

Abstract
Transient receptor potential (TRP) channels are abundant in the brain where they regulate transmission of sensory signals. The expression patterns of different TRPC subunits (TRPC1, 4, and 5) are consistent with their potential role in fear-related behaviors. Accordingly, we found recently that mutant mice lacking a specific TRP channel subunit, TRPC5, exhibited decreased innate fear responses. Both TRPC5 and another member of the same subfamily, TRPC4, form heteromeric complexes with the TRPC1 subunit (TRPC1/5 and TRPC1/4, respectively). As TRP channels with specific subunit compositions may have different functional properties, we hypothesized that fear-related behaviors could be differentially controlled by TRPCs with distinct subunit arrangements. In this study, we focused on the analysis of mutant mice lacking the TRPC4 subunit, which, as we confirmed in experiments on control mice, is expressed in brain areas implicated in the control of fear and anxiety. In behavioral experiments, we found that constitutive ablation of TRPC4 was associated with diminished anxiety levels (innate fear). Furthermore, knockdown of TRPC4 protein in the lateral amygdala via lentiviral-mediated gene delivery of RNAi mimicked the behavioral phenotype of constitutive TRPC4-null (TRPC4−/−) mouse. Recordings in brain slices demonstrated that these behavioral modifications could stem from the lack of TRPC4 potentiation in neurons in the lateral nucleus of the amygdala through two Gαq/11 protein-coupled signaling pathways, activated via Group I metabotropic glutamate receptors and cholecystokinin 2 receptors, respectively. Thus, TRPC4 and the structurally and functionally related subunit, TRPC5, may both contribute to the mechanisms underlying regulation of innate fear responses.
Keywords: amygdala, anxiety, cholecystokinin 4, fear, TRP channel, TRPC4
Introduction
Transient receptor potential channels (TRPC) are tetrameric complexes, formed by the subunits (TRPC1–TRPC7) belonging to two subfamilies, TRPC1/4/5 and TRPC3/6/7 (Clapham, 2007). TRPCs are nonselective cationic channels, activated by Gαq/11-coupled receptors (Clapham, 2003, 2007; Ramsey et al., 2006), which are expressed in the brain (with the exception of the TRPC2 subunit), where they can mediate transmission of different forms of sensory information (Clapham, 2003; Birnbaumer, 2009; Nilius and Owsianik, 2011). Recently, we obtained evidence that the TRPC5 is present in brain areas controlling fear-related behavioral responses, including the amygdala and the auditory thalamic and cortical areas transmitting conditioned stimulus (CS) information to the amygdala during auditory fear conditioning, as well as in the somatosensory cortex and the parietal insular cortex relaying nociceptive unconditioned stimuli (US) to fear circuits (LeDoux, 2000; Davis and Whalen, 2001; Fanselow and Poulos, 2005; Riccio et al., 2009). Consistent with this expression pattern, TRPC5 was found to have an essential function in innate fear, and contribute to conditioned fear under certain conditions (Riccio et al., 2009). The observed decreases in innate fear in TRPC5-null mice were accompanied by significant reductions in the magnitude of Group I mGluR-mediated synaptic responses in cortical and thalamic inputs to the lateral nucleus of the amygdala (LA) and cholecystokinin 2 receptor (CCK2)-triggered currents in LA neurons. Notably, Group I mGluRs or CCK2 receptors, linked to activation of TRPC5 subunit-containing channels (Faber et al., 2006; Meis et al., 2007), were repeatedly implicated in control of fear-related behaviors in previous studies (Frankland et al., 1997; Rodrigues et al., 2002; Pietraszek et al., 2005; Wang et al., 2005).
