

Abstract
Protoporphyrinogen IX oxidase (PPO), the last common enzyme of haem and chlorophyll biosynthesis, catalyses the oxidation of protoporphyrinogen IX to protoporphyrin IX. The membrane-embedded flavoprotein is the target of a large class of herbicides. In humans, a defect in PPO is responsible for the dominantly inherited disease variegate porphyria. Here we present the crystal structure of mitochondrial PPO from tobacco complexed with a phenyl-pyrazol inhibitor. PPO forms a loosely associated dimer and folds into an FAD-binding domain of the p-hydroxybenzoate-hydrolase fold and a substrate-binding domain that enclose a narrow active site cavity beneath the FAD and an α-helical membrane-binding domain. The active site architecture suggests a specific substrate-binding mode compatible with the unusual six-electron oxidation. The membrane-binding domains can be docked onto the dimeric structure of human ferrochelatase, the next enzyme in haem biosynthesis, embedded in the opposite side of the membrane. This modelled transmembrane complex provides a structural explanation for the uncoupling of haem biosynthesis observed in variegate porphyria patients and in plants after inhibiting PPO.
Keywords: crystal structure, FAD cofactor, membrane binding, protoporphyrinogen IX oxidase, tetrapyrrole biosynthesis
Introduction
Protoporphyrinogen IX oxidase (PPO; EC 1.3.3.4) catalyses the oxidation of protoporphyrinogen IX to protoporphyrin IX by molecular oxygen (Figure 1A). PPOs represent a highly conserved family of enzymes, which are membrane associated supposedly by a hydrophobic conserved region (Arnould et al, 1999). In plants, there exist two isoforms of PPO, the plastidic PPO1 and the mitochondrial PPO2 (Lermontova et al, 1997); PPO1 is located in the thylakoid and in the envelope membranes of chloroplasts, and PPO2 is situated on the outer surface of the inner mitochondrial membrane (Ferreira et al, 1988). Ferrochelatase (Fc, EC 4.99.1.1) that inserts iron into the PPO product protoporphyrin IX is located on the inner surface of the inner mitochondrial membrane and in the thylakoid membrane of chloroplasts (Wu et al, 2001). It has been proposed that substrate channelling occurs between PPO and Fc (Ferreira et al, 1988).
Figure 1.
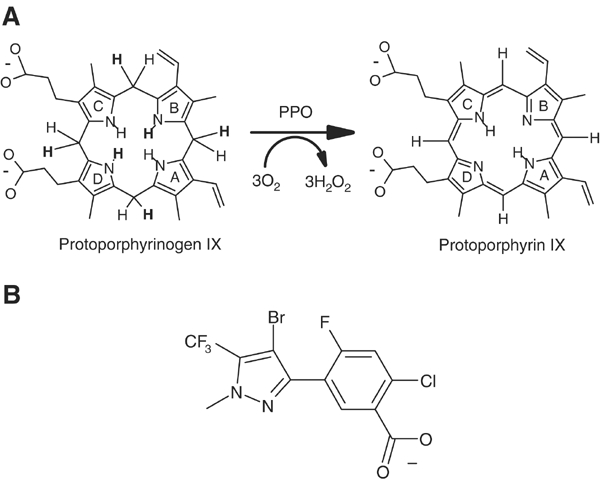
Reaction and inhibition of PPO. (A) Reaction scheme of PPO. Protoporphyrinogen IX is oxidised to protoporphyrin IX by molecular oxygen that is reduced to hydrogen peroxide. Hydrogen atoms eliminated during the reaction are written in bold letters. (B) Inhibitor INH: 4-Bromo-3-(5′-carboxy-4′-chloro-2′-fluoro-phenyl)-1-methyl-5-trifluoromethyl-pyrazol.
PPO is the main target of light-dependent peroxidising herbicides like the diphenylether acifluoren (Lermontova et al, 1997). Inhibition of PPO leads to nonenzymatical formation of protoporphyrin IX, which is generated from protoporphyrinogen IX spontaneously by reaction with molecular oxygen. The uncoupling of protoporphyrin IX generation and iron insertion by ferrochelatase lead to a loss of feedback control of haem biosynthesis by the final product haem. The photosensitising protoporphyrin IX generates singlet oxygen that causes lipid peroxidation and cell death, leading to the observed bleaching of photosynthetically active parts of plants (Lermontova et al, 1997). Interestingly, humans become more sensitive to light, when the activity of PPO is reduced by mutations. The subsequent protoporphyrin IX enrichment causes the dominantly inherited metabolic disease variegate porphyria (Maneli et al, 2003).
We have solved the crystal structure of the mitochondrial PPO2 from common tobacco (Nicotiana tabacum), a dimeric yellow protein with a molecular mass of ca. 55 kDa per monomer, at 2.9 Å resolution by selenium single anomalous diffraction. The protein was cocrystallised with the inhibitor 4-bromo-3-(5′-carboxy-4′-chloro-2′-fluoro-phenyl)-1-methyl-5-trifluoromethyl-pyrazol (INH, Figure 1B) and with a noncovalently bound FAD cofactor.
