

Abstract
During development, experience plays a crucial role in sculpting neuronal connections. Patterned neural activity guides formation of functional neural circuits through the selective stabilization of some synapses and the pruning of others. Activity-regulated factors are fundamental to this process, but their roles in synapse stabilization and maturation is still poorly understood. CPG15, encoded by the activity-regulated gene candidate plasticity gene 15, is a small, glycosylphosphatidylinositol (GPI)-linked, extracellular protein that promotes synapse stabilization. Here we show that global knock-out of cpg15 results in abnormal postnatal development of the excitatory network in visual cortex and an associated disruption in development of visual receptive field properties. In addition, whereas repeated stimulation induced potentiation and depression in wild-type mice, the depression was slower in cpg15 knock-out mice, suggesting impairment in short-term depression-like mechanisms. These findings establish the requirement for cpg15 in activity-dependent development of the visual system and demonstrate the importance of timely excitatory network development for normal visual function.
Keywords: depression, plasticity, potentiation, visual cortex
Introduction
During development, neuronal processes explore their environment to identify appropriate partners before establishing presynaptic and postsynaptic contacts leading to formation of stable synapses. Strong evidence suggests that sensory experience and activity play critical roles in synapse formation and stabilization, and multiple molecular candidates have been proposed to mediate these processes (Flavell and Greenberg, 2008; Leslie and Nedivi, 2011). One such molecule is candidate plasticity gene 15 (cpg15, neuritin), first identified in a screen for activity-regulated genes in rat (Nedivi et al., 1993). CPG15 is a downstream target of the classic synaptic plasticity signaling cascade, involving the NMDA receptor, MAPK, CaMK and CREB (Fujino et al., 2003). cpg15 encodes an extracellular protein attached to the cell surface and is highly conserved between mouse and human (Fujino et al., 2008). During development, cpg15 expression is correlated with synapse formation and activity-dependent plasticity (Nedivi et al., 1996; Corriveau et al., 1999; Lee and Nedivi, 2002). CPG15 overexpression has been shown to enhance or delay dendritic and axonal elaboration as well as synapse formation and maturation (Nedivi et al., 1998; Cantallops et al., 2000; Javaherian and Cline, 2005). Conversely, in its absence, the development of excitatory neuron arbors and synaptic properties is delayed (Fujino et al., 2011).
In the rodent visual cortex, the onset of cpg15 transcription coincides with eye opening and increases throughout the following 2 weeks, peaking during the period of highest plasticity for receptive field properties (Gordon and Stryker, 1996; Lee and Nedivi, 2002). At the same time, visual cortical neurons exhibit an increase in the frequency and a decrease in the amplitude of spontaneous miniature EPSCs (mEPSCs) (Desai et al., 2002). This is accompanied by an increase in pyramidal neuron dendritic arbor complexity (Miller, 1981; Juraska, 1982) and spine numbers (Oray et al., 2006). These developmental changes occur concomitant with the acquisition of proper adult responses to external stimuli and tuning for orientation and motion (Wang et al., 2010; Rochefort et al., 2011). Despite the concurrence of activity-regulated cpg15 expression with emergence of mature excitatory network features in visual cortex, the requirement for cpg15 during these well characterized milestones of visual cortex development has never been established. Moreover, there has been no assessment of CPG15's contribution to the development of cellular functional properties and plasticity in vivo.
To address this, we combined a multilevel analysis of visual cortical circuits in cpg15 knock-out (KO) mice to determine the requirement for CPG15 in the development of visual properties. We found that CPG15 is required for normal morphological and synaptic development of visual cortical pyramidal neurons, and for the maturation of their receptive field properties and rapid plasticity mechanisms in vivo.
Materials and Methods
NIH guidelines for the use and care of vertebrate animals were followed, and all procedures were approved by the Institutional Animal Care and Use Committee at Boston Children's Hospital and the Massachusetts Institute of Technology Committee on Animal Care. The cpg15 KO mouse was generated at MIT by Dr. T. Fujino (Fujino et al., 2011). All control animals were WT age-matched littermates of the mutant mice. Mice of either sex were used in this study.
