that Asp644 is the Brønsted base.
Figure 3. The active center of Bt3990 and catalysis with inversion of anomeric configuration.
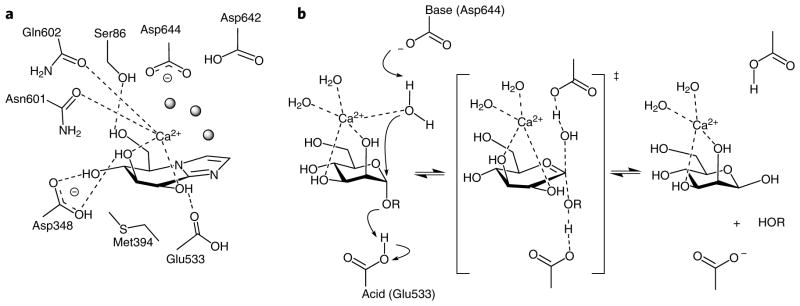
(a) Schematic diagram of the interactions of Bt3990 with Ca2+/mannoimidazole (6) highlighting the interactions of the −1 subsite and the distortion of the ligand toward a B2,5 conformation. (b) Putative reaction mechanism for an inverting GH92 α-mannosidase in which Asp644 acts as catalytic base, Ca2+ bridges the O2 and O3 hydroxyls while also interacting with the attacking water, and Glu533 provides brønsted acid assistance to leaving group departure.
Consistent with the catalytic function of Asp644, Asp642 and Glu533, alanine variants of each residue have a substantial impact on catalytic efficiency against α-1,2-mannobioside (8), which lacks a good leaving group (Supplementary Table 4). Further support for the role of Glu533 as the catalytic acid comes from the kinetics of the E533A variant on the activated aryl mannoside, DNP-Man (9), with kcat/Km increased to 370% of the wild-type enzyme, consistent with literature values for acid/base variants of inverting enzymes on substrates in which the leaving group does not require protonation19. Other enzyme variants, in which active site residues have been altered, also displayed substantially reduced catalytic efficiency (Supplementary Table 4). Of particular note is E585A, which behaves very similarly to the catalytic acid mutant E533A. The kcat/Km of E585A on unactivated substrates is reduced by 36,000-fold but is increased on the activated substrate DNP-Man. Consistent with work elsewhere19, this may suggest a role for Glu585 in maintaining the pKa of Glu533 in Bt3990. Glu585, however, also plays a role in binding the leaving group of the natural 2-linked mannoside, described below.
Substrate conformation and the role of Ca2+
Nucleophilic substitution at the acetal center of an α-mannoside is chemically difficult, requiring the alleviation of the 1,2-diaxial interactions between the incoming nucleophile and the C2 hydroxyl group, and it is widely believed that α-mannosidases achieve this through various distortions of the substrate from its ground-state 4C1 chair conformation and that different enzymes use different pathways14,20 (Supplementary Fig. 5). The two best studied α-mannosidase families are GH38 and GH47. These are structurally21 and mechanistically22 distinct, performing mannoside catalysis with retention and inversion of anomeric configuration, respectively, in a metal ion–dependent manner. GH38 and GH47 enzymes require Zn2+ and Ca2+, respectively. In each of these enzymes, the divalent metal ion plays a role similar to that observed here: bridging the O2 and O3 hydroxyl groups of the −1 subsite mannoside. The GH47 α-mannosidases are believed to perform catalysis via a pathway in which the mannoside is initially distorted to a 3S1 skew, with catalysis occurring via a 3H4 half chair transition state, implying a 1C4 conformation for the reaction product. Evidence for this 3S1 → 3H4 → 1C4 pathway derives from the 1C4 conformation seen for complexes with deoxymannojirimycin (10) and kifunensine21, and from the 3S1 conformation for the −1 subsite mannoside of the thiomannobioside Michaelis complex on GH47 enzymes23. In contrast, GH38 α-mannosidases perform catalysis via a OS2 → B2,5 → 1S5 pathway to the glycosyl-enzyme intermediate, proposed on the basis of the 1S5 conformation observed22 for the latter species. Such a pathway is consistent with experimental work on β-mannosidase pathways24,25,26 and with computational studies27.
