

Abstract
The general effects of cocaine are not well understood at the molecular level. What is known is that the dopamine D1 receptor plays an important role. Here we show that a key mechanism may be cocaine's blockade of the histamine H3 receptor-mediated inhibition of D1 receptor function. This blockade requires the σ1 receptor and occurs upon cocaine binding to σ1-D1-H3 receptor complexes. The cocaine-mediated disruption leaves an uninhibited D1 receptor that activates Gs, freely recruits β-arrestin, increases p-ERK 1/2 levels, and induces cell death when over activated. Using in vitro assays with transfected cells and in ex vivo experiments using both rats acutely treated or self-administered with cocaine along with mice depleted of σ1 receptor, we show that blockade of σ1 receptor by an antagonist restores the protective H3 receptor-mediated brake on D1 receptor signaling and prevents the cell death from elevated D1 receptor signaling. These findings suggest that a combination therapy of σ1R antagonists with H3 receptor agonists could serve to reduce some effects of cocaine.
Introduction
The striatum is involved in the learning and elicitation of reward-related behaviors. GABAergic striatal medium sized spiny neurons (MSNs) comprise ∼95% of striatal neurons and receive two main extrinsic inputs, cortical-limbic-thalamic glutamatergic and mesencephalic dopaminergic, converging in the dendritic spines of the MSNs (Ferré et al., 2007). There are two distinct MSNs, giving rise to two efferent pathways of the basal ganglia, the striatonigral MSN or direct pathway and the striatopallidal MSN or the indirect pathway. These neurons selectively express dopamine D1 (D1R) and D2 (D2R) receptors, respectively. The initiation of movement leads to dual input of these two pathways (Cui et al., 2013). Despite the dual input, it is the balance of both types of MSNs that determines the final striatal output and the facilitation and inhibition of specific motor responses involved in reward-related behavior (Gerfen and Surmeier, 2011). Drugs of abuse are able to subvert these balanced inputs by altering the cell signaling of striatopallidal and striatonigral MSN. In the case of cocaine, it binds to and inhibits the dopamine transporter (DAT) producing a large increase in extracellular dopamine (Williams and Galli, 2006). This is associated with an increase in D1R signaling while D2R density is decreased, tipping the balance of signaling toward the direct pathway (Pascoli et al., 2012; Volkow et al., 2013). Our work suggests that the role of D1R in cocaine's effects depends on the ability of σ1 receptors (σ1R) to bind and differentially modulate D1R and D2R in both MSNs (Navarro et al., 2010, 2013). Cocaine, especially in the form of crack, is associated with toxic consequences such as seizures and death. The biochemical alterations that follow the intake of cocaine are not well understood, but several studies indicate that D1R is involved in cocaine's effects (Ritz and George, 1997; Aksenov et al., 2006; Lepsch et al., 2009).
In addition to glutamatergic and dopaminergic inputs, the striatum receives hypothalamic histaminergic input, which releases histamine from asynaptic varicosities (Takagi et al., 1986). Histamine H3 receptors (H3R) are highly expressed presynaptically and postsynaptically in the striatal spine module (Ellenbroek, 2013; Panula and Nuutinen, 2013) and mostly localized postsynaptically in both types of MSN where they can control the D1R signaling through the formation of D1R-H3R receptor heteromers (Moreno et al., 2011a; Ellenbroek, 2013; Panula and Nuutinen, 2013). This heteromer acts as a relay where activated H3R can serve as a “molecular brake” for D1R signaling. This effect is reached through a molecular protein-protein interaction between receptors in the heteromer. This is a common biochemical property of receptor heteromers, and it is defined as an intermolecular interaction by which the presence of one receptor, or the ligand binding to one receptor unit in the heteromer, changes positively or negatively the binding and/or the functional properties of another receptor unit in the heteromer (Ferré et al., 2009). These interprotomer interactions or cross-talk have been described for D1R-H3R heteromers upon heteromer coactivation with agonists(Ferrada et al., 2009). Thus, through a negative cross-talk between receptors, H3R agonist decreases the D1R agonist affinity and signaling. Some receptor heteromers, including D1R-H3R heteromers, have been found to display cross-antagonism, the ability of an antagonist of one receptor to also antagonize the signaling of the partner receptor (Ferrada et al., 2009; Moreno et al., 2011b; González et al., 2012). Cross-antagonism requires heteromer formation for any cross-receptor effects as antagonists do not signal on their own. Thus, cross-antagonism can be used as a fingerprint for identification of the presence of the heteromer (Ferré et al., 2009). In this frame here we explore a new physiological role for D1R-H3R heteromers with the idea of using the H3R-mediated inhibitory effects of D1R signaling to dampen cocaine's effects. We found that H3R activation blocks D1R agonist-mediated cell death in striatal organotypic cultures. We discovered a potentially key biochemical mechanism involved in the effects of cocaine. Cocaine strongly disrupts D1R-H3R heteromer function, including the neuroprotective effects of H3R activation in the D1R-H3R heteromer. We thus propose the association of σ1R antagonists with H3 receptor agonists as a therapeutic approach for cocaine abuse.
Materials and Methods
Animals, cocaine acute treatment, and cocaine self-administration.
All animal protocols were in accordance with the European Communities Council Directive 86/609/EEC and approved by the Ethics Committee for Human and Animal Research of the Universitat Autònoma de Barcelona. A maximal effort was made to minimize the number and suffering of animals. Male Sprague Dawley rats were obtained from the Animal Service, Universitat Autònoma de Barcelona, Bellaterra, Spain. To obtain rats self-administered with cocaine, our protocol exhaustively described previously (Hoffmann et al., 2012) was used. To obtain rats acutely treated with cocaine, rats (6–11 weeks old) were conditioned to handling during previous days to a single 15 mg/kg cocaine intraperitoneal injection (free base) dissolved in saline. As naive rats, 9- to 11-week-old rats (±350 g) were housed in a temperature (21 ± 1°C) and humidity-controlled (55 ± 10%) room with a 12 h light/dark cycle with food and water ad libitum. In all cases, 4% isoflurane (2-chloro-2- (difluoromethoxy)-1,1,1-trifluoro-ethane) anesthetized animals were killed by decapitation to obtain rat brains. Brains from wild-type (WT) littermates and σ1R knock-out (KO) CD1 albino Swiss male mice (8 weeks old, 25 g) were generously provided by Laboratorios Esteve (Barcelona, Spain). The characterization of these mice has been previously described (Langa et al., 2003). Cocaine-HCl was purchased from Spanish Agencia del Medicamento #2003C00220.
Striatal slices preparation.
Mouse or rat brains were rapidly removed and placed in ice-cold oxygenated (O2/CO2: 95%/5%) Krebs-HCO3− buffer (124 mm NaCl, 4 mm KCl, 1.25 mm KH2PO4, 1.5 mm MgCl2, 1.5 mm CaCl2, 10 mm glucose and 26 mm NaHCO3, pH 7.4). The brains were sliced perpendicularly to the long axis of the striatum at 4°C using a brain matrix (Zivic Instruments). Slices (500 μm thick) were kept at 4°C in Krebs-HCO3− buffer during the dissection. Each striatal slice was transferred into an incubation tube containing 1 ml of ice-cold Krebs-HCO3− buffer. The temperature was raised to 23°C and after 30 min, the media was replaced by 2 ml of fresh Krebs-HCO3− buffer (23°C). The slices were incubated under constant oxygenation (O2/CO2: 95%/5%) at 30°C for 4–5 h in an Eppendorf Thermomixer (5 Prime). The media was replaced by 200 μl of fresh Krebs-HCO3− buffer and incubated for 30 min before the addition of any agent. Slices were treated or not with the indicated ligand for the indicated time. After the indicated incubation period, the solution was discarded, and slices were frozen on dry ice and stored at −80°C.
Organotypic striatal slice cultures and cell death determination.
Coronal striatal slices (500 μm thickness) from 7- to 8-week-old rats were obtained using a brain matrix and cultured in Neurobasal medium supplemented with 20% horse serum, 0.5% B27, 2 mm l-glutamine, 100 μg/ml sodium pyruvate, MEM Non-Essential Amino Acids Solution (1/100), and 100 U/ml penicillin/streptomycin (all supplements were from Invitrogen). After 24 h, culture medium was replaced by fresh medium containing no ligands or the indicated concentrations of the σ1R ligands cocaine or PRE-084. After 1 h, medium, SCH 23390, imetit, or thioperamide were added at the indicated concentrations and incubated for an additional 1 h before the addition of D1R agonist. Slices were maintained 48 h more in culture. When the σ1R antagonist PD 144418 was used it was added 1 h before the addition of cocaine. After the total 72 h of culture, 10 μm propidium iodide (PI) was added and maintained at 37°C for 1 h, then slices were washed twice in cold-PBS and fixed with 4% paraformaldehyde for 1 h at 4°C. Slices were stained with Hoechst 1:1000 and total and PI-positive nuclei were quantified using Leica SP2 confocal microscope (20×; UV, 561 lasers) and the quantification program Image-based Tool for Counting Nuclei for ImageJ.
Cell culture, expression vectors, and transient transfection.
Human neuroblastoma SK-N-MC cell clone expressing human D1R and H3R was developed and grown as previously described (Ferrada et al., 2009) Human embryonic kidney (HEK293T) cells were grown in DMEM supplemented with 2 mm l-glutamine, 100 μg/ml sodium pyruvate, 100 U/ml penicillin/streptomycin, MEM Non-Essential Amino Acids Solution (1/100), and 5% (v/v) heat-inactivated fetal bovine serum. For transfection, sequences encoding fusion proteins H3R-Rluc, D1R-Rluc, σ1R-YFP, H3R-YFP, D1R-YFP, and σ1R-GFP2, which correspond with the human receptors fused to Renilla luciferase (Rluc) or to fluorescent proteins YFP or GFP2 on the C-terminal end of the receptor, were obtained and characterized as previously described (Ferrada et al., 2009; Navarro et al., 2010, 2013). Cells were transiently transfected with the corresponding fusion protein cDNA by the ramified polyethylenimine (PEI; Sigma) method (Ferrada et al., 2009; Navarro et al., 2010, 2013) or by the lipofectamine (Invitrogen) method, in transfections where siRNA was present, following the instructions of the supplier. siRNA that targets both human and rodent σ1R RNA and a scrambled control siRNA were purchased from Invitrogen (catalog #HSS 145543). To control the cell number, sample protein concentration was determined using a Bradford assay kit (Bio-Rad) using bovine serum albumin (BSA) dilutions as standards.