Results and discussion
The overall structure
The 503 amino acids of PPO2 fold into a compact structure (Figure 2) in three domains, an FAD-binding, a substrate-binding and a membrane-binding domain (Figure 3A, Supplementary Figure 1). The first two domains display a p-hydroxybenzoate-hydroxylase (PHBH)-fold-like topology (Schreuder et al, 1992; Pawelek et al, 2000). The parts of the structure responsible for FAD binding (Supplementary Figure 2) show significant amino acid (aa) sequence and structural homologies to other flavoenzymes (Ghisla and Massey, 1989), like human monoamine oxidase B (Binda et al, 2002) (MAO B, 1.68 Å rmsd over 256 Cα atoms with 15.1% aa sequence identity), polyamine oxidase (Binda et al, 1999) (PAO, 1.71 Å rmsd over 190 Cα atoms with 14.9% aa sequence identity) and D-amino acid oxidase (Mizutani et al, 1996) (DAO, 1.98 Å rmsd over 148 Cα atoms with 14.9% aa sequence identity) and, furthermore, phytoene desaturase (PDS) (Norris et al, 1995) with 15.2% aa sequence identity, a plant enzyme involved in carotenoid biosynthesis (Supplementary Figure 3).
Figure 2.
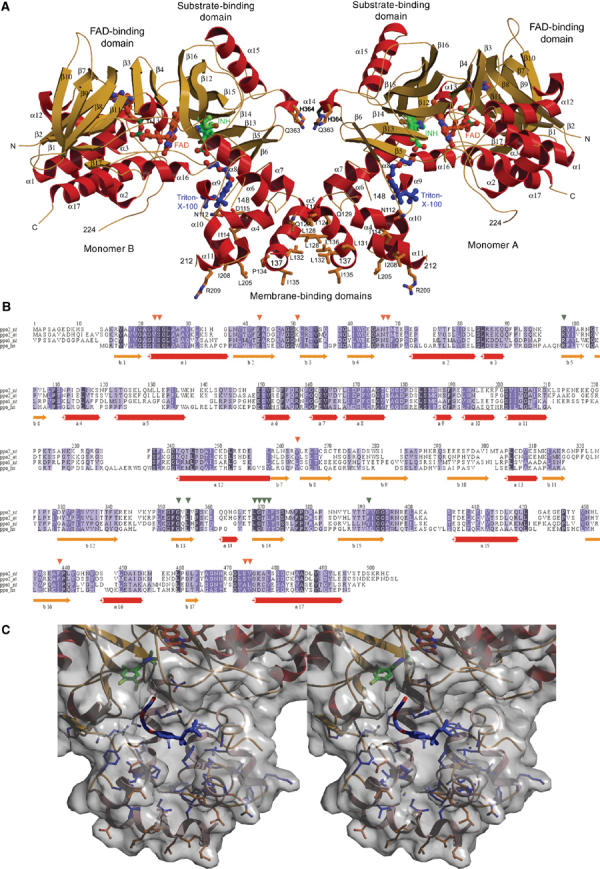
Structure and sequence of mitochondrial PPO from N. tabacum. (A) Dimeric structure of PPO from N. tabacum. The three-domain monomer consists of an FAD-binding, a substrate-binding and a membrane-binding domain. The inhibitor INH (green) binds into the active site cavity near the cofactor FAD (red–orange). The product channel in the membrane-binding domain starts at the bound Triton X-100 detergent molecule (blue). In addition, residues involved in crystal contacts are shown. (B) Multiple amino-acid sequence alignment of eukaryotic PPOs: mitochondrial PPO2 from N. tabacum (NT, tobacco) and Arabidopsis thaliana (AT, Mouse Ear Cress), chloroplast PPO1 from N. tabacum and human mitochondrial PPO (HS). Red arrows mark residues in contact with the cofactor FAD and green arrows significant inhibitor-binding residues. The highly conserved membrane-binding residues 112–136 and 150–213 are surrounded by two black boxes. Below the alignment, the secondary structure elements (SSE) of PPO (in red and orange–yellow) are shown. The alignment was compiled with CLUSTALW (Thompson et al, 1994). (C) Stereo view of the membrane channel. The membrane-bound channel below the active site cavity (FAD—orange, INH—green) is shown with the lining residues (blue) behind the molecular surface and the bound detergent molecule (blue). Residues involved in crystal contacts are shown in orange.
Figure 3.
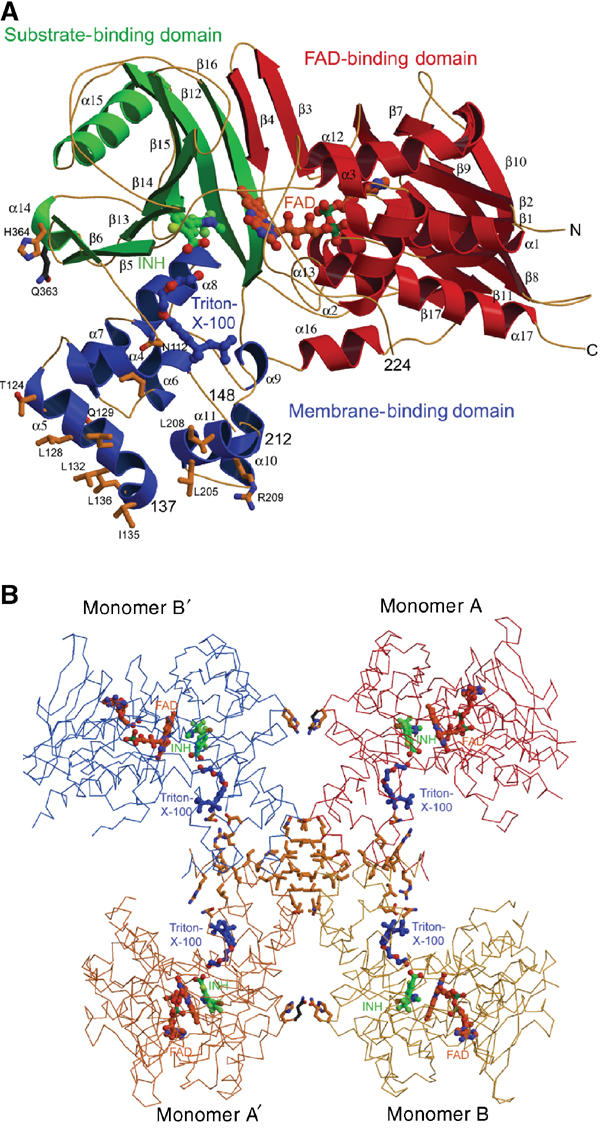
Arrangement of domains and subunits in the PPO structure. (A) Three-domain structure of PPO from N. tabacum (PPO2NT). The FAD-binding domain is shown in red, the substrate-binding domain is painted in green and the membrane-binding domain helices are shown in blue. Ligands are coloured as in Figure 1A. Helices α5 and α14 form the dimer interface, and the residues shown provide the crystal contacts. (B) Tetrameric assembly of PPO monomers in the crystal. The noncrystallographically related monomers A (red) and B (yellow) are shown with two of their crystallographically related partners monomers A′ (orange) and B′ (blue). Residues involved in crystal contacts are shown at the respective interfaces: the large interfaces of about 800 A2 between A and B (B′ and A′) and the small interfaces of about 400 A2 between A and B′ (A′ and B) building up the physiological dimer. Ligands are shown in the same colours as in Figure 1A.