On the GH92 enzyme Bt3990, mannoimidazole and kifunensine are observed in approximate 1S5/B2,5 and 1,4B/1S5 conformations, although in solution, these inhibitors have the “ring-flipped” conformations 6H7 (ref. 28) (equating to 3H4 for a regular mannoside) and 1C4 (ref. 29), respectively. These two structures imply a catalytic conformational itinerary that centers around the B2,5 conformation, and thus is more likely to be in the region of a OS2 → B2,5 → 1S5 pathway. The absence of calcium in one of the thiomannobioside substrate complexes, and the conformational changes reflecting sulfur incorporation in these complexes, hinder unambiguous assignment of the conformational itinerary, but they contribute to our insight into the actual role of calcium. Chemically, Ca2+ could act as a Lewis base, generating a hydroxide ion that, if loosely bound to the metal, would be a strong nucleophile. Alternatively, and fully consistent with the observed geometry, Asp644 (with Asp642) is likely sufficient to deprotonate the nucleophilic water, and Ca2+ may simply orientate the water to direct the remaining lone pair for optimal nucleophilic attack at C1. Though this chemical role for Ca2+ is reasonable, the presence of metal ions in both retaining (GH38) and inverting (GH4723 and now GH92) α-mannosidases is likely highly significant. Currently, there are over 100 GH families and, apart from these three enzyme families, no other families use a metal ion to bridge the O2 and O3 hydroxyls and/or interact with the nucleophilic group. Why then, if it is generally unnecessary, have three α-mannosidase families converged independently on the same solution?
We propose that, although other strategies may pertain to other systems, the divalent metal ions on the GH38, GH47 and GH92 exo-acting α-mannosidases aid distortion of a ground-state 4C1 mannoside toward the skewed boat or boat of the transition state—a problem that is more acute for exo-α-mannosidases than for β-mannosidases. In the case of a β-mannosidase, the interactions of the leaving group with the enzyme and the establishment of an anomeric effect provide driving forces for ring distortion; in addition, placing the leaving group axial opens a trajectory for nucleophilic attack. No such stereoelectronic effect is available to distort an α-mannoside away from a 4C1 conformation, as the α-configured leaving group is already axial. We therefore propose that these exo-α-mannosidases have converged on an alternative strategy in which the distortion of the ring from 4C1 toward both the Michaelis complex and appropriate transition states is facilitated by coordination of a divalent metal ion between O2 and O3, thereby contracting the ground-state O2-C2-C3-O3 torsion angle of the 4C1 conformation of ~60° toward the 0–15° angle expected at the transition state, as also envisaged in chemical glycosylation strategies30. Consistent with these proposals, the O2-C2-C3-O3 torsional angle observed in the mannoimidazole, kifunensine and swainsonine complexes is close to 15°. In further support of these proposals, the only complex obtained without calcium bridging these two hydroxyls, the thiomannobioside complex, is not distorted, and the −1 subsite mannoside is in its 4C1 conformation with a 55° O2-C2-C3-O3 torsion angle. A possible further benefit of a Ca2+ ion in the case of inverting α-mannosidases (GH4723 and GH92, this work) is that the more flexible coordination geometry of Ca2+ (which may be octa- or hepta-coordinate) not only allows for easier ground-state to transition-state migrations but also more easily accommodates the loss of the ligation to the nucleophilic water once the first-formed product departs the active center.