Lentivirus production and cell transduction.
Silencing lentiviral vectors were produced by cotransfecting HEK293 cells with lentiviral silencing plasmids GIPZ Human SIGMAR1 shRNA (Clone Id:V3LHS_377499 or V3LHS_406026; Thermo Scientific) with packing plasmid psPAX2 and envelope coding plasmid pMD2.G (Addgene #12260 and #12259, respectively) using the calcium phosphate method. For production of control nonsilencing lentiviral particles the σ1R silencing plasmids were substituted with GIPZ Non-silencing Lentiviral shRNA Control (RHS4346; Thermo Scientific). Infectious lentiviral particles were harvested at 48 h post-transfection, centrifuged 10 min at 3000 rpm to get rid of cell debris, and then filtered through 0.45 μm cellulose acetate filters. The titer of recombinant lentivirus was determined by serial dilution on HEK293T cells. For lentivirus transduction, SK-N-MC cells were subcultured to 50% confluence, cells were transduced with σ1R-shRNA-expressing lentivirus obtained with plasmid V3LHS_377499 (LV sh) or control-shRNA-expressing lentivirus (LV control) at a multiplicity of infection of 10 in the presence of polybrene 5 μg/ml. Virus-containing supernatant was removed after 3 h. Puromycin was added to the culturing media at the final concentration of 1 μg/ml 2 d after infection. Five days after puromycin selection cells were transduced with the second σ1R-shRNA-expressing lentivirus obtained with plasmid V3LHS_406026 to improve the level of silencing achieved. LV control-infected cells were reinfected with control-shRNA-expressing lentivirus. The second infection was performed as the first one. Cells were tested 72 h after the second transduction was performed.
RNA and real-time PCR.
RNA was extracted using TRIzol Reagent (Molecular Research Center). cDNA was synthesized from 2 μg total RNA (High Capacity cDNA Reverse Transcription Kit; Applied Biosystems). The mRNAs of GAPDH and σ1R were measured by real-time (RT)-PCR with 1 μL cDNA and power SYBER green PCR Master Mix (Applied Biosystems). Primer sequences are as follows: hGAPDH For: GGCTGGGGCTCATTTGCAGGG, hGAPDH Rev: TGACCTTGGCCAGGG GTGCT, σ1R For: TTCCAGCGCGAAGAGATAGC, and σ1R Rev: CCGTGTACTA CCGTCTCCC. Thermal cycling conditions for amplification were set at 50°C for 30 min and 95°C for 10 min, respectively. PCR denaturing was set at 95°C for 15 s and annealing/extending at 60°C for 60 s for 40 cycles. mRNA levels normalized for GAPDH are expressed as fold change relative to control cells. The results were quantified with the comparative Ct method (known as the 2−δδCt method).
Bioluminescent resonance energy transfer assays.
HEK293T cells were transiently cotransfected with a constant amount of cDNA encoding for the receptor fused to Rluc and with increasingly amounts of cDNA corresponding to the receptor fused to YFP (see figure legends). To quantify receptor-YFP expression, fluorescence of cells (20 μg protein) was read in a Fluoro Star Optima Fluorimeter (BMG Labtechnologies) equipped with a high-energy xenon flash lamp, using a 10 nm bandwidth excitation filter at 400 nm reading. For bioluminescent resonance energy transfer (BRET) or BRET with BiLFC measurements, 5 μm coelenterazine H (Invitrogen) was added to the equivalent of 20 μg of cell suspension. After 1 min for BRET or after 5 min for BRET with BiLFC after adding coelenterazine H, the readings were collected using a Mithras LB 940 that allows the integration of the signals detected in the short-wavelength filter at 485 nm and the long-wavelength filter at 530 nm. To quantify receptor-Rluc expression, luminescence readings were also performed after 10 min of adding 5 μm coelenterazine H. The net BRET is defined as [(long-wavelength emission)/(short-wavelength emission)] − cf., where cf. corresponds to [(long-wavelength emission)/(short-wavelength emission)] for the donor construct expressed alone in the same experiment. Data were fitted to a nonlinear regression equation, assuming a single phase saturation curve with GraphPad Prism software. BRET is expressed as mili BRET units (mBU; net BRET × 1000).
Sequential resonance energy transfer assays.
For sequential resonance energy transfer (SRET) assays, HEK293T cells growing in six-well plates were transiently cotransfected with constant amounts of cDNAs encoding for both receptor fused to Rluc and GFP2 proteins and with increasing amounts of cDNA corresponding to the receptor fused to YFP protein. In SRET, the oxidation of the Rluc substrate DeepBlueC by receptor-Rluc triggers receptor-GFP2 excitation (BRET), which triggers a subsequent excitation of receptor-YFP (FRET). Emission of YFP after addition of DeepBlueC is only possible if the three fusion proteins are in close proximity (<10 nm), allowing sequential bioluminescent and fluorescent resonance energy transfer to occur. Cells were used 48 h post-transfection. Using aliquots of transfected cells (20 μg of protein), different determinations were performed in parallel. (1) Quantification of protein-YFP expression was performed as indicated in BRET experiments. (2) For quantification of protein-Rluc expression cells were distributed in 96-well microplates (Corning 3600, white plates with white bottom), and luminescence was determined 10 min after addition of 5 μm coelenterazine H in a Mithras LB 940 multimode reader. (3) For SRET, cells were distributed in 96-well microplates (Corning 3600, white plates with white bottom), and 5 μm DeepBlueC (Invitrogen) was added. The SRET signal was collected using a Mithras LB 940 reader with detection filters for short wavelength (410 nm) and long wavelength (530 nm). Analogous with BRET, net SRET is defined as [(long wavelength emission)/(short wavelength emission)] − cf., where cf. corresponds to long wavelength emission/short wavelength emission for cells expressing protein-Rluc and protein-GFP2. Linear unmixing was done for SRET quantification, taking into account the spectral signature to separate the two fluorescence emission spectra (Zimmermann et al., 2002). SRET is expressed as mili SRET units, mSU (net SRET × 1000). Data were fitted as in BRET experiments.
Striatal membrane preparation and coimmunoprecipitation experiments.
Mouse or rat striatal tissue was homogenized in 50 mm Tris-HCl buffer, pH 7.4, containing a protease inhibitor cocktail (1/1000; Sigma). The cellular debris was removed by centrifugation at 13,000 × g for 5 min at 4°C, and membranes were obtained by centrifugation at 105,000 × g for 1 h at 4°C. Membranes were washed three more times in the same conditions and were used for ligand binding assays or were solubilized for coimmunoprecipitation experiments with a Dynabeads Protein G kit (Invitrogen) using rat anti-D1 receptor antibody (1:1000; Sigma). As negative control anti-calnexin antibody (1:1000; BD Biosciences PharMingen) was used. Immunoprecipitates were separated on SDS-polyacrylamide gel and transferred onto PVDF membranes to perform Western blot assays as previously described (Navarro et al., 2010; Moreno et al., 2011a) using as primary antibodies guinea pig anti-D1R antibody (1:1000; Frontier Institute), rabbit anti-H3R antibody (1:1000; Alpha Diagnostic), or mouse anti-σ1R antibody B-5 (sc-137075) (1:800; Santa Cruz Biotechnology).
In situ proximity ligation assays.
Mouse or rat striatal slices were mounted on slide glass, thawed at 4°C, washed in 50 mm Tris-HCl, 0.9% NaCl, pH 7.8 buffer (TBS), permeabilized with TBS containing 0.01% Triton X-100 for 10 min, and successively washed with TBS. Heteromers were detected using the Duolink II in situ PLA detection Kit (OLink Bioscience) following the instructions of the supplier. A mixture of equal amounts of the primary antibodies–guinea pig anti-D1R antibody (see above) and rabbit anti-H3R antibody (see above) or mouse anti-σ1R antibody B-5 (see above) and guinea pig anti-D1R antibody or mouse anti-σ1R antibody B-5 and rabbit anti-H3R antibody–were used to detect D1R-H3R, σ1R-D1R and σ1R-H3R heteromers. respectively. Slices were mounted using the mounting medium with DAPI. The samples were observed in a Leica SP2 confocal microscope (Leica Microsystems). Images were opened and processed with ImageJ confocal.
Cell signaling.
The cAMP concentration was determined by HTRF methodology using the LANCE Ultra cAMP kit (PerkinElmer) and a PHERAstar Flagship Microplate Reader (BMG Labtechnologies) following the instructions of the supplier. To determine ERK 1/2 phosphorylation, cells and mouse or rat striatal slices were treated or not with the indicated ligand for the indicated time and were lysed by the addition of 500 μl of ice-cold lysis buffer (50 mm Tris-HCl, pH 7.4, 50 mm NaF, 150 mm NaCl, 45 mm β-glycerophosphate, 1% Triton X-100, 20 μm phenyl-arsine oxide, 0.4 mm NaVO4, and protease inhibitor cocktail). The cellular debris was removed by centrifugation at 13,000 × g for 5 min at 4°C and the protein was quantified by the bicinchoninic acid method using BSA dilutions as standard. ERK1/2 phosphorylation were then detected as described previously (Moreno et al., 2011a). Arrestin recruitment was determined by BRET experiments (see above) in HEK293T cells 48 h after transfection with the indicated amounts of cDNA corresponding to D1R, β-arrestin 1-Rluc, and H3R-YFP. Cells (20 μg of cell suspension protein per well in a 96-well microplates) were not treated or treated for 30 min with 30 μm cocaine or 1 μm of the antagonists SCH 23390 or thioperamide alone or in combination, and 5 μm coelenterazine H was added before stimulation with the agonists SKF 38393 (100 nm) or imetit (100 nm) for 12 min and BRET between β-arrestin 1-Rluc and H3R-YFP was determined as above.
