

Abstract
The eucaryotic translation initiation factor 4B (eIF4B) stimulates the helicase activity of the DEAD box protein eIF4A to unwind inhibitory secondary structure in the 5′ untranslated region of eucaryotic mRNAs. Here, using phosphopeptide mapping and a phosphospecific antiserum, we identify a serum-responsive eIF4B phosphorylation site, Ser422, located in an RNA-binding region required for eIF4A helicase-promoting activity. Ser422 phosphorylation appears to be regulated by the S6Ks: (a) Ser422 phosphorylation is sensitive to pharmacological inhibitors of phosphoinositide-3 kinase and the mammalian target of rapamycin; (b) S6K1/S6K2 specifically phosphorylate Ser422 in vitro; and (c) rapamycin-resistant S6Ks confer rapamycin resistance upon Ser422 phosphorylation in vivo. Substitution of Ser422 with Ala results in a loss of activity in an in vivo translation assay, indicating that phosphorylation of this site plays an important role in eIF4B function. We therefore propose that eIF4B may mediate some of the effects of the S6Ks on translation.
Keywords: eIF4B, phosphorylation, S6K, signaling, translation
Introduction
Translational control is primarily exerted at the initiation phase, a complex process mediated by a number of eucaryotic translation initiation factors (eIFs), during which ribosomes are recruited to the 5′ end of an mRNA and positioned at a start codon (reviewed in Gingras et al, 1999b; Hershey and Merrick, 2000). The mRNA 5′ end is distinguished by the presence of a ‘cap' structure (m7GpppN, where m is a methyl group and N is any nucleotide), which is specifically bound by the cap-binding protein eIF4E. eIF4E, via an interaction with the large scaffolding protein eIF4G, guides the translation machinery to the 5′ end of mRNA. eIF4E and eIF4G function as components of the dynamic, heterotrimeric eIF4F complex, along with the DEAD box RNA helicase eIF4A. Optimal ribosome binding is thought to require a region of single-stranded mRNA (Gingras et al, 1999b). Thus, it has been proposed that one critical function of eIF4F is to melt cap-proximal inhibitory secondary structure to provide a single-stranded RNA ‘landing pad' for the 40S ribosomal subunit (Gingras et al, 1999b).
eIF4A alone displays only low levels of RNA helicase activity (Rogers et al, 1999), but this activity is significantly stimulated by a cofactor, eucaryotic translation initiation factor 4B (eIF4B) (Lawson et al, 1989). eIF4B is an RNA-binding protein first purified as an activity capable of stimulating translation and promoting ribosome binding to mRNA (Trachsel et al, 1977; Benne and Hershey, 1978). More recent studies utilizing a ribosome toe-printing assay have validated this role (Pestova et al, 1996; Morino et al, 2000). eIF4B possesses no autonomous catalytic activity (Grifo et al, 1984), but enhances the affinity of eIF4A for ATP and mRNA (Rogers et al, 1999), and increases the processivity of the helicase (Rogers et al, 2001).
eIF4B is a phosphoprotein that migrates as multiple isoelectric variants in the two-dimensional isoelectric focusing (IEF)/SDS–PAGE system (Duncan and Hershey, 1984). eIF4B hyperphosphorylation, as elicited by mitogens and phorbol esters, correlates with the growth status of the cell (Duncan and Hershey, 1985) and an increase in the translation rates of mRNAs possessing long, structured 5′ untranslated regions (5′UTRs) (Manzella et al, 1991). Importantly, eIF4B is required for the formation of 48S translation initiation complexes in vitro on 5′UTRs with even only moderate amounts of secondary structure (Dmitriev et al, 2003). Interestingly, recombinant eIF4B cannot substitute for purified native eIF4B in this assay (Dmitriev et al, 2003), suggesting that a post-translational modification that occurs only in mammalian cells is important for eIF4B activity.
We and others have demonstrated that the activity of several translation regulatory factors is modulated by the phosphoinositide-3 kinase (PI3K) and mammalian target of rapamycin (mTOR) signaling modules (reviewed in Gingras et al, 1999b; Fumagalli and Thomas, 2000; Raught et al, 2000b; Dennis and Thomas, 2002). The PI3Ks are a family of mitogen-responsive lipid kinases that play a role in many critical cellular processes (reviewed in Cantley, 2002). The TOR proteins are evolutionarily conserved protein kinases thought to function in a nutrient-sensing checkpoint control capacity. Amino-acid deprivation results in a TOR-mediated shift from anabolic to catabolic processes, including alteration in amino-acid transporter expression levels, a dramatic shift in the transcription levels of genes involved in amino-acid utilization, and the activation of autophagy (reviewed in Schmelzle and Hall, 2000; Raught et al, 2001). Inactivation of the TOR proteins, or treatment with an inhibitor of TOR signaling, rapamycin, mimics nutrient deprivation in yeast, Drosophila and mammalian cells (e.g. Barbet et al, 1996; Kimball and Jefferson, 2000, and references therein; Oldham et al, 2000; Zhang et al, 2000). A current model for TOR checkpoint signaling proposes that a permissive signal is relayed to downstream translational targets only in the presence of sufficient nutrients to fuel protein synthesis. In signaling to translation effectors, the TOR proteins appear to function in a coregulatory capacity with other signaling pathways (such as the PI3K signaling module): In this way, a passive nutrient sufficiency signal is combined with stimulatory signaling from a mitogen-responsive pathway, to coordinate a process that requires an abundance of energy and the uptake of extracellular amino acids.
Two key downstream targets of PI3K/TOR signaling are the ribosomal S6 kinases S6K1 and S6K2. The S6Ks play critical roles in the modulation of cell growth and cell cycle progression (reviewed in Fumagalli and Thomas, 2000). S6K inactivation inhibits or slows cell cycle progression in many eucaryotic cell types (e.g. Chung et al, 1992), and Drosophila melanogaster mutants harboring loss-of-function alleles of the S6K ortholog dS6K are developmentally delayed and significantly smaller than their wild–type (wt) counterparts (Montagne et al, 1999). Similarly, deletion of the murine S6K1 ortholog yields a small mouse (Shima et al, 1998), possessing significantly smaller pancreatic β-cells (Pende et al, 2000).
How S6K signaling regulates translation is unknown. S6K1 was one of the first mitogen-activated protein kinases to be characterized (Jenö et al, 1988), yet the only S6K substrate involved in translation identified to date was the S6 ribosomal protein. Notably, S6 phosphorylation levels are normal in S6K1 null murine embryo fibroblasts (presumably due to the presence of S6K2; Shima et al, 1998), suggesting that a lack of S6 phosphorylation is not responsible for the observed small mouse phenotype. Thus, S6 phosphorylation does not appear to mediate all of the effects of the S6Ks on cell growth and proliferation.