In the brain, however, both the TRPC5 and TRPC4 subunits form heteromeric complexes with TRPC1 (Clapham, 2003, 2007). As the biophysical and functional properties of heteromeric channels are determined by their subunit composition (Strübing et al., 2001), we asked whether distinct TRPC subunits and their specific combinations in the brain might differentially control the functions of the amygdala and fear-related behaviors. To this end, we generated mice lacking TRPC4 and assayed the effects of this mutation on functional properties of LA neurons and amygdala-based behavioral processes. TRPC4 is closely related to TRPC5, sharing ∼70% sequence homology. Similar to TRPC5, TRPC4 is activated by phospholipase C and/or by micromolar concentration of lanthanides (La3+ or gadolinium, Gd3+), independently of G-protein-coupled receptors and exhibits an identical doubly rectifying current–voltage (I–V) relation (Schaefer et al., 2000; Strübing et al., 2001). Here we show that, similar to TRPC5−/− mice, constitutive TRPC4-null mice were less anxious than control littermates when presented with stimuli triggering innate fear responses. Likewise, the knockdown of TRPC4 in the LA reproduced the same fear-related behavior deficit in mice. The deficits in innate fear were associated with diminished Group I mGluR-mediated synaptic responses and CCK2 receptor-activated signaling in LA neurons. These findings indicate that TRPC4, along with the genetically and functionally related TRPC5 subunit (Riccio et al., 2009), may contribute to regulation of the mechanisms underlying anxiety-driven behaviors.
Materials and Methods
Generation of TRPC4−/− mice.
TRPC4−/− mice were generated by recombineering (Liu et al., 2002). The targeting construct was linearized and electroporated into embryonic stem (ES) cells derived from 129/SvJ1 mice. Southern blot using probes flanking the targeting construct sequence detected clones with successful homologous recombination. ES cells harboring the targeting construct were transfected with pOG231, a plasmid for transient Cre expression, to excise the neomycin cassette and create the constitutive knock-out (KO) allele. Chimeric mice were generated by injection of the ES cells into C57BL/6 mouse blastocysts. The chimeric mice were bred with 129/SvImJ mice. The F2 heterozygous mice were backcrossed to 129/SvImJ mice for eight generations. Heterozygotes were then crossed to generate paired littermates for all studies. Mice were treated in accordance with guidelines approved by Boston Children's Hospital Animal Care and Use Committee (IACUC).
PCR analysis of mouse tail DNA.
DNA was isolated from mouse tails according to standard protocols. A set of three primers was designed for genotyping. Primer sequences were as follows: F, 5′-gagaaacccatgcatgtgtacatatgat-3′; R, 5′-caagctgtggttcacctgat cttagag-3′; and R1, 5′-ctatcaacttccttgccgtaatgtttc-3′. The F and R amplified primers were designed to yield a 731 bp PCR fragment from wild-type (wt) mTRPC4 gene; the F and R1 primers amplified a 492 bp PCR fragment from the disrupted targeted TRPC4 gene.
Reverse transcription PCR analysis.
One microgram of total RNA from brain was used to generate first-strand cDNA (Superscript III; Invitrogen). The KOF and KOR primers, spanning exon 3 to exon 5, amplified fragments of 711 and 374 bp from wt and TRPC4−/− cDNAs, respectively. Primer sequences: KOF, 5′-cctttccttactgcctttcagttaagttg-3′; and KOR, 5′-cttcaaggagattgttgccagatacaag-3′. For quantitative reverse-transcription (RT)-PCR, primers for mouse TRPC1, TRPC3, TRPC5, TRPC6, TRPC7, and β-actin were added to SYBR Green 2× Master Mix (Applied Biosystems) to a final concentration of 300 nm. qRT-PCR was performed as described previously (Riccio et al., 2009). Data were captured using Sequence Detector Software (7500 Real Time PCR System; Applied Biosystems) and each sample was normalized by dividing the quantity (the threshold cycle value) of target gene cDNA by the quantity (the threshold cycle value) of a housekeeping cDNA (β-actin) to correct for differences in RNA quantity and quality. Data were expressed as arbitrary units on a scale from 0 to 1. This method avoids the comparison of absolute expression levels between different genes of interest, which is difficult to justify due to potential differences in amplification efficiency and sensitivity of PCR primers (Riccio et al., 2002).
In situ hybridization.
Brains were isolated from 4-week-old mice and frozen in powdered dry ice. Cryostat sections (18–20 μm) were incubated with anti-digoxigenin-AP antibody overnight followed by nitroblue tetrazolium (340 μg/ml) and BCIP (170 μg/ml) for 40 min in the dark. Color development was stopped, and the sections were placed on coverslips in buffered 50% glycerol. The mouse TRPC4-mRNA-specific antisense riboprobe was directed against nucleotides 3321–3436 of the mTRPC4 sequence. Control experiments with sense probe did not label brain sections.
Immunoprecipitation and immunohistochemistry.
Immunoprecipitation (IP) buffer contained 20 mm HEPES-NaOH, pH 7.5, 1% Triton X-100, 150 mm NaCl, and protease inhibitors. Brain microsomes (4-week-old mouse) were solubilized in IP buffer; 1 mg was immunoprecipitated with 5 μg of anti-TRPC4 antibody (NeuroMab, University of California (UC) Davis) or 5 μg of anti-TRPC5 antibody (NeuroMab, UC Davis) and 10 μg of protein A Sepharose (GE Healthcare). Antibodies for Western blots included the following: 5 μg/ml anti-TRPC4 (NeuroMab, UC Davis), 5 μg/ml anti-TRPC5 (NeuroMab, UC Davis), GAPDH (1:5000; Abcam), and anti-Na+,K+-ATPase-α (NKA-α; 1:5000; Thermo Scientific); and 1:10,000 dilution of secondary goat anti-rabbit IgG conjugated with horseradish peroxidase (HRP; Pierce). For Western blotting of LA lysates, punches containing the LA were obtained using a 1 mm punch tool (Fine Science Tools) from 400-μm-thick sections taken on a freezing microtome (Leica VT1000S). Punches were dounced in 70 μl of ice-cold lysate buffer (20 mm HEPES-NaOH, pH 7.5), 1% Triton X-100, 150 mm NaCl, and protease inhibitors). Densitometry was conducted using ImageJ software; optical densities were normalized to either GAPDH protein (1:5000; Abcam) or NKA-α (1:5000; Thermo Scientific). Data were normalized to the average value of controls and analyzed using Student's t test.
For immunohistochemistry, slides were soaked in xylene, passed through graded alcohols, and placed in distilled water. Slides were then pretreated with 10 mm citrate, pH 6.0 (Zymed) in a steam pressure cooker (Decloaking Chamber; Biocare Medical), followed by washing in distilled water. All subsequent steps were performed at room temperature in a hydrated chamber. Slides were pretreated with Peroxidase Block (DAKO) for 45 min to quench endogenous peroxidase activity. Slides were then washed in 50 mm Tris-Cl, pH 7.4, and incubated in Background Sniper (Biocare Medical) for 10 min to reduce nonspecific background staining. Primary antibody mixtures consisted of either rabbit monoclonal antibody to CaMKIIα (1:1000; clone EP1829Y, Abcam), rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP; 1:2000; Abcam), or rabbit polyclonal antibody to Gad67 (1:100; AnaSpec) combined with mouse monoclonal to TRPC4 (1:500; clone: N77/15, NeuroMab, UC Davis) and diluted in DaVinci Green diluent (Biocare Medical) applied for 1 h. Mouse monoclonal antibody to CaMKIIα (1:1000; Abcam) was combined with rabbit polyclonal antibody to CCK8 (1:200; ImmunoStar). Rabbit polyclonal antibody to GFP (1:200; Abcam) was used to detect GFP in mice infused with virus. For double labeling, a mixture of secondary antibodies (Alexa 555-conjugated goat anti-rabbit diluted 1:200, Invitrogen, plus Envision anti-mouse, DAKO) was applied for 30 min. After washing, the Cy5-tyramide Signal Amplification System (PerkinElmer Life Science Products) was applied to couple Cy5 dyes to the HRP-conjugated Envision secondary antibodies. Coverslips were sealed to slides with Prolong Gold Antifade Reagent with DAPI (Invitrogen) to visualize nuclei. Slides were detected with a confocal laser scanning biological microscope Olympus FluoView 1000. Immunofluorescence quantification of CCKergic fibers was performed using ImageJ software. The confocal images were split into three channels (green for CCK, red for CaMKIIα, and DAPI for nuclei) to obtain one image per channel. The integrated density values (IDV) for the blue and red channels were assessed separately. To normalize the CCK fluorescence to CaMKIIα fluorescence, the red channel IDV was divided by blue channel IDV. This procedure was repeated for six different images taken from different stainings (two images from each of three controls and three KOs). Normalized data were analyzed using Student's t test.
Virus production.
Short-hairpin (sh) oligonucleotides designed to target TRPC4-mRNA were cloned into BamHI/SalI sites of the pHAGE-CMV-eGFPW-SC3 vector (Mostoslavsky et al., 2005), in which GFP was under the pCMV promoter and anti-TRPC4 shRNA under the pU6 promoter. The previously used oligonucleotide sequences (Puram et al., 2011) were as follows: shTRPC4i.1-F, 5′-ggtggaatctaatggactttcca agttaaccaaagtccattagattccaccctttttg-3′ and shTRPC4i.1-R, 5′-caaaaagggtggaatcta atggactttggttaacttggaaagtccattagattccacc-3′. The lentiviruses (LV-shTRPC4-GFP and LV-SCRM-GFP) were produced by the Harvard Gene Therapy Initiative Core. Titers of LV-shTRPC4-GFP and LV-SCRM-GFP were 6.9 × 108 and 7.1 × 108 infectious U/mg, respectively.
Stereotaxic surgery and viral injections.
All surgical procedures were performed using aseptic techniques and conducted in accordance with Boston Children's Hospital's IACUC rules. The mice were anesthetized using isoflurane (3–4%) in oxygen and the head fur shaved. The mice were then mounted into a stereotaxic frame and the head stabilized. All surgical work proceeded under a maintained concentration of isoflurane (1.5–2.5%) in oxygen. After sterilizing the skin with Betadine and alcohol, an incision was made and the skin parted to reveal the skull. The connective tissue was removed, revealing the cranial bone and holes were drilled bilaterally above the lateral amygdala. The stereotaxic coordinates to locate the LA (based on Franklin and Paxinos, 2007) were as follows: −1.7 mm caudal to bregma, ±3.4 mm lateral to midline, and −3.4 mm ventral to dura. The virus was delivered using a glass microsyringe (Hamilton) and injection needle (33G), mounted in a microsyringe pump (Elite 11 Nanomite; Harvard Instruments) attached to the arm of the stereotaxic frame, which delivered 0.25 μl of LV solution over 10 min. Following injection, the microsyringe was left in place for an additional 10 min and the holes were sealed with bone wax. Following completion of bilateral injections, the animals, which were housed singly, were moved to a recovery cage, provided with warmth, and observed until fully ambulatory and able to take food and water, followed by their return to the home cage. Four weeks after the surgery, the mice underwent behavioral tests. To determine the location of the virus after the behavioral tests, mice were killed, perfused with 4% paraformaldehyde in 0.1 m phosphate buffer (pH 7.4, w/v), and brains were sectioned coronally (40 μm). Brain sections were examined for GFP fluorescence in the LA regions. Only mice with bilateral transfections confined to the borders of the LA were included in the behavioral analysis.
Electrophysiological recordings.
Vibratome slices of the amygdala (250–300 μm) were prepared from TRPC4−/− mice or control littermates (males). Slices were continuously superfused in solution containing the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.0 MgSO4, 1.25 NaH2PO4, 26.0 NaHCO3, 10 glucose, and 0.1 picrotoxin and equilibrated with 95% O2 and 5% CO2, pH 7.3–7.4, at 22–23°C. Whole-cell recordings of compound or miniature EPSCs (mEPSCs) were obtained from pyramidal neurons in the LA under visual guidance (DIC/infrared optics) with an EPC-10 amplifier and Pulse v8.67 software (HEKA Elektronik). Cells were classified as principal neurons based on spike frequency adaptation in response to prolonged depolarizing current injections (Shin et al., 2006, 2010). In current-clamp experiments, the recording patch electrodes (3–5 MΩ resistance) contained the following (in mm): 135 K-gluconate, 5 NaCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 MgATP, and 0.1 NaGTP (adjusted to pH 7.2 with KOH). In voltage-clamp experiments, 135 mm Cs-methane-sulfonate was used instead of potassium gluconate. In the experiments involving CCK4 applications, free Ca2+ concentration in the pipette solution was buffered to 100 nm with 5 mm EGTA/1.97 mm CaCl2 as described previously (Riccio et al., 2009). Synaptic responses were evoked by field stimulation of the fibers in either the internal capsule (thalamic input) or the external capsule (cortical input) with a fine-tipped (∼2 μm) silver-painted glass stimulation pipette (Cho et al., 2012). Currents were filtered at 1 kHz and digitized at 5 kHz. The EPSC or EPSP amplitudes were measured as the difference between the mean amplitude during a prestimulus baseline and the mean amplitude over a 1–2 ms window at the response peak. For induction of long-term potentiation (LTP), 80 presynaptic stimuli were delivered at 2 Hz to either the cortical or thalamic inputs. The EPSPs were paired with action potentials (APs) evoked in a postsynaptic neuron with a controlled delay (4–8 ms) from the onset of each EPSP (Shumyatsky et al., 2005; Riccio et al., 2009). In LTP experiments, the stimulus intensity was adjusted to evoke baseline synaptic responses with the amplitudes that were ∼20–25% of the maximum amplitude response. Summary LTP graphs were constructed by normalizing data in 60 s epochs to the mean value of the baseline EPSPs recorded at a holding potential of –70 mV. mEPSCs (recorded in the presence of 1 μm tetrodotoxin, TTX) and spontaneous IPSCs (sIPSCs) were analyzed with the Mini Analysis Program v6.0.7 (Synaptosoft).
Behavioral assays.
All behavioral tests were conducted with counterbalanced groups (wt and null mice; littermate male adults; Riccio et al., 2009). All electrophysiological and behavioral studies were performed blind to mouse genotype. The same mice were used in all behavioral experiments, except for the open field tests, which were performed on a different cohort of mice. Different tests were performed on different days. Experimental procedures were approved by the Boston Children's Hospital and the McLean Hospital's IACUC.
Elevated-plus maze.
The maze consisted of four interconnected arms raised 85 cm above the floor and illuminated (30 lux) by directional overhead lighting. The maze floor was made of white Plexiglas and the enclosed arms were made of gray Plexiglas. The arms were 30 × 5 cm. Two of the arms were open while the other two were enclosed by a 20 cm high perimeter wall. Animals were placed in the center of the elevated plus maze facing an open arm and were allowed to freely explore the maze for 5 min. The number of open and enclosed arm entries, time on the open and enclosed arms, time on the central platform, and the total distance traveled were measured. Sessions were analyzed using the Noldus EthoVision XT video tracking system.
Open field test.
The apparatus was an evenly lit (100 lux) clear Plexiglas box measuring 42 × 42 × 31 cm. When placed in the open field, subjects will readily explore the periphery (thigmotaxis) but largely avoid the center area (38 × 38 cm), a known anxiety-provoking location; the center area was ethologically defined as beginning one body length (4 cm) from the edge of the testing box. Subjects were placed along the periphery of the apparatus and allowed to explore for 30 min. The entire session was analyzed using the Noldus EthoVision XT video tracking system.
Open field test under red light conditions.
Mice were tested in the same apparatus as described above but under red light (<1 lux), nonanxiogenic conditions. The same measurements were recorded.
Single-trial fear-conditioning.
On the training day, the mouse (control or KO) was placed in the conditioning chamber for 2 min before the onset of the CS, a tone that lasted for 30 s at 2800 Hz at 85 dB (Riccio et al., 2009). The last 2 s of the CS was paired with the US of 0.7 mA of continuous footshock. After an additional 30 s in the chamber, the mouse was returned to its home cage. Mice were tested at 24 h after training. For testing, mice were placed in a novel environment (cage) in which the tone (60 s) that had been presented during training was given after a 1 min habituation period. Freezing was captured by ANY-maze software (Stoelting). Freezing scores were calculated as the fraction (percentage) of the total CS duration the mouse remained immobile (frozen).
Contextual fear conditioning.
Similar to a previously published study (Crestani et al., 2002), mice on the training day were placed individually in the conditioning chamber for 5 min before being exposed to three consecutive footshocks (0.7 mA, 2 s duration, 1 min apart). For testing, mice were placed in the same environment (cage) and freezing in the same context was captured 24 h after conditioning during an 8 min period.
Tail suspension test.
Mice were suspended for a 6 min test session by taping the tail to the edge of a table (height, 70 cm; Vollenweider et al., 2011). Subjects were videotaped and the latency to first immobility and total time spent immobile were manually scored. Immobility was defined as the complete cessation of movement while suspended.
Acoustic startle.
Animals were placed in a startle chamber for 5 min for habituation (Kinder Scientific). After this acclimation period, the test session was automatically started. To test the amplitude of startle response, animals experienced acoustic stimuli ranging from 90 to 105 dB presented in a random order at 30 s intervals. The duration of the noise stimulus was 40 ms. Responses were recorded for 150 ms from the startle onset and were sampled every ms (full scale = 4.0 N). The transducer created an AC signal (a positive voltage when the subject pushed and a negative voltage value when the subject retracted), which was converted to a digital response and calculated as the maximum force in Newtons. The maximum force (either due to a push or a retraction) during the 150 ms after the startle stimulus was recorded and stored by the software for statistical analysis.
Beam walking assay.
Balance beam tests of motor coordination were performed as described previously (Carter et al., 1999; Puram et al., 2011). Mice were trained to walk on a balance beam (20 mm × 0.75 m) for three trials. All mice traversed this wide beam without foot slips. Mice were then trained on a narrow (4 mm width × 0.75 m length) beam for three trials. Mice were videotaped as they performed three test trials of three beam walks, for a total of nine runs per animal. Videotaped walks were scored for number of foot slips and time to cross.
Gait analysis.
Gait parameters were measured using the automated DigiGait analysis system (Mouse Specifics; Puram et al., 2011). Using this system, mice were imaged ventrally with a high-frame-rate camera while running on a transparent treadmill. Software analysis was used to identify individual paw prints and calculate gait metrics based on the position, area, and timing of paw steps. All mice were run at 30 cm/s.
Results
Expression of TRPC4 in the mouse brain
As an initial step in exploring the functional roles of TRPC4 in fear-related behavioral processes, we characterized its expression in the brain using RNA in situ hybridization. Similar to TRPC5, TRPC4 expression was detected in brain areas known to be important for both innate and learned fear (Shumyatsky et al., 2005; Riccio et al., 2009). Specifically, TRPC4 expression was observed in the amygdala (in lateral and basolateral nuclei) and the hippocampus, including the CA1, CA2, and CA3 areas (Fig. 1A). TRPC4 mRNA was also present in both the auditory cortex (AuD1, Au1, and AuV) and auditory thalamus (Fig. 1B), which relay auditory CS information to the LA during the acquisition and recall of conditioned fear memory (LeDoux, 2000; Maren and Quirk, 2004; Shumyatsky et al., 2005). The US areas, including somatosensory cortex (S1 and S2 regions) and perirhinal cortex (Shi and Davis, 1999; Lanuza et al., 2004; Shumyatsky et al., 2005), also expressed TRPC4 mRNA (Fig. 1A). Notably, we previously found that TRPC5 is also expressed in the dentate gyrus but not in auditory thalamic areas (Riccio et al., 2009), suggesting that expression patterns of TRPC4 and TRPC5 may not completely overlap in the mouse brain and exhibit a certain degree of regional expression specificity.
Figure 1.