Radioligand binding experiments and binding data analysis.
Ligand binding was performed with rat striatal membranes (0.2 mg of protein/ml) in 50 mm Tris-HCl buffer, pH 7.4, containing 10 mm MgCl2, at 25°C. For competition experiments, membranes were incubated with free 2 nm [3H]SCH 23390 (PerkinElmer) or free 3 nm [3H]R-a-methyl histamine ([3H]RAMH; GE Healthcare) or free 2 nm [3H] YM-09151-02 (PerkinElmer) in the presence or the absence of increasing free concentrations of SKF 38393 (0.01 nm–30 μm; Tocris Bioscience) with or without 2 nm RAMH (Sigma), increasing free concentrations of SCH 23390 (0.01 nm–10 μm; Sigma), increasing free concentrations of RAMH (0.01 nm–30 μm), or increasing free concentrations of PRE-084 (0.01 nm to 10 μm; Tocris Bioscience). In all cases, membranes were incubated with ligands providing enough time to achieve stable equilibrium for the lower ligand concentrations. Nonspecific binding was determined in the presence of 30 μm nonlabeled ligand and confirmed that the value was the same as calculated by extrapolation of the competition curves. Free and membrane bound ligands were separated by rapid filtration of 500 μl aliquots in a cell harvester (Brandel) through Whatman GF/C filters embedded in 0.3% PEI that were subsequently washed for 5 s with 5 ml of ice-cold Tris-HCl buffer. The filters were incubated overnight with 10 ml of Ecoscint H scintillation mixture (National Diagnostics) at room temperature and radioactivity counts were determined using a Tri-Carb 1600 scintillation counter (PerkinElmer) with an efficiency of 62%. Protein was quantified by the bicinchoninic acid method (Pierce Chemical) using BSA dilutions as standard.
Competition curves were analyzed by nonlinear regression, using the commercial GraFit software (Erithacus software), by fitting the binding data to the two-state dimer receptor model (Franco et al., 2005, 2006) To calculate the macroscopic equilibrium dissociation constants Equation 1 deduced by Casadó et al. (2007) was used.
 |
where A represents the radioligand concentration, RT is the total amount of receptor dimers, and KDA1 and KDA2 are the macroscopic dissociation constants describing the binding of the first and the second radioligand molecule (A) to the dimeric receptor; B represents the assayed competing compound concentration, and KDB1 and KDB2 are, respectively, the equilibrium dissociation constants of the first and second binding of B. KDAB can be described as a hybrid equilibrium radioligand/competitor dissociation constant, which is the dissociation constant of B binding to a receptor dimer semi-occupied by A.
To quantify cooperativity, the dimer cooperativity index for the competing ligand B is defined by Casadó et al. (2007) as follows:
Depending on the characteristics of the ligands (the radioligand A and the competitor B) the following simplifications of the Equation 1 were considered:
For A ([3H]SCH 23390) noncooperative and B (SKF 38393) cooperative (biphasic competition curves; Table 1): Equation 1 can be simplified to Equation 3 due to the fact that KDA2 = 4KDA1 and, therefore, KDA1 is enough to characterize the binding of the radioligand A (Casadó et al., 2007):
 |
For A ([3H] YM-09151–02) and B (Bmax determination for σ1R in Fig. 7): Equation 1 was simplified to Equation 4 due to the fact that KDA2 = 4KDA1 and KDB2 = 4KDB1 (Casadó et al., 2007):
 |
For A ([3H]SCH 23390 or [3H]RAMH) and B (SCH 23390 or RAMH) noncooperative being the A and B the same compound (Bmax determination for D1R or H3R in Fig. 7): Equation 1 was simplified to Equation 5 due to the fact that KDA2 = 4KDA1, KDB2 = 4KDB1, KDA1 = KDB1, KDA2 = KDB2 and KDAB = 4KDA1 (Casadó et al., 2007):
 |
Goodness of fit was tested according to reduced χ2 value given by the nonlinear regression program.
Table 1.
Cocaine blocks the positive allosteric interaction between D1R and H3R.
| Competition experiment ([3H]SCH vs SKF38393) | Parameters |
|
|---|---|---|
| KD1 (nm) | KD2 (nm) | |
| Not treated rats in the absence of cocaine | ||
| Control | 31 ± 4 | 1100 ± 200 |
| +RAMH | 14 ± 1* | 320 ± 10* |
| Not treated rats in the presence of cocaine | ||
| Control | 28 ± 2 | 1000 ± 100 |
| +RAMH | 30 ± 4 | 1100 ± 200 |
| Cocaine acutely treated rats | ||
| Control | 28 ± 4 | 440 ± 60 |
| +RAMH | 19 ± 4 | 390 ± 40 |
| Sham rats | ||
| Control | 26 ± 4 | 700 ± 100 |
| +RAMH | 3 ± 2** | 130 ± 40** |
| Cocaine self-administered rats | ||
| Control | 33 ± 6 | 800 ± 200 |
| +RAMH | 37 ± 9 | 1900 ± 600 |
Competition curves of the D1R antagonist [3H]SCH 23390 (2 nm) binding versus increasing concentrations of the D1R agonist SKF 38393 were performed in the absence or the presence of 2 nm of the H3R agonist RAMH using striatal membranes from control rats treated or not with 30 μm cocaine for 30 min, rats acutely treated with cocaine, or cocaine self-administered rats. KD1 and KD2 are, respectively, the equilibrium dissociation constants of SKF 38393 binding to D1R and were determined by fitting binding data to Equation 3. Parameters are mean ± SEM (n = 3). For each group, significant differences with respect to control were calculated by an unpaired Student's t test (*p < 0.05, **p < 0.01).
Figure 7.

Receptor expression in the striatum of cocaine acutely treated rats compared with sham rats. The expression of H3R (white bars), D1R (gray bars), and σ1R (black bars) in striatal membranes from a pool (n = 5) of sham rats or rats acutely treated (24 h) with cocaine (cocaine) was determined by competition experiments of [3H]RAMH (3 nm) versus RAMH, [3H]SCH 23390 (2 nm) versus SCH 23390, or [3H] YM-09151-02 (2 nm) versus PRE-084, respectively. Competition curves were monophasic and binding data were fitted to Equations 4 or 5 (see Materials and Methods) to calculate the maximum binding.
Results
D1R-H3R heteromers are signaling units that can be modulated by cocaine
It has previously been described that the formation of D1R-H3R heteromers in neuroblastoma cells leads to a change in the D1R G-protein coupling from the Gs to the Gi protein, to which H3R is coupled. In fact, in the presence of the H3R, D1R was no longer coupled to Gs, and could not activate adenylyl cyclase, yet the heteromer remains coupled to Gi (Ferrada et al., 2009). This change in G-protein coupling was identified as a specific characteristic of the heteromer. We first examined whether cocaine is able to influence the G-protein coupling to D1R-H3R heteromers by measuring cAMP production, which is tied to the activation of G-proteins. In SK-N-MC neuroblastoma cells expressing D1R and H3R, the D1R agonist SKF 38393 was able to reduce the forskolin-stimulated cAMP production in accordance with a Gi protein coupling (Fig. 1a). Interestingly, in the presence of 30 μm cocaine, SKF 38393 increases cAMP production and did not affect forskolin-induced cAMP, in agreement with a Gs coupling (Fig. 1a). In contrast with D1R agonist, the H3R agonist imetit induced a decrease in cAMP production in the presence or in the absence of cocaine in accordance with a Gi-protein coupling (Fig. 1a). Similar results were obtained using the H3R agonist RAMH and the D1R agonist SKF 81297 (Fig. 1b). These data suggest that cocaine may alter the G-proteins associated with the heteromer or at a minimum alter D1R-H3R signaling. Next, we examined whether cocaine could disrupt the cross-antagonism between both receptors in the heteromer previously described (Moreno et al., 2011a). In SK-N-MC neuroblastoma cells expressing D1R and H3R, the D1R agonist SKF 38393 induced ERK 1/2 phosphorylation that was similar in the absence or the presence of cocaine suggesting that this signal was G-protein independent. SKF 38393-induced ERK 1/2 phosphorylation was blocked by its own antagonist SCH 23390 as well as by the H3R antagonist thioperamide, but this cross-antagonism was not seen in cells treated with 30 μm cocaine (Fig. 1c). Analogously, the H3R agonist imetit-induced ERK 1/2 phosphorylation was reverted by both thioperamide and the D1R antagonist SCH 23390. Again, the cross-antagonism was lost in the presence of cocaine and, in these conditions, the signaling induced by imetit was very low (Fig. 1c) in agreement with the requirement of D1R-H3R heteromers for imetit to signal via the ERK 1/2 pathway (Moreno et al., 2011a).
Figure 1.