To better understand the role of eIF4B phosphorylation in the control of translation initiation, we sought to identify the location of the phosphorylation sites within the eIF4B protein, and to characterize the intracellular signaling pathways that regulate eIF4B phosphorylation. Here, we unambiguously identify a serum and mitogen-responsive phosphorylation site, and present strong evidence that phosphorylation of this site is modulated by S6K signaling. Since eIF4B plays a critical role in the loading of ribosomes onto the mRNA 5′UTR, we suggest that eIF4B may mediate some of the effects of S6K signaling on translation.
Results
eIF4B phosphopeptide mapping
eIF4B phosphorylation is stimulated by serum or mitogen addition to quiescent cells in culture, and eIF4B hyperphosphorylation correlates with increased translation rates (Duncan and Hershey, 1985; Duncan and Hershey, 1987). To further characterize the regulation of eIF4B by phosphorylation, immunoprecipitated eIF4B labeled in vivo with [32P]orthophosphate was subjected to two-dimensional phosphopeptide mapping (see Materials and methods). eIF4B isolated from serum-deprived 293 cells yields a single major phosphopeptide (Figure 1A), while eIF4B immunoprecipitated from serum-stimulated cells yields two major phosphopeptides (Figure 1B). Thus, while the intensity of peptide 1 is unaffected by serum stimulation, a dramatic increase in the phosphorylation state of peptide 2 is observed in response to serum treatment, indicating a change in the phosphorylation status of one or more amino-acid residues. Phosphopeptides 1 and 2 were eluted from the chromatography plate and subjected to phosphoamino-acid analysis (van der Geer et al, 1994). These phosphopeptides contain only phosphoserine (data not shown). Analysis of the entire endogenous eIF4B protein isolated from 32P-labeled 293 or HeLa cells also yielded only phosphoserine (not shown).
Figure 1.
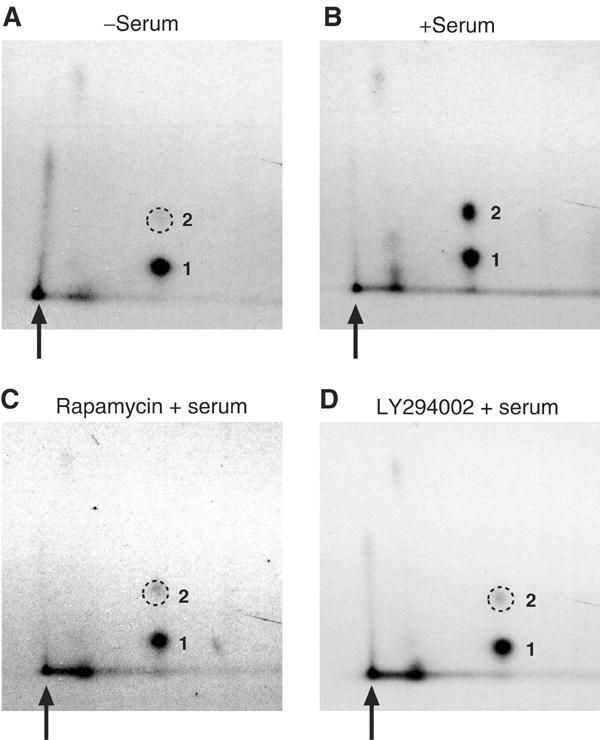
Phosphorylation of a single major eIF4B peptide is modulated by serum, and is sensitive to pharmacological inhibitors of PI3K and mTOR. Phosphopeptide mapping of endogenous eIF4B isolated from 293 cells, (A) starved of serum for 36 h; (B) starved of serum, and then serum stimulated for 30 min; (C) starved of serum, pretreated with 5 μM LY294002 for 30 min, and then serum stimulated for 30 min; or (D) starved of serum, pretreated with 50 nM rapamycin for 30 min, and then serum stimulated for 30 min. The two major phosphopeptides are numbered, and the loading origin is indicated by an arrow. The position of the diminished phosphopeptide 2 is indicated with a dashed circle. A vertical streaked signal near the loading origin is also observed, but this signal does not appear to be responsive to serum stimulation nor sensitive to kinase inhibitors.
PI3K and mTOR signaling modulate eIF4B phosphorylation
The PI3K and mTOR signaling modules have been implicated in translational control, and several translation regulatory factors have been identified as targets of PI3K and mTOR signaling (reviewed in Fumagalli and Thomas, 2000; Raught et al, 2000b). It was thus pertinent to test whether the serum-stimulated phosphorylation of eIF4B is also regulated via these pathways. To this end, serum-starved 293 cells metabolically labeled with [32P]orthophosphate were treated with various kinase inhibitors for 30 min, followed by serum stimulation for 30 min. eIF4B was then immunoprecipitated and subjected to phosphopeptide mapping, as above. Pretreatment with the PI3K inhibitors LY294002 (5 μM; Figure 1C) or wortmannin (100 nM, data not shown), or with an inhibitor of mTOR signaling, rapamycin (50 nM; Figure 1D), significantly decreased the serum-stimulated phosphorylation of peptide 2. These inhibitors had no apparent effect on the phosphorylation state of peptide 1. The serum-stimulated phosphorylation of peptide 2 therefore appears to require PI3K and mTOR signaling.
S6Ks phosphorylate eIF4B in vitro on a physiologically relevant site
Our mapping data suggested that the serum-stimulated phosphorylation of eIF4B phosphopeptide 2 is effected primarily via a rapamycin-sensitive signaling module. Three rapamycin-sensitive kinases previously demonstrated to play important roles in the regulation of translation, mTOR, S6K1 and S6K2, were therefore tested for their ability to phosphorylate eIF4B in vitro. Recombinant eIF4B protein possessing an N-terminal glutathione S-transferase (GST) tag was expressed in Escherichia coli and purified. GST-eIF4B was then incubated with [γ-32P]ATP and GST-tagged S6K1 or S6K2 proteins (purified from 293T cell extracts, as in Weng et al, 1995; Dennis et al, 1998), or an immunoprecipitate of endogenous mTOR from rat brain (as in Brunn et al, 1996). While GST-eIF4B alone did not incorporate significant levels of 32P in this assay, GST-S6K1 (Figure 2A) and GST-S6K2 (data not shown) readily phosphorylated eIF4B in vitro. GST-eIF4B was also incubated with a kinase-dead version of GST-S6K1. As in the absence of kinase, only background levels of 32P were incorporated into GST-eIF4B, and no resolvable phosphopeptides were observed via two-dimensional phosphopeptide mapping (not shown). Finally, while the mTOR immunoprecipitate exhibited kinase activity toward recombinant 4E-BP1 protein, it did not display significant eIF4B kinase activity (data not shown).
Figure 2.