The dimer interface and membrane binding
PPO2 is tightly bound to the inner mitochondrial membrane. The solubilisation and crystallisation of the recombinant protein are only possible after detergent extraction with Triton X-100. The apparent molecular mass of PPO2 in solution is 130 kDa as determined by size exclusion chromatography, indicating a dimer with a calculated molecular mass of ca. 110 kDa (data not shown). We suggest that the domain encompassing the eight helices α4–11 with the conserved sequence regions (Figure 2B) from residue 112 to 136 and from 150 to 213 is the membrane anchor and is monotopically (Blobel, 1980) inserted into the membrane reminiscent of the monotopic membrane proteins squalene-hopene cyclase (Wendt et al, 1999) and prostaglandine-H2 synthase (Loll et al, 1995). In PPO2 helices α4, α5 and α11 form the base of this domain with mostly apolar residues, which would face the lipid portion of the membranes. The wall of the cylindrical putative membrane-binding domain is lined up by a couple of negatively and positively charged residues, which may interact with positively and negatively charged head groups of the mitochondrial membrane phospholipids. In the crystals of PPO2, the putative membrane-binding domains of four protomers contact each other, forming two contacts that each bury 800 Å2 accessible surface area and perpendicular to these two more contacts with interface areas of 400 Å2 (Figure 3B). We suppose that the smaller interface corresponds to the natural PPO2 dimer, possibly further stabilised by phospholipids of the surrounding membrane in vivo. In the dimeric assembly of PPO2, the openings of the active site cavities are positioned on the outside of the dimer near the membrane leading to two independent active sites. The dimer interface is formed by the helices α14 and α5, with major contributions made by leucine residues of helix α5 that interact with the corresponding leucine residues of the second monomer (Figure 2A). Despite high overall structural homology with MAO B (Binda et al, 2002), dimerisation takes place at completely different surface parts in the two enzymes. It is unclear whether PPO2 is inserted into the membrane as a monomer or dimer. Both insertion modes are structurally possible.
Putative complex with ferrochelatase
The membrane-binding domain contains a U-shaped channel opened at one side leading from the active site into the membrane bilayer (Figure 2C). We suggest that protoporphyrin IX is translocated to dimeric Fc through this channel (Fc, Figure 4A). Fc has been proposed to be monotopically inserted into the membrane as a dimer with an arrangement seen in the crystal structure of human ferrochelatase (HFc) (Wu et al, 2001). In our model of the heterodimeric complex of the PPO2 and HFc dimers, the twofold axes of the two dimers were aligned and the protomers were rotated and shifted until the best fit is obtained (Figure 4B). The residues that form the interface match very well; residues 195–212 (α10 and α11) of PPO2 interact with residues 99–122 of HFc (large lip, helix from 104 to 122) and residues 131–137 (α5, PPO2) with residues 304–310 (small lip loop, HFc) (Wu et al, 2001). Interestingly, these interface residues of PPO are identical to those that give rise to the 800 Å2 crystal contact described above (Figure 4A). The part of the complex that is supposed to span the inner mitochondrial membrane has a length of 28 Å, corresponding well to the assumed width of 30 Å for the lipidic part of a membrane. An appealing feature of the model is the overlap of the channel openings of PPO2 and HFc, facilitating uninterrupted product/substrate diffusion from the active sites of the PPO2 dimer right into those of the dimeric HFc in the complex. In the complex, the uptake of protoporphyrinogen IX could occur from the periplasmic side through the opening beneath the active site cavity of PPO2, perhaps via a complex with coproporphyrinogen III oxidase that provides the substrate of PPO2 in animal mitochondria. After oxidation of protoporphyrinogen IX, the product could thus be channelled to the active site of Fc and after iron insertion released into the membrane via the lateral opening of the combined channels. The observation that iron insertion by Fc requires the presence of PPO (Ferreira et al, 1988) argues for a physiological complex between the two components.
Figure 4.