Cocaine alters the functional characteristics of D1R-H3R heteromers in transfected cells. a–c, f, Neuroblastoma SK-N-MC cells stable expressing D1R and H3R were not treated or treated overnight with 30 μm cocaine. a, b, cAMP production was determined after 10 min stimulation with D1R agonist SKF 38393 (1 μm) or the H3R agonist imetit (1 μm) (a) or with D1R agonist SKF 81297 (100 nm) or the H3R agonist RAMH (100 nm) (b) in the absence or in the presence of 0.5 μm forskolin. Values (cAMP produced in each condition minus basal stimulation in the absence of forskolin or agonists) represent mean ± SEM, n = 4–8. One-way ANOVA followed by Bonferroni post hoc test showed a significant (*p < 0.05, **p < 0.01, ***p < 0.001) effect over basal (cells not stimulated) or (#p < 0.05, ###p < 0.001) over forskolin effect (cells stimulated with forskolin). c, f, Cells infected with the empty virus vector (c) or infected with lentiviral particles expressing shRNA (shσ1R) to silence the receptor (f) were treated for 30 min with the D1R antagonist SCH 23390 (1 μm) or the H3R antagonist thioperamide (1 μm) before the addition of SKF 38393 (100 nm) or imetit (100 nm) for an additional incubation period of 10 min and ERK 1/2 phosphorylation was determined by Western blot. Values represent mean ± SEM (n = 3–5) of percentage of phosphorylation relative to basal levels found in untreated cells. One-way ANOVA followed by Bonferroni post hoc tests showed a significant (*p < 0.05, **p < 0.01) effect over basal or of the antagonist plus agonist treatment over the agonist treatment (#p < 0.05). d, β-Arrestin 1 recruitment was measured by BRET experiments in HEK293T cells 48 h post-transfection with 2 μg of cDNA corresponding to D1R, 1 μg of cDNA corresponding to β-arrestin 1-Rluc, and 3 μg of cDNA corresponding to H3R-YFP. Cells were not treated (BRET < 10) or treated for 30 min with 30 μm cocaine, 1 μm SCH 23390, or 1 μm thioperamide alone or in combination before 10 min stimulation with SKF 38393 (100 nm) or imetit (100 nm). Values represent mean ± SEM, n = 4. One-way ANOVA followed by Bonferroni post hoc tests showed a significant effect over basal (**p < 0.01, ***p < 0.001) or of the antagonist plus agonist treatment over the agonist treatment (##p < 0.01, ###p < 0.001). e, σ1R expression was determined after silencing of σ1R by shRNA. Neuroblastoma SK-N-MC cells stable expressing D1R and H3R were infected with lentiviral particles containing either control (LV control) or shRNA specific for σ1R (LV sh σ1R) and the relative expression levels were tested using quantitative RT-PCR. Results are expressed as an average of three replicates.
Apart from G-protein-mediated signaling, many GPCRs are able to signal by an arrestin recruitment-dependent mechanism with p-ERK 1/2 being one of the signaling pathways downstream of arrestin (Shenoy and Lefkowitz, 2003; Shenoy et al., 2006; DeWire et al., 2007). If cocaine is able to alter the D1R-H3R heteromer-driven activation of ERK 1/2 then one possibility is that it also alters arrestin recruitment. Using BRET to measure β-arrestin 1 recruitment we investigated if cocaine is able to modulate the ligand-induced β-arrestin 1 recruitment to these receptors. In HEK293 cells transiently expressing D1R, β-arrestin 1-Rluc, and H3R-YFP, a positive BRET signal between β-arrestin 1 and H3R was detected after stimulation with H3R agonist imetit or with D1R agonist SKF 38393 (Fig. 1d). Interestingly, the BRET signal was diminished when cells were coactivated with both agonists, indicating a negative cross-talk between both receptors and in line with that seen in neurons (Arias-Montaño et al., 2001). In addition, we could also detect cross-antagonism in the β-arrestin 1 recruitment assays, that is, the BRET induced by SKF 38393 was blocked by the H3R antagonist thioperamide and the BRET induced by imetit was blocked by the D1R antagonist SCH 23390 (Fig. 1d). These results match what was seen with the p-ERK 1/2, suggesting that the earlier signaling changes may be due to changes in β-arrestin 1 recruitment. This negative cross-talk and cross-antagonism at the level of β-arrestin 1 recruitment were not observed in cells treated with 30 μm cocaine (Fig. 1d), supporting the hypothesis that cocaine can alter the function of D1R-H3R heteromers. In the presence of cocaine, the imetit-induced β-arrestin 1 recruitment was very low (Fig. 1d) indicating the need for functional D1R-H3R heteromers for imetit to induce arrestin recruitment as well as ERK 1/2 signaling.
We and others have shown that one of the targets of cocaine is the chaperone-like protein, σ1R, which can interact with and modulate D1R (Hiranita et al., 2010, 2013; Navarro et al., 2010; Katz et al., 2011; Kourrich et al., 2012, 2013). From these studies it is tempting to hypothesize that σ1R can mediate cocaine's effect by molecularly interacting with D1R-H3R heteromers. To test this, we first repeated the signaling in neuroblastoma cells using cells in which σ1R was silenced by shRNA infection (Fig. 1e). In these cells, cocaine did not affect the cross-antagonism or the imetit-induced signaling (Fig. 1f), demonstrating that σ1R are indeed mediating cocaine's effects on D1R-H3R signaling. These results show that σ1R is required for cocaine's effects but does not demonstrate a molecular interaction between σ1R and D1R-H3R heteromers. To test first if σ1R can directly interact with H3R we performed BRET experiments in HEK293T cells expressing a constant amount of H3R-Rluc and increasing amounts of σ1R-YFP. As shown in Figure 2a, we did not detect energy transfer in these cells nor in cells treated with 30 μm cocaine for 30 min, suggesting a lack of interaction between H3R and σ1R. Next we used SRET (Carriba et al., 2008) to study if H3R and σ1R can interact in the presence of D1R. For this, Rluc was fused to H3R to act as a BRET donor, GFP2 was fused to D1R to act as a BRET acceptor and as a FRET donor, and YFP was fused to σ1R to act as a FRET acceptor (Fig. 2b). In HEK293T cells expressing a constant amount of H3R-Rluc and D1R-GFP2 and increasing amounts of σ1R-YFP, a positive SRET saturation curve was obtained (Fig, 2b, red curve) with a SRETmax of 114 ± 18 mSU and SRET50 of 41 ± 18. These results indicate that σ1Rs can complex with D1R-H3R heteromers through an interaction with D1R in the heteromer. As cocaine can bind σ1R at pharmacological relevant concentrations (Matsumoto et al., 2003; Hayashi and Su, 2005; Hiranita et al., 2010, 2013; Kourrich et al., 2012) we next wanted to examine whether cocaine is able to modulate the D1R-H3R heteromer at the molecular level via σ1R. When the SRET experiments described above were performed in cells treated 30 min with 30 μm cocaine (Fig. 2b, black curve), a positive SRET saturation curve was obtained with a slight increase in the maximum energy transfer (SRETmax = 145 ± 19 mBU and SRET50 = 48 ± 17), suggesting that cocaine binding changes the quaternary structure of the heteromer decreasing the distance or favoring the orientation between the donor and acceptor proteins. The cocaine-induced structural changes were also observed in BRET saturation curves for the D1R-Rluc-H3R-YFP pair in cells expressing a constant amount of D1R-Rluc and increasing amounts of H3R-YFP where there was an increase in energy transfer after cells were treated with 30 μm cocaine for 30 min (Fig. 2c, red curve; BRETmax = 52 ± 2 mBU and BRET50 = 14 ± 2 vs black curve, BRETmax = 68 ± 6 mBU and BRET50 = 16 ± 4). This effect was reverted in cells whose endogenous σ1R expression was silenced using siRNA previously described (Navarro et al., 2010; Fig. 2c, green curve; BRETmax = 49 ± 4 mBU and BRET50 = 11 ± 3). Similar results were obtained in cells treated with 30 μm cocaine for 24 h (Fig. 2d). These results suggest that cocaine binding to σ1R is not disrupting D1R-H3R heteromers, but is inducing structural changes in D1R-H3R heteromers. Thus, it is clear that in transfected cells cocaine, by binding to σ1R, alters the D1R-H3R heteromer such that it can diminish H3R receptor ability to modify D1R signaling.
Figure 2.

σ1R-D1R-H3R complexes detected by energy transfer experiments. a, c, d, BRET saturation experiments were performed using HEK293T cells 48 h post-transfection with 0.2 μg of cDNA corresponding to H3R-Rluc (a) or 0.15 μg of cDNA corresponding to D1R-Rluc (c, d) and increasing amounts of cDNA corresponding to σ1R-YFP (a; 0.1–0.5 μg cDNA) or H3R-YFP (c, d; 0.2–1.5 μg cDNA). Cells were treated for 30 min (a, c) or 24 h (d) with medium (red lines) or with 30 μm cocaine (black lines) prior BRET determination. c, d, Cells were also transfected with siRNA for σ1R (see Materials and Methods; green line). Both fluorescence and luminescence of each sample were measured before every experiment to confirm similar donor expressions (∼110,000 bioluminescence units) while monitoring the increase in acceptor expression (10,000–40,000 fluorescence units). b, SRET saturation experiments were performed using HEK293T cells 48 h post-transfection with 0.3 μg of cDNA corresponding to H3R-Rluc, 0.23 μg of cDNA corresponding to σ1R-GFP2, and increasing amounts of cDNA corresponding to D1R-YFP (0.05–0.6 μg). Cells were treated for 30 min with medium (red lines) or with 30 μm cocaine (black lines) prior to SRET determination. The relative amount of BRET or SRET is given as a function of 100 × the ratio between the fluorescence of the acceptor (YFP) and the luciferase activity of the donor (Rluc). BRET and SRET are expressed as mBU or mSU, respectively, and values are means ± SEM; three to four different experiments grouped as a function of the amount of acceptor. a, b, Top, A schematic representation of the BRET (a) and SRET (b) techniques is shown.