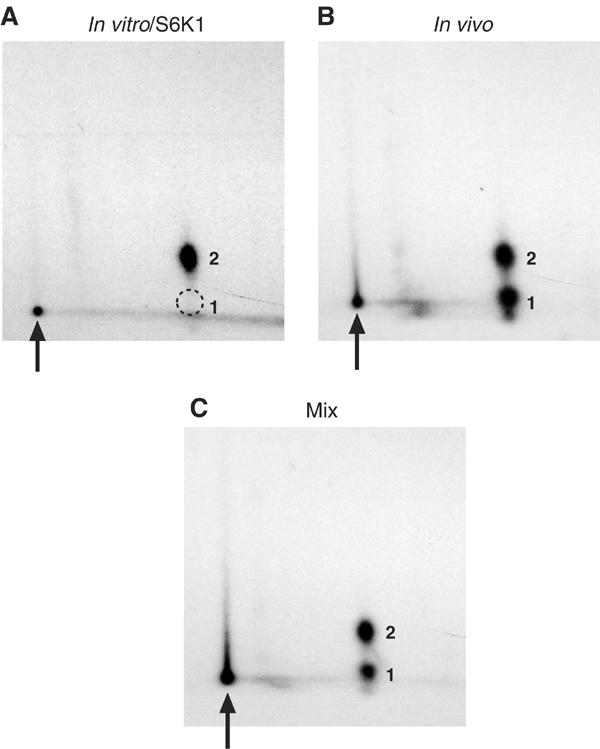
S6K1 phosphorylates eIF4B in vitro on a phosphopeptide comigrating with the endogenous serum-stimulated phosphopeptide 2. (A) Recombinant eIF4B was labeled in vitro with S6K1; or (B) immunoprecipitated from 32P-labeled 293 cells, and subjected to phosphopeptide mapping. (C) The in vitro- and in vivo-labeled samples were then mixed, and subjected to phosphopeptide mapping.
To ascertain whether the site(s) phosphorylated in vitro by S6K1/2 corresponds to a bona fide in vivo phosphorylation site, recombinant GST-eIF4B protein labeled in vitro with S6K1 was subjected to phosphopeptide mapping. The full-length, in vitro-labeled eIF4B protein yields a single phosphopeptide (Figure 2A) that migrates in the same region as the in vivo phosphopeptide 2 (Figure 2B). Mixing the recombinant eIF4B protein labeled in vitro with immunoprecipitated endogenous eIF4B protein labeled in vivo yielded a phosphopeptide pattern identical to the endogenous protein alone (Figure 2C). The phosphopeptide generated in vitro thus comigrates with the serum-responsive, rapamycin-sensitive phosphopeptide 2, strongly suggesting that the phosphopeptide generated in vitro is identical to that generated in vivo.
Localization of eIF4B phosphorylation sites
To delineate the region(s) of the eIF4B protein phosphorylated by S6K1 in vitro, six contiguous (His6)-tagged eIF4B fragments, each comprised of ∼100 aa (Figure 3A), were expressed in E. coli, purified via Ni2+ affinity chromatography, and then subjected to SDS–PAGE and Coomassie blue staining (Figure 3B). The fragments were also incubated with [γ-32P]ATP and tagged S6K1 protein (as above), and then subjected to SDS–PAGE and autoradiography (Figure 3C). Only fragment 5, encompassing aa 380–480, incorporated significant amounts of 32P in this assay. Two serines within a consensus S6K1 phosphorylation site (Flotow and Thomas, 1992) are present in fragment 5, Ser406 and Ser422.
Figure 3.
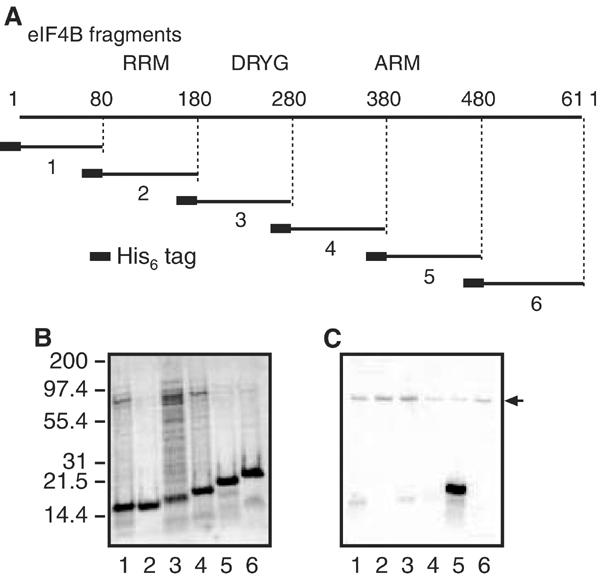
(A) Schematic representation of His6-eIF4B fragments. Six contiguous hexahistidine-tagged fragments comprised of ∼100 residues each were generated. The RRM, DRYG domain and ARM RNA-binding domain are indicated. (B) Recombinant fragments were purified by Ni2+ affinity chromatography, and then subjected to SDS–PAGE and Coomassie blue staining. (C) Equal amounts of the same fragments were phosphorylated by S6K1 in vitro. Lane numbers indicate eIF4B fragment number. The arrow indicates autophosphorylated S6K1.
Ser422 is phosphorylated by S6K1 in vitro
Ser406 and Ser422 were mutated to Ala, Asp or Thr residues in the context of the full-length His6-eIF4B protein. These proteins were expressed in E. coli, purified, 32P-labeled in vitro with GST-S6K1, and subjected to phosphopeptide mapping. The 32P-labeled wt His6-eIF4B protein yielded a single major phosphopeptide, as above (Figure 4A). The Ser406Ala mutation had no effect on the phosphopeptide map of in vitro-phosphorylated eIF4B (not shown). However, mutation of Ser422 to Ala or Asp abrogated the phosphorylation of this peptide (not shown). Surprisingly, mutation of Ser422 to Thr also abrogated phosphorylation of this residue by S6K1 (Figure 4B). Identical results were observed with S6K2 (data not shown). These data thus strongly suggest that Ser422 is phosphorylated by S6K1/2 in vitro. Interestingly, these results also indicate a marked preference for serine at this site, as replacement of Ser422 with threonine, even in the context of an S6K consensus phosphorylation sequence, abrogated phosphorylation of this residue in vitro.
Figure 4.