A potential exploitation of similar active-center reaction geometry is in the use of small-molecule intervention strategies to tackle an entire multigene family. To determine how known α-mannosidase inhibitors might be developed in exploring the functional significance of GH92s expressed by B. thetaiotaomicron, we assessed the affinity of several candidate inhibitors for selected α-mannosidases (Supplementary Table 5). Mannoimidazole, for example, displays a very broad range of affinities that varies by up to three orders of magnitude across the GH92 family. As the inhibitor cannot make significant interactions with the leaving group subsites, the variation in affinity across the enzyme landscape may reflect the different contributions of the −1 subsite toward substrate binding and perhaps subtly different transition state poise between the different enzymes. There is a reasonable correlation (r value of 0.76) between the affinity of the enzyme for mannoimidazole and its activity against 4NP-Man, a substrate that interacts primarily with the −1 subsite. These data provide a platform for the synthesis of more elaborate generic inhibitors of GH92 α-mannosidases that will use the full binding potential afforded by the complex structure of N-glycans, in combination with the transition state mimicry exemplified here.
Structural basis for α1–2man specificity in family GH92
To explore the structural basis for the α-1,2-mannosidase activity displayed by Bt3990, we determined the structure of the enzyme in complex with the α-1,2-linked disaccharide thiomannobioside at 2.25 Å (co-crystallization) and 2.1 Å (soaking ligand into crystals) resolution. Both structures reveal unambiguous density for the disaccharide in the −1 and +1 subsites (Fig. 4). Neither complex shows distortion of the sugar away from the 4C1 chair conformation, which likely reflects the steric bulk of the introduced interglycosidic sulfur atom and the subsequent conformational rearrangements that arise. In the cocrystallized complex, the bridging Ca2+ is lost from the active center. In contrast, Ca2+-containing apo-crystals that were soaked with thiomannobioside retain their calcium, but there is movement of Glu533 away from the glycosidic sulfur, breaking its interaction with the O2 hydroxyl of the −1 subsite, accompanied by a concerted movement of Asn601 out of the active center. Asn601 (Fig. 4c) positioned in this way interacts with neither O2 nor the Ca2+ as it had in complexes with inhibitors 5, 6 and 7. Despite these conformational changes at −1, these complexes allow description of the +1 subsite, and hence the residues that contribute to specificity. At the +1 subsite, Trp88 (from the N-terminal domain) forms an aromatic platform “above” the sugar, while Glu585 makes hydrogen bonds with O3 and O4 of the mannoside, and His584 likewise hydrogen bonds to the O3 hydroxyl. The methyl group at C1 points into solvent, consistent with the ability of Bt3990 to hydrolyze the α-1,2 linkages in Man9GlcNAc2.
Figure 4. Interactions of thiomannobioside with GH92 α-1,2-mannosidase Bt3990.
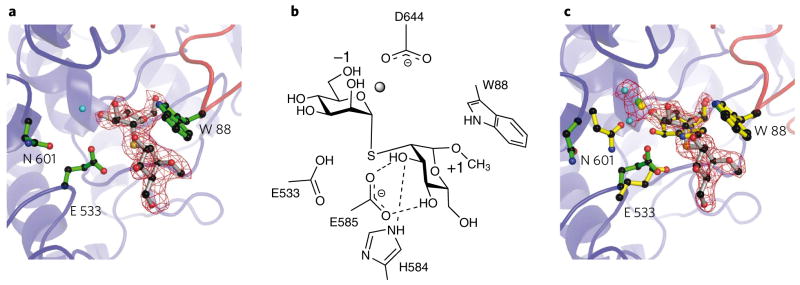
(a) Structure of the Bt3990 complex cocrystallized with thiomannobioside, with the acid (Glu533), the putative base (Asp644) and the α-1,2 signature motifs (Trp88, His584 and Glu585) shown. The main chain is colored red for the N-terminal domain and blue for the C-terminal domain, showing how the active center is cradled by residues from both domains. (b) Schematic diagram showing the interactions of the aforementioned residues with the thiomannobioside. (c) Structure of the Bt3990 complex, soaked with thiomannobioside (green protein, gray ligand) overlaid with the Bt3990 mannoimidazole (6) complex (yellow). a and c include the maximum-likelihood/σA weighted 2Fobs − Fcalc density contoured at 1σ.