The σ1R mediates cocaine's effects on D1R-H3R heteromer function in the striatum
Although the σ1R-mediated ability of cocaine to modulate the structure and signaling of D1R-H3R heteromers in cells is suggestive of a novel cocaine mechanism, it is important to show that similar events can occur in tissue. First we tested whether σ1R-D1R-H3R complexes are expressed in WT and σ1R KO mice striatum by coimmunoprecipitation experiments and by proximity ligation assays (PLA) using specific antibodies we described previously (Moreno et al., 2011a; Navarro et al., 2013). By Western blot experiments we found the expression of both D1R and H3R in the striatum of WT and KO mice and the expression of σ1R receptors in WT but not in the striatum of σ1R KO mice (Fig. 3a). In coimmunoprecipitation experiments (Fig. 3b–d) the antibody against D1R could indeed coprecipitate D1R (Fig. 3b) and H3R (Fig. 3c) in WT or KO mice striatal membranes, and this antibody also coimmunoprecipitates σ1R from WT striatum but not from KO striatum (Fig. 3d). It is interesting to note that the D1R-H3R or D1R-σ1R coimmunoprecipitation increased after treatment with 30 μm cocaine (Fig. 3c and d). This could be due to a cocaine-induced higher σ1R expression at the membrane (Hayashi and Su, 2005; Navarro et al., 2010) where heterocomplex formation might be favored. The coimmunoprecipitation results are in line with the BRET experiments above and highly suggestive that σ1R-D1R-H3R complexes are expressed in the striatum of WT animals and that D1R-H3R heteromer expression is maintained in the striatum of σ1R KO mice. Interestingly, the coprecipitation of H3R by anti-D1R is more efficient in the presence of cocaine, suggesting a change in the strength of interaction, consistent with the increase in the SRET detected in Figure 2b. Furthermore we used the PLA to explore whether D1R-H3R heteromer expression was maintained in σ1R KO mice and whether σ1R-D1R-H3R complexes could be detected in WT mice. PLA requires that both receptors be close enough to allow the two different antibody-DNA probes to be able to ligate (<17 nm; Söderberg et al., 2008; Trifilieff et al., 2011). If the receptors are within sufficient proximity, a punctate fluorescent signal can be detected by confocal microscopy. Using striatal slices from both WT (Fig. 3e–g) and σ1R KO (Fig. 3h–j), a red punctate fluorescent stain surrounding DAPI-positive nuclei was observed for the H3R-D1R (Fig. 3e), D1R-σ1R (Fig. 3f), or H3R-σ1R (Fig. 3g) pairs in WT mouse striatum but only H3R-D1R heteromers were detected in σ1R KO animals (Fig. 3h). These data confirm the coprecipitation experiments above. As a negative control we checked that the red spots were not seen in slices treated with only one primary antibody and both secondary antibody-DNA probes (Fig. 3 k–m). To determine the cocaine effect on the striatal D1R-H3R heteromerization we performed PLA experiments in the presence of cocaine. In these conditions, results from PLA experiments were very similar to the ones obtained in the absence of cocaine (results not shown), indicating that σ1R, D1R, and H3R are indeed in a complex in mice striatum in the presence or absence of cocaine. Next, by using the coimmunoprecipitation and PLA experiments described above, we observed that σ1R-D1R-H3R complexes are expressed in the rat striatum since the antibody against D1R could indeed coprecipitate D1R, H3R, and σ1R in the absence or the presence of cocaine and we detected red spots in the PLA experiments for the pairs H3R-D1R, D1R-σ1R, or H3R-σ1R (Fig. 4). These results suggest that σ1R-D1R-H3R complexes are expressed in rat striatum and that cocaine, by binding to σ1R, does not disrupt the heteromer per se.
Figure 3.
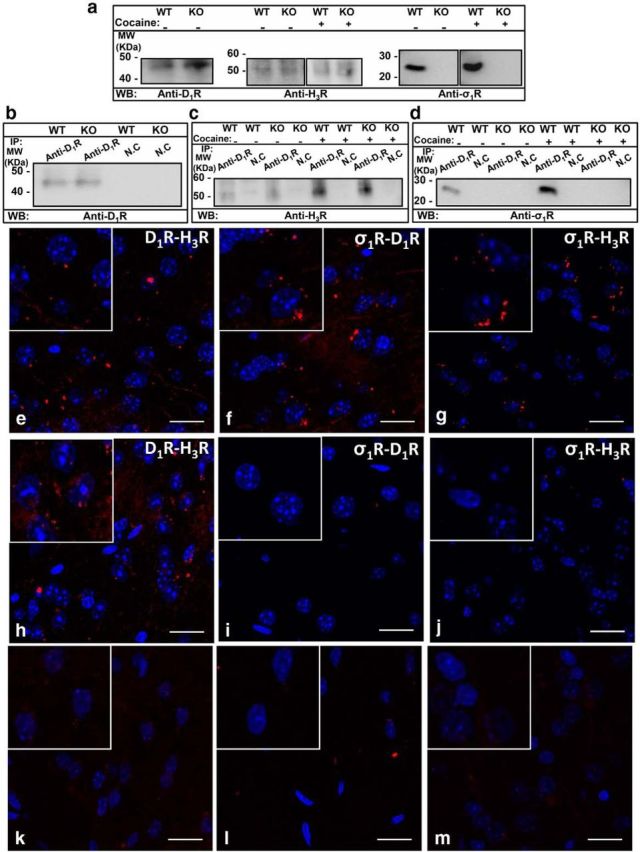
Expression of σ1R-D1R-H3R complexes in the mouse striatum detected by coimmunoprecipitation and PLA experiments. a–d, Coimmunoprecipitation experiments are shown. Striatal slices from WT and σ1R KO mice were untreated or treated with 150 μm cocaine for 30 min (cocaine). Solubilized striatal membranes (a) and immunoprecipitates with anti-D1R antibody or anti-calnexin antibody as negative control (NC; b–d) were analyzed by SDS-PAGE and immunoblotted using anti-D1R antibody (b), anti-H3R antibody (c), or anti-σ1R antibody (c). IP, immunoprecipitation; WB, Western blotting; MW, molecular mass. e–m, PLAs were performed as indicated in Materials and Methods, using WT (e–g) or σ1R KO mice (h–j) striatal slices. D1R-H3R (e, h), σ1R-D1R (f, i), and σ1R-H3R (g, j) heteromers were visualized as red spots around blue-colored DAPI-stained nucleus. k–m, Negative controls were performed doing the PLA experiments in WT mouse striatal slices incubated with only anti D1R (k), anti-σ1R (l), or anti-H3R (m) antibody as primary antibodies. Cell nuclei were stained with DAPI (blue). For each part a magnification is shown. Scale bar, 20 μm.
Figure 4.

Expression of σ1R-D1R-H3R complexes in the rat striatum detected by coimmunoprecipitation and PLA experiments. a–d, Coimmunoprecipitation experiments are shown. Rat striatal slices were untreated or treated with 150 μm cocaine for 30 min (cocaine). Solubilized striatal membranes (a) and immunoprecipitates with anti-D1R antibody or anti-calnexin antibody as negative control (NC; b–d) were analyzed by SDS-PAGE and immunoblotted using anti-D1R antibody (b), anti-H3R antibody (c), or anti-σ1R antibody (c). IP, immunoprecipitation; WB, Western blotting; MW, molecular mass. e–g, PLAs were performed as indicated in Materials and Methods, using rat striatal slices. D1R-H3R (e), σ1R-D1R (f), and σ1R-H3R (g) heteromers were visualized as red spots around blue–colored DAPI-stained nucleus. Scale bar, 20 μm.
To test the cocaine effect on H3R-D1R heteromer signaling in the striatum we determined the receptor agonists induced ERK 1/2 phosphorylation using striatal slices. To test if cocaine effect is mediated by σ1R we performed the signaling experiments in WT and σ1R KO mice. If cocaine is indeed acting via σ1R to modify the D1R-H3R heteromer in the striatum, then we should see no effect of cocaine in the σ1R KO mice. The striatal level of cocaine reached after pharmacologically significant doses is 15 μm (Pettit et al., 1990). To allow diffusion into the tissue in brain slices we used a 10-fold higher cocaine concentration, 150 μm. We determined the cross-antagonism in ERK 1/2 phosphorylation using striatal slices from WT and σ1R KO mice in the absence or the presence of cocaine. In the absence of cocaine the ERK 1/2 phosphorylation induced by the D1R agonist SKF 38393 was reverted by the H3R antagonist thioperamide in striatal slices from WT (Fig. 5a) and from σ1R KO mice (Fig. 5b). These data indicate that functional D1R-H3R heteromers are expressed in the striatum of both WT and σ1R KO mice in agreement to the PLA results above. In addition, the presence of cocaine did not affect H3R signaling and the cross-antagonism in striatal slices from σ1R KO mice (Fig. 5b). In contrast, cocaine blocked the H3R signaling and the D1R-H3R cross-antagonism in slices from WT mice (Fig. 5a). These results indicate that cocaine binding to σ1R blocks the H3R-mediated modulation of D1R signaling in the striatum.
Figure 5.

Cocaine inhibits the H3R signaling and the D1R-H3R cross-antagonism in ERK 1/2 phosphorylation in mouse striatum. WT (a) and σ1R KO (b) mouse striatal slices were treated or not with 150 μm cocaine for 2 h and were preincubated for 20 min with medium, the D1R antagonist SCH 23390 (10 μm), or the H3R antagonist thioperamide (10 μm) before the addition of the D1R agonist SKF 38393 (1 μm) or the H3R agonist imetit (1 μm) for an additional incubation period of 10 min. ERK 1/2 phosphorylation was determined by Western blot. Immunoreactive bands from three to eight slices obtained from eight WT or KO animals were quantified for each condition. Values represent mean ± SEM percentage of phosphorylation relative to basal levels found in untreated slices. No significant differences were obtained between the basal levels of the WT and the σ1R KO mice. One-way ANOVA followed by Bonferroni post hoc tests showed a significant (*p < 0.05) effect over basal or of the antagonist plus agonist treatment over the agonist treatment (#p < 0.05, ##p < 0.01).
Short or long treatment with cocaine disrupts the allosteric interaction between D1R and H3R in the striatum
One of the striking effects of cocaine is its ability to show prolonged effects even after a single exposure. We next wanted to examine whether and how D1R-H3R heteromer function might change in acute versus prolonged exposure to cocaine. For this we used a rat model of self-administration and compared the results to acute treatment. First, we confirmed that the effect of cocaine on the heteromer signaling in ex vivo rat striatal slices was equivalent to that of those in mice striatal slices seen above. In the absence of cocaine, the activation of slices with the D1R agonist SKF 38393 or the H3R agonist imetit induced ERK 1/2 phosphorylation, in line with the results found in mice. In addition, the coactivation with both agonists inhibited this signaling (Fig. 6a) indicating a negative cross-talk between both receptors. Importantly, when slices were treated with 150 μm cocaine, imetit was not able to induce ERK 1/2 phosphorylation and the negative cross-talk was not observed (Fig. 6a). The ERK 1/2 phosphorylation induced by the D1R agonist SKF 38393 or the H3R agonist imetit was blocked by both the H3R antagonist thioperamide and the D1R antagonist SCH 23390 (Fig. 6b). This cross-antagonism was not observed when slices were treated with 150 μm cocaine again matching the results found in mice.
Figure 6.

Cocaine inhibits the H3R-mediated modulation of D1R-promoted ERK 1/2 phosphorylation in rat striatum. Striatal slices from naive rats treated or not with 150 μm cocaine for 2 h (a, b) and striatal slices from sham rats or rats acutely treated (24 h) with cocaine (c, d), or striatal slices from sham rats or 9 weeks cocaine self-administered rats (e, f) were preincubated for 20 min with medium, the D1R antagonist SCH 23390 (10 μm), or the H3R antagonist thioperamide (10 μm) before the addition of the D1R agonist SKF 38393 (1 μm) or the H3R agonist imetit (1 μm) for an additional incubation period of 10 min and ERK 1/2 phosphorylation was determined by Western blot. Immunoreactive bands from 3 to 10 slices obtained from four animals were quantified for each condition. Values represent mean ± SEM percentage of phosphorylation relative to basal levels found in untreated slices. One-way ANOVA followed by Bonferroni post hoc tests showed a significant (*p < 0.05, **p < 0.01, ***p < 0.001) effect over basal or of the antagonist plus agonist treatment over the agonist treatment (#p < 0.05, ##p < 0.01, ###p < 0.001).
Next we examined whether a single exposure in vivo to cocaine could influence the function of D1R-H3R heteromers. Using rats acutely injected with one dose of cocaine, we analyzed the ability of H3R to influence D1R-induced ERK 1/2 phosphorylation using striatal slices 24 h after injection. Unlike the sham rats, in the cocaine-treated rats the ERK 1/2 phosphorylation induced by the D1R agonist SKF 38393 was not reverted by the H3R antagonist thioperamide (Fig. 6c). The ERK 1/2 phosphorylation induced by the H3R agonist imetit was reverted by the D1R antagonist SCH 23390 in the sham rats, but imetit was not able to signal in cocaine-treated rats (Fig. 6d), suggesting that 24 h after cocaine treatment the heteromer is not functional or disrupted, an effect not due to changes in the receptor expression by cocaine treatment (Fig. 7). Next, we wanted to test whether the molecular changes observed in the presence of cocaine were short term or whether they might persist in models of long-term addiction. To explore this we tested the effects of cocaine in long-term cocaine self-administered rats using striatal slices obtained 1 d after the last session of rats exposed to a cocaine self-administration regime of 6–11 weeks. Comparing signaling in striatal slices from rats with cocaine self-administration and sham rats we again observed a reduced cross-antagonism in the cocaine-treated animals (Fig. 6e,f). Together, it is clear that short and long cocaine treatment allows D1R to signal freely by hindering the dampening effect that H3R normally provides in the brain striatum.
One of the mechanisms by which heteromers function is through the direct modulation of the ligand affinity of the partner receptor (González et al., 2012). We sought to test this property for rat striatal D1R-H3R heteromers and whether cocaine could modulate it. We performed radioligand binding experiments using striatal membranes obtained 1 d after the last session of rats exposed to cocaine self-administration for 6–11 weeks, rats acutely treated with one dose of cocaine, or sham rats and we compared the results with the ones obtained with naive rat striatal membranes exogenously treated with 30 μm cocaine for 30 min. Competition curves of D1R antagonist [3H]SCH 23390 binding versus increasing concentrations of D1R agonist SKF 38393 in the absence or the presence of the H3R agonist RAMH were performed as indicated (see Materials and Methods). In all cases the competition curves were biphasic and binding data were fitted to the two-state dimer receptor model (see Materials and Methods) to calculate the macroscopic equilibrium dissociation constants using the equations described (see Materials and Methods). The values appear in Table 1. In striatal membranes from naive rats not treated with cocaine or from sham rats, the H3R agonist induced a decrease of the D1R macroscopic equilibrium dissociation constant values, indicating an allosteric interaction between both receptors. However, treating the striatal membranes from naive rats with cocaine or using striatal membranes from cocaine self-administered rats or from rats acutely treated with cocaine, this allosteric interaction was not observed (Table 1). Since the effect was observed after a short treatment with cocaine, these results suggest that cocaine binding to σ1R is blocking the allosteric interaction between H3R and D1R in the σ1R-D1R-H3R complex.
Effect of cocaine in the H3R-mediated modulation of D1R-induced cell death in the striatum
The results above described indicate that H3R can act as a “molecular brake” of D1R function by forming heteromers in the rat striatum, blocking the ability of D1R to signal and that cocaine can remove this brake and allow D1R to signal unimpeded by H3R. Ideally we would like to have a physiological readout of these changes in the D1R-H3R heteromer function. It has been shown that the dopamine increase due to drug intake is neurotoxic (Cunha-Oliveira et al., 2008) and it has been described that striatal cells are especially vulnerable to dopamine D1R activation by a nonoxidative process (Paoletti et al., 2008). Here, using organotypic rat striatal cultures we tested the hypothesis that blocking D1R signaling via H3R could lead to a change in D1R overactivation-induced cell death. We first tested the H3R ligands effect on D1R-induced cell death in organotypic rat striatal cultures. As indicated in Materials and Methods, we determined cell death by comparing DAPI and PI staining. The D1R agonist SKF 38393, but not H3R agonist imetit or the antagonist thioperamide, induced cell death, an effect that was reverted by the D1R antagonist SCH 23390 (Fig. 8a). When SKF 38393 and imetit were added together to the organotypic cultures, imetit blocked the D1R-mediated cell death (Fig. 8a) and the imetit effect was specific for D1R-mediated cell death as it was not observed in glutamate-induced cell death (Fig. 8b). Similar blocking of cell death was observed when SKF 38393 and thioperamide were added together (Fig. 8a). Thus, reducing D1R signaling via H3R can serve to reduce downstream physiological effects in our organotypic models, indicating that H3R indeed serves as a “molecular brake” or control point for D1R function. We next examined whether cocaine could remove this brake performing the same experiments in organotypic cultures treated with 150 μm cocaine. As shown in Figure 8c, cocaine was able to disrupt the imetit inhibition of the SKF 38393-promoted cell death without altering the effect of SKF 38393 alone and also blocked the cross-antagonism exerted by the H3R antagonist thioperamide on SKF 38393-promoted cell death (Fig. 8c).
Figure 8.

Cocaine inhibits the H3R-mediated modulation of D1R-promoted cell death in organotypic striatal slice cultures. Organotypic cultures of rat striatal slices were prepared as indicated (see Materials and Methods) and after 24 h, culture medium was replaced by fresh medium containing no ligands (a) or 150 μm cocaine (c). After 1 h, vehicle or 10 μm SCH 23390, imetit, or thioperamide were added and incubated for an additional 1 h before the addition of the D1R agonist SKF 38393 (50 μm) and slices were maintained 48 h more in culture. As a control, cell death induced by 1 mm glutamate in the presence or in the absence of 10 μm imetit was analyzed (b). Cell death was determined by DAPI and PI staining as indicated (see Materials and Methods). Values represent mean ± SEM percentage of PI stained cells versus total DAPI-stained cells determined in six to eight fields from two to three independent organotypic cultures. Bifactorial ANOVA showed a significant (***p < 0.001) effect over basal corresponding to nontreated organotypic cultures. One-way ANOVA followed by Bonferroni post hoc tests showed a significant (***p < 0.001) effect over basal corresponding to nontreated organotypic cultures or of organotypic cultures treated with two ligands respect to the cultures treated with SKF 38393 (###p < 0.001).
The results above suggest that cocaine's disruption of D1R-H3R signaling is an important enough disruption that can translate into pathological consequences. Thus, in cases where altering D1R signaling might be desired, it could be advantageous to target D1R-H3R heteromers via σ1R. To test this, we analyzed the effect of H3R ligands on the D1R agonist SKF 38393-induced cell death in organotypic cultures pretreated with σ1R agonists and antagonists. The pretreatment with the agonist PRE-084 (1 μm) mimicked the effect of cocaine in the H3R ligand modulation of D1R-induced cell death (Fig. 9a) and the pretreatment with the antagonist PD 144418 (10 μm), which had no effect in this process (Fig. 9b), could block the 150 μm cocaine's effects on the D1R-H3R heteromer in our cell death model (Fig. 9c). We also analyzed the D1R-H3R cross-talk and cross-antagonism in signaling in rat striatal slices pretreated with 1 μm PRE-084 or 10 μm PD 144418 in the absence or in the presence of 150 μm cocaine. As shown in Figure 9d, PRE-084 mimicked cocaine's effect by blocking the negative cross-talk between SKF 38393 and imetit in ERK 1/2 phosphorylation and the cross-antagonism between both receptors. When striatal slices where pretreated with the σ1 receptor antagonist PD 144418 (10 μm) cocaine could no longer block H3 receptor's control of D1 receptor signaling (Fig. 9f). PD 144418 had no effect in the H3R ligand modulation of D1R signaling (Fig. 9e) suggesting that it was indeed through blockage of cocaine binding. These data strongly suggest that H3R control of D1R signaling can be pharmacologically influenced with σ1R compounds and that σ1R antagonists may prove effective at reducing the effects of cocaine.
Figure 9.

