

Abstract
Dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel causes cystic fibrosis, while inappropriate activity of this channel occurs in secretory diarrhea and polycystic kidney disease. Drugs that interact directly with CFTR are therefore of interest in the treatment of a number of disease states. This review focuses on one class of small molecules that interacts directly with CFTR, namely inhibitors that act by directly blocking chloride movement through the open channel pore. In theory such compounds could be of use in the treatment of diarrhea and polycystic kidney disease, however in practice all known substances acting by this mechanism to inhibit CFTR function lack either the potency or specificity for in vivo use. Nevertheless, this theoretical pharmacological usefulness set the scene for the development of more potent, specific CFTR inhibitors. Biophysically, open channel blockers have proven most useful as experimental probes of the structure and function of the CFTR chloride channel pore. Most importantly, the use of these blockers has been fundamental in developing a functional model of the pore that includes a wide inner vestibule that uses positively charged amino acid side chains to attract both permeant and blocking anions from the cell cytoplasm. CFTR channels are also subject to this kind of blocking action by endogenous anions present in the cell cytoplasm, and recently this blocking effect has been suggested to play a role in the physiological control of CFTR channel function, in particular as a novel mechanism linking CFTR function dynamically to the composition of epithelial cell secretions. It has also been suggested that future drugs could target this same pathway as a way of pharmacologically increasing CFTR activity in cystic fibrosis. Studying open channel blockers and their mechanisms of action has resulted in significant advances in our understanding of CFTR as a pharmacological target in disease states, of CFTR channel structure and function, and of how CFTR activity is controlled by its local environment.
Keywords: Cystic fibrosis, Cystic fibrosis transmembrane conductance regulator, Chloride channel, Open channel block, Channel pore, Permeation, Anion secretion, Potentiators
Core Tip: This review summarizes our understanding of small molecules that inhibit the cystic fibrosis transmembrane conductance regulator (CFTR) by blocking the channel pore. It describes how such inhibitors could be used in the treatment of diarrhea and hereditary kidney disease; how studying these inhibitors’ mechanisms of action has led to advances in our understanding of CFTR channel structure and function; and how substances acting via this mechanism could contribute to the physiological control of CFTR function in epithelial cells. Ironically, studying channel inhibitors has recently led to the discovery of a new class of CFTR potentiators that could be used to treat cystic fibrosis.
INTRODUCTION
Cystic fibrosis (CF) is the most common fatal autosomal recessive disease affecting Caucasians, with around 80000 CF sufferers in the world today. CF is caused by mutations that cause loss of function of the CF transmembrane conductance regulator (CFTR) protein[1]. Over 1900 different mutations that affect the transcription, synthesis, trafficking, turnover, or function of CFTR have been shown to cause CF. CFTR is expressed in the apical membrane of many different epithelial tissues, where it plays a central role in epithelial Cl-, HCO3-, and fluid transport[2]. As a consequence, CF is associated with respiratory, pancreatic, gastrointestinal, and reproductive disease that results from deficient salt and fluid secretion in these epithelia[1,3]. Conversely, inappropriately elevated CFTR function results in excessive intestinal fluid secretion in secretory diarrhoeas such as that associated with cholera[4]. CFTR-mediated Cl- transport by renal epithelial cells also underlies fluid accumulation and growth of renal cysts in autosomal dominant polycystic kidney disease (ADPKD), the most common hereditary kidney disease[5]. The involvement of CFTR in such common and serious disease states makes it an attractive target for therapeutic intervention. Many different small molecules interact directly with the CFTR protein, and these have proven useful experimental tools. The therapeutic potential of drugs that act directly with CFTR is also receiving increasing interest. This review focuses on one class of small molecules interacting with CFTR-those that directly block Cl- movement through the open channel pore.
OVERVIEW OF CFTR ARCHITECTURE
CFTR is a member of a large family of membrane proteins, the adenosine triphosphate (ATP)-binding cassette (ABC) proteins, most members of which function as active transport ATPases[6,7]. CFTR appears to be unique within the ABC family in functioning instead as an ATP-dependent Cl- channel[8]. The structure and function of CFTR has been reviewed in detail recently[8-12] and will be described only briefly here. In common with other ABC proteins, CFTR has a modular architecture, consisting of two membrane-spanning domains (MSDs) each comprising six transmembrane α-helices (TMs) (Figure 1). Each MSD is followed by a cytoplasmic nucleotide binding domain (NBD), and the two MSD-NBD modules are joined by a cytoplasmic regulatory domain (R domain) that is unique to CFTR. The modular architecture of CFTR also corresponds with its defining functional features. The R domain contains multiple consensus phosphorylation sites for protein kinase A and protein kinase C, allowing the channel to be regulated physiologically by hormones that act through these protein kinases. Phosphorylation of the R domain is a prerequisite for channel activity. Following R domain phosphorylation, CFTR channel gating (opening and closing) is controlled by ATP binding and hydrolysis at a dimer of the two NBDs. The NBDs also make physical contact with the long intracellular loops (ICLs) that join individual TMs (Figure 1). The channel pore that forms the transmembrane pathway for the movement of Cl- ions is formed by a pseudo-symmetrical arrangement of the two MSDs. Recent evidence suggests that the ICLs form a functional link that allows a conformational rearrangement initiated by ATP interaction with the NBDs to be transmitted to the TMs, controlling the opening and closing the channel pore.
Figure 1.
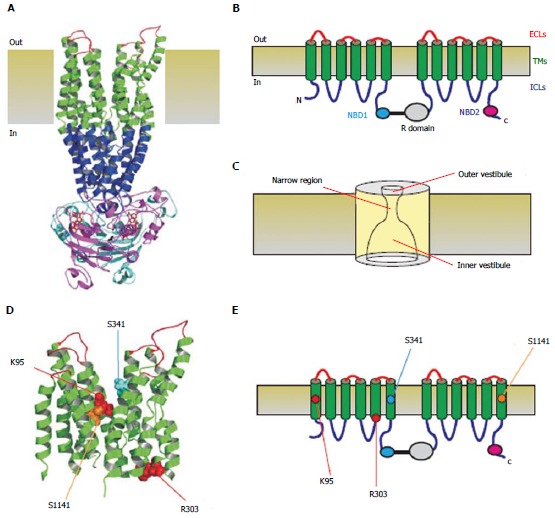
Three-dimensional and two-dimensional representations of cystic fibrosis transmembrane conductance regulator structure. A: Atomic homology model of cystic fibrosis transmembrane conductance regulator in a so-called “channel like” conformation[20]. Different colours are used to illustrate the approximate extent of the extracellular loops (ECLs, red), transmembrane domains (TMs, green), intracellular loops (ICLs, blue), and two nucleotide binding domains (NBD1, cyan; NBD2, magenta). The cytoplasmic R domain is not included in this homology model; B: Schematic representation of these different domains (and the R domain), using the same colour scheme; C: Functional model of pore architecture. As described in the text, experimental evidence suggests that the pore has a narrow region that is connected to the cytoplasmic and extracellular solutions by a wide inner vestibule and a narrower outer vestibule, respectively; D: Location of putative blocker-interacting residues in the TMs (K95-TM1; R303-TM5; S341-TM6; S1141-TM12) within the same homology model shown in A. E: Location of these same residues in the same schematic model shown in B.
The channel pore itself has been studied using a combination of structural[10,13], functional[8,14], substituted cysteine accessibility[8,15,16] and molecular modeling[17-21] approaches. A simple model of the proposed overall functional architecture of the pore is shown in Figure 1C. The pore is thought to have a relatively narrow region over which discrimination between different anions is predominantly determined. This region is flanked by outer and inner vestibules, with functional evidence suggesting that the inner vestibule is both deeper and wider. Of the 12 TMs (Figure 1), TM6 appears to play a dominant role in determining functional interactions between the narrow pore region and permeating anions[15,22]. TM1, TM6, TM11 and TM12 all contribute to the inner vestibule[15,23-29], while TM1, TM6, TM11, TM12, and the extracellular loops (ECLs) adjacent to these TMs contribute to the outer vestibule[16,30-33]. As described in detail below (see “Biophysical Relevance”), residues from TM1 (K95), TM5 (R303), TM6 (S341) and TM12 (S1141) have all been proposed to interact with CFTR open channel blockers (Figure 1D and E).