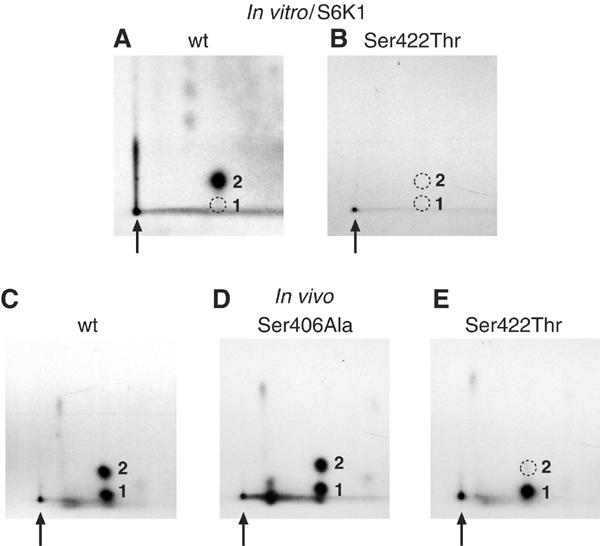
Ser422 is phosphorylated in vivo, and is phosphorylated in vitro by S6K1. Recombinant (A) wt and (B) Ser422Thr eIF4B proteins were phosphorylated in vitro with S6K1, and then subjected to phosphopeptide mapping. Similarly, Flag-tagged eIF4B (C) wt, (D) Ser406Ala and (E) Ser422Thr proteins were expressed in 293T cells, 32P-labeled in vivo and subjected to phosphopeptide mapping.
Ser422 is phosphorylated in vivo
To determine whether eIF4B Ser422 is phosphorylated in vivo, various Flag-tagged eIF4B proteins were expressed in 293T cells. Phosphopeptide mapping of wt Flag-eIF4B yielded a pattern identical to the endogenous eIF4B protein (Figure 4C). Mutation of Ser406 to Asp (not shown) or Ala (Figure 4D) had no effect on the phosphopeptide pattern. However, mutation of Ser422 to Ala or Asp (not shown) eliminated phosphopeptide 2. Reminiscent of the in vitro data, mutation of Ser422 to Thr also abrogated phosphorylation of peptide 2 in vivo (Figure 4E). Thus, eIF4B Ser422 also appears to be phosphorylated in vivo.
It remained possible that amino-acid substitutions at Ser422 alter eIF4B structure to prevent phosphorylation on another residue. To verify that phosphorylation occurs at this site, antisera directed against the phosphopeptide (C)ERSRTGpSESSQT (corresponding to eIF4B residues 416-427, with an N-terminal Cys) were prepared, as described in Materials and methods. The Ser422 phosphospecific antiserum weakly detected an ∼80 kDa protein in serum-starved HeLa cells (Figure 5A, lane 1). This signal was dramatically enhanced, however, in serum-stimulated cells (lane 2). The phosphospecific antiserum did not recognize the Ser422Ala, Ser422Asp or Ser422Thr mutant Flag-tagged eIF4B proteins expressed in 293 cells (not shown). The signal detected by the Ser422 phosphospecific antiserum was specifically depleted by preincubation with the eIF4B 416–427 phosphopeptide (Figure 5A, lanes 3 and 4), but not by the corresponding unphosphorylated peptide (lanes 5 and 6), indicating that this antisera specifically recognizes the phosphorylated form of the protein.
Figure 5.
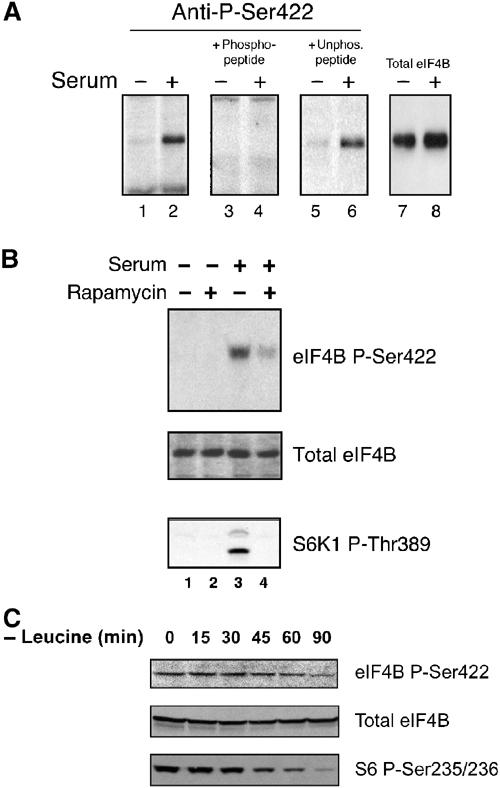
A phosphospecific antiserum confirms that phosphorylation of Ser422 is serum responsive, and is sensitive to rapamycin treatment and leucine deprivation. (A) Cells were starved of serum (odd-numbered lanes), or starved of serum and then serum stimulated (even-numbered lanes). Lysates were subjected to Western analysis using the phosphospecific eIF4B Ser422 antiserum (lanes 1 and 2). Preincubation of the antiserum with the phosphopeptide against which it was raised (lanes 3 and 4), or with the same peptide containing an unphosphorylated Ser422 (lanes 5 and 6). eIF4B expression levels were not affected by these treatments (lanes 7 and 8). (B) Cells were pretreated with rapamycin, and subjected to Western analysis using the phosphospecific antiserum (top panel) and antisera directed against total eIF4B (middle panel), as above. The same lysates were subjected to Western analysis using a phosphospecific antibody directed against S6K1 Thr389 (lower panel). (C) Cells were subjected to leucine-free (but otherwise complete) culture media for the indicated time periods. Lysates were subjected to Western blotting using the eIF4B Ser422 phosphospecific antiserum (top panel), a phosphorylation-independent antiserum directed against eIF4B (middle panel) or a phosphospecific antiserum directed against ribosomal S6 protein Ser235/236 (lower panel).
Consistent with the phosphopeptide mapping analyses, pretreatment with rapamycin significantly diminished the serum-stimulated phosphorylation of eIF4B Ser422 (Figure 5B, lane 4, upper panel). Western analysis of an identical blot using a phosphorylation state-independent eIF4B antibody demonstrated that this treatment regimen does not affect eIF4B expression levels (middle panel). The same lysates were also subjected to Western blotting using a phosphospecific antibody directed against S6K1 Thr389 (a rapamycin-sensitive phosphoresidue in the catalytic domain; bottom panel). As expected, serum addition dramatically increased the phosphorylation state of this residue, and rapamycin treatment abrogated Thr389 phosphorylation, indicating that Thr389 phosphorylation, which is required for S6K activity, is correlated with eIF4B Ser422 phosphorylation in these cells. Thus, a phosphospecific antibody directed against eIF4B Ser422 confirmed that this residue is phosphorylated, and that the phosphorylation state of this site is modulated by serum addition, and is sensitive to rapamycin treatment (lane 4, upper panel).