For in vitro studies, pyramidal neurons were all selected from L2/3 of primary visual cortex (V1) identified according to the Paxinos mouse brain atlas (Paxinos and Franklin, 2004). For the diolistics and spine analysis, neurons were selected only when a clear vertical apical dendritic process was visible. Neurons with oblique apical dendrites were avoided. Neurons at the very top and bottom of the slice were also avoided to capture as complete a dendritic arbor as possible, as well as to clearly differentiate the pyramidal morphology and to distinguish apical from basal dendrites. When selecting neurons for EPSC recording, superficial neurons at the border of L2/3 and L1, where oblique pyramidal neurons are located, were avoided.
Diolistic labeling.
Mice were perfused with 4% paraformaldehyde, and brains were removed and postfixed in 4% paraformaldehyde. V1 coronal sections (150 μm) were prepared and stored in 30% sucrose in PBS. Diolistic labeling was performed as described previously (Grutzendler et al., 2003; Fujino et al., 2011). Sections were postfixed in 4% paraformaldehyde/30% sucrose overnight and mounted on glass slides with Fluoromount-G. Stacks (50 μm thick) were acquired at 2 μm intervals using a 20× objective lens. Dendritic arbors were then traced and analyzed with Neurolucida and Neurolucida Explorer software (MBF Bioscience).
In vitro mEPSC recordings.
As described previously (Fujino et al., 2011), acute 300 μm V1 slices were prepared on a vibratome in a cold cutting solution, placed in artificial CSF, and allowed to recover at 32°C for 30 min and then at room temperature for 30 min. Whole-cell patch-clamp recordings of mEPSCs were performed. A total of 0.2% biocytin (Sigma-Aldrich) was included in the patch pipette for some recordings. At least 200 mEPSCs, with a threshold set at 6 pA, were recorded at −70 mV from each cell.
In utero electroporations and spine analysis.
E16.5 embryos were electroporated with dio-YFP (1 μl) and Cre recombinase containing plasmids at a ratio of 10:1 (final concentration, 2 μg/μl). Mice were anesthetized using 2% isoflurane, and the uterine horns were exposed for DNA injection using a 32 gauge Hamilton syringe. Using tweezers with round plate electrodes, five pulses (50 ms, 36 V) were delivered from an ECM830 electroporator. Pups were perfused on P28 with 4% paraformaldehyde. V1 coronal sections were prepared and mounted on glass slides with Fluoromount (Southern Biotechnology Associates). Images were obtained with a 63× objective using an Olympus FluoView 300 laser-scanning confocal microscope with FluoView 500 acquisition software.
Images were analyzed in ImageJ. Spines were qualitatively classified as thin (long neck and small head or thin protrusions), stubby (large head without a neck), or mushroom (large head with a neck). All spines projecting laterally from segments of basal dendrites were counted. Analysis was performed blind to the genotype.
In vivo single-unit recordings.
In vivo recordings were performed between P28 and P32 or in adulthood (P60–P110), under Nembutal (50 mg/kg, i.p.)/chlorprothixene (0.025 mg/kg, i.m.) anesthesia using standard techniques (Gordon and Stryker, 1996; Hensch et al., 1998). For each mouse, two to four penetrations were performed across the mediolateral extent of binocular V1. Responses were recorded using multichannel probes (A1x13-3mm50-177; Neuronexus Technologies), and the signal was amplified, thresholded, and discriminated (SortClient; Plexon Technologies). Full-screen sine wave gratings (100%contrast, 0.025 cpd, 2 Hz) were presented on a screen (mean luminance, 32 cd/m2). Orientations varying between 0 and 360° (12 steps, 30° spacing) were presented in random order (3 s each, eight repeats). A uniform gray screen of intermediate luminance was used to monitor the spontaneous activity (3 s, eight repeats). Rapid plasticity was then assessed by presenting 100 successive stimulations at the best orientation (drifting bars, 100% contrast, 0.025 cpd, 2 Hz).