CFTR CHANNEL BLOCKERS
The first kinds of CFTR inhibitors to be identified were those that act as open channel blockers[34,35] (Figure 2). These are substances that enter into the open channel pore and physically occlude it, temporarily preventing the flow of Cl- ions until the blocker molecule dissociates from the pore. Many diverse substances share this mechanism of CFTR channel block, the best known (and best studied) of which are sulfonylureas such as glibenclamide[36-42] and related substances[36,42-44], arylaminobenzoates such as 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) and diphenylamine-2-carboxylate[23,45-48], and disulfonic stilbenes such as 4,4’-diisothiocyanostilbene-2,2’-disulfonic acid and 4,4’-dinitrostilbene-2,2’-disulfonic acid (DNDS)[49]. Detailed biophysical analysis of the blocking effects of these groups of negatively charged substances reveal a number of common features that may reflect a common mechanism of action. In each case the blocker enters the pore only from its cytoplasmic end to reach its binding site inside the channel pore (Figure 2); block is voltage-dependent, being stronger at more hyperpolarized voltages that favour entry of negatively charged substances into the pore from its cytoplasmic end; and block is sensitive to the extracellular Cl- concentration, being stronger at low Cl- and weaker at higher Cl-. Each of these defining features tells us something about the mechanism of inhibition and the location of the blocker binding site. Inhibition from the cytoplasmic side of the membrane was originally used to suggest that the open CFTR channel pore is structurally asymmetric, with a wide inner vestibule that is easily accessible from the cytoplasm[35,49], and a narrower extracellular entrance that prevents the entry of large substances from the extracellular solution (Figure 2). Voltage-dependent block suggests that the blocker binding site is located within the transmembrane electric field, such that the blocker apparently experiences at least part (generally about 20%-50%) of this electric field as it moves between the cytoplasm and its binding site inside the pore. While the relationship between distance across the transmembrane electric field and physical distance across the membrane itself is neither direct nor straightforward, this voltage-dependence is consistent with the blocker moving into the membrane-spanning parts of CFTR to access the blocker binding site. Finally, sensitivity of block to the extracellular Cl- concentration is usually ascribed to repulsive electrostatic interactions between Cl- and the negatively charged blocker molecule that take place when both are bound simultaneously within the open channel pore.
Figure 2.
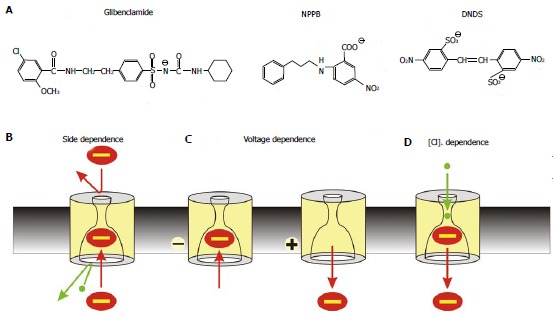
Mechanism of open channel blocker action. A: Chemical structures of three well-known voltage-dependent cystic fibrosis transmembrane conductance regulator (CFTR) channel blockers: the sulfonylurea glibenclamide, the aryl amino benzoate 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), and the disulfonic stilbene 4,4’-dinitrostilbene-2,2’-disulfonic acid (DNDS). B-D: Characteristic functional properties of block shared by these and other CFTR open channel blockers: block is side-dependent, voltage-dependent, and sensitive to extracellular Cl- concentration; B: Blockers enter the pore only from its cytoplasmic end, likely because the extracellular entrance to the pore is too small and/or the narrow pore region prevents them from accessing their binding site in the wide inner vestibule; C: Block is relatively strengthened at hyperpolarized membrane potentials that favour entry of negative substances into the pore from the cytoplasm, and weakened at depolarized membrane potentials that favour anion retention in the cytoplasm; D: Block is weakened at higher extracellular Cl- concentrations; this is usually ascribed to a “knock-off” mechanism whereby Cl- entering the pore from its extracellular end electrostatically repels negatively charged blockers back into the cytoplasm, destabilizing blocker binding inside the pore.
Many or all of these features of the blocking reaction have been observed with other, unrelated blocking anions, including substrates of related ABC proteins[50,51] such as the conjugated bile salt taurolithocholate-3-sulfate (TLCS) and the conjugated steroid β-estradiol 17-(β-d-glucuronide), indazole compounds such as lonidamine[52,53], short-chain fatty acids butyrate and 4-phenylbutyrate[54], the fluorescein derivative phloxine B[55], and even commonly used experimental compounds such as the pH buffer 3-(4-morpholino) propane sulfonic acid[56] and the negatively charged cysteine-reactive reagent (2-sulfatoethyl) methanesulfonate (MTSES)[57]. Together these open channel blocking substances represent a large and structurally diverse group of organic anions, suggesting that entry of anions into the CFTR Cl- channel pore from its cytoplasmic end is a process that shows little specificity or size discrimination. Furthermore, most of these blockers show relatively low potency (dissociation constants usually in the micromolar to millimolar range, depending on voltage). At the single channel level, these blockers may cause discrete “flickers” in the open channel current (due to resolved individual blocking events)[37,38,44,45,47,49,51,52], or an apparent reduction in single channel current amplitude where blocking and unblocking are too fast to be resolved at the bandwidth used for patch clamp recording[44,48,50,54,56-58]. These effects reflect kinetically “intermediate” and “fast” blocking and unblocking reactions, respectively, according to the scheme proposed by Hille[59]. The low apparent affinity of CFTR open channel blockers limits their potential for use in vivo. Furthermore, many of these substances also block other classes of Cl- channels[34,60], perhaps reflecting some structural similarity amongst Cl- channel pores that results in similar sensitivity to block by organic anions[14].
In more recent years, high-throughput screening technologies have been used to identify more potent CFTR inhibitors[61]. The thiazolidinone CFTRinh-172 inhibits CFTR from the cytoplasmic side of the membrane at sub-micromolar concentrations[62], due to a voltage-independent effect on channel gating[63,64]. Glycine hydrazides such as GlyH-101 cause voltage-dependent block from the extracellular side of the membrane at low micromolar concentrations[65]. These substances appear to inhibit CFTR by different mechanisms than that described above and in Figure 2 for intracellular open channel blockers. CFTRinh-172 has been shown to bind preferentially to open channels, perhaps triggering a conformational change to an “inactivated” nonconducting state[64]. GlyH-101 does appear to act as an open channel blocker, however acting from the extracellular side of the membrane[65], perhaps becoming lodged close to the narrow pore region to occlude Cl- permeation[66]. These two potent and relatively selective inhibitors have become drugs of choice for experimental inhibition of CFTR activity; because of their different sidedness of action, CFTRinh-172 is preferred when applied to the intracellular side of the membrane, and GlyH-101 for extracellular application.
Finally, a 3.7 kDa peptide toxin isolated from scorpion venom and named GaTx1 inhibits CFTR channels from the cytoplasmic side of the membrane at sub-micromolar concentrations[67]. Although the molecular mechanism of GaTx1 inhibition is not well defined, this substance has been described as acting as a non-competitive inhibitor of channel gating[67,68], with no demonstrated open channel blocking action. Currently GaTx1 is the only known peptide inhibitor of CFTR.
PHARMACOLOGICAL RELEVANCE
Because of the inviolable relationship between loss of CFTR function and CF, there is tremendous current interest in the identification and development of small molecules that directly interact with the CFTR protein to increase its function (known as CFTR “potentiators”)[1,69-72]. On the other hand, it has long been suggested that CFTR channel blockers could (at least in theory) be used in the treatment of secretory diarrhoea and ADPKD[35,73]. CFTR inhibitors have also been suggested as potential male contraceptives[53,74]. As described above, known intracellular-active open channel blockers lack either the potency or the specificity for in vivo use. However, the higher affintiy CFTR inhibitors CFTRinh-172 and GlyH-101 have been shown to be effective in in vitro and in vivo models of secretory diarrhea[62,65,75]. Moreover, non-absorbable lectin conjugated forms of GlyH-101 were active against cholera-induced fluid secretion and mortality in mice when administered orally[76]. Similarly, CFTRinh-172 and GlyH-101 (or closely related substances) have been shown to retard cyst growth in in vitro[77,78] and in vivo[78] models of ADPKD. The therapeutic potential of potent and specific CFTR inhibitors has been discussed in several recent reviews[61,79,80].