Ser422 phosphorylation is responsive to amino-acid deprivation
Translation initiation is extremely sensitive to environmental amino-acid supply (Kimball and Jefferson, 2000). In particular, decreased levels of the essential branched-chain amino acid leucine elicit a potent inhibition of S6K activity (Shigemitsu et al, 1999; Anthony et al, 2000). To determine whether eIF4B Ser422 phosphorylation is also modulated by environmental amino-acid levels, cells were maintained in culture media lacking only leucine, and the phosphospecific antiserum was utilized to monitor Ser422 phosphorylation levels (Figure 5C). A gradual decrease in Ser422 phosphorylation was observed, such that after 90 min of leucine deprivation it was significantly diminished (upper panel). Cells maintained in complete media containing leucine demonstrated no loss of Ser422 phosphorylation over the same time course (not shown), and leucine deprivation had no effect on eIF4B expression levels (middle panel). As expected, leucine deprivation also elicited a decrease in ribosomal S6 protein Ser235/236 phosphorylation (lower panel). Thus, consistent with the amino-acid-responsive S6Ks effecting eIF4B phosphorylation, Ser422 phosphorylation is sensitive to leucine deprivation.
S6Ks modulate eIF4B Ser422 phosphorylation in vivo: rapamycin-resistant S6K proteins confer rapamycin resistance to Ser422
Activation of the S6K1/2 kinases is a complex process, involving multiple phosphorylation events (reviewed in Fumagalli and Thomas, 2000). When several Ser/Thr residues in the C-terminal autoinhibitory domain are mutated to Asp or Glu, and Thr389 is mutated to Glu, the resulting mutant kinase (D3E-E389) acquires resistance to rapamycin. Similarly, deletion of the C-terminal autoinhibitory domain yields a kinase (ΔCT104) that displays rapamycin resistance (Cheatham et al, 1995; Pearson et al, 1995; Weng et al, 1995; Dennis et al, 1996).
To determine whether S6K signaling can modulate eIF4B Ser422 phosphorylation in vivo, various S6K1 constructs were cotransfected with Flag-tagged wt eIF4B, and eIF4B phosphorylation was examined via phosphopeptide mapping. Cotransfected 293 cells were starved of serum for 36 h, and then metabolically labeled with [32P]orthophosphate. Labeled cells were pretreated with rapamycin for 30 min, and then stimulated with 10% FBS, as above. Flag-tagged wt eIF4B cotransfected with an empty vector (Figure 6A and B) or coexpressed with wt S6K1 protein (C and D) responded normally to serum addition, and remained sensitive to rapamycin treatment. Strikingly, however, eIF4B cotransfected with the rapamycin-resistant D3E-E389 (Figure 6E and F) or ΔCT104 S6K1 mutants (G and H) acquired resistance to rapamycin. Thus, as evidenced by its ability to confer rapamycin resistance upon eIF4B phosphorylation, S6K1 alone is sufficient to modulate eIF4B Ser422 phosphorylation in vivo.
Figure 6.
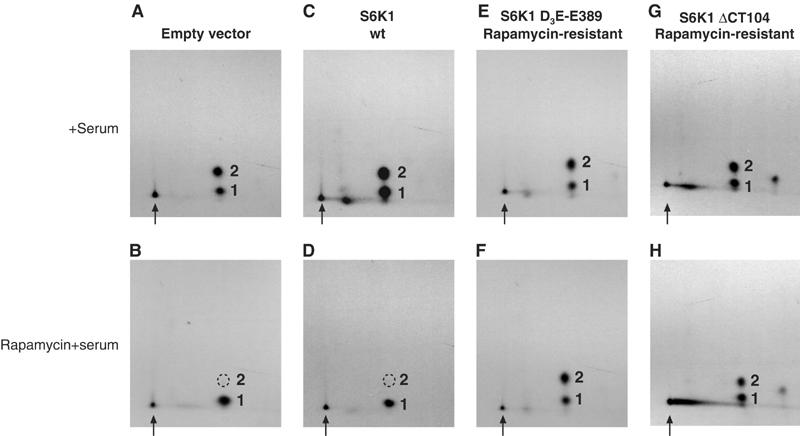
S6K1 mutants confer rapamycin resistance to eIF4B phosphorylation in vivo. Wt Flag-eIF4B was cotransfected into 293 cells with empty vector (A, B), wt S6K1 (C, D) or two different rapamycin-resistant S6K1 proteins (E–H), as indicated. Cells were starved of serum for 36 h, and then stimulated with serum for 30 min (A, C, E, G), or pretreated for 30 min with rapamycin (B, D, F, H) prior to serum stimulation. Flag-eIF4B was isolated and subjected to phosphopeptide mapping, as above.
A Ser422 substitution interferes with eIF4B activity in vivo
To determine whether Ser422 phosphorylation affects the ability of eIF4B to modulate translation initiation, 293T cells were transiently transfected to overexpress various mutant forms of Flag-tagged eIF4B with substitutions at Ser422 designed to prevent (Ser422Ala) or mimic (Ser422Glu) phosphorylation at this residue. Cells were cotransfected with vectors coding for secretable growth hormone (GH). As shown in Figure 7A, overexpression of wt eIF4B resulted in a notable inhibition of GH protein synthesis (compare lanes 1 and 4, lower panel). Inhibition of protein synthesis in vivo by high levels of eIF4B has been reported previously (Milburn et al, 1990), and is also observed when exogenous eIF4B is added to a reticulocyte lysate (data not shown). When the eIF4B Ser422Glu mutant was overexpressed, a similar extent of inhibition of GH synthesis was observed (lane 3, lower panel). However, when the nonphosphorylatable Ser422Ala mutant was overexpressed, the inhibition of GH synthesis was significantly attenuated (lane 2, lower panel), even though comparable amounts of eIF4B protein were produced (middle panel). Similar results were obtained with a reporter construct expressing PABP (results not shown).
Figure 7.
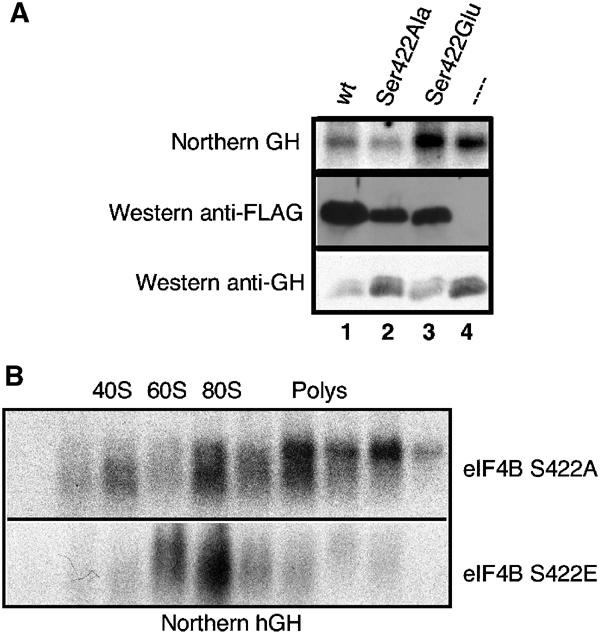
Ser422 phosphorylation (or phosphorylation mimic) is required to inhibit translation. 293T cells were cotransfected with a reporter construct encoding human GH and various Flag eIF4B constructs. (A; top) Northern blot of total RNA probed with a human GH DNA segment. (Middle panel) Western analysis using the anti-Flag antibody. (Bottom panel) Western analysis using an anti-hGH antiserum. (B) Polysome profile analysis of human GH mRNA from cells transiently transfected with pS16-GH and Flag-eIF4B S422A (top panel) or eIF4B S422E (bottom panel). Locations in the gradient of 40S, 60S and 80S ribosomes, and polysomes are indicated.