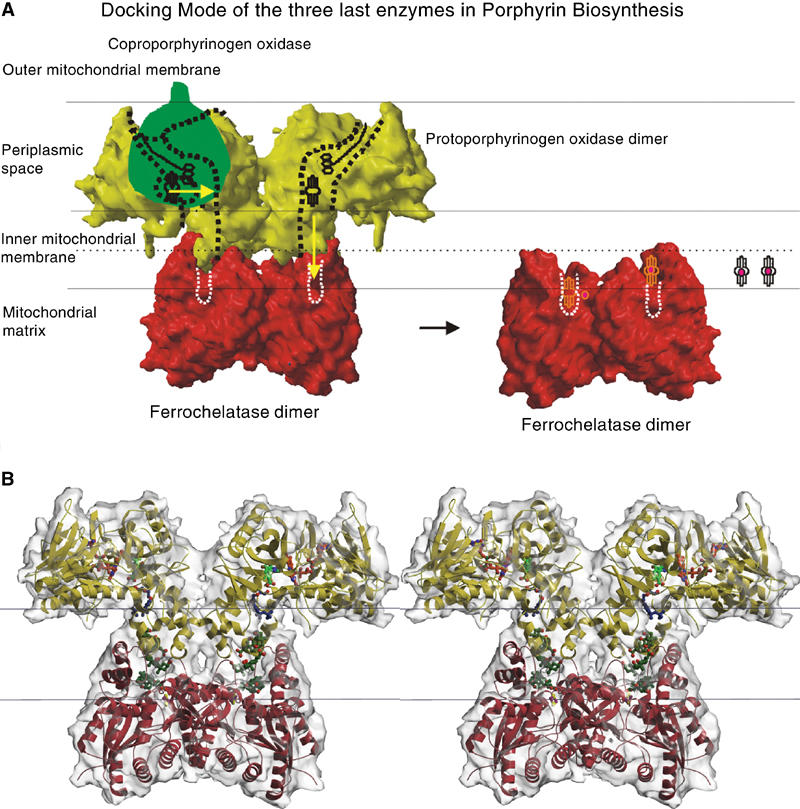
Supposed interaction mode of the last three enzymes in the human mitochondrial haem biosynthesis. (A) Localisation of coproporphyrinogen III oxidase (Santana et al, 2002) (CPO, EC 1.3.3.3, green), PPO (surface of the tobacco structure in yellow) and human ferrochelatase (Wu et al, 2001) (HFc, red) in the mitochondrial periplasm. CPO binds to the inner surface of the outer mitochondrial membrane in human mitochondria. PPO binds to the outer surface of the inner mitochondrial membrane and HFc to the opposite surface of the inner mitochondrial membrane. The dashed lines symbolise the channel within the PPO and HFc molecules where ligands can bind and the protoporphyrinogen IX and protoporphyrin IX are processed from PPO to HFc. From left to right, the reaction sequence is shown starting with protoporphyrinogen IX uptake by PPO from CPO (1), reaction to protoporphyrin IX in PPO and channelling to the active site of HFc (2), insertion of the iron into protoporphyrin IX resulting in protohaem or haem b (3) and its release into the membrane (4). The channel from PPO to HFc is opened towards the membrane at the PPO side of the PPO–HFc complex, so here it is possible for molecules to enter or leave the complex. One possibility how the final reaction step, that is, the release of haem, takes place is that haem leaves through the channel opening of the assembled complex as shown here. Alternatively, the complex that has been described as not very stable by Ferreira et al (1988) could disassemble after step2. (B) Stereo-view of the putative PPO–HFc complex, the PPO is shown in yellow, HFc in red. The channel in the complex is filled with the PPO inhibitor (INH, green) below the cofactor FAD (PPO, red–orange), the detergent molecules Triton X-100 (PPO-part of the channel, blue) and three cholate molecules (HFc, olive-green). The inner mitochondrial membrane is symbolised by black lines.
The active site and structural basis of catalysis
The active site of PPO is located between the FAD- and the substrate-binding domain. The electron density map (Figure 5A) shows the binding of the inhibitor INH in the active site cavity (Figure 5B) near the FAD cofactor in its extended conformation. The substrate-binding site beneath the FAD is a flat cavity that is formed by a number of aromatic and aliphatic amino acids as well as by Asn67 and Arg98 (Figure 5C). Below the inhibitor, there is well-defined electron density for a Triton X-100 molecule. It is located in the product channel, with the polar diethyleneoxide moiety directed towards the active site. Two further electron density maxima are found deeper in the channel, which could represent two less well-defined detergent molecules. This channel may also be a putative binding site for ubiquinone when it inhibits the PPO reaction in concentrations above 30 μM as described previously (Ferreira and Dailey, 1988). Ubiquinones, which are membrane-bound, could enter the channel from the membrane and inhibit the PPO reaction by steric hindrance of substrate binding and by blocking the channelling of the product.
Figure 5.