The general significance of His584 and Glu585 (the “His-Glu” motif) and Trp99 (in a “Pro-Trp” motif) in recognition of an α-1,2-linked mannose at the +1 subsite was established through the three-dimensional structure of a second α-1,2-mannosidase: Bt2199 (Supplementary Fig. 6). The protein displays an identical fold to Bt3990, and the residues involved in substrate recognition and catalysis, confirmed by mutagenesis (Supplementary Table 4), are invariant in the two enzymes (Supplementary Fig. 4). The helical insertion in Bt3990 containing His584 and Glu585 is also present in Bt2199. The Bt2199 structure also highlights active site conformational changes. In the Ca2+-free enzyme, the catalytic residues are not in the active site pocket. Upon binding swainsonine, conformational changes partially restore the catalytic apparatus to the active center (Supplementary Fig. 6). The Bt3990 and 2199 structures, in light of sequence data, suggest that the His-Glu motif and the N-terminal domain tryptophan might present a signature motif for α-1,2-mannosidase activity that will have useful predictive value. Inspection of the B. thetaiotaomicron GH92 enzymes supports this view with sequences having the His-Glu motif also containing the physically distant Pro-Trp motif (Supplementary Fig. 4). All the GH92 enzymes that display preferential α-1,2-mannosidase activity contain both the His-Glu and Pro-Trp motifs. Indeed, phylogenetic analysis of all the B. thetaiotaomicron GH92s (Supplementary Fig. 7) shows that the α-1,2-mannosidases cluster into a highly defined clade, while α-1,3-mannosidases segregate into two clades. The enzymes that display low or no disaccharidase activity also form a discrete clade. This tree therefore may be of value in classifying the likely catalytic activity of GH92s identified through genome and metagenome sequencing programs.
DISCUSSION
The natural substrates likely targeted by the B. thetaiotaomicron GH92 enzymes are the α-mannosides present in the N-glycans that are present on host and dietary glycoproteins (it is unlikely that the human microbiota will contain significant numbers of yeast, another potential source of α-linked mannosides). The targeting of N-glycans is consistent with the α-1,2-mannosidase and α-1,3-mannosidase activities displayed by several of the GH92 enzymes. While one of the GH92 enzymes hydrolyzes an α-1,6-mannoside linkage in N-glycans (Fig. 1), this bond may also be targeted by the seven B. thetaiotaomicron GH76 (ref. 8) enzymes—a family known to contain α-1,6-mannanases. Currently, we are unable to explain the activity of some GH92 enzymes (Bt3965, Bt4073 and Bt4093) against α-1,4-mannosides. The α-1,4 linkage between two mannose residues is unknown in human and plant glycans. It is known to exist in some fungal and lichen mannans, and it is formally possible that the microbiota themselves may contain bacteria that synthesize complex carbohydrates, as part of cell-associated polysaccharides, that contain α-1,4-mannosidic linkages.
The transcriptome of B. thetaiotaomicron exposed to different glycans provides some insight into the expanded repertoire of GH92 enzymes that is not evident from their activities described above. Thus, different GH92s appear to be expressed in response to distinct environmental cues within the large bowel of the mouse. Researchers31,32 have shown that 50% of the GH92s are upregulated in response to suckling, post weaning (fed a diet rich in plant polysaccharides), and when the bacterium is cultured in a fermentor in the presence of yeast mannan (a proposed mimic of high-mannose N-glycans). Indeed, many of the GH92 genes are located in close proximity to genes encoding GH76s and GH18s—the other enzymes required to fully metabolize high-mannose N-glycans. It would appear, therefore, that B. thetaiotaomicron contains several loci that encode N-glycan–degrading enzyme systems, although the biological significance of these multiple loci is unclear.