Diminished yields of GH protein observed with the wt and Ser422Glu eIF4B proteins were due to an inhibition in the initiation phase of protein synthesis, as demonstrated by polysome analysis of GH mRNA (Figure 7B): GH mRNA was found in heavy polysomes in cells expressing the Ser422Ala mutant protein (upper panel), whereas cells expressing the Ser422Glu variant (lower panel) exhibited only modest amounts of GH mRNA in the polysome region of the gradient. Thus, a nonphosphorylatable eIF4B mutant effected a significantly lower level of translation inhibition on two different reporter mRNAs than did the wt and phosphoserine mimic (Ser422Glu) proteins, suggesting that phosphorylation of Ser422 plays an important role in eIF4B function.
Discussion
In the present study, using phosphopeptide mapping and a phosphospecific antiserum, we have unambiguously identified a serum- and mitogen-responsive phosphorylation site in the eIF4B, Ser422. Treatment of cells with pharmacological inhibitors of the PI3K and mTOR signaling modules inhibited Ser422 phosphorylation, and S6K1/2 were demonstrated to phosphorylate Ser422 in vitro. Ser422 phosphorylation is sensitive to leucine deprivation, and expression of rapamycin-resistant S6K1 proteins conferred rapamycin resistance upon Ser422 phosphorylation. Thus, the S6Ks appear to be the primary effectors of eIF4B Ser422 phosphorylation. Finally, substitution of Ser422 for Ala to create a nonphosphorylatable eIF4B protein results in a loss of activity in an in vivo translation inhibition assay, suggesting that phosphorylation of this site plays an important role in eIF4B function.
eIF4B is an avid RNA-binding protein, possessing two sequence-nonspecific RNA-binding domains: an RNA recognition motif (RRM) near the N-terminus, and an arginine-rich motif (ARM) in the C-terminal region (Methot et al, 1994; Naranda et al, 1994). The ARM binds RNA more strongly than the RRM, and is essential for eIF4A helicase-promoting activity (Methot et al, 1994). Ser422 lies at the C-terminal end of the ARM. While it is tempting to postulate that phosphorylation of Ser422 may affect RNA binding, recombinant proteins harboring various Ser422 mutations do not show significant changes in RNA-binding affinity in vitro (F Peiretti, unpublished results).
It has been extremely challenging to study eIF4B function in vivo—overexpression of eIF4B inhibits translation, and depletion of eIF4B via RNAi elicits cell death (B Raught, A-C Gingras and D Shahbazian, unpublished observations). In addition, expression of any recombinant protein in vivo has the potential to quench or otherwise interfere with the normal intracellular signaling processes modulating the activity of the endogenous protein. Nevertheless, we were able to obtain evidence that Ser422 phosphorylation is biologically relevant: while a significant inhibition of reporter mRNA translation was achieved in response to overexpression of the wt or Ser422Glu eIF4B proteins, a nonphosphorylatable eIF4B mutant, Ser422Ala, had little or no effect on the translation of the two reporter mRNAs. Similar data were observed for reporter constructs possessing relatively structured or unstructured 5′UTRs. Although this experimental protocol does not mimic the normal behavior of eIF4B in vivo (and we cannot rule out subtle effects of the single amino-acid substitution on protein folding), Ser422 phosphorylation does appear to be critical for the inhibition of translation initiation observed in this type of assay.
eIF4B does not possess detectable catalytic activity (Grifo et al, 1984), but significantly enhances the affinity of the DEAD box helicase eIF4A for both ATP and mRNA (Rogers et al, 1999). eIF4B also increases eIF4A processivity (Rogers et al, 2001), thereby enhancing the ability of eIF4F to melt cap-proximal mRNA secondary structure. mRNA 5′UTR unwinding is likely a critical regulatory step in translation. Whereas global changes in protein synthesis following mitogenic stimulation are modest (∼2-fold), a dramatic increase in the translation rates of a relatively small number of mRNAs is observed. This phenomenon has been termed ‘translational discrimination' (Lodish, 1976). One explanation to account for translational discrimination is that differences in the degree of 5′UTR secondary structure lead to a significant disparity in the requirement of individual mRNAs for eIF4F helicase activity. According to this model, when eIF4F helicase activity is low, translation of mRNAs that do not possess significant 5′UTR secondary structure would be affected to a lesser extent than those possessing more extensive secondary structure. This prediction has been borne out in several different ways: (a) The degree of 5′UTR secondary structure correlates with the requirement for eIF4A activity; that is, the translation of mRNAs possessing a higher degree of 5′UTR secondary structure is more sensitive to inhibition by an eIF4A dominant-negative mutant than comparable mRNAs with less secondary structure (Svitkin et al, 2001). (b) In Saccharomyces cerevisiae lacking eIF4B, the translation of reporter mRNAs possessing structured 5′UTRs is preferentially inhibited (Altmann et al, 1993). (c) eIF4E overexpression preferentially enhances the translation of mRNAs with structured 5′UTRs (Koromilas et al, 1992). (d) Whereas insulin stimulation increases global translation rates ∼2-fold, the increase in translation of the ornithine decarboxylase (ODC) mRNA, which possesses a long, structured 5′UTR, is 20- to 50-fold (Manzella et al, 1991). The ODC translational block is relieved by removing structured 5′UTR elements (Manzella and Blackshear, 1990), or by overexpression of eIF4E (Shantz et al, 1996). (e) Finally, a recent study established that eIF4B is absolutely required for the formation of 48S translation initiation complexes on 5′UTRs with even moderate secondary structure (Dmitriev et al, 2003). Interestingly, recombinant protein substituted only poorly for purified endogenous eIF4B in this assay, suggesting that a post-translational modification is required for full activity.
Many mRNAs coding for proteins involved in cell cycle progression possess long, putatively structured 5′UTRs (Kozak, 1987), and are translated inefficiently. In response to various types of extracellular stimuli, translation of these mRNAs is dramatically enhanced. We propose that one component of this modulation may occur via eIF4B phosphorylation, to enhance the processivity of the eIF4F helicase. Further study will be required to validate this model.