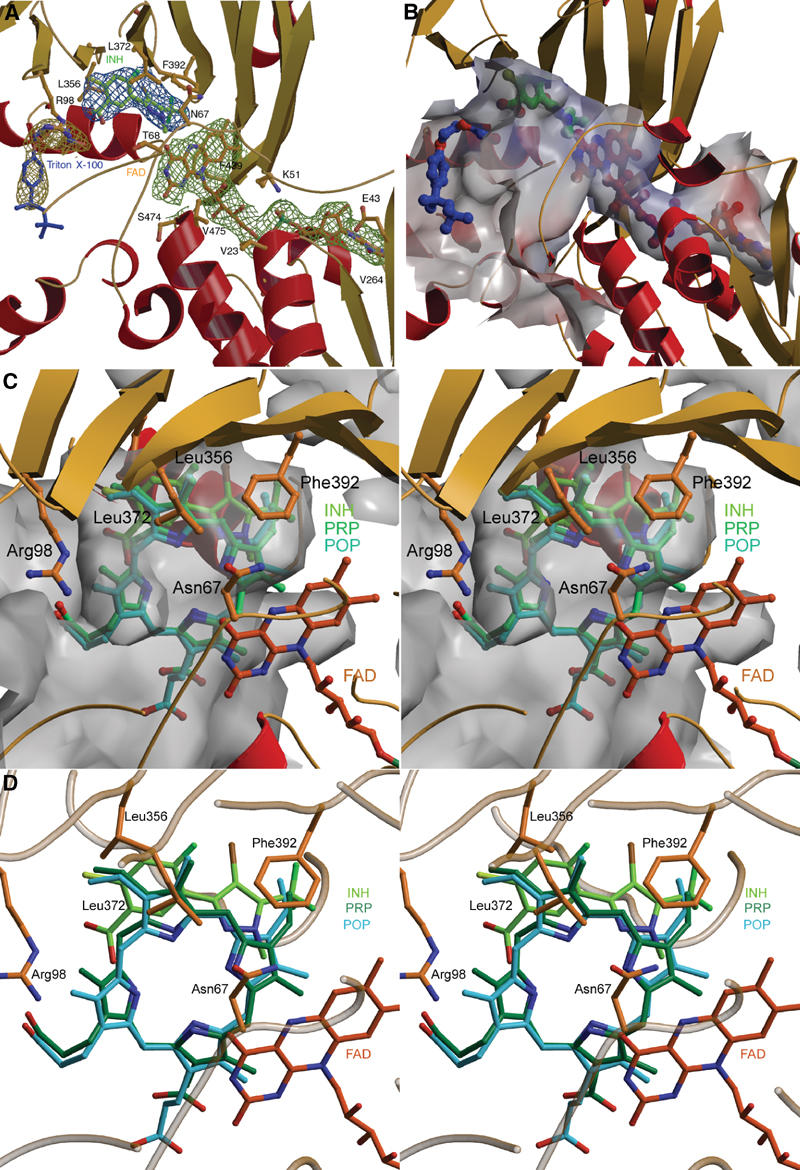
Active site of PPO. (A) The active site cavity with all ligands. The inhibitor INH (green) is shown with its electron density (steel blue) and is positioned by the residues Arg98, Leu356, Leu372 and Phe392. The coordination of the cofactor FAD (red–orange) surrounded by the electron density (grass-green) resembles that of MAO (Binda et al, 2002) and PAO (Binda et al, 1999). The detergent molecule Triton X-100 (blue) has bound into the entrance of the product channel; the aliphatic trimethylethyl group of the octyl group is not defined in the electron density (gold–yellow). The omit electron density map is displayed at 1.0σ contour level. (B) Binding tunnel for FAD, substrate and Triton X-100. The adenosyl moiety of FAD (red–orange) is exposed to solvent, the active site cavity is opened towards the membrane. The FAD cofactor (red–orange), the inhibitor INH (green) and a Triton X-100 detergent molecule (blue), which bind into the tunnel, are shown. (C) Surface representation of the active site cavity. The active site cavity is very narrow and the substrate is held tightly by ionic interaction of one propionyl oxygen from ring C with NH2 of Arg98, by stacking of ring B between Leu356 and Leu372 and by aromatic stacking interaction of ring A with Phe392, thus rotation of the substrate during the reaction is unlikely. (D) Active site cavity with bound FAD (red–orange), inhibitor INH (light green) and the modelled substrate protoporphyrinogen IX (PRP, green) and the product protoporphyrin IX (POP, cyan). The FAD-N5 atom, responsible for the hydride abstraction from the methylene bridge between ring A and D (C20 atom), is close to the C20 atom.
We have modelled the binding of protoporphyrinogen IX and protoporphyrin IX guided by the INH-binding site (Figure 5D). The negatively charged propionyl groups are oriented towards the solvent-exposed parts of the active site cavity. Arg98 being highly conserved over all eukaryotic PPOs provides a counter charge for the carboxylate group of the inhibitor INH and most likely also for the propionate group of ring C of protoporphyrin(ogen) IX. The pyrazole ring of INH serves as a model for ring A of protoporphyrin(ogen) IX, and is kept in position by aromatic stacking with residue Phe392 conserved in the mitochondrial plant enzymes. In the chloroplast isoenzymes, a tyrosine residue replaces it. The phenylring of the INH mimicking ring B of protoporphyrin(ogen) IX is sandwiched between the conserved residues Leu356 and Leu372. The methylene bridge (C20) between rings A and D of the modelled protoporphyrinogen IX is oriented towards the reactive N5 atom of the FAD, giving rise to the assumption that this position is initially oxidised by the FAD. Further oxidation can proceed from this position only as the narrow active site cavity allows binding of protoporphyrinogen IX only in this orientation and precludes rotations of the substrate. By hydrogen rearrangements through imine–enamine tautomerisations, all hydride abstraction reactions occur from C20. The atom C10 between rings B and C cannot be oriented close enough to the N5 atom of the FAD because of the interfering vinyl group on the C10 side of the molecules. The FAD is reoxidised three times by molecular oxygen, yielding three hydrogen peroxide molecules (Jordan, 1991) (Figure 6). No conserved residue is observed near the substrate that could serve as a base. A possible mechanism of deprotonation would include reduction of molecular oxygen by a negatively charged FADH− to yield a peroxide anion, which could then serve as a base and this could abstract a proton from the substrate molecule.
Figure 6.