The analysis of the GH92 family of enzymes from B. thetaiotaomicron adds to our growing knowledge of the diverse hydrolytic enzymes that act on mannosidic substrates. Both α- and β-mannosidases provide model systems25 for the study of substitution reactions at the anomeric center of mannose, one of the most challenging reactions in carbohydrate chemistry30,33,34. α- and β-mannosidases are of considerable importance in cellular biology. Whereas β-mannosides are important substrates in the environmental and industrially recognized hydrolysis of plant polysaccharides, the enzymes active on α-mannosides provide a medically relevant set of enzymes implicated in fungal cell wall degradation and in the maturation and processing of N-glycans. One example of the role of cellular mannosidases in cellular events is the reprogramming of N-glycan synthesis during metastatic progression35—a sequence of reactions for which α-mannosidase II action is an important prerequisite and one that has added considerable impetus to the study of α-mannosidase chemistry, conformational analysis and inhibition (see for recent examples ref. 36 and refs. therein). The analysis of the complete repertoire of GH92 enzymes presented here therefore not only provides detailed insight into the specificity and evolution of one of the major enzyme families secreted by a large bowel microorganism, but also adds to our fundamental understanding of the mechanistic chemistry of mannoside hydrolysis. This knowledge should have an impact on the design and application of small-molecule inhibitors both in therapeutic contexts and in approaches to the study of the human microbiota.
METHODS
Gene cloning and expression
The B. thetaiotaomicron genes encoding the GH92 enzymes were amplified by PCR from genomic DNA using the primers listed in Supplementary Table 6. The DNA products were cloned into the NheI and XhoI sites of pET21a. The resultant plasmids encode GH92s containing a C-terminal His6 tag. Site-directed mutagenesis was carried out using the QuikChange mutagenesis kit (Stratagene), using pBt3990 as the template and primers listed in Supplementary Table 6.
Protein purification
The conditions used to culture Escherichia coli Tuner or B834 cells harboring appropriate GH92-encoding plasmids and the recombinant gene expression regime were as described previously25. Bt3990 was purified by immobilized metal ion affinity chromatography (IMAC) using Talon resin (Clontech), anion exchange chromatography and size exclusion chromatography (Superdex 200 16/60 Hiload column; Amersham) using a method described previously25. The other GH92 enzymes were purified only by IMAC. Selenomethionine protein was produced using standard techniques in the methionine auxotroph E. coli strain B834.
Enzyme assays
The GH92 enzymes were screened for enzyme activity using 1 mM of substrate (disaccharides and arylglycosides) in 100 mM sodium HEPES, pH 7.0, containing 1 mg ml−1 of bovine serum albumin (BSA) and 2 mM calcium chloride. Activity against arylglycosides was monitored as described previously25. The release of mannose from disaccharides was determined as follows: the mannose released was phosphorylated to mannose-6-phosphate by hexokinase. The phosphorylated mannose was then converted to fructose-6-phosphate, through the action of phosphomannose isomerase, which was then isomerized to glucose-6-phosphate by phosphoglucose isomerase. Finally, the phosphorylated glucose was oxidized to gluconate-6-phosphate by glucose-6-phosphate dehydrogenase with concomitant reduction of NADP+ to NADPH, which was monitored at 340 nm using an extinction coefficient of 6,223 M−1 cm−1. The assay, carried out at 37 °C in 100 mM MOPS buffer, pH 7.0, containing 2 mM calcium chloride, is based largely on the Megazyme International kit for mannose detection deploying ATP and NADP+ at 1 mM. The enzymes were individually obtained from Sigma Chemical Company, and the concentrations of the four linker enzymes were modified such that the release of mannose by the mannosidase was the rate-limiting step in the reaction. Activity was measured at seven substrate concentrations that straddled the Km. Quantification of mannose release from yeast mannan and ovalbumin (contains primarily high-mannose N-glycans) involved incubation of 1 mg ml−1 substrate with enzymes at 2 μM in the HEPES buffer described above. After 1 and 16 h an aliquot was removed, the enzyme was inactivated by boiling for 10 min, and the amount of mannose released was determined using Dionex HPLC37. The analysis of the anomeric configuration of mannose was performed using 1H NMR38.
Inhibition studies were carried out with at least five concentrations of the inhibitor at a substrate concentration ≪ Km for the specific enzyme. The intercept of the x axis of a plot of kcat/Km against inhibitor concentration revealed the Ki value. Mannoimidazole28 and “thio α-1,2 mannobioside”23 were synthesized as described previously. Swainsonine and kifunensine were purchased from Oxford Glycosystems. Compounds 1 and 10 were purchased from Sigma-Aldrich. Compounds 2, 3 and 8 were purchased from Dextra Laboratories. Compounds 5 and 7 were purchased from GlycoFineChem. Compound 9 was a kind gift from S. Withers (University of British Columbia).
Crystallization, data collection, structure solution and refinement
Crystals of recombinant Bt3990, selenomethionine derivative, were grown by hanging drop vapor diffusion at 19 °C in equal volumes protein (16 mg ml−1 in H2O) and reservoir solution (0.1 M malonic acid, imidazole, borate buffer system, pH 6.0, 10% w/v polyethylene glycol (PEG) 3350). Bt3990 crystals complexed with mannoimidazole were obtained in 25% 1500 w/v PEG, 0.1 M MIBs (pH 6.0), with swainsonine in 20% w/v 6000 PEG, 0.2 M NH4Cl, tris(hydroxymethyl) aminomethane (pH 8.0), with kifunensine in 20% w/v 3350, 0.1 M MIBs (pH 6.0), and thiol-linked mannobiose in 20% 3350 PEG, 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.0), 0.15 M K2SO4, with ~5 mM ligand. The structure of calcium/thiomannobioside was obtained from a crystal grown in 15% v/v PEG 3350, 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid pH 7.0, 0.1 M CaCO3 that was subsequently soaked with 5 mM 4 for 30 min. Crystals were cryo-protected by serially increasing glycerol and ligand to 20% (v/v) and 2.5 mM, respectively. Bt2199 crystals were grown by hanging drop method in equal volumes of protein (18 mg ml−1 in H2O) and 10% v/v 2-methyl-2,4-pentanediol, 1.2 M C3H2O4Na2 and 0.1 M 1,3- bis(tris(hydroxymethyl)methylamino) proponate (pH 7.5). Crystals were cryoprotected by increasing C3H2O4Na2 to 1.5 M and addition of 10% v/v glycerol. Crystals of the mixed occupancy swainsonine complex form were obtained by soaking Bt2199 crystals for 16 h in 5 mM swainsonine.
Bt3990 selenomethionine derivative, mannoimidazole, swainsonine, kifunensine and thiol-linked mannobiose diffraction data were collected at European Synchrotron Radiation Facility (ESRF) beamlines, and data were processed with either the HKL suite39 or MOSFLM/SCALA from the CCP4 suite40. The structure of Bt3990 was solved by MAD phasing at the peak wavelength 12.6607 keV of a selenomethionine derivative using 0.5° oscillation for 360° and at a remote wavelength (12.7107 keV) for 180°. SOLVE/RESOLVE41 were used for phasing and initial model building. Cycles of maximum-likelihood refinement using REFMAC (CCP4 suite40) were interspersed with manual corrections of the models using COOT42. Complexed structures of Bt3990 were solved by molecular replacement using the apo coordinates as the search model. Bt2199 data were collected at the ESRF and solved by molecular replacement, as above. Structural figures were drawn with PyMol (DeLano Scientific LLC, http://pymol.sourceforge.net/).
Supplementary Material
Acknowledgments
We thank S. Withers (University of British Columbia) for compound 9. The Biotechnology and Biological Sciences Research Council of the UK is thanked for funding. M.D.L.S. is a European Molecular Biology Laboratory long-term fellowship holder, and G.J.D. is a Royal Society/Wolfson Research Merit Award recipient. C.D. was supported by a Marie Curie European Reintegration grant within the 7th European Community Framework Programme. Y.Z. is supported by the Department for Innovation, Universities and Skills (DIUS), UK, Newcastle University and the Chinese Scholarship Council. S.J.W. thanks the Australian Research Council for funding support. K.W.M. acknowledges the support of US National Institutes of Health grants GM047533, RR005351 and DK075322. S.C. is supported by a postdoctoral fellowship from the Agence Nationale de Recherche sur le Sida et les Hépatities Virales (ANRS). A.S. is supported by Centre National de la Recherche Scientifique.
Footnotes
Accession codes. Protein Data Bank: The crystal structures of Bt3990 by itself, and with compounds 6, 7, 5, 4, and 4 with calcium, have been deposited under accession codes 2WVX, 2WZS, 2WVZ, 2WW0, 2WW1 and 2WW3, respectively. The crystal structures of Bt2199 by itself and with compound 5 have been deposited under accession codes 2WVY and 2WW2, respectively.
Author contributions
H.J.G. and G.J.D. designed the experiments. Y.Z., M.D.L.S., A.J.T., C.D., N.S., Y.X. and K.W.M. performed or supervised molecular biology, kinetics, glycan analysis and X-ray crystallography. S.C., A.S., Z.D. and S.J.W. synthesized ligands. H.J.G., G.J.D. and S.J.W., aided by the other authors, wrote the paper.
Additional information
Supplementary information and chemical compound information is available online at http://www.nature.com/naturechemicalbiology/.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 2.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 3.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 4.Wen L, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 6.Kim YS, Milner JA. Dietary modulation of colon cancer risk. J Nutr. 2007;137:2576S–2579S. doi: 10.1093/jn/137.11.2576S. [DOI] [PubMed] [Google Scholar]
- 7.Cerdeno-Tarraga AM, et al. Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science. 2005;307:1463–1465. doi: 10.1126/science.1107008. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gill SR, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, et al. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maruyama Y, Nakajima T, Ichishima E. A 1,2-alpha-D-mannosidase from a Bacillus sp.: purification, characterization, and mode of action. Carbohydr Res. 1994;251:89–98. doi: 10.1016/0008-6215(94)84278-7. [DOI] [PubMed] [Google Scholar]
- 13.Davies GJ, Wilson KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J. 1997;321:557–559. doi: 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vocadlo D, Davies GJ. Mechanistic insights into glycosidase chemistry. Curr Opin Chem Biol. 2008;12:539–555. doi: 10.1016/j.cbpa.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 16.Heikinheimo P, et al. The structure of bovine lysosomal alpha-mannosidase suggests a novel mechanism for low-pH activation. J Mol Biol. 2003;327:631–644. doi: 10.1016/s0022-2836(03)00172-4. [DOI] [PubMed] [Google Scholar]
- 17.Gibson RP, et al. Molecular basis for trehalase inhibition revealed by the structure of trehalase in complex with potent inhibitors. Angew Chem Int Ed. 2007;46:4115–4119. doi: 10.1002/anie.200604825. [DOI] [PubMed] [Google Scholar]
- 18.Nurizzo D, Nagy T, Gilbert HJ, Davies GJ. The structural basis for catalysis and specificity of the Pseudomonas cellulosa α-glucuronidase, GlcA67A. Structure. 2002;10:547–556. doi: 10.1016/s0969-2126(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 19.Damude HG, Withers SG, Kilburn DG, Miller RC, Jr, Warren RAJ. Site-directed mutation of the putative catalytic residues of endoglucanase CenA from Cellulomonas fimi. Biochemistry. 1995;34:2220–2224. doi: 10.1021/bi00007a016. [DOI] [PubMed] [Google Scholar]
- 20.Davies GJ, Ducros VMA, Varrot A, Zechel DL. Mapping the conformational itinerary of β-glycosidases by X-ray crystallography. Biochem Soc Trans. 2003;31:523–527. doi: 10.1042/bst0310523. [DOI] [PubMed] [Google Scholar]
- 21.Vallee F, Karaveg K, Herscovics A, Moremen KW, Howell PL. Structural basis for catalysis and inhibition of N-glycan processing class I alpha 1,2-mannosidases. J Biol Chem. 2000;275:41287–41298. doi: 10.1074/jbc.M006927200. [DOI] [PubMed] [Google Scholar]
- 22.Numao S, Kuntz DA, Withers SG, Rose DR. Insights into the mechanism of Drosophila melanogaster Golgi alpha-mannosidase II through the structural analysis of covalent reaction intermediates. J Biol Chem. 2003;278:48074–48083. doi: 10.1074/jbc.M309249200. [DOI] [PubMed] [Google Scholar]
- 23.Karaveg K, et al. Mechanism of class 1 (Glycosylhydrolase family 47) alpha-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J Biol Chem. 2005;280:16197–16207. doi: 10.1074/jbc.M500119200. [DOI] [PubMed] [Google Scholar]
- 24.Ducros V, et al. Substrate distortion by a β-mannanase: snapshots of the Michaelis and covalent intermediate complexes suggest a B2,5 conformation for the transition-state. Angew Chem Int Ed. 2002;41:2824–2827. doi: 10.1002/1521-3773(20020802)41:15<2824::AID-ANIE2824>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 25.Tailford LE, et al. Structural and biochemical evidence for a boat-like transition state in β-mannosidases. Nat Chem Biol. 2008;4:306–312. doi: 10.1038/nchembio.81. [DOI] [PubMed] [Google Scholar]
- 26.Offen WA, Zechel DL, Withers SG, Gilbert HJ, Davies GJ. Structure of the Michaelis complex of β-mannosidase, Man2A, provides insight into the conformational itinerary of mannoside hydrolysis. Chem Commun (Camb) 2009:2484–2486. doi: 10.1039/b902240f. [DOI] [PubMed] [Google Scholar]
- 27.Whitfield DM. DFT studies of the ionization of alpha and beta glycopyranosyl donors. Carbohydr Res. 2007;342:1726–1740. doi: 10.1016/j.carres.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Terinek M, Vasella A. Synthesis and evaluation of two mannosamine-derived lactone-type inhibitors of snail beta-mannosidase. Tetrahedron Asymmetry. 2005;16:449–469. [Google Scholar]
- 29.Kayakiri H, et al. Structure of kifunensine, a new immunomodulator isolated from an actinomycete. J Org Chem. 1989;54:4015–4016. [Google Scholar]
- 30.Crich D, Chandrasekera NS. Mechanism of 4,6–0-benzylidene-directed beta-mannosylation as determined by alpha-deuterium kinetic isotope effects. Angew Chem Int Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 31.Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem. 2006;281:36269–36279. doi: 10.1074/jbc.M606509200. [DOI] [PubMed] [Google Scholar]
- 32.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito Y, Ogawa T. A novel-approach to the stereoselective synthesis of beta-mannosides. Angew Chem Int Edn Engl. 1994;33:1765–1767. [Google Scholar]
- 34.Gridley JJ, Osborn HMI. Recent advances in the construction of β-D-mannose and β-D-mannosamine linkages. J Chem Soc Perkin Trans 1. 2000;10:1471–1491. [Google Scholar]
- 35.Granovsky M, et al. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 2000;6:306–312. doi: 10.1038/73163. [DOI] [PubMed] [Google Scholar]
- 36.Kuntz D, Zhong W, Guo J, Rose D, Boons G. The molecular basis of inhibition of Golgi alpha-mannosidase II by mannostatin A. Chem Bio Chem. 2009;10:268–277. doi: 10.1002/cbic.200800538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hogg D, et al. Crystal structure of mannanase 26A from Pseudomonas cellulosa and analysis of residues involved in substrate binding. J Biol Chem. 2001;276:31186–31192. doi: 10.1074/jbc.M010290200. [DOI] [PubMed] [Google Scholar]
- 38.Bolam D, et al. Mannanase A from Pseudomonas fluorescens ssp cellulosa is a retaining glycosyl hydrolase in which E212 and E320 are the putative catalytic residues. Biochemistry. 1996;35:16195–16204. doi: 10.1021/bi961866d. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 41.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.