Single-unit isolation was ensured by analyzing the cell waveforms and discriminated on the basis of their individual characteristics (Offline Sorter; Plexon Technologies). Activity analysis was done using Matlab via SigTOOL (Lidierth, 2009). The preferred orientation was determined as the one giving the maximum response (Rmax). The orientation selectivity index (OSI) was calculated as (Rmax − Rorth)/(Rmax + Rorth), where Rorth was the mean response of the two orientations orthogonal to the preferred orientation. Among the cells with OSI > 0.3, the direction selectivity index (DSI) was calculated as (Rmax − Ropp)/(Rmax + Ropp), where Ropp is the response in the direction opposite to the preferred direction. Spontaneous activity was not subtracted from maximal evoked response. The coefficient of variation, measuring the response variability to the preferred gratings, was calculated according to Ko et al. (2013).
Criteria were imposed on the orientation tuning curves to exclude neurons with weak visual responses. Cells with a maximal evoked response more than 0.3 spike/s and significantly different from the spontaneous activity (Wilcoxon signed-rank test) were kept in the analysis. Based on their waveforms (Niell and Stryker, 2008), only excitatory cells were further analyzed. Cells were assigned to superficial layers according to the electrode's depth and the level of spontaneous activity. Cells in superficial layers are characterized by a sparse background activity, which strongly increases in deeper layers (Niell and Stryker, 2008; Durand et al., 2012).
Results
Dendritic growth and elaboration is deficient in the visual cortex of cpg15 KO mice
cpg15 is known to promote dendritic arbor development in the hippocampal dentate gyrus (Fujino et al., 2011). Therefore, we first examined L2/3 pyramidal cell dendritic arbor maturation in the visual cortex in relation to visual developmental milestones: at P10, before eye opening; at P14, shortly after eye opening and onset of cpg15 expression; and at P28, the peak of cpg15 expression and of visual cortex plasticity (Fig. 1).
Figure 1.

Abnormal growth of L2/3 pyramidal neuron basal dendrites in cpg15 KO mice. A, Example of a diolistically labeled neuron in slice (left) and after tracing (right) from a P14 mouse. B, Average number of primary dendrites per neuron. C, Total length of basal dendrites in KO and WT littermates. D–F, Scholl analysis of basal dendrites at P10 (D), P14 (E), and P28 (F). P10, n = 24 WT and KO cells; P14, n = 23 WT cells and n = 21 KO cells; P28, n = 22 WT cells and n = 25 KO cells. **p < 0.01; ***p < 0.001, Kruskal–Wallis with Dunn's post-test.
As described previously, in WT mice the number of primary branches was already established by P10 (Fig. 1B; Miller, 1981; Juraska, 1982). Higher-order branches were then added, and dendritic length gradually increased until P28 (Fig. 1C). The developmental dynamics of branch growth was different in KO cells. The number of primary branches significantly increased between P10 and P14, whereas no further change in total dendritic length was observed between P14 and P28. These changes resulted in altered complexity of dendritic arborization as shown by Scholl analysis at all ages (Kruskal–Wallis ANOVA for all ages, p < 0.0001; Fig. 1D–F).
Excitatory synapse development is abnormal in the visual cortex of cpg15 KO mice
To examine whether the abnormal dendritic arbor development in the cpg15 KO was accompanied by changes in synaptic transmission, we next analyzed synaptic function in cortical slices by recording mEPSCs from L2/3 pyramidal cells. At P10, we found no difference in mEPSC amplitudes between KO and WT mice. However, by P14, KO mice displayed a significant reduction in mEPSC amplitudes compared with controls, which was also observed at P28 (Fig. 2A–C), indicating that KO neurons received weaker synaptic input at these ages. Similarly, no difference was observed in mEPSC frequency early in development, with both KO and WT neurons showing a similar increase in mEPSC frequency between P10 and P14. However, WT mice exhibited another large increase in mEPSC frequency between P14 and P28, which KO mice did not. Consequently, at P28, mEPSC frequency was significantly lower in the KO compared with WT mice (Fig. 2D,E). The absence of a deficit in mEPSC frequency between P10 and P14 in the KO mouse despite the large amount of synaptogenesis that normally occurs during this period, together with weaker synapses at both P14 and P28, is consistent with previous studies showing that CPG15 is not required for synapse formation but is instead important for synapse maturation.
Figure 2.
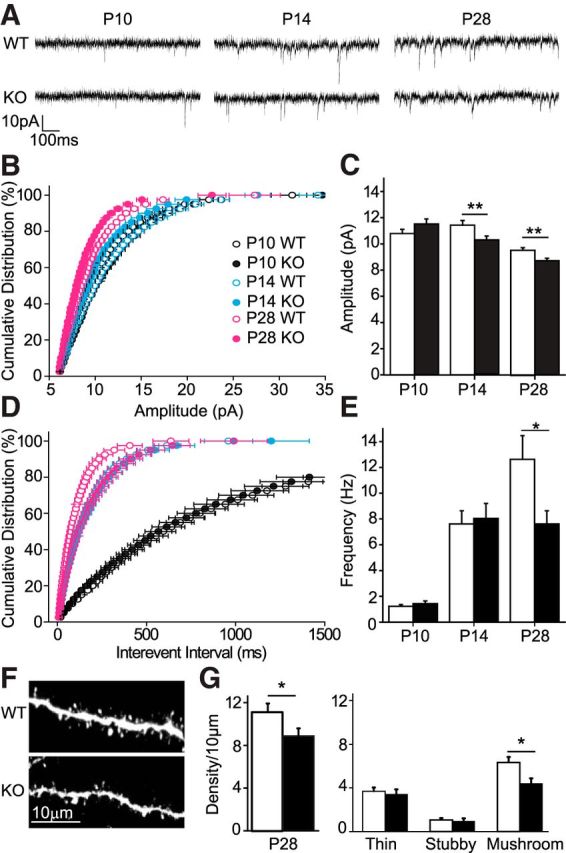
Reduced mEPSC amplitudes and frequencies in cpg15 KO mice associated with reduced mushroom spine density. A, Examples of whole-cell patch-clamp mEPSC recordings from neurons of KO and WT littermates. B, Cumulative distribution of mEPSC amplitudes. C, Average mEPSC amplitudes. D, Cumulative distribution of mEPSC interevent intervals. E, Average mEPSC frequencies (P10, n = 14 WT cells and n = 15 KO cells; P14, n = 17 WT and KO cells; P28, n = 13 WT cells and n = 14 KO cells; Mann–Whitney U test, *p < 0.05 and **p < 0.01). F, Representative images of spines on basal dendrites from L2/3 pyramidal neurons. G, Quantification of total spine density [n = 10 cells (1124 spines) in WT and n = 9 cells (726 spines) in KO] and classification as thin, stubby, or mushroom at P28 (t test, *p < 0.05).
To validate that the reduction in mEPSC frequency was attributable to a reduced number of excitatory synapses, we examined spine number and morphology on the basal dendrites of L2/3 neurons in V1 at P28. We found that the density of spine protrusions was lower in KO than in WT mice, because of a decrease in the density of mushroom-type spines, whereas the densities of thin and stubby protrusions were unaffected (Fig. 2F,G). This decrease in mushroom spine number is consistent with the reduced mEPSC frequency measured at P28, suggesting a failure of normal excitatory synapse maturation.
In vivo single-unit responses and receptive field properties are disrupted in cpg15 KO mice
To understand the functional consequences of the aberrant dendritic growth trajectories and excitatory synapse development in the cpg15 KO, we analyzed in vivo visual function. Spontaneous and maximal evoked responses of pyramidal cells were recorded in V1 during the developmental period of high plasticity (P28–P32) (Fig. 3A) and in adulthood (P60–P110). To allow direct comparison with the anatomical and in vitro data, we focused our analysis only on pyramidal neurons in superficial layers. In contrast to WT mice, where all visual parameters were mature by P28–P32 (Fig. 3 B–G), in KO mice the maximal evoked response was dramatically reduced at P28–P32 but eventually reached the WT level at adulthood (Fig. 3C). Spontaneous activity and signal-to-noise ratio were not significantly affected across ages (Fig. 3B,D). Strikingly, the coefficient of variation of response was higher at P28–P32 and decreased to the WT level at adulthood, suggesting an immature response early in development (Ko et al., 2013). The deficit in maximal evoked responses observed at P28–P32 may reflect a decrease in excitatory drive onto pyramidal cells, consistent with the reduced mEPSC frequency and mushroom spine numbers.
Figure 3.
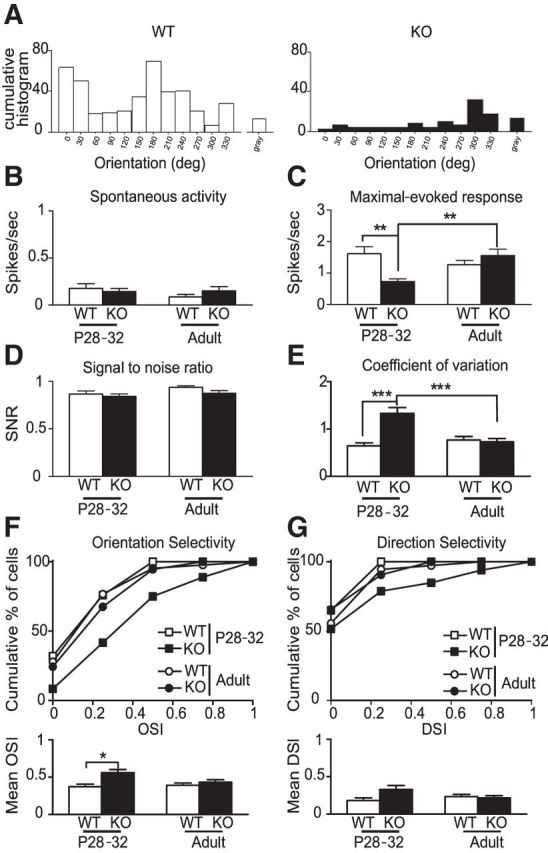
Visual responses and development of receptive field properties in cpg15 KO mice. A, Representative WT and KO cumulative histograms of spikes in response to 12 orientated gratings or a uniform gray stimulus at P28–P32. B–E, Averages of spontaneous activity (B), maximal evoked response (C), signal-to-noise ratio (D), and coefficient of variation (E) (WT, n = 24 and 36 cells; KO, n = 43 and 37 cells at P28–P32 and adult, respectively). F, Cumulative percentage of cells and average OSI. G, Cumulative percentage of cells and average DSI among cells tuned for the orientation. *p < 0.05; **p < 0.01; ***p < 0.001, Kruskal–Wallis with Dunn's post-test.
We next analyzed the expression of orientation selectivity (OS) and direction selectivity (DS), two receptive field properties that show a clear developmental profile (Wang et al., 2010; Rochefort et al., 2011; Kang et al., 2013). In WT, OS was already mature by P28–P32 with no further modification in adulthood (Fig. 3F). In contrast, KO mice displayed a significantly higher OS at P28–P32, as shown by the shift of the cumulative percentage of cell distribution toward higher OSI values and mean OSI, but decreased to the WT level in adulthood (Fig. 3F). During postnatal development, there is a significant decrease in the proportion of cells that are direction selective among orientation-selective cells without modification of the mean DSI (Rochefort et al., 2011). Here, we saw no change in DS in WT and KO mice over development.
In vivo rapid plasticity is disrupted in cpg15 KO mice
Finally, we tested whether cpg15 was required for functional plasticity of V1 neurons by examining rapid changes in their firing rates in response to repeated visual stimulation. We recorded neuronal responses throughout 100 successive stimuli at preferred orientation and compared the responses between the first block of 20 stimuli and the following blocks (Fig. 4A). For each cell, a linear regression was calculated allowing their classification into two groups: cells displaying an increased response (with a positive slope of linear regression; cells with potentiation) and ones with a decreased response (with a negative slope of linear regression; cells with depression). The slope of the linear regression describes the rate of modification over the entire stimulus train.
Figure 4.

In vivo rapid adaptation to repeated stimulation is disrupted in cpg15 KO mice. A, Experimental design and representative WT spike trains at P28–P32. B, Percentage of cells (D, cell with depression; P, cell with potentiation; =, no modification). C, Time-course responses of cells from WT and KO mice displaying an increase to repeated stimulation (*p < 0.05; **p < 0.01; ***p < 0.001, Friedman test and Dunn's post-test). D, Time-course responses of cells displaying a decrease to repeated stimulation (Friedman test and Dunn's post-test; WT, *p < 0.05, **p < 0.01, ***p < 0.001; KO, ϕp < 0.05, ϕϕp < 0.01). E, F, Average of the slope calculated over 100 stimulations (E) and the 40 first stimulations (F) (*p < 0.05; **p < 0.01, Kruskal-Wallis with Dunn's post-test). Cells with potentiation: n = 10 in WT and 9 in KO at P28–P32; n = 4 in adult KO. Cells with depression: n = 7 in WT and KO at P28–P32; n = 13 in WT and 6 in KO at adulthood.
In P28–P32 WT mice, we found both types of cells: 10 of 18 cells significantly potentiated their response over 100 stimulations (Fig 4C), whereas 8 of 18 cells depressed (Fig. 4D). In adulthood, only cells with depression were recorded (and one without modification; Fig. 4B). Although they depressed at the same level as P28–P32 (Fig 4D), this depression was faster as shown by the slope calculated over the first 40 stimulations (Fig. 4F). In KO mice, cells with potentiation significantly increased their response only after 80 stimulations at P28–P32 compared with 60 in WT. However, the magnitude or the rate of potentiation was similar to WT cells (Fig 4C). Interestingly, cells with potentiation were still present at adulthood (Fig. 4B,C). At P28–P32, KO cells displayed a significant depression of response only after 60–80 stimulations (Fig 4D). Whereas there was no difference in the relative magnitude of depression at each stimulus block between WT and KO, the rate of the decrease was significantly lower in the KO group (Fig. 4E). At adulthood, cells with depression displayed a similar profile to P28–P32 WT cells, confirming that the adaptation to repeated stimulation was slower in KO compared with WT across ages.
Discussion
Here we demonstrate that during the classic developmental peak of V1 plasticity, absence of CPG15 results in abnormal dendritic arbor elaboration and excitatory synapse maturation, accompanied by delayed maturation of receptive field properties. Once adulthood is reached, receptive field parameters normalize to the WT level, but rapid plasticity mechanisms remain significantly impaired.
Consistent with overexpression studies in the Xenopus retinotectal system demonstrating a role for CPG15 in the conversion of silent synapses to functional AMPA receptor-containing synapses (Cantallops et al., 2000), knocking out cpg15 in the mammalian visual cortex leads to abnormal synaptic function at P14 and P28, a time when AMPA receptors are being inserted into the maturing excitatory synapses in L2/3 cortical neurons (Rumpel et al., 2004). Both the electrophysiological and morphological data support a deficit in this process in the KO, leading to fewer mature, AMPA receptor-containing synapses and reduced numbers of mushroom spines at P28.
In dendritic arbor development, there is an early period of exuberant primary branch growth, followed by continued development of some and loss of others, with an eventual stabilization of the number of primary branches (Miller, 1981). This process occurs simultaneously with synapse formation and maturation. It is interesting that, whereas the WT mouse had already established a set number of primary branches by P10, in the KO there was a significant increase in the number of primary dendritic branches between P10 and P14. According to the synaptotrophic hypothesis, synapse formation and maintenance is important for the elaboration of the dendritic arbor (Cline and Haas, 2008). Therefore, impairments in synapse maturation may contribute to the deficits in the establishment of primary dendrites as well as dendrite elongation observed in the KO mouse. These data suggest that the lack of CPG15 has a destabilizing effect on dendritic branches that may be secondary to the initial deficit in synapse maturation. This reduced structural stability appears to continue through adulthood in the absence of CPG15, manifested in reduced spine stabilization in the visual cortex of adult cpg15 KO mice relative to WT (Fujino et al., 2011).
Single-unit recordings in vivo revealed direct functional effects of the abnormalities observed in vitro. The decrease in maximal evoked response at P28–P32 may be directly related to the diminished excitatory drive onto pyramidal cells. Moreover, the variability in the response and the altered orientation selectivity at P28–P32 support an important role for CPG15 in the development and stabilization of the visual cortical circuit early in development.
Whereas the involvement of the inhibitory system is not directly investigated in this study, it has been shown that orientation selectivity is mainly driven by the excitatory NMDA pathway and not by the inhibitory system (Fagiolini and Hensch, 2000). This suggests that the changes we observed in KO mice are most likely caused by excitatory network impairments. However, we cannot completely exclude a contribution of the inhibitory network to our results.
Our paradigm of short plasticity/adaptation provides additional insight regarding the role of CPG15 in neuronal circuit function. We found that in WT mice, responses to repeated visual stimulation were either potentiated or depressed similar to the stimulus-specific response potentiation described in awake mice over days by Frenkel et al. (2006) or short-term depression (STD) mechanisms in vitro. However, the potentiation in awake mice does not occur in a single session but requires several days. The underlying mechanisms may thus be very different. Unexpectedly, whereas the visual properties were normalized at adulthood, deletion of cpg15 has still a dramatic effect on the short plasticity paradigm at this age. In contrast to the fast depression in WT, depression in KO cells occurred at a slower rate and amplitude. This longer time scale is consistent with previous data showing that KO mice display poor learning skills that can be overcome with repeated training (Fujino et al., 2011). Moreover these data suggest that CPG15 may be necessary in processes comparable with STD but perhaps not in short-term potentiation (STP)-like mechanisms. Additional studies are needed to investigate why CPG15 differentially affects these two processes and how the ratio between STP/STD can affect visual circuit processing and plasticity.
To conclude, we present evidence that CPG15 is involved in the maturation of visual cortical function and in the modulation of sensory processing in vivo. These results provide new insights on how synaptic stabilization and maturation influence circuit function in the brain, which is critical for the understanding of neurodevelopmental disorders that may result from abnormal synaptic development.
Footnotes
This work was supported by National Eye Institute Grant RO1 EY011894 (E.N.), National Research Service Award Grant F31-NS0695 (J.H.L.), and a Harvard/MIT Joint Research Grant Program in Neuroscience (E.N. and M.F.).
The authors declare no competing financial interests.
References
- Cantallops I, Haas K, Cline HT. Postsynaptic CPG15 promotes synaptic maturation and presynaptic axon arbor elaboration in vivo. Nat Neurosci. 2000;3:1004–1011. doi: 10.1038/79823. [DOI] [PubMed] [Google Scholar]
- Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol. 2008;586:1509–1517. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau RA, Shatz CJ, Nedivi E. Dynamic regulation of cpg15 during activity-dependent synaptic development in the mammalian visual system. J Neurosci. 1999;19:7999–8008. doi: 10.1523/JNEUROSCI.19-18-07999.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A, Carninci P, Hensch TK, Fagiolini M. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron. 2012;76:1078–1090. doi: 10.1016/j.neuron.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Hensch TK. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 2000;404:183–186. doi: 10.1038/35004582. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel MY, Sawtell NB, Diogo AC, Yoon B, Neve RL, Bear MF. Instructive effect of visual experience in mouse visual cortex. Neuron. 2006;51:339–349. doi: 10.1016/j.neuron.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Fujino T, Lee WC, Nedivi E. Regulation of cpg15 by signaling pathways that mediate synaptic plasticity. Mol Cell Neurosci. 2003;24:538–554. doi: 10.1016/S1044-7431(03)00230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T, Wu Z, Lin WC, Phillips MA, Nedivi E. cpg15 and cpg15–2 constitute a family of activity-regulated ligands expressed differentially in the nervous system to promote neurite growth and neuronal survival. J Comp Neurol. 2008;507:1831–1845. doi: 10.1002/cne.21649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T, Leslie JH, Eavri R, Chen JL, Lin WC, Flanders GH, Borok E, Horvath TL, Nedivi E. CPG15 regulates synapse stability in the developing and adult brain. Genes Dev. 2011;25:2674–2685. doi: 10.1101/gad.176172.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Tsai J, Gan WB. Rapid labeling of neuronal populations by ballistic delivery of fluorescent dyes. Methods. 2003;30:79–85. doi: 10.1016/S1046-2023(03)00009-4. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaherian A, Cline HT. Coordinated motor neuron axon growth and neuromuscular synaptogenesis are promoted by CPG15 in vivo. Neuron. 2005;45:505–512. doi: 10.1016/j.neuron.2004.12.051. [DOI] [PubMed] [Google Scholar]
- Juraska JM. The development of pyramidal neurons after eye opening in the visual cortex of hooded rats: a quantitative study. J Comp Neurol. 1982;212:208–213. doi: 10.1002/cne.902120210. [DOI] [PubMed] [Google Scholar]
- Kang ED, Sr, LeBlanc JJ, Hensch T, Chen C, Fagiolini M. Visual acuity development and plasticity in the absence of sensory experience. J Neurosci. 2013;33:17789–17796. doi: 10.1523/JNEUROSCI.1500-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H, Cossell L, Baragli C, Antolik J, Clopath C, Hofer SB, Mrsic-Flogel TD. The emergence of functional microcircuits in visual cortex. Nature. 2013;496:96–100. doi: 10.1038/nature12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Nedivi E. Extended plasticity of visual cortex in dark-reared animals may result from prolonged expression of cpg15-like genes. J Neurosci. 2002;22:1807–1815. doi: 10.1523/JNEUROSCI.22-05-01807.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JH, Nedivi E. Activity-regulated genes as mediators of neural circuit plasticity. Prog Neurobiol. 2011;94:223–237. doi: 10.1016/j.pneurobio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidierth M. sigTOOL: a MATLAB-based environment for sharing laboratory-developed software to analyze biological signals. J Neurosci Methods. 2009;178:188–196. doi: 10.1016/j.jneumeth.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Miller M. Maturation of rat visual cortex. I. A quantitative study of Golgi-impregnated pyramidal neurons. J Neurocytol. 1981;10:859–878. doi: 10.1007/BF01262658. [DOI] [PubMed] [Google Scholar]
- Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential Cdna cloning. Nature. 1993;363:718–722. doi: 10.1038/363718a0. [DOI] [PubMed] [Google Scholar]
- Nedivi E, Fieldust S, Theill LE, Hevron D. A set of genes expressed in response to light in the adult cerebral cortex and regulated during development. Proc Natl Acad Sci U S A. 1996;93:2048–2053. doi: 10.1073/pnas.93.5.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Wu GY, Cline HT. Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science. 1998;281:1863–1866. doi: 10.1126/science.281.5384.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell CM, Stryker MP. Highly selective receptive fields in mouse visual cortex. J Neurosci. 2008;28:7520–7536. doi: 10.1523/JNEUROSCI.0623-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oray S, Majewska A, Sur M. Effects of synaptic activity on dendritic spine motility of developing cortical layer v pyramidal neurons. Cereb Cortex. 2006;16:730–741. doi: 10.1093/cercor/bhj019. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates, compact 2nd edition. Boston: Elsevier Academic; 2004. [Google Scholar]
- Rochefort NL, Narushima M, Grienberger C, Marandi N, Hill DN, Konnerth A. Development of direction selectivity in mouse cortical neurons. Neuron. 2011;71:425–432. doi: 10.1016/j.neuron.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Rumpel S, Kattenstroth G, Gottmann K. Silent synapses in the immature visual cortex: layer-specific developmental regulation. J Neurophysiol. 2004;91:1097–1101. doi: 10.1152/jn.00443.2003. [DOI] [PubMed] [Google Scholar]
- Wang BS, Sarnaik R, Cang J. Critical period plasticity matches binocular orientation preference in the visual cortex. Neuron. 2010;65:246–256. doi: 10.1016/j.neuron.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]