Materials and methods
Constructs
For expression in mammalian cells, the coding sequence of human eIF4B was amplified by PCR and subcloned in-frame into the pcDNA3-Flag vector (a kind gift of Dr S Morino), generating an N-terminal fusion protein. Ser406 and Ser422 point mutants were generated by PCR mutagenesis, and inserted in-frame into pcDNA3-Flag. All inserts were sequenced. Wt and mutant eIF4B coding regions in pcDNA3-Flag were transferred into pGEX-6p1 for expression in bacteria. To generate His6-tagged human eIF4B, the eIF4B coding region was PCR amplified from pGEM3-4B (Milburn et al, 1990) and ligated into pET28c (Novagen), to yield pET-H4B. The pRK5 S6K1 constructs (a kind gift of Dr G Thomas) have been described (Pearson et al, 1995).
Cell culture/transfection
293, 293T and HeLa cells were obtained from ATCC. Cell culture and [32P]orthophosphate metabolic labeling were carried out as described (Gingras et al, 1998). Pharmacologic inhibitors were purchased from Calbiochem. Cells were transfected using Lipofectamine Plus (Invitrogen), according to the manufacturer's instructions. For identification of the in vivo eIF4B phosphorylation site, 10 μg of the Flag-eIF4B constructs was transfected into one 100 cm2 plate of 80% confluent 293T cells. For eIF4B/S6K1 cotransfection studies, 10 μg of each of the empty pRK5 vector or S6K1 construct was cotransfected with 2 μg of pcDNA3 Flag-eIF4B into a 100 cm2 plate of 80% confluent 293 cells.
Antibodies/immunoprecipitation/Western blotting
Endogenous eIF4B was immunoprecipitated with a previously characterized rabbit antiserum (Figures 1 and 2; Methot et al, 1996). For Figures 1 and 2, immunoprecipitation was conducted as in Raught et al (2000a). Flag-tagged proteins were immunoprecipitated with anti-Flag–sepharose (Sigma). Immunoprecipitation of myc-tagged S6K proteins, and kinase assay were conducted as in Dennis et al (1996). The phosphospecific S6K1 Thr389 and ribosomal protein S6 Ser235/236 antisera were developed at Cell Signaling Technology (Beverly, MA), and used as per the manufacturer's instructions. The antibody directed against mTOR was a kind gift of Dr RT Abraham. Phospho-eIF4B(Ser422) polyclonal antibodies were produced by immunizing rabbits with a synthetic KLH-coupled phosphopeptide (CERSRTGpSESSQT), as previously described (Weng et al, 1998). Immunoglobulin was first purified using protein A–sepharose, and then subjected to stepwise peptide affinity chromatography to obtain the phosphospecific antibodies.
Phosphorylation studies
Recombinant His6-tagged eIF4B fragments were purified with the Ni-NTA Spin Kit (Qiagen), and phosphorylated in vitro by S6K1, as described (Flotow and Thomas, 1992). Tryptic mapping of eIF4B was conducted essentially as described for eIF4GI (Raught et al, 2000a), except that plastic-backed Machery-Nagel plates were used. For in vivo studies, HEK 293 cells were starved of serum for 36 h, and then metabolically labeled with [32P]orthophosphate for 3 h. Labeled cells were treated with 10% FBS for 30 min, lysed, and eIF4B was immunoprecipitated with a previously characterized polyclonal antiserum (Methot et al, 1994). Immunoprecipitated material was subjected to SDS–PAGE and transferred to a nitrocellulose membrane. Following autoradiography, the region of the membrane harboring 32P-labeled eIF4B was excised. Membrane pieces were subjected to tryptic digestion, and liberated phosphopeptides were visualized by two-dimensional phosphopeptide mapping as in Gingras et al (1999a).
Acknowledgments
We thank R Duncan and B Fabbri for early experiments on eIF4B phosphorylation, RT Abraham for mTOR antiserum, N Méthot for eIF4B antiserum and constructs, S Morino for the pcDNA3-Flag vector, G Thomas for the S6K constructs, and S MacMillan and C Lister for invaluable technical assistance. BR was supported by a fellowship from the Canadian Institutes for Health Research (CIHR). FP was supported by a fellowship from the Fondation pour la Recherche Médicale of France. A-CG was a recipient of a CIHR studentship. Work in NS's laboratory was funded by grants from the CIHR and the Howard Hughes Medical Institute. Work in JWBH's laboratory was funded by NIH grant GM22135.
References
- Altmann M, Muller PP, Wittmer B, Ruchti F, Lanker S, Trachsel H (1993) A Saccharomyces cerevisiae homologue of mammalian translation initiation factor 4B contributes to RNA helicase activity. EMBO J 12: 3997–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony JC, Anthony TG, Kimball SR, Vary TC, Jefferson LS (2000) Orally administered leucine stimulates protein synthesis in skeletal muscle of postabsorptive rats in association with increased eIF4F formation. J Nutr 130: 139–145 [DOI] [PubMed] [Google Scholar]
- Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN (1996) TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell 7: 25–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benne R, Hershey JW (1978) The mechanism of action of protein synthesis initiation factors from rabbit reticulocytes. J Biol Chem 253: 3078–3087 [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JCJ, Abraham RT (1996) Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J 15: 5256–5267 [PMC free article] [PubMed] [Google Scholar]
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655–1657 [DOI] [PubMed] [Google Scholar]
- Cheatham I, Monfar M, Chou M, Blenis J (1995) Structural and functional analysis of pp70S6K. Proc Natl Acad Sci USA 92: 11696–11700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J (1992) Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69: 1227–1236 [DOI] [PubMed] [Google Scholar]
- Dennis PB, Pullen N, Kozma SC, Thomas G (1996) The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol Cell Biol 16: 6242–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PB, Pullen N, Pearson RB, Kozma SC, Thomas G (1998) Phosphorylation sites in the autoinhibitory domain participate in p70(s6k) activation loop phosphorylation. J Biol Chem 273: 14845–14852 [DOI] [PubMed] [Google Scholar]
- Dennis PB, Thomas G (2002) Quick guide: target of rapamycin. Curr Biol 12: R269. [DOI] [PubMed] [Google Scholar]
- Dmitriev S, Terenin I, Dunaevsky Y, Merrick W, Shatsky IN (2003) Assembly of 48S translation initiation complexes from purified components with mRNAs that have some base pairing within their 5′ untranslated regions. Mol Cell Biol 23: 8925–8933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R, Hershey JW (1984) Heat shock-induced translational alterations in HeLa cells. Initiation factor modifications and the inhibition of translation. J Biol Chem 259: 11882–11889 [PubMed] [Google Scholar]
- Duncan R, Hershey JW (1985) Regulation of initiation factors during translational repression caused by serum depletion. Covalent modification. J Biol Chem 260: 5493–5497 [PubMed] [Google Scholar]
- Duncan RF, Hershey JW (1987) Initiation factor protein modifications and inhibition of protein synthesis. Mol Cell Biol 7: 1293–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H, Thomas G (1992) Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J Biol Chem 267: 3074–3078 [PubMed] [Google Scholar]
- Fumagalli S, Thomas G (2000) S6 phosphorylation and signal transduction. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews M (eds) pp 695–718. Plainview, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Gingras A-C, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N (1999a) Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13: 1422–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A-C, Kennedy SG, O'Leary MA, Sonenberg N, Hay N (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 12: 502–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A-C, Raught B, Sonenberg N (1999b) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68: 913–963 [DOI] [PubMed] [Google Scholar]
- Grifo JA, Abramson RD, Satler CA, Merrick WC (1984) RNA-stimulated ATPase activity of eukaryotic initiation factors. J Biol Chem 259: 8648–8654 [PubMed] [Google Scholar]
- Hershey JWB, Merrick WC (2000) Pathway and mechanism of initiation of protein synthesis. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Merrick WC (eds) pp 33–88. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Jenö P, Ballou LM, Novak-Hofer I, Thomas G (1988) Identification and characterization of a mitogenic-activated S6 kinase. Proc Natl Acad Sci USA 85: 406–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR, Jefferson LS (2000) Regulation of translation initiation in mammalian cells by amino acids. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds) pp 561–580. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Koromilas AE, Lazaris-Karatzas A, Sonenberg N (1992) mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E [published erratum appears in EMBO J 1992 Dec;11(13):5138]. EMBO J 11: 4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 15: 8125–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson TG, Lee KA, Maimone MM, Abramson RD, Dever TE, Merrick WC, Thach RE (1989) Dissociation of double-stranded polynucleotide helical structures by eukaryotic initiation factors, as revealed by a novel assay. Biochemistry 28: 4729–4734 [DOI] [PubMed] [Google Scholar]
- Lodish HF (1976) Translational control of protein synthesis. Annu Rev Biochem 45: 39–72 [DOI] [PubMed] [Google Scholar]
- Manzella JM, Blackshear PJ (1990) Regulation of rat ornithine decarboxylase mRNA translation by its 5′-untranslated region. J Biol Chem 265: 11817–11822 [PubMed] [Google Scholar]
- Manzella JM, Rychlik W, Rhoads RE, Hershey JW, Blackshear PJ (1991) Insulin induction of ornithine decarboxylase. Importance of mRNA secondary structure and phosphorylation of eucaryotic initiation factors eIF-4B and eIF-4E. J Biol Chem 266: 2383–2389 [PubMed] [Google Scholar]
- Methot N, Pause A, Hershey J, Sonenberg N (1994) The translation initiation factor eIF-4B contains an RNA-binding region that is distinct and independent from its ribonucleoprotein consensus sequence. Mol Cell Biol 14: 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot N, Song M, Sonenberg N (1996) A region rich in aspartic acid, arginine, tyrosine, and glycine (DRYG) mediates eukaryotic initiation factor 4B (eIF4B) self-association and interaction with eIF3. Mol Cell Biol 16: 5328–5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milburn S, Hershey J, Davies M, Kelleher K, Kaufman R (1990) Cloning and expression of eukaryotic initiation factor 4B cDNA: sequence determination identifies a common RNA recognition motif. EMBO J 9: 2783–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G (1999) Drosophila S6 kinase: a regulator of cell size [see comments]. Science 285: 2126–2129 [DOI] [PubMed] [Google Scholar]
- Morino S, Imataka H, Svitkin YV, Pestova TV, Sonenberg N (2000) Eukaryotic translation initiation factor 4E (eIF4E) binding site and the middle one-third of eIF4GI constitute the core domain for cap-dependent translation, and the C-terminal one-third functions as a modulatory region. Mol Cell Biol 20: 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranda T, Strong WB, Menaya J, Fabbri BJ, Hershey JW (1994) Two structural domains of initiation factor eIF-4B are involved in binding to RNA. J Biol Chem 269: 14465–14472 [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E (2000) Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 14: 2689–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RB, Dennis PB, Han J-W, Williamson NA, Kozma SC, Wettenhall REH, Thomas G (1995) The principal target of rapamycin-induced p70S6K inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J 14: 5279–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, Klumperman J, Thorens B, Thomas G (2000) Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 408: 994–997 [DOI] [PubMed] [Google Scholar]
- Pestova TV, Hellen CU, Shatsky IN (1996) Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol 16: 6859–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B, Gingras A-C, Gygi SP, Imataka H, Morino S, Gradi A, Aebersold R, Sonenberg N (2000a) Serum-stimulated, rapamycin sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J 19: 434–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B, Gingras A-C, Sonenberg N (2000b) Regulation of ribosomal recruitment in eukaryotes. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds) pp 245–294. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Raught B, Gingras AC, Sonenberg N (2001) The target of rapamycin (TOR) proteins. Proc Natl Acad Sci USA 98: 7037–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GW, Richter NJ, Lima WF, Merrick WC (2001) Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J Biol Chem 276: 30914–30922 [DOI] [PubMed] [Google Scholar]
- Rogers J, GW, Richter NJ, Merrick WC (1999) Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J Biol Chem 274: 12236–12244 [DOI] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN (2000) TOR, a central controller of cell growth. Cell 103: 253–262 [DOI] [PubMed] [Google Scholar]
- Shantz LM, Hu RH, Pegg AE (1996) Regulation of ornithine decarboxylase in a transformed cell line that overexpresses translation initiation factor eIF-4E. Cancer Res 56: 3265–3269 [PubMed] [Google Scholar]
- Shigemitsu K, Tsujishita Y, Hara K, Nanahoshi M, Avruch J, Yonezawa K (1999) Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J Biol Chem 274: 1058–1065 [DOI] [PubMed] [Google Scholar]
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC (1998) Disruption of the p70S6k/p85S6k gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 17: 6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell GW, Belsham GJ, Sonenberg N (2001) The requirement for eukaryotic initiation factor 4A (eIF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7: 382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachsel H, Erni B, Schreier MH, Staehelin T (1977) Initiation of mammalian protein synthesis. II. The assembly of the initiation complex with purified initiation factors. J Mol Biol 116: 755–767 [DOI] [PubMed] [Google Scholar]
- van der Geer P, Luo K, Sefton BM, Hunter T (1994) Phosphopeptide mapping and phosphoamino acid analysis on cellulose thin-layer plates. In Cell Biology: A Laboratory Handbook, Celis JE (ed) Vol. 3, pp 422–448. Academic Press: San Diego, CA [Google Scholar]
- Weng Q-P, Andrabi K, Kozlowski MT, Grove JR, Avruch J (1995) Multiple independent inputs are required for activation of the p70 S6 kinase. Mol Cell Biol 15: 2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng QP, Kozlowski M, Belham C, Zhang A, Comb MJ, Avruch J (1998) Regulation of the p70 S6 kinase by phosphorylation in vivo. Analysis using site-specific anti-phosphopeptide antibodies. J Biol Chem 273: 16621–16629 [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock J, Ng J, Reinhard C, Neufeld T (2000) Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 14: 2712–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]