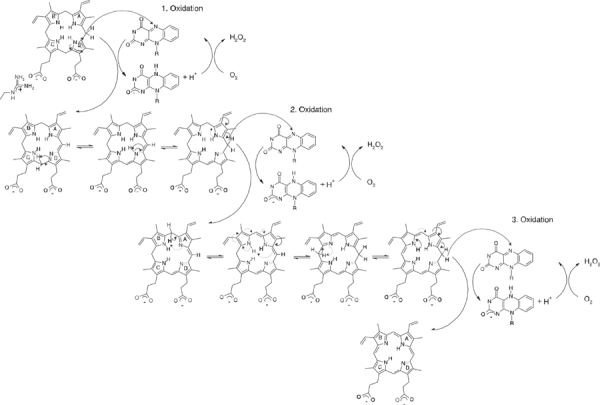
Proposed reaction mechanism of PPO. The oxidation of protoporphyrinogen IX to protoporphyrin IX occurs in three steps, whereby each time the FAD cofactor is reduced by the tetrapyrrol ring and reoxidised by an oxygen molecule that is reduced to hydrogen peroxide. The reaction always starts at the C20 atom of the tetrapyrrol ring by hydride transfer, followed by hydrogen rearrangements that take place in the whole ring system by enamine–imine tautomerisations (Jordan, 1991).
Variegate porphyria
In humans, a defect in PPO resulting in decreased enzymatic activity is responsible for the dominantly inherited disorder variegate porphyria, whose most obvious sign is the light sensitivity of the patient's skin. In more than 94% of the patients, Arg59 is mutated to Trp (Maneli et al, 2003). The residue in the tobacco mitochondrial enzyme that corresponds to human Arg59 conserved in mouse and Drosophila is Asn67 conserved in all known plant enzymes (Figure 2B). Asn67 is positioned on a loop between the isoalloxazine ring of FAD and the substrate-binding site. A bulky tryptophane residue at this position may disturb the protein structure in the active site and interfere with FAD and substrate binding. An Arg59Lys mutant of human PPO showed more than one-third of the wild-type enzyme activity, while Arg59Ser and Arg59Ile mutants were nearly inactive (Maneli et al, 2003). In animal PPOs including human PPO, the conserved Arg59 plays an important role. In the N. tabacum structure of PPO, Ser374 that is conserved between various plant species (Figure 2B) is in a position opposite to Asn67. The corresponding residue in animal PPOs is the conserved Asp349 that is likely to form a salt bridge with Arg59. This salt bridge is stabilising the active site structure. Mutating Arg59 to an uncharged residue disrupts this salt bridge, destabilising the active site structure of animal PPO.
Materials and methods
Protein expression, crystallisation, crystal transformation and structure solution
Recombinant thioredoxin- and his-tagged protein was produced with Escherichia coli strain B834(DE3) containing the vector pET32a-ppo2nt(M1-, K224Q), and the mass was verified by mass spectroscopy. SeMet-Protein was expressed in New Minimal Media (NMM, Budisa et al, 1995) as described (Budisa et al, 1997). The three-step purification included Ni-NTA, proteolytic cleavage of the tags, size exclusion chromatography (S-200) and anion exchange chromatography via ResourceQ. SeMet-Protein in 5 mM Tris/HCl, pH 8.0, 0.1% Triton X-100 and 50 mM sodium chloride was crystallised at 18°C using the vapour diffusion method and 10% polyethylene glycol 1000, 0.1 M sodium citrate/HCl, pH 4.0, 0.2 M sodium chloride, as crystallisation solution. Long yellow crystals (150 × 100 × 100 μm3) appeared within 2 days and belonged to space group C2221 with cell parameters a=119.1 Å, b=147.3 Å and c=127.0 Å, corresponding to two monomers per asymmetric unit. Crystals only showed reflections up to 8 Å, but by transforming them with a free-mounting system (Kiefersauer et al, 2000) (Proteros biostructures GmbH, Martinsried) they diffracted up to 2.9 Å using synchrotron radiation. A complete single-wavelength anomalous diffraction experiment was performed with the SeMet crystals at the high-brillance beamline ID29 at the European Radiation Synchrotron Facility (ESRF). Direct methods using SnB (Weeks and Miller, 1999) yielded 14 out of the 18 theoretical selenium sites. Phases were calculated by the single anomalous diffraction technique with SHARP (delaFortelle and Bricogne, 1997). Data statistics are shown in Table I.
Table 1.