Effect of σ1R agonist and antagonists on the H3R-mediated modulation of the D1R-promoted cell death and signaling in the striatum. Rat striatal organotypic cultures (a–c) or striatal slices (d–f) were prepared as indicated (see Materials and Methods) and were treated for 1 h with medium (a, d) or PD 144418 (10 μm; b, c, e, f) before adding 1 μm PRE-084 (a, d), vehicle (b, e), or 150 μm cocaine (c, f). At 24 h of organotypic culture (a–c) or after 2 h of slice incubation (d–f) the D1R antagonist SCH 23390 (50 μm, a–c or 10 μm, d–f) or the H3R antagonist thioperamide (10 μm) were added 20 min before the addition of the D1R agonist SKF 38393 (50 μm, a–c or 1 μm, d–f) or the H3R agonist imetit (10 μm, a–c or 1 μm, d–f) alone or in combination. Striatal organotypic cultures were cultured for an additional 48 h and slices were incubated for an additional 10 min and were processed to determine cell death or ERK 1/2 phosphorylation, respectively, as indicated in Materials and Methods. a–c, Values represent mean ± SEM percentage of PI stained cells versus total DAPI-stained cells determined in four to six fields from two to three independent organotypic cultures. d–f, Values represent mean ± SEM phosphorylation relative to basal levels found in untreated slices, determined in 3–10 slices obtained from four animals. One-way ANOVA followed by Bonferroni post hoc tests showed a significant (*p < 0.05, ***p < 0.001) effect over basal (nontreated organotypic cultures or slices) or of the two ligands treatment over the agonist treatment (#p < 0.05, ###p < 0.001).
Discussion
From the data presented here four major conclusions can be made. First, σ1R binds D1R-H3R heteromers in transfected cells as well as in mouse and rat striatum. Second, σ1R agonists, such as cocaine, modify the structure and counteract the biochemical properties of the D1R-H3R heteromer, composing of heteromer signaling through Gi protein, the ability of H3R activation to signal through MAPK, and the ability of H3R ligands to inhibit the effects of D1R-mediated signaling, including cell death. Therefore, a third important general conclusion is that blockade of H3R-mediated inhibition of D1R function in the σ1R-D1R-H3R complexes plays a key role in the effects of cocaine. Finally, σ1R-D1R-H3R complexes might provide a new target for the treatment of cocaine abuse.
Cocaine binds with somewhat higher affinity to DAT than to σ1R; therefore, the role of σ1R in the addictive properties of cocaine has been a matter of debate. More accepted has been their role in the acute neurotoxic effects of cocaine (for review, see Robson et al., 2012). In fact, σ1R antagonists provide a protective effect in cocaine-induced convulsions and lethality in rodents (Ritz and George, 1997; Robson et al., 2012). Nevertheless, an important amount of experimental data demonstrates the involvement of σ1R in many pharmacologic, including rewarding, effects of cocaine (Maurice and Su, 2009; Robson et al., 2012). Repeated cocaine exposure increases the levels of σ1R in the brain, including the striatum, by a process that depends on the cocaine binding σ1R (Liu and Matsumoto, 2008), and does not occur in mice with genetic deletion of D1R (Zhang et al., 2005). Upregulation of σ1R might underlie the ability of cocaine to induce self-administration of selective σ1R agonists, which has been invoked as a possible mechanism that contributes to the intractability of psychostimulant abuse (Hiranita et al., 2013). Indeed, we have demonstrated the importance of the σ1R and D1R receptor interaction on the initial events upon cocaine exposure (Navarro et al., 2010).
We previously described that in the striatum D1R can form heteromers with H3R and that H3R agonist binding to the D1R-H3R heteromers are negatively modulating D1R function (Ferrada et al., 2009; Moreno et al., 2011a). The modulation of D1R signaling by H3R is thought to serve as a control point that could set the tone of D1R signaling from a given neuron (Moreno et al., 2011a; Ellenbroek, 2013; Panula and Nuutinen, 2013). However, previous studies using the H3R ligand imetit have shown that it was not effective at preventing drug seeking in the presence of cocaine, but does prevent drug seeking in the absence of cocaine (Ortiz De Pablo and Self, 2011). Our work here clearly explains the molecular mechanism behind these results and highlights the pleiotropic effects of σ1R. As stated in Figure 10, H3R and D1R form heteromers to which σ1R can bind. Under normal conditions (in the absence of cocaine), H3R can serve to control the levels of D1R signaling altering its coupling to G-proteins. Activating only the D1R protomer in the heteromer promotes a decrease of cAMP levels in accordance with Gi coupling to the heteromer. In addition, D1R activation induces β-arrestin recruitment and promotes ERK 1/2 phosphorylation (Fig. 10a). However, upon dual stimulation of the heteromer with both H3R and D1R agonists in the absence of cocaine, a negative cross-talk between receptors in the heteromer occurs, inhibiting β-arrestin recruitment and subsequently reducing ERK 1/2 phosphorylation from the heteromer (Fig. 10b). When cocaine (Fig. 10c) binds to σ1R in the heteromeric complex, it induces structural changes in the heteromer, and disrupts the negative cross-talk. Thus, in the presence of cocaine the H3R-mediated brake on D1R is lost. Importantly, cocaine also blocks the dopamine transporter, DAT, in presynaptic terminals. This leads to an increase in extracellular levels of dopamine. This dopamine then can bind and activate an uninhibited D1R allowing D1R to increase cAMP by coupling to Gs protein and increasing p-ERK 1/2, presumably via recruitment of β-arrestin. This uninhibited D1R is normally balanced by inputs from D2R-containing neurons. However, we and others have shown that this balance becomes undone in the presence of cocaine (Navarro et al., 2013; Park et al., 2013).
Figure 10.

Scheme representing the D1R signaling in the D1R-H3R heteromer in the absence or presence of cocaine. In GABAergic neurons (gray), in the absence of cocaine, H3R (blue) and D1R (red) form heteromers to which σ1R (yellow) can bind. Activation of only the D1R protomer (a) promotes a decrease of cAMP levels in accordance with Gi (orange) coupling of both receptors in the heteromer. Moreover, D1R activation induces β-arrestin (purple) recruitment and promotes ERK 1/2 phosphorylation. Upon dual stimulation of the heteromer with both H3R and D1R agonists in the absence of cocaine (b), a negative cross-talk (the ability of one receptor agonist to negatively modulate the partner receptor ligand binding or signaling) between receptors in the heteromer occurs, inhibiting β-arrestin recruitment and subsequently reducing ERK 1/2 phosphorylation from the heteromer. When cocaine (brown) binds to σ1R (c) this induces structural changes in the heteromer disrupting the negative cross-talk. Thus, the H3R-mediated brake on D1R is lost, allowing D1R (green) to couple to Gs (pale green) and increasing cAMP. Importantly, β-arrestin recruitment and ERK 1/2 phosphorylation are also detected in the presence of cocaine.
The present study supports the use of σ1R antagonists as a treatment for cocaine abuse, particularly in the frame of σ1R modulation of D1R-H3R heteromers. In fact, we showed that blockade of σ1R restores the protective H3R-mediated brake on D1R signaling involved in cocaine-induced cell death. Although at the experimental level D1R seems to be clearly involved in the pharmacological effects of cocaine (Xu et al., 1994; Weiss et al., 2001; Wolf et al., 2003; Bertran-Gonzalez et al., 2008), conflicting results of D1R antagonists have been observed at the clinical level (Nann-Vernotica et al., 2001), which might be related to the inability to reach therapeutic doses without secondary effects. Antagonists of σ1R receptors can block cocaine's ability to modulate D1-H3 receptor heteromers, providing a means to decrease the overdose-related side effects of orthosteric D1 receptor antagonists. Our results indicate that a potentially good therapeutic approach for the pharmacologic effects of cocaine could include the association of σ1R antagonists with H3 receptor agonists.
Footnotes
This study was supported by grants from the Spanish Ministerio de Ciencia y Tecnología (SAF2010-18472, SAF2011-23813, SAF2006-08240, SAF2009-12510, and Red de Trastornos Adictivos RD06/0001/0015) and by Intramural Funds of the National Institute on Drug Abuse to S. Ferré. P.J.M. is a Ramón y Cajal Fellow. We thank Jasmina Jiménez for technical help (University of Barcelona). We thank Laboratorios Esteve (Barcelona, Spain) for providing σ1 receptor KO and WT mice brains.
The authors declare no competing financial interests.
References
- Aksenov MY, Aksenova MV, Nath A, Ray PD, Mactutus CF, Booze RM. Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 dopamine receptor. Neurotoxicology. 2006;27:217–228. doi: 10.1016/j.neuro.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Arias-Montaño JA, Floran B, Garcia M, Aceves J, Young JM. Histamine H(3) receptor-mediated inhibition of depolarization-induced, dopamine D(1) receptor-dependent release of [(3)H]-gamma-aminobutyric acid from rat striatal slices. Br J Pharmacol. 2001;133:165–171. doi: 10.1038/sj.bjp.0704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriba P, Navarro G, Ciruela F, Ferré S, Casadó V, Agnati L, Cortés A, Mallol J, Fuxe K, Canela EI, Lluís C, Franco R. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat Methods. 2008;5:727–733. doi: 10.1038/nmeth.1229. [DOI] [PubMed] [Google Scholar]
- Casadó V, Cortés A, Ciruela F, Mallol J, Ferré S, Lluis C, Canela EI, Franco R. Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol Ther. 2007;116:343–354. doi: 10.1016/j.pharmthera.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–242. doi: 10.1038/nature11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Oliveira T, Rego AC, Oliveira CR. Cellular and molecular mechanisms involved in the neurotoxicity of opioid and psychostimulant drugs. Brain Res Rev. 2008;58:192–208. doi: 10.1016/j.brainresrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. β-Arrestins and Cell Signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA. Histamine H3 receptors, the complex interaction with dopamine and its implications for addiction. Br J Pharmacol. 2013;170:46–57. doi: 10.1111/bph.12221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C, Moreno E, Casadó V, Bongers G, Cortés A, Mallol J, Canela EI, Leurs R, Ferré S, Lluís C, Franco R. Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br J Pharmacol. 2009;157:64–75. doi: 10.1111/j.1476-5381.2009.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Agnati LF, Ciruela F, Lluis C, Woods AS, Fuxe K, Franco R. Neurotransmitter receptor heteromers and their integrative role in “local modules”: the striatal spine module. Brain Res Rev. 2007;55:55–67. doi: 10.1016/j.brainresrev.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pin JP, Volkow ND, Waldhoer M, Woods AS, Franco R. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferré S, Fuxe K, Cortés A, Ciruela F, Lluis C, Canela EI. Dimer-based model for heptaspanning membrane receptors. Trends Biochem Sci. 2005;30:360–366. doi: 10.1016/j.tibs.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferrada C, Ferré S, Fuxe K, Cortés A, Ciruela F, Lluis C, Canela EI. The two-state dimer receptor model: a general model for receptor dimers. Mol Pharmacol. 2006;69:1905–1912. doi: 10.1124/mol.105.020685. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González S, Moreno-Delgado D, Moreno E, Pérez-Capote K, Franco R, Mallol J, Cortés A, Casadó V, Lluís C, Ortiz J, Ferré S, Canela E, McCormick PJ. Circadian-related heteromerization of adrenergic and dopamine D4 receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol. 2012;10:e1001347. doi: 10.1371/journal.pbio.1001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Su T. The sigma receptor: evolution of the concept in neuropsychopharmacology. Curr Neuropharmacol. 2005;3:267–280. doi: 10.2174/157015905774322516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiranita T, Soto PL, Tanda G, Katz JL. Reinforcing Effects of σ-Receptor Agonists in Rats Trained to Self-Administer Cocaine. J Pharmacol Exp Ther. 2010;332:515–524. doi: 10.1124/jpet.109.159236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiranita T, Mereu M, Soto PL, Tanda G, Katz JL. Self-administration of cocaine induces dopamine-independent self-administration of sigma agonists. Neuropsychopharmacology. 2013;38:605–615. doi: 10.1038/npp.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann HM, Nadal R, Vignes M, Ortiz J. Chronic cocaine self-administration modulates ERK1/2 and CREB responses to dopamine receptor agonists in striatal slices. Addict Biol. 2012;17:565–575. doi: 10.1111/j.1369-1600.2011.00353.x. [DOI] [PubMed] [Google Scholar]
- Katz JL, Su TP, Hiranita T, Hayashi T, Tanda G, Kopajtic T, Tsai SY. A role for sigma receptors in stimulant self administration and addiction. Pharmaceuticals. 2011;4:880–914. doi: 10.3390/ph4060880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Su TP, Fujimoto M, Bonci A. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012;35:762–771. doi: 10.1016/j.tins.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Hayashi T, Chuang JY, Tsai SY, Su T-P, Bonci A. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell. 2013;152:236–247. doi: 10.1016/j.cell.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langa F, Codony X, Tovar V, Lavado A, Giménez E, Cozar P, Cantero M, Dordal A, Hernández E, Pérez R, Monroy X, Zamanillo D, Guitart X, Montoliu L. Generation and phenotypic analysis of sigma receptor type I (sigma 1) knockout mice. Eur J Neurosci. 2003;18:2188–2196. doi: 10.1046/j.1460-9568.2003.02950.x. [DOI] [PubMed] [Google Scholar]
- Lepsch LB, Munhoz CD, Kawamoto EM, Yshii LM, Lima LS, Curi-Boaventura MF, Salgado TM, Curi R, Planeta CS, Scavone C. Cocaine induces cell death and activates the transcription nuclear factor kappa-B in PC12 cells. Mol Brain. 2009;2:3. doi: 10.1186/1756-6606-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Matsumoto RR. Alterations in fos-related antigen 2 and sigma1 receptor gene and protein expression are associated with the development of cocaine-induced behavioral sensitization: time course and regional distribution studies. J Pharmacol Exp Ther. 2008;327:187–195. doi: 10.1124/jpet.108.141051. [DOI] [PubMed] [Google Scholar]
- Matsumoto RR, Liu Y, Lerner M, Howard EW, Brackett DJ. Sigma receptors: potential medications development target for anti-cocaine agents. Eur J Pharmacol. 2003;469:1–12. doi: 10.1016/S0014-2999(03)01723-0. [DOI] [PubMed] [Google Scholar]
- Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Hoffmann H, Gonzalez-Sepúlveda M, Navarro G, Casadó V, Cortés A, Mallol J, Vignes M, McCormick PJ, Canela EI, Lluís C, Moratalla R, Ferré S, Ortiz J, Franco R. Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J Biol Chem. 2011a;286:5846–5854. doi: 10.1074/jbc.M110.161489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Vaz SH, Caí NS, Ferrada C, Quiroz C, Barodia SK, Kabbani N, Canela EI, McCormick PJ, Lluis C, Franco R, Ribeiro JA, Sebastião AM, Ferré S. Dopamine-galanin receptor heteromers modulate cholinergic neurotransmission in the rat ventral hippocampus. J Neurosci. 2011b;31:7412–7423. doi: 10.1523/JNEUROSCI.0191-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nann-Vernotica E, Donny EC, Bigelow GE, Walsh SL. Repeated administration of the D1/5 antagonist ecopipam fails to attenuate the subjective effects of cocaine. Psychopharmacology. 2001;155:338–347. doi: 10.1007/s002130100724. [DOI] [PubMed] [Google Scholar]
- Navarro G, Moreno E, Aymerich M, Marcellino D, McCormick PJ, Mallol J, Cortés A, Casadó V, Canela EI, Ortiz J, Fuxe K, Lluís C, Ferré S, Franco R. Direct involvement of sigma-1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc Natl Acad Sci U S A. 2010;107:18676–18681. doi: 10.1073/pnas.1008911107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G, Moreno E, Bonaventura J, Brugarolas M, Farré D, Aguinaga D, Mallol J, Cortés A, Casadó V, Lluís C, Ferre S, Franco R, Canela E, McCormick PJ. Cocaine inhibits dopamine D2 receptor signaling via sigma-1-D2 receptor heteromers. PLoS One. 2013;8:e61245. doi: 10.1371/journal.pone.0061245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz De Pablo J, Self DW. Use of H3 histaminergic agonists for the treatment of addiction to drugs of abuse. [Accessed June 10, 2013];2011 [Google Scholar]
- Panula P, Nuutinen S. The histaminergic network in the brain: basic organization and role in disease. Nat Rev Neurosci. 2013;14:472–487. doi: 10.1038/nrn3526. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Vila I, Rifé M, Lizcano JM, Alberch J, Ginés S. Dopaminergic and glutamatergic signaling crosstalk in Huntington's disease neurodegeneration: the role of p25/cyclin-dependent kinase 5. J Neurosci. 2008;28:10090–10101. doi: 10.1523/JNEUROSCI.3237-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Volkow ND, Pan Y, Du C. Chronic cocaine dampens dopamine signaling during cocaine intoxication and unbalances D1 over D2 receptor signaling. J Neurosci. 2013;33:15827–15836. doi: 10.1523/JNEUROSCI.1935-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- Pettit HO, Pan HT, Parsons LH, Justice JB., Jr Extracellular concentrations of cocaine and dopamine are enhanced during chronic cocaine administration. J Neurochem. 1990;55:798–804. doi: 10.1111/j.1471-4159.1990.tb04562.x. [DOI] [PubMed] [Google Scholar]
- Ritz MC, George FR. Cocaine toxicity: concurrent influence of dopaminergic, muscarinic and sigma receptors in mediating cocaine-induced lethality. Psychopharmacology. 1997;129:311–321. doi: 10.1007/s002130050198. [DOI] [PubMed] [Google Scholar]
- Robson MJ, Noorbakhsh B, Seminerio MJ, Matsumoto RR. Sigma-1 receptors: potential targets for the treatment of substance abuse. Curr Pharm Des. 2012;18:902–919. doi: 10.2174/138161212799436601. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375:503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Söderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45:227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Takagi H, Morishima Y, Matsuyama T, Hayashi H, Watanabe T, Wada H. Histaminergic axons in the neostriatum and cerebral cortex of the rat: a correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Res. 1986;364:114–123. doi: 10.1016/0006-8993(86)90992-3. [DOI] [PubMed] [Google Scholar]
- Trifilieff P, Rives ML, Urizar E, Piskorowski RA, Vishwasrao HD, Castrillon J, Schmauss C, Slättman M, Gullberg M, Javitch JA. Detection of antigen interactions ex vivo by proximity ligation assay: endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. Biotechniques. 2011;51:111–118. doi: 10.2144/000113719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Tomasi D, Baler RD. Unbalanced neuronal circuits in addiction. Curr Opin Neurobiol. 2013;23:639–648. doi: 10.1016/j.conb.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss F, Ciccocioppo R, Parsons LH, Katner S, Liu X, Zorrilla EP, Valdez GR, Ben-Shahar O, Angeletti S, Richter RR. Compulsive drug-seeking behavior and relapse. Ann N Y Acad Sci. 2001;937:1–26. doi: 10.1111/j.1749-6632.2001.tb03556.x. [DOI] [PubMed] [Google Scholar]
- Williams JM, Galli A. The dopamine transporter: a vigilant border control for psychostimulant action. Handb Exp Pharmacol. 2006:215–232. doi: 10.1007/3-540-29784-7_11. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Mangiavacchi S, Sun X. Mechanisms by which dopamine receptors may influence synaptic plasticity. Ann N Y Acad Sci. 2003;1003:241–249. doi: 10.1196/annals.1300.015. [DOI] [PubMed] [Google Scholar]
- Xu M, Hu XT, Cooper DC, Moratalla R, Graybiel AM, White FJ, Tonegawa S. Elimination of cocaine-induced hyperactivity and dopamine-mediated neurophysiological effects in dopamine D1 receptor mutant mice. Cell. 1994;79:945–955. doi: 10.1016/0092-8674(94)90026-4. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhang L, Tang Y, Zhang Q, Lou D, Sharp FR, Zhang J, Xu M. Repeated cocaine administration induces gene expression changes through the dopamine D1 receptors. Neuropsychopharmacology. 2005;30:1443–1454. doi: 10.1038/sj.npp.1300680. [DOI] [PubMed] [Google Scholar]
- Zimmermann T, Rietdorf J, Girod A, Georget V, Pepperkok R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett. 2002;531:245–249. doi: 10.1016/S0014-5793(02)03508-1. [DOI] [PubMed] [Google Scholar]