BIOPHYSICAL RELEVANCE
Since open channel blockers bind to specific sites within the channel pore with relatively high affinity (compared to Cl- and other permeant anions), they have proven invaluable probes of the structure and function of the Cl- permeation pathway. Mutations in TM6 and TM12 have been shown to alter the affinity of block by arylaminobenzoates[23,46], sulfonylureas[42,81] and lonidamine[52], consistent with functional evidence[25-28,82] and molecular models[17-21] that suggest that these two TMs make substantial contributions to the inner vestibule of the pore where the blocker binding site is thought to reside (Figures 1 and 2). Because open channel blockers are anions, and because positively charged amino acid side chains in the CFTR channel pore are known to play important roles in electrostatic attraction of Cl- ions[24,30,31,82,83], much attention has also been placed on the role of such fixed positive charges in interactions with blockers. In particular, mutations that remove the positive charge at lysine residue K95 in TM1 (Figure 1D and E) dramatically reduce the channel blocking affinity of glibenclamide, DNDS, lonidamine, NPPB and TLCS[24,82]. This finding suggests that these structurally diverse open channel blockers share a common molecular mechanism of block - they are attracted into the wide inner vestibule by electrostatic attraction between the negative charge on the blocker molecule and the positive charge on the lysine side chain at K95, and once in the inner vestibule they bind tightly enough to occlude the pore and temporarily prevent Cl- permeation (Figure 3). This model of blocker binding in the pore inner vestibule is also supported by a recent in silico investigation of blocker docking inside the pore of an atomic homology model of CFTR[20]. Neutralization of fixed positive charge in the inner vestibule by mutagenesis of K95 also decreases single channel Cl- conductance by about 85%[22,29,82], suggesting that this positive charge also plays an important role in the normal Cl- permeation mechanism, most likely due to electrostatic attraction of Cl- ions. The functional importance of the positive charge on the side chain of K95 may explain the sensitivity of CFTR to broad range of intracellular anionic blockers: a positive charge in the inner vestibule is necessary to attract Cl- ions and so maximize the rate of Cl- permeation, however, this fixed positive charge also attracts all anions in the cytoplasm, many of which reside within the wide inner vestibule for long enough to temporarily block the passage of Cl- ions beyond into the narrow pore region. Mutagenesis of all positively charged lysine and arginine residues within the TMs suggests that K95 plays a unique role within the pore inner vestibule in attracting permeant and blocking ions[31,83], although other positive charges may also play somewhat analogous roles in attracting cytoplasmic ions to more superficial parts of the pore close to its intracellular mouth.
Figure 3.
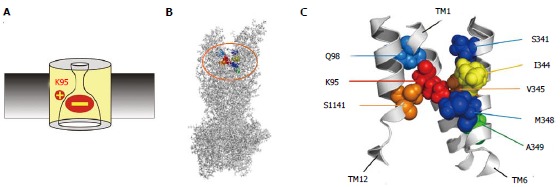
Location of amino acid residues key for blocker interactions in the pore inner vestibule. A: The positively charged side chain of lysine residue K95 is essential for block, due to electrostatic attraction between this positive charge and the negatively charged blocker. However, this important charge can also be supported by other amino acid side chains that line the pore inner vestibule. B, C: Sites that have been shown to host positive charge that can support block are shown in an atomic homology model of the whole cystic fibrosis transmembrane conductance regulator protein (B) and in a detailed view of the central portions of TMs 1, 6 and 12 (C) the area highlighted in (B). The endogenous positively charged side chain of K95 is shown in red; those residues that were deemed best able to support this functionally important positive charge in orange (V345, S1141) or yellow (I344); and those that were able to host this positive charge to a lesser extent in blue (Q98, S341, M348) or green (A349). The homology model used here is the “channel like” conformation presented by ref[20] and shown in Figure 1A; other models give similar relative positions of these pore-lining side chains.
If K95 does play a unique role in attracting anions into the pore inner vestibule - suggesting that it might be the only fixed positive charge located close to the blocker binding site within this vestibule[82] then what would be the effect of adding a second positive charge to the walls of this vestibule? This question has been addressed by using mutagenesis to introduce additional positively charged lysine residues at positions that have been shown to donate pore-lining side chains to the pore inner vestibule. Initially it was demonstrated that the unique, important role played by the positive charge at K95 could be “moved” from TM1 to TM12. Thus, while the charge-neutralizing K95S mutation dramatically decreased both Cl- conductance and sensitivity to open channel blockers, the double mutant K95S/S1141K showed similar single channel conductance and open channel blocker binding properties as wild type CFTR[82]. This “rescue” of channel function suggests that these two amino acids play interchangeable roles within the pore inner vestibule, in that either could effectively host the positive charge that supports interactions with Cl- and blocking anions[82] (Figure 4). Substituted cysteine accessibility mutagenesis and disulfide cross-linking experiments indicated that the amino acid side chains at these two positions line the inner vestibule in open channels and that these side chains are in close physical proximity[82]. Subsequent experiments showed that the positive charge from K95 could similarly be transplanted to different pore-accessible positions in TM6 (I344, V345, M348, A349), as well as a site closer to the extracellular end of TM1 (Q98)[29]. Thus it appears that the exact location of the positive charge in the pore inner vestibule is not critical to support channel function. The ability of other sites in TMs 1, 6 and 12 to accommodate the positive charge that normally resides at K95 then allowed investigation of the effects of introducing a second positive charge at these sites (by mutagenesis to lysine) while retaining the positive charge at K95 - in effect, increasing the number of positive charges located deep in the pore inner vestibule from one to two (Figure 4). Interestingly, at no site tested (Q98K, I344K, V345K, M348, A349K, S1141K) did the addition of a second positive charge increase single channel conductance[29,82]. This suggests that, while the presence of one positive charge is essential for normal Cl- conductance, a second positive charge provides no additional benefit. However, a second fixed positive charge (in S1141K) increased the strength of block by cytoplasmic NPPB, and also induced apparent voltage-dependent channel block by polyvalent anions present in the experimental solutions (ATP, pyrophosphate)[82], suggesting that the number of positive charges was correlated with open channel blocker potency (Figure 4). This suggestion was most strongly supported using the small divalent anion Pt(NO2)42-, which also causes voltage-dependent open channel block in a K95-dependent manner[84,85]. Addition of a second positive charge to nearby pore-lining sites in TM6 (I344K, V345K) or TM12 (S1141K) led to a dramatic (40-100 fold) increase in the apparent affinity of intracellular Pt(NO2)42- block[29,82], suggesting that increasing the number of positive charges in the pore has a greater impact on interactions with multivalent anions (such as Pt(NO2)42- and ATP) than monovalent anions such as Cl-. Positive charges introduced at other sites within the pore inner vestibule (Q98K, S341K, M348K, A349K) also supported strengthened Pt(NO2)42- block, albeit to a lesser extent. These findings, summarized in Figure 4, led to the hypothesis that one positive charge in the inner vestibule (as in wild type CFTR) was optimum for CFTR channel function[82]. Thus, removal of the one endogenous positive charge (as in K95Q or K95S) decreases channel function due to reduced electrostatic attraction of Cl- ions and a resulting dramatic decrease in channel conductance. This effect can be “rescued” by introducing a positive charge at other, nearby positions (as in K95S/S1141K and K95Q/V345K). Conversely, addition of a second positive charge (as in I344K, V345K, S1141K and other lysine substitutions) results in no further increase in Cl- conductance but increases the electrostatic attraction of multivalent anions that block the pore, resulting in an overall decrease in channel function. Thus the greatest importance of a single fixed positive charge in the inner vestibule may be in conferring preference for monovalent anions, including the physiological channel transport substrates Cl- and HCO3-.
Figure 4.
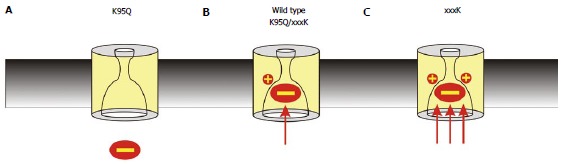
Importance of the number of fixed positive charges in the pore inner vestibule. The importance of electrostatic interactions with the pore inner vestibule is demonstrated by the finding that the strength of block can be decreased or increased by mutations that decrease or increase, respectively, the number of positively charged amino acid side chains in the pore inner vestibule area shown in Figure 3C. A: Block is relatively weak in when the endogenous positive charge is removed, for example as in the K95Q mutation; B: Block is of similar strength to that observed in wild type cystic fibrosis transmembrane conductance regulator when the positive charge is “transplanted” to other, nearby sites, for example as in the double mutants K95Q/I344K, K95Q/V345K, and K95S/S1141K; C: Block is relatively strong when a second positive charge is introduced, for example as in I344K, V345K and S1141K.
If one is the optimum number of positive charges in the inner vestibule to maximize channel function, where is the optimal location for this charge? Since normal channel function can be rescued by moving the positive charge to other, nearby sites in TM1, TM6 or TM12, the exact location of this charge does not appear to be critical[29]. Of all sites tested as hosts of the positive charge (K95, Q98 in TM1; S341, I344, V345, M348, A349 in TM6; S1141 in TM12), K95 appears optimal in terms of maximizing single channel conductance[29]. In terms of both single channel conductance and blocker binding properties, I344, V345 and S1141 appeared to be the best locations for a positive charge to reproduce wild type properties, with Q98, S341, M348 and A349 also being able to host this positive charge to some extent[29]. Similarly, a second positive charge at I344, V345 or S1141 had the greatest impact on divalent Pt(NO2)42- block[29]. These ideas are presented graphically, within the framework of a recent atomic homology model of CFTR, in Figure 3C. Within this model, the side chains of I344, V345 and S1141 appear to be at approximately the same “depth” into the channel pore as K95; with Q98 and S341 being located more deeply into the pore from its cytoplasmic end, and M348 and A349 closer to the cytoplasmic mouth of the pore. This relative location of amino acids is also supported by experimental evidence that disulfide bonds can be formed between cysteine side chains substituted for K95 and S1141[82], as well as between K95C and I344C and between Q98C and I344C[86]. This model suggests that it is location along the axis of the channel pore that is most important in defining the functional effects of a positive charge in the pore inner vestibule: residues close to the endogenous site at K95 are best able to substitute and rescue pore function, while residues either further from, or closer to, the cytoplasmic entrance of the pore are less able to host this important positive charge. This is consistent with molecular modeling studies that show open channel blockers docked within the pore inner vestibule and with their negative charges close to the positive charge of K95[20].
As described above, interaction with the positive charge at K95 and occlusion of the pore inner vestibule appears to be the molecular mechanism of many different kinds of CFTR open channel blockers, including those shown in Figure 2. However, a second blocker binding site was identified using the large, polyvalent organic anion suramin[87]. Suramin causes potent, voltage-independent block of CFTR channels exclusively from the intracellular side of the membrane[87,88]. Furthermore, block by intracellular suramin is independent of extracellular Cl- concentration, and completely unaffected by removal of the key positive charge in the inner vestibule in the K95Q mutant[87]. This suggests that suramin does not enter deeply enough into the pore inner vestibule to experience electrostatic interaction with K95, perhaps because the suramin molecule is simply too big to pass into this restricted pore region. In contrast, suramin block was weakened in an electrostatic fashion by mutagenesis of another positively charged amino acid, R303 at the cytoplasmic end of TM5[87] (Figure 1D and E). This result was consistent with the previous finding that the positive charge of R303 attracts intracellular Cl- ions to the cytoplasmic entrance of the pore[83] and suggests that the large suramin molecule may be able to occlude the cytoplasmic mouth of the pore to prevent Cl- movement into or out of the pore. As shown in Figure 5, this proposed molecular mechanism of suramin action is consistent with observed biophysical differences between suramin block and block by other (smaller) open channel blockers that interact with K95 (see above). Because of its size, suramin does not penetrate deeply into the inner vestibule; as a result, it does not traverse enough of the transmembrane electric field to generate measurable voltage-dependence of block, it does not reside in close proximity to Cl- ions bound within the channel pore (perhaps in the narrow pore region or close to the outer extent of the inner vestibule) and so does not experience the kind of repulsive electrostatic interactions that are thought to underlie extracellular Cl- dependence, and it does not approach close enough to K95 to experience attractive electrostatic interactions with this positively charged residue (Figure 5). Electrostatic interaction with R303 near the cytoplasmic mouth of the pore may also contribute to the inhibitory effects of other substances on CFTR, for example arachidonic acid[89].
Figure 5.
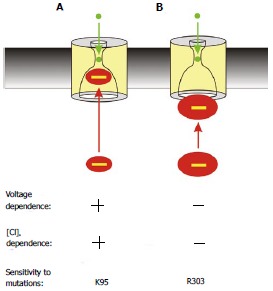
Two distinct proposed mechanisms of block by cytoplasmic anions. A: The effects of most blockers are voltage- and Cl- dependent (as described in Figure 2) and are sensitive to mutations that remove the positive charge at K95; B: The large multivalent anion suramin blocks the channel in a voltage- and Cl- independent fashion, and its effects are dependent on a positive charge at R303 but independent of K95. This is interpreted as the large suramin molecule blocking the cytoplasmic entrance to the pore; at a site that does not involve entering significantly into the transmembrane electric field or approaching close enough to Cl- ions inside the pore to experience repulsive electrostatic interactions.
PHYSIOLOGICAL RELEVANCE
CFTR channel currents are routinely studied in excised, inside-out membrane patches, where the current-voltage relationship is uniformly linear[90,91] (Figure 6). Conversely, CFTR channel currents in intact cells, including native epithelial cells[92-94], cardiac myocytes[95,96], and many different heterologous expression systems[82,97-99], exhibit outward rectification of the current-voltage relationship, such that outward currents (carried by Cl- influx) show greater conductance than inward currents (carried by Cl- efflux) (Figure 6). This rectification - and its disappearance in cell-free membrane patches - led to the longstanding suggestion that CFTR channels in intact cells are subject to voltage-dependent block by unknown cytosolic anions[58,97,100]. This appears to reflect predominantly a voltage-dependent flickery block by cytosolic anions that is lost when the membrane patch is excised from the cell[94,97,98,100], although differences in single channel conductance in cell-attached and excised patches have also been reported[92,95,97]. Detailed single channel recording experiments from cell-attached membrane patches suggested that the flickery blocking mechanism was functionally analogous to that generated by exogenous voltage-dependent blocking anions with intermediate blocking and unblocking kinetics[100]. Open channel block as the mechanism of outward rectification was further supported by the more recent finding that inhibition of currents in intact cells was reduced in K95Q-CFTR and (to a lesser extent) R303Q-CFTR[101]. This indicates that the unknown cytosolic blocking anions interact with these positively charged residues in the CFTR pore in intact cells, much as had previously been shown for exogenous channel blockers.
Figure 6.
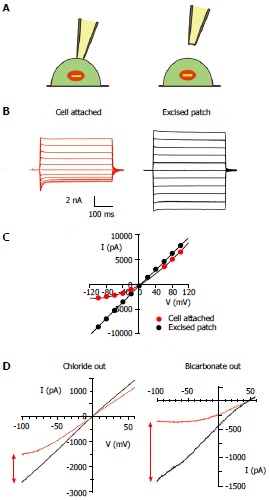
Channel block by cytoplasmic anions in intact cells and its dependence on extracellular anions. A: During patch clamp recording from intact cells, cystic fibrosis transmembrane conductance regulator (CFTR) channels in the cell membrane are subject to block anions present in the cytoplasm of the cell (left). This blocking effect is lost when the patch of membrane is excised into the inside-out patch configuration (right); B: Example of this effect during macroscopic CFTR current recording from a baby hamster kidney cell expressing human CFTR, as described in detail[82]. Currents were recorded before (red) and after (black) excision of the patch from the cell, during voltage steps to between -100 mV and +100 mV in 20 mV increments from a holding potential of 0 mV. Dotted line represents the zero current level; C: Current-voltage relationships for the currents shown in (B). Note the outward rectification of the relationship in cell-attached recording (red) due to voltage-dependent channel block, and loss of this blocking effect following patch excision (black); D: Similar example current-voltage relationships from baby hamster kidney cell membrane patches when the extracellular solution contained 150 mmol/L NaCl (left) or 150 mmol/L NaHCO3- (right), as described in detail[101]. Note that the apparent degree of block in cell attached patches (red) is stronger when the extracellular solution contains HCO3- compared to Cl-, an effect quantified in detail in ref. [101].
While outward rectification of CFTR currents in intact cells-and the weak form of voltage-dependence it confers on CFTR channel currents (Figure 6) has long been recognized, only recently has it been suggested that the voltage-dependent channel block that underlies this voltage-dependence might fulfil some kind of channel regulatory role. Just as block by exogenously applied open channel blockers is sensitive to extracellular Cl- (Figure 2D), so too is block by endogenous cytosolic anions in intact cells[82,100,101] (Figures 6D and Figure 7). This is not surprising if, as described above, these two intracellular voltage-dependent blocking effects share a common molecular mechanism. Recently it was proposed that this Cl- dependence might be one mechanism that allows CFTR conductance to be regulated by the composition of secreted fluid bathing the extracellular face of epithelial cells[101]. Most CFTR-expressing epithelia secrete substantial amounts of Cl- and HCO3- (up to 140 mmol/L HCO3- in the case of the pancreas[102]) in a CFTR-dependent fashion[2,103,104]. Furthermore, in many epithelia the concentrations of Cl- and HCO3- in secreted fluid vary greatly under physiological conditions[102,104-110]. Interestingly, it was shown that voltage-dependent block of CFTR in intact cells was significantly stronger under high extracellular (HCO3-) conditions than under high extracellular Cl- conditions[101]. This suggests that extracellular HCO3- is unable to substitute for Cl- in relieving the blocking effects of endogenous cytoplasmic blocking anions. As a result, overall CFTR activity will be increased under high extracellular Cl- conditions (i.e., during periods of epithelial Cl- secretion) and decreased under high HCO3- conditions (i.e., during periods of secretion of relatively HCO3--rich fluid)[101] (Figure 7). These findings led to the suggestion that endogenous cytoplasmic blocking anions are physiologically relevant regulators of CFTR channel function, in that they confer upon the channel sensitivity to physiologically relevant changes in extracellular fluid composition[101]. In epithelial cells, this may be one mechanism by which CFTR channel function is fine-tuned by the concentration of its transport substrates Cl- and HCO3- at the apical face of these cells[111-113].
Figure 7.
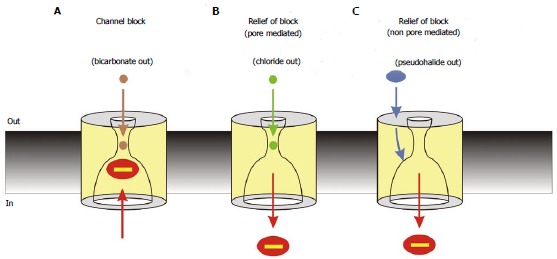
Interactions between cytoplasmic blocking anions and extracellular anions. Cytoplasmic block is modified by extracellular anions by different mechanisms, leading to different degrees of block under different conditions. A: Block is strong in the absence of modulation of block by extracellular anions; physiologically, such a condition may occur during periods of epithelial HCO3- secretion, leading to strong block of cystic fibrosis transmembrane conductance regulator (CFTR) channel currents under these conditions[101]; B: Block is weakened by extracellular anions that can enter the channel pore, such as Cl-, due to an electrostatic “knock-off” mechanism. This may lead to increased CFTR channel currents during periods of epithelial Cl- secretion[101]; C: Block is weakened by extracellular anions that interact with an extracellular part of the protein involving extracellular loop 4. This is presumed to result in a long-range conformational change in CFTR that decreases the affinity of the cytoplasmic blocker binding site. This mechanism may allow pharmacological manipulation of CFTR activity by compounds that interact with the extracellular anion binding site[114]. Note that Cl- ions may also interact with intracellular blocking anions by the non-pore mediated effect shown in (C)[114].
While extracellular Cl- may be an endogenous substance regulating CFTR channel function via modulation of the blocking effects of cytoplasmic anions, this mechanism of channel regulation may also be subject to pharmacological manipulation. Thus, millimolar concentrations of extracellular multivalent psuedohalide anions (Co(CN)63-, Co(NO2)63-, Fe(CN)63-, IrCl63-, Fe(CN)64-) were shown to mimic the effects of high extracellular Cl- on channel block in intact cells, leading to an overall stimulation of CFTR channel function[114] (Figure 7). It was suggested that these anions represent the founder members of a new class of CFTR potentiators, and that their effects identify a novel mechanism by which CFTR function could potentially be increased therapeutically in the treatment of CF. Interestingly, these pseudohalide anions do not enter into the CFTR channel pore[114] and as such presumably do not interact electrostatically with blocking anions inside the channel pore; such an electrostatic “knock off” mechanism is commonly proposed to underlie the effect of extracellular Cl- ions on intracellular open channel blockers[115,116] (Figure 7), as well as permeant ion effects on blocker binding in many other ion channel types[59]. Instead, pseudohalide anions were shown to exert their effects via interation with extracellular parts of CFTR, in particular ECL4[114,117]. The molecular mechanism of action of these substances, acting on extracellular parts of the protein, is therefore distinct from those of known CFTR potentiators, perhaps allowing additive or synergistic effects with other types of potentiators. Furthermore, the suggestion that a novel potentiator “binding site” might exist on ECL4 raises the possibility that this externally-accessible part of the CFTR protein could in future be targetted by drugs that can manipulate CFTR function therapeutically.
CONCLUSION
The architecture and Cl- permeation mechanism of CFTR likely results in a susceptibility to relatively low affinity, voltage dependent open channel block by a very broad range of structurally diverse organic anions, including unidentified anions that the channel normally encounters in the cytoplasm of the cell. Because the channel is normally involved in the secretion of Cl- and HCO3- ions at hyperpolarized cell membrane potentials, the channel has a relatively large intracellular vestibule that contains fixed positive charges to allow it to capture these anions from the cytoplasm by the process of electrostatic attraction. As the vestibule narrows toward the centre of the pore (Figure 1C and D), a single, functionally unique positive charge (K95 in TM1) ensures efficient attraction of monovalent anions (Figure 3). Beyond this point, permeating anions pass into a narrow, uncharged pore region that may allow some level of discrimination between different anions, and also acts as a size selectivity filter to prevent larger organic anions from escaping the cell. While this architecture appears efficient at maximizing channel Cl- conductance (Figure 4), it also probably results in some degree of channel inhibition by cytoplasmic anions that are attracted by the positive charge at K95, but which are too large to pass into the narrow pore region (Figures 2, 3, 4 and 5). CFTR experimentalists have long taken advantage of these voltage dependent blocking anions to investigate CFTR-dependent processes, to think about the possible advantages of inhibiting CFTR function in disease states associated with inappropriately elevated CFTR function, and as relatively high-affinity probes to investigate the structure and function of the wide inner vestibule of the channel pore. This has allowed the development of functional (Figures 3, 4 and 5) and structural[20] models of the pore. More recently, it has been suggested that endogenous substances that act in this fashion may in fact play a role in tying CFTR function to the content of epithelial cell secretions (Figures 6 and 7), perhaps allowing CFTR activity to be fine-tuned directly by the amount of its substrate(s) being secreted from epithelial cells. In the future, this mechanism of CFTR regulation could be targetted by new drugs that act at an extracellular site on the CFTR protein to reduce the voltage-dependent blocking effects of endogenous cytoplasmic anions and so increase overall CFTR function in CF patients.
Footnotes
P- Reviewers: Fujiwara N, Jana SS, Rampoldi L S- Editor: Qi Y L- Editor: A E- Editor: Wu HL
References
- 1.Lubamba B, Dhooghe B, Noel S, Leal T. Cystic fibrosis: insight into CFTR pathophysiology and pharmacotherapy. Clin Biochem. 2012;45:1132–1144. doi: 10.1016/j.clinbiochem.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 2.Frizzell RA, Hanrahan JW. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med. 2012;2:a009563. doi: 10.1101/cshperspect.a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 4.Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931–943. doi: 10.1172/JCI18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terryn S, Ho A, Beauwens R, Devuyst O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1314–1321. doi: 10.1016/j.bbadis.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 6.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 7.Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang TC, Kirk KL. The CFTR ion channel: gating, regulation, and anion permeation. Cold Spring Harb Perspect Med. 2013;3:a009498. doi: 10.1101/cshperspect.a009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chong PA, Kota P, Dokholyan NV, Forman-Kay JD. Dynamics intrinsic to cystic fibrosis transmembrane conductance regulator function and stability. Cold Spring Harb Perspect Med. 2013;3:a009522. doi: 10.1101/cshperspect.a009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunt JF, Wang C, Ford RC. Cystic fibrosis transmembrane conductance regulator (ABCC7) structure. Cold Spring Harb Perspect Med. 2013;3:a009514. doi: 10.1101/cshperspect.a009514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patrick AE, Thomas PJ. Development of CFTR Structure. Front Pharmacol. 2012;3:162. doi: 10.3389/fphar.2012.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jih KY, Hwang TC. Nonequilibrium gating of CFTR on an equilibrium theme. Physiology (Bethesda) 2012;27:351–361. doi: 10.1152/physiol.00026.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg MF, O’Ryan LP, Hughes G, Zhao Z, Aleksandrov LA, Riordan JR, Ford RC. The cystic fibrosis transmembrane conductance regulator (CFTR): three-dimensional structure and localization of a channel gate. J Biol Chem. 2011;286:42647–42654. doi: 10.1074/jbc.M111.292268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linsdell P. Mechanism of chloride permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. Exp Physiol. 2006;91:123–129. doi: 10.1113/expphysiol.2005.031757. [DOI] [PubMed] [Google Scholar]
- 15.Wang W, El Hiani Y, Rubaiy HN, Linsdell P. Relative contribution of different transmembrane segments to the CFTR chloride channel pore. Pflugers Arch. 2014;466:477–490. doi: 10.1007/s00424-013-1317-x. [DOI] [PubMed] [Google Scholar]
- 16.Gao X, Bai Y, Hwang TC. Cysteine scanning of CFTR’s first transmembrane segment reveals its plausible roles in gating and permeation. Biophys J. 2013;104:786–797. doi: 10.1016/j.bpj.2012.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mornon JP, Lehn P, Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci. 2008;65:2594–2612. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mornon JP, Lehn P, Callebaut I. Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci. 2009;66:3469–3486. doi: 10.1007/s00018-009-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalton J, Kalid O, Schushan M, Ben-Tal N, Villà-Freixa J. New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J Chem Inf Model. 2012;52:1842–1853. doi: 10.1021/ci2005884. [DOI] [PubMed] [Google Scholar]
- 21.Norimatsu Y, Ivetac A, Alexander C, Kirkham J, O’Donnell N, Dawson DC, Sansom MS. Cystic fibrosis transmembrane conductance regulator: a molecular model defines the architecture of the anion conduction path and locates a “bottleneck” in the pore. Biochemistry. 2012;51:2199–2212. doi: 10.1021/bi201888a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge N, Muise CN, Gong X, Linsdell P. Direct comparison of the functional roles played by different transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem. 2004;279:55283–55289. doi: 10.1074/jbc.M411935200. [DOI] [PubMed] [Google Scholar]
- 23.McDonough S, Davidson N, Lester HA, McCarty NA. Novel pore-lining residues in CFTR that govern permeation and open-channel block. Neuron. 1994;13:623–634. doi: 10.1016/0896-6273(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 24.Linsdell P. Location of a common inhibitor binding site in the cytoplasmic vestibule of the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem. 2005;280:8945–8950. doi: 10.1074/jbc.M414354200. [DOI] [PubMed] [Google Scholar]
- 25.El Hiani Y, Linsdell P. Changes in accessibility of cytoplasmic substances to the pore associated with activation of the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem. 2010;285:32126–32140. doi: 10.1074/jbc.M110.113332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai Y, Li M, Hwang TC. Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J Gen Physiol. 2010;136:293–309. doi: 10.1085/jgp.201010480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian F, El Hiani Y, Linsdell P. Functional arrangement of the 12th transmembrane region in the CFTR chloride channel pore based on functional investigation of a cysteine-less CFTR variant. Pflugers Arch. 2011;462:559–571. doi: 10.1007/s00424-011-0998-2. [DOI] [PubMed] [Google Scholar]
- 28.Bai Y, Li M, Hwang TC. Structural basis for the channel function of a degraded ABC transporter, CFTR (ABCC7) J Gen Physiol. 2011;138:495–507. doi: 10.1085/jgp.201110705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El Hiani Y, Linsdell P. Tuning of CFTR chloride channel function by location of positive charges within the pore. Biophys J. 2012;103:1719–1726. doi: 10.1016/j.bpj.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith SS, Liu X, Zhang ZR, Sun F, Kriewall TE, McCarty NA, Dawson DC. CFTR: covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J Gen Physiol. 2001;118:407–431. doi: 10.1085/jgp.118.4.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou JJ, Fatehi M, Linsdell P. Identification of positive charges situated at the outer mouth of the CFTR chloride channel pore. Pflugers Arch. 2008;457:351–360. doi: 10.1007/s00424-008-0521-6. [DOI] [PubMed] [Google Scholar]
- 32.Fatehi M, Linsdell P. Novel residues lining the CFTR chloride channel pore identified by functional modification of introduced cysteines. J Membr Biol. 2009;228:151–164. doi: 10.1007/s00232-009-9167-3. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Linsdell P. Relative movements of transmembrane regions at the outer mouth of the cystic fibrosis transmembrane conductance regulator channel pore during channel gating. J Biol Chem. 2012;287:32136–32146. doi: 10.1074/jbc.M112.385096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultz BD, Singh AK, Devor DC, Bridges RJ. Pharmacology of CFTR chloride channel activity. Physiol Rev. 1999;79:S109–S144. doi: 10.1152/physrev.1999.79.1.S109. [DOI] [PubMed] [Google Scholar]
- 35.Hwang TC, Sheppard DN. Molecular pharmacology of the CFTR Cl- channel. Trends Pharmacol Sci. 1999;20:448–453. doi: 10.1016/s0165-6147(99)01386-3. [DOI] [PubMed] [Google Scholar]
- 36.Sheppard DN, Welsh MJ. Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J Gen Physiol. 1992;100:573–591. doi: 10.1085/jgp.100.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schultz BD, DeRoos AD, Venglarik CJ, Singh AK, Frizzell RA, Bridges RJ. Glibenclamide blockade of CFTR chloride channels. Am J Physiol. 1996;271:L192–L200. doi: 10.1152/ajplung.1996.271.2.L192. [DOI] [PubMed] [Google Scholar]
- 38.Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl- channels expressed in a murine cell line. J Physiol. 1997;503(Pt 2):333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Hu S, Hwang TC. Probing an open CFTR pore with organic anion blockers. J Gen Physiol. 2002;120:647–662. doi: 10.1085/jgp.20028685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang ZR, Zeltwanger S, McCarty NA. Steady-state interactions of glibenclamide with CFTR: evidence for multiple sites in the pore. J Membr Biol. 2004;199:15–28. doi: 10.1007/s00232-004-0672-0. [DOI] [PubMed] [Google Scholar]
- 41.Zhang ZR, Cui G, Zeltwanger S, McCarty NA. Time-dependent interactions of glibenclamide with CFTR: kinetically complex block of macroscopic currents. J Membr Biol. 2004;201:139–155. doi: 10.1007/s00232-004-0712-9. [DOI] [PubMed] [Google Scholar]
- 42.Cui G, Song B, Turki HW, McCarty NA. Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers. Pflugers Arch. 2012;463:405–418. doi: 10.1007/s00424-011-1035-1. [DOI] [PubMed] [Google Scholar]
- 43.Venglarik CJ, Schultz BD, DeRoos AD, Singh AK, Bridges RJ. Tolbutamide causes open channel blockade of cystic fibrosis transmembrane conductance regulator Cl- channels. Biophys J. 1996;70:2696–2703. doi: 10.1016/S0006-3495(96)79839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai Z, Lansdell KA, Sheppard DN. Inhibition of heterologously expressed cystic fibrosis transmembrane conductance regulator Cl- channels by non-sulphonylurea hypoglycaemic agents. Br J Pharmacol. 1999;128:108–118. doi: 10.1038/sj.bjp.0702748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarty NA, McDonough S, Cohen BN, Riordan JR, Davidson N, Lester HA. Voltage-dependent block of the cystic fibrosis transmembrane conductance regulator Cl- channel by two closely related arylaminobenzoates. J Gen Physiol. 1993;102:1–23. doi: 10.1085/jgp.102.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh KB, Long KJ, Shen X. Structural and ionic determinants of 5-nitro-2-(3-phenylprophyl-amino)-benzoic acid block of the CFTR chloride channel. Br J Pharmacol. 1999;127:369–376. doi: 10.1038/sj.bjp.0702562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang ZR, Zeltwanger S, McCarty NA. Direct comparison of NPPB and DPC as probes of CFTR expressed in Xenopus oocytes. J Membr Biol. 2000;175:35–52. doi: 10.1007/s002320001053. [DOI] [PubMed] [Google Scholar]
- 48.Scott-Ward TS, Li H, Schmidt A, Cai Z, Sheppard DN. Direct block of the cystic fibrosis transmembrane conductance regulator Cl(-) channel by niflumic acid. Mol Membr Biol. 2004;21:27–38. doi: 10.1080/09687680310001597758. [DOI] [PubMed] [Google Scholar]
- 49.Linsdell P, Hanrahan JW. Disulphonic stilbene block of cystic fibrosis transmembrane conductance regulator Cl- channels expressed in a mammalian cell line and its regulation by a critical pore residue. J Physiol. 1996;496(Pt 3):687–693. doi: 10.1113/jphysiol.1996.sp021719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linsdell P, Hanrahan JW. Substrates of multidrug resistance-associated proteins block the cystic fibrosis transmembrane conductance regulator chloride channel. Br J Pharmacol. 1999;126:1471–1477. doi: 10.1038/sj.bjp.0702458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diena T, Melani R, Caci E, Pedemonte N, Sondo E, Zegarra-Moran O, Galietta LJ. Block of CFTR-dependent chloride currents by inhibitors of multidrug resistance-associated proteins. Eur J Pharmacol. 2007;560:127–131. doi: 10.1016/j.ejphar.2007.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gong X, Burbridge SM, Lewis AC, Wong PY, Linsdell P. Mechanism of lonidamine inhibition of the CFTR chloride channel. Br J Pharmacol. 2002;137:928–936. doi: 10.1038/sj.bjp.0704932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong XD, Linsdell P, Cheung KH, Leung GP, Wong PY. Indazole inhibition of cystic fibrosis transmembrane conductance regulator Cl(-) channels in rat epididymal epithelial cells. Biol Reprod. 2002;67:1888–1896. doi: 10.1095/biolreprod.102.007450. [DOI] [PubMed] [Google Scholar]
- 54.Linsdell P. Direct block of the cystic fibrosis transmembrane conductance regulator Cl(-) channel by butyrate and phenylbutyrate. Eur J Pharmacol. 2001;411:255–260. doi: 10.1016/s0014-2999(00)00928-6. [DOI] [PubMed] [Google Scholar]
- 55.Cai Z, Sheppard DN. Phloxine B interacts with the cystic fibrosis transmembrane conductance regulator at multiple sites to modulate channel activity. J Biol Chem. 2002;277:19546–19553. doi: 10.1074/jbc.M108023200. [DOI] [PubMed] [Google Scholar]
- 56.Ishihara H, Welsh MJ. Block by MOPS reveals a conformation change in the CFTR pore produced by ATP hydrolysis. Am J Physiol. 1997;273:C1278–C1289. doi: 10.1152/ajpcell.1997.273.4.C1278. [DOI] [PubMed] [Google Scholar]
- 57.Li MS, Demsey AF, Qi J, Linsdell P. Cysteine-independent inhibition of the CFTR chloride channel by the cysteine-reactive reagent sodium (2-sulphonatoethyl) methanethiosulphonate. Br J Pharmacol. 2009;157:1065–1071. doi: 10.1111/j.1476-5381.2009.00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linsdell P, Hanrahan JW. Flickery block of single CFTR chloride channels by intracellular anions and osmolytes. Am J Physiol. 1996;271:C628–C634. doi: 10.1152/ajpcell.1996.271.2.C628. [DOI] [PubMed] [Google Scholar]
- 59.Hille B. Ion channels of excitable membranes. 3rd ed. Sunderland (MA): Sinauer Assoc; 2001. [Google Scholar]
- 60.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164 Suppl 1:S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJ. Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett. 2004;558:52–56. doi: 10.1016/S0014-5793(04)00011-0. [DOI] [PubMed] [Google Scholar]
- 64.Kopeikin Z, Sohma Y, Li M, Hwang TC. On the mechanism of CFTR inhibition by a thiazolidinone derivative. J Gen Physiol. 2010;136:659–671. doi: 10.1085/jgp.201010518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Norimatsu Y, Ivetac A, Alexander C, O’Donnell N, Frye L, Sansom MS, Dawson DC. Locating a plausible binding site for an open-channel blocker, GlyH-101, in the pore of the cystic fibrosis transmembrane conductance regulator. Mol Pharmacol. 2012;82:1042–1055. doi: 10.1124/mol.112.080267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuller MD, Thompson CH, Zhang ZR, Freeman CS, Schay E, Szakács G, Bakos E, Sarkadi B, McMaster D, French RJ, et al. State-dependent inhibition of cystic fibrosis transmembrane conductance regulator chloride channels by a novel peptide toxin. J Biol Chem. 2007;282:37545–37555. doi: 10.1074/jbc.M708079200. [DOI] [PubMed] [Google Scholar]
- 68.Fuller MD, Zhang ZR, Cui G, McCarty NA. The block of CFTR by scorpion venom is state-dependent. Biophys J. 2005;89:3960–3975. doi: 10.1529/biophysj.105.060731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Derichs N. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev. 2013;22:58–65. doi: 10.1183/09059180.00008412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanrahan JW, Sampson HM, Thomas DY. Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol Sci. 2013;34:119–125. doi: 10.1016/j.tips.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 71.Rowe SM, Verkman AS. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb Perspect Med. 2013;3 doi: 10.1101/cshperspect.a009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sermet-Gaudelus I. Ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation. Eur Respir Rev. 2013;22:66–71. doi: 10.1183/09059180.00008512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Super M. CFTR and disease: implications for drug development. Lancet. 2000;355:1840–1842. doi: 10.1016/S0140-6736(00)02282-0. [DOI] [PubMed] [Google Scholar]
- 74.Chen H, Ruan YC, Xu WM, Chen J, Chan HC. Regulation of male fertility by CFTR and implications in male infertility. Hum Reprod Update. 2012;18:703–713. doi: 10.1093/humupd/dms027. [DOI] [PubMed] [Google Scholar]
- 75.Thiagarajah JR, Broadbent T, Hsieh E, Verkman AS. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology. 2004;126:511–519. doi: 10.1053/j.gastro.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 76.Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. Lectin conjugates as potent, nonabsorbable CFTR inhibitors for reducing intestinal fluid secretion in cholera. Gastroenterology. 2007;132:1234–1244. doi: 10.1053/j.gastro.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 77.Li H, Findlay IA, Sheppard DN. The relationship between cell proliferation, Cl- secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int. 2004;66:1926–1938. doi: 10.1111/j.1523-1755.2004.00967.x. [DOI] [PubMed] [Google Scholar]
- 78.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:1300–1310. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H, Sheppard DN. Therapeutic potential of cystic fibrosis transmembrane conductance regulator (CFTR) inhibitors in polycystic kidney disease. BioDrugs. 2009;23:203–216. doi: 10.2165/11313570-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 80.Thiagarajah JR, Verkman AS. CFTR inhibitors for treating diarrheal disease. Clin Pharmacol Ther. 2012;92:287–290. doi: 10.1038/clpt.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gupta J, Evagelidis A, Hanrahan JW, Linsdell P. Asymmetric structure of the cystic fibrosis transmembrane conductance regulator chloride channel pore suggested by mutagenesis of the twelfth transmembrane region. Biochemistry. 2001;40:6620–6627. doi: 10.1021/bi002819v. [DOI] [PubMed] [Google Scholar]
- 82.Zhou JJ, Li MS, Qi J, Linsdell P. Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J Gen Physiol. 2010;135:229–245. doi: 10.1085/jgp.200910327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aubin CN, Linsdell P. Positive charges at the intracellular mouth of the pore regulate anion conduction in the CFTR chloride channel. J Gen Physiol. 2006;128:535–545. doi: 10.1085/jgp.200609516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gong X, Linsdell P. Mutation-induced blocker permeability and multiion block of the CFTR chloride channel pore. J Gen Physiol. 2003;122:673–687. doi: 10.1085/jgp.200308889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou JJ, Fatehi M, Linsdell P. Direct and indirect effects of mutations at the outer mouth of the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Membr Biol. 2007;216:129–142. doi: 10.1007/s00232-007-9056-6. [DOI] [PubMed] [Google Scholar]
- 86.Wang W, El Hiani Y, Linsdell P. Alignment of transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Gen Physiol. 2011;138:165–178. doi: 10.1085/jgp.201110605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.St Aubin CN, Zhou JJ, Linsdell P. Identification of a second blocker binding site at the cytoplasmic mouth of the cystic fibrosis transmembrane conductance regulator chloride channel pore. Mol Pharmacol. 2007;71:1360–1368. doi: 10.1124/mol.106.031732. [DOI] [PubMed] [Google Scholar]
- 88.Bachmann A, Russ U, Quast U. Potent inhibition of the CFTR chloride channel by suramin. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:473–476. doi: 10.1007/s002109900096. [DOI] [PubMed] [Google Scholar]
- 89.Zhou JJ, Linsdell P. Molecular mechanism of arachidonic acid inhibition of the CFTR chloride channel. Eur J Pharmacol. 2007;563:88–91. doi: 10.1016/j.ejphar.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 90.McCarty NA. Permeation through the CFTR chloride channel. J Exp Biol. 2000;203:1947–1962. doi: 10.1242/jeb.203.13.1947. [DOI] [PubMed] [Google Scholar]
- 91.Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gray MA, Harris A, Coleman L, Greenwell JR, Argent BE. Two types of chloride channel on duct cells cultured from human fetal pancreas. Am J Physiol. 1989;257:C240–C251. doi: 10.1152/ajpcell.1989.257.2.C240. [DOI] [PubMed] [Google Scholar]
- 93.Tabcharani JA, Low W, Elie D, Hanrahan JW. Low-conductance chloride channel activated by cAMP in the epithelial cell line T84. FEBS Lett. 1990;270:157–164. doi: 10.1016/0014-5793(90)81257-o. [DOI] [PubMed] [Google Scholar]
- 94.Haws C, Krouse ME, Xia Y, Gruenert DC, Wine JJ. CFTR channels in immortalized human airway cells. Am J Physiol. 1992;263:L692–L707. doi: 10.1152/ajplung.1992.263.6.L692. [DOI] [PubMed] [Google Scholar]
- 95.Ehara T, Matsuura H. Single-channel study of the cyclic AMP-regulated chloride current in guinea-pig ventricular myocytes. J Physiol. 1993;464:307–320. doi: 10.1113/jphysiol.1993.sp019636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Overholt JL, Hobert ME, Harvey RD. On the mechanism of rectification of the isoproterenol-activated chloride current in guinea-pig ventricular myocytes. J Gen Physiol. 1993;102:871–895. doi: 10.1085/jgp.102.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tabcharani JA, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation-regulated Cl- channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- 98.Fischer H, Machen TE. CFTR displays voltage dependence and two gating modes during stimulation. J Gen Physiol. 1994;104:541–566. doi: 10.1085/jgp.104.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Overholt JL, Saulino A, Drumm ML, Harvey RD. Rectification of whole cell cystic fibrosis transmembrane conductance regulator chloride current. Am J Physiol. 1995;268:C636–C646. doi: 10.1152/ajpcell.1995.268.3.C636. [DOI] [PubMed] [Google Scholar]
- 100.Zhou Z, Hu S, Hwang TC. Voltage-dependent flickery block of an open cystic fibrosis transmembrane conductance regulator (CFTR) channel pore. J Physiol. 2001;532:435–448. doi: 10.1111/j.1469-7793.2001.0435f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li MS, Holstead RG, Wang W, Linsdell P. Regulation of CFTR chloride channel macroscopic conductance by extracellular bicarbonate. Am J Physiol Cell Physiol. 2011;300:C65–C74. doi: 10.1152/ajpcell.00290.2010. [DOI] [PubMed] [Google Scholar]
- 102.Argent BE, Gray MA, Steward MC, Case RM. Cell physiology of pancreatic ducts. In: Physiology of the gastrointestinal tract., editor. 5th edition. Johnson L, editor. San Diego: Elsevier; 2012. pp. 1399–1423. [Google Scholar]
- 103.Tang L, Fatehi M, Linsdell P. Mechanism of direct bicarbonate transport by the CFTR anion channel. J Cyst Fibros. 2009;8:115–121. doi: 10.1016/j.jcf.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 104.Bridges RJ. Mechanisms of bicarbonate secretion: lessons from the airways. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cuthbert AW. Bicarbonate secretion in the murine gallbladder--lessons for the treatment of cystic fibrosis. JOP. 2001;2:257–262. [PubMed] [Google Scholar]
- 106.Wang XF, Zhou CX, Shi QX, Yuan YY, Yu MK, Ajonuma LC, Ho LS, Lo PS, Tsang LL, Liu Y, et al. Involvement of CFTR in uterine bicarbonate secretion and the fertilizing capacity of sperm. Nat Cell Biol. 2003;5:902–906. doi: 10.1038/ncb1047. [DOI] [PubMed] [Google Scholar]
- 107.Allen A, Flemström G. Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am J Physiol Cell Physiol. 2005;288:C1–19. doi: 10.1152/ajpcell.00102.2004. [DOI] [PubMed] [Google Scholar]
- 108.Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12:3496–3511. doi: 10.3748/wjg.v12.i22.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fischer H, Widdicombe JH. Mechanisms of acid and base secretion by the airway epithelium. J Membr Biol. 2006;211:139–150. doi: 10.1007/s00232-006-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chan HC, Ruan YC, He Q, Chen MH, Chen H, Xu WM, Chen WY, Xie C, Zhang XH, Zhou Z. The cystic fibrosis transmembrane conductance regulator in reproductive health and disease. J Physiol. 2009;587:2187–2195. doi: 10.1113/jphysiol.2008.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O’Reilly CM, Winpenny JP, Argent BE, Gray MA. Cystic fibrosis transmembrane conductance regulator currents in guinea pig pancreatic duct cells: inhibition by bicarbonate ions. Gastroenterology. 2000;118:1187–1196. doi: 10.1016/s0016-5085(00)70372-6. [DOI] [PubMed] [Google Scholar]
- 112.Shcheynikov N, Kim KH, Kim KM, Dorwart MR, Ko SB, Goto H, Naruse S, Thomas PJ, Muallem S. Dynamic control of cystic fibrosis transmembrane conductance regulator Cl(-)/HCO3(-) selectivity by external Cl(-) J Biol Chem. 2004;279:21857–21865. doi: 10.1074/jbc.M313323200. [DOI] [PubMed] [Google Scholar]
- 113.Wright AM, Gong X, Verdon B, Linsdell P, Mehta A, Riordan JR, Argent BE, Gray MA. Novel regulation of cystic fibrosis transmembrane conductance regulator (CFTR) channel gating by external chloride. J Biol Chem. 2004;279:41658–41663. doi: 10.1074/jbc.M405517200. [DOI] [PubMed] [Google Scholar]
- 114.Li MS, Cowley EA, Linsdell P. Pseudohalide anions reveal a novel extracellular site for potentiators to increase CFTR function. Br J Pharmacol. 2012;167:1062–1075. doi: 10.1111/j.1476-5381.2012.02041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Linsdell P, Tabcharani JA, Hanrahan JW. Multi-Ion mechanism for ion permeation and block in the cystic fibrosis transmembrane conductance regulator chloride channel. J Gen Physiol. 1997;110:365–377. doi: 10.1085/jgp.110.4.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gong X, Linsdell P. Coupled movement of permeant and blocking ions in the CFTR chloride channel pore. J Physiol. 2003;549:375–385. doi: 10.1113/jphysiol.2002.038216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou JJ, Linsdell P. Evidence that extracellular anions interact with a site outside the CFTR chloride channel pore to modify channel properties. Can J Physiol Pharmacol. 2009;87:387–395. doi: 10.1139/y09-023. [DOI] [PubMed] [Google Scholar]