Data collection, phasing and refinement statisticsa
| Data set | PPO2NT SeMet |
| Data collection | |
| Resolution (Å)b | 20–2.9 (3.0–2.9) |
| Unit cell (Å) | 119.09/147.25/127.04 |
| Wavelength (Å) | 0.9790 |
| I/σ(I) | 11.6 (2.8) |
| Completeness of data (%) | 99.3 (100.0) |
| Multiplicity | 3.8 (3.8) |
| Rmerge (%)c | 7.9 (52.8) |
| Phasing procedure | |
| Anomalous scatterer | 14 Se |
| Phasing power anod | 1.13 |
| Rcullis anoe | 0.79 |
| Figure of merit | 0.51 |
| Model refinement | |
| Resolution (Å) | 20–2.9 |
| Number of reflections Rcryst/Rfree | 23 234/1187 |
| Number of protein atoms/ligand atoms/solvent molecules | 6962/192/143 |
| Mean temperature factors (protein/ligand/solvent) | 67.7/60.7/57.2 |
| Rmsd (bonds (Å)/angles (°)/bonded Bs (Å2))f | 0.008/1.3/1.5 |
| Ramachandran most favoured/disallowed (%) | 82.6/0.0 |
|
R-factors: Rcryst/Rfree (%)g |
22.7/29.3 |
| bData of the last resolution shell are given in brackets. | |
| cRmerge=∑h ∑i∣Ii(h)−〈I(h)〉∣/∑h∑iIi(h). | |
| dPhasing power is the mean value of the heavy atom derivative structure factor amplitude divided by the residual lack of closure error. | |
| eRcullis=∑h∣ ∣Fph∣−∣Fp∣−Fh,calc∣/∑h∣Fph−Fp∣. | |
| fRoot mean square deviations (rmsd) of temperature factors of bonded atoms. | |
| gRcryst=∑h∣∣Fobs (h)∣−k ∣Fcalc (h)∣ ∣/∑hkl ∣Fobs (h)∣ for the working set of reflections; Rfree is the R-value for 5% of the reflections excluded from refinement. | |
aAll values refer to anomalous diffraction. The diffraction data were processed and scaled with the program XDS (Kabsch, 1993). Refinement of heavy atom parameters, phase calculation and solvent flattening were done with SnB (Weeks and Miller, 1999), SHARP (delaFortelle and Bricogne, 1997) and SOLVE (http://www.ccp14.ac.uk/solution/macromolecular-software/). The stereochemistry of the model was validated with PROCHECK (Laskowski et al, 1993).
Model building and refinement
The initial electron density map was two-fold averaged using AVE (Jones, 1992). Model building was carried out in the improved map with O (http://xray.bmc.uu.se/alwyn). Energy-restrained crystallographic refinement was carried out with maximum likelihood algorithms implemented in CNS (Brunger et al, 1998). Refinement proceeded in several cycles, which were interrupted for manual rebuilding with the program O (Jones et al, 1991). After the addition of solvent molecules, the refinement converged at an R-factor of 22.7% (Rfree=29.3%, Table I). Two regions are not defined in the electron density, namely residues 138–147 and residues 213–223. Four loops are not well defined, namely residues 148–149, 224–230, 272–278 and 286–292. All graphical representations were prepared using the programs MOLSCRIPT (Kraulis, 1991), BOBSCRIPT (Esnouf, 1999), RASTER3D (Merrit and Bacon, 1997), GRASP (Nicholls et al, 1993) and ALSCRIPT (Barton, 1993). Final Ramachandran plot analysis with the program PROCHECK (Laskowski et al, 1993) resulted in 82.6% residues in the most favoured regions, 15.3% residues in the additionally allowed regions, 2.1% residues in the generously allowed regions and no residues in the disallowed regions. For the superposition of the different 3D structures, the program LSQMAN (Kleywegt and Jones, 1994) was applied.
Docking of the PPO–HFc complex
The two-fold axes of the two dimers of PPO and HFc were aligned and the protomers were manually rotated and shifted until the best visual fit was obtained. The dimensions of the two channel parts and the two-fold axes in both enzymes fit very well (Supplementary Figure 4A and B), except for residues 209–213 from PPO, whose conformations are presumably influenced by crystal packing and that are not very well defined in the electron density.
Molecular modelling of substrate and product binding
Molecular modelling was pursued with modules Viewer, Builder and Discover3 of program Insight II (Version 98.0; Molecular Dimensions LtD, Los Angeles, 1998). The PPO2 X-ray structure was chosen as the starting point for the modelling and energy minimisation of the modelled complexes. The protoporphyrinogen IX and protoporphyrin IX molecules were generated with module Builder. The narrow substrate/product-binding pocket with its hydrophobic bottom permits the insertion of substrate/product molecule in such a way only that the vinyl groups of the molecule lie at the bottom of the pocket and methylene bridge atom C20 is close to N5 of the FAD cofactor. The propionate group of ring C may form a salt bridge with the strictly conserved Arg98. The Consistent Valence Forcefield (CVFF) of the Insight II program was used for the energy minimisations with Discover3, which was run over 1000 steps until convergence (0.1 kcal/mol tolerance). The protein ligand complexes were minimised simultaneously. The enzyme product complex was minimised first to a total energy of 1247 kcal/mol. This minimised complex served as a starting point for the energy minimisation of the enzyme substrate complex. The bond structure of the product was changed to be consistent with that of the substrate before the respective minimisation, which yielded a total energy of 764 kcal/mol. The positive total energy values are due to relatively high repulsive van der Waals energy terms caused by the narrow substrate/product-binding pocket of PPO2.
Accesssion numbers
The atomic coordinates of the structure and the structure factors have been deposited with the Protein Data Bank (Accession No. 1SEZ).
Supplementary Material
Supplemental Figures
Acknowledgments
We thank G Auerbach, O Boscheinen, C Haussmann and C Krebs from Bayer CropScience for initiating the project and initial cloning experiments, M Fischer from the TU München and S Marinkovic and S Macieira, MPI Martinsried, for help with further cloning and S Monaco for assistance during data collection at beamline ID29 (ESRF, Grenoble), T Krojer for help with the programs XDS and SHARP and L Weyher-Stingel for mass-spectroscopic analysis.
References
- Arnould S, Takahashi M, Camadro JM (1999) Acylation stabilizes a protease-resistant conformation of protoporphyrinogen oxidase, the molecular target of diphenyl ether-type herbicides. Proc Natl Acad Sci USA 96: 14825–14830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GJ (1993) Alscript—a tool to format multiple sequence alignments. Prot Eng 6: 37–40 [DOI] [PubMed] [Google Scholar]
- Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A (1999) A 30 angstrom long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Struct Fold Des 7: 265–276 [DOI] [PubMed] [Google Scholar]
- Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A (2002) Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol 9: 22–26 [DOI] [PubMed] [Google Scholar]
- Blobel G (1980) Intracellular protein topogenesis. Proc Natl Acad Sci USA—Biol Sci 77: 1496–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Budisa N, Karnbrock W, Steinbacher S, Humm A, Prade L, Neuefeind T, Moroder L, Huber R (1997) Bioincorporation of telluromethionine into proteins: a promising new approach for X-ray structure analysis of proteins. J Mol Biol 270: 616–623 [DOI] [PubMed] [Google Scholar]
- Budisa N, Steipe B, Demange P, Eckerskorn C, Kellermann J, Huber R (1995) High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur J Biochem 230: 788–796 [DOI] [PubMed] [Google Scholar]
- delaFortelle E, Bricogne G (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. In Methods in Enzymology, Vol. 276, Macromolecular crystallography, Part B, Carter CW, Sweet RM (eds) pp 472–494. London: Academic Press [DOI] [PubMed] [Google Scholar]
- Esnouf RM (1999) Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D 55: 938–940 [DOI] [PubMed] [Google Scholar]
- Ferreira GC, Andrew TL, Karr SW, Dailey HA (1988) Organization of the terminal 2 enzymes of the heme biosynthetic-pathway—orientation of protoporphyrinogen oxidase and evidence for a membrane complex. J Biol Chem 263: 3835–3839 [PubMed] [Google Scholar]
- Ferreira GC, Dailey HA (1988) Mouse protoporphyrinogen oxidase—kinetic-parameters and demonstration of inhibition by bilirubin. Biochem J 250: 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisla S, Massey V (1989) Mechanisms of flavoprotein-catalyzed reactions. Eur J Biochem 181: 1–17 [DOI] [PubMed] [Google Scholar]
- Jones TA (1992) A, yaap, asap, @**?: a set of averaging programs. In Molecular Replacement (CCP4), Dodson EJ, Gover S, Wolf W (eds) pp 99–105. Warrington: Daresbury Laboratory [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr Section A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Jordan PM (1991) Biosynthesis of Tetrapyrroles. New York: Elsevier [Google Scholar]
- Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Kiefersauer R, Than ME, Dobbek H, Gremer L, Melero M, Strobl S, Dias JM, Soulimane T, Huber R (2000) A novel free-mounting system for protein crystals: transformation and improvement of diffraction power by accurately controlled humidity changes. J Appl Crystallogr 33: 1223–1230 [Google Scholar]
- Kleywegt GJ, Jones TA (1994) A super position. ESF/CCP4 Newslett 31: 9–14 [Google Scholar]
- Kraulis PJ (1991) Molscript—a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM (1993) Procheck—a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lermontova I, Kruse E, Mock HP, Grimm B (1997) Cloning and characterization of a plastidal and a mitochondrial isoform of tobacco protoporphyrinogen IX oxidase. Proc Natl Acad Sci USA 94: 8895–8900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loll PJ, Picot D, Garavito RM (1995) The structural basis of aspirin activity inferred from the crystal-structure of inactivated prostaglandin H-2 synthase. Nat Struct Biol 2: 637–643 [DOI] [PubMed] [Google Scholar]
- Maneli MH, Corrigall AV, Klump HH, Davids LM, Kirsch RE, Meissner PN (2003) Kinetic and physical characterisation of recombinant wild-type and mutant human protoporphyrinogen oxidases. Biochim Biophys Acta—Proteins Proteomics 1650: 10–21 [DOI] [PubMed] [Google Scholar]
- Merrit EA, Bacon DJ (1997) Raster 3d-photorealistic molecular graphics. In Methods in Enzymology, Vol. 277, Macromolecular crystallography, Part B, Carter CW, Sweet RM (eds) pp 505–524. London: Academic Press [DOI] [PubMed] [Google Scholar]
- Mizutani H, Miyahara I, Hirotsu K, Nishina Y, Shiga K, Setoyama C, Miura R (1996) Three-dimensional structure of porcine kidney D-amino acid oxidase at 3.0 angstrom resolution. J Biochem 120: 14–17 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Bharadwaj R, Honig B (1993) Grasp—graphical representation and analysis of surface-properties. Biophys J 64: A166–A166 [Google Scholar]
- Norris SR, Barrette TR, DellaPenna D (1995) Genetic dissection of carotenoid synthesis in Arabidopsis defines plastoquinone as an essential component of phytoene desaturation. Plant Cell 7: 2139–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A (2000) The structure of L-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. EMBO J 19: 4204–4215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana MA, Tan FC, Smith AG (2002) Molecular characterisation of coproporphyrinogen oxidase from Glycine max and Arabidopsis thaliana. Plant Physiol Biochem 40: 289–298 [Google Scholar]
- Schreuder HA, Vanderlaan JM, Swarte MBA, Kalk KH, Hol WGJ, Drenth J (1992) Crystal-structure of the reduced form of P-hydroxybenzoate hydroxylase refined at 2.3-a resolution. Proteins—Struct Funct Genet 14: 178–190 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) Clustal-W—improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks CM, Miller R (1999) The design and implementation of SnB version 2.0. J Appl Crystallogr 32: 120–124 [Google Scholar]
- Wendt KU, Lenhart A, Schulz GE (1999) The structure of the membrane protein squalene-hopene cyclase at 2.0 angstrom resolution. J Mol Biol 286: 175–187 [DOI] [PubMed] [Google Scholar]
- Wu CK, Dailey HA, Rose JP, Burden A, Sellers VM, Wang BC (2001) The 2.0 angstrom structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat Struct Biol 8: 156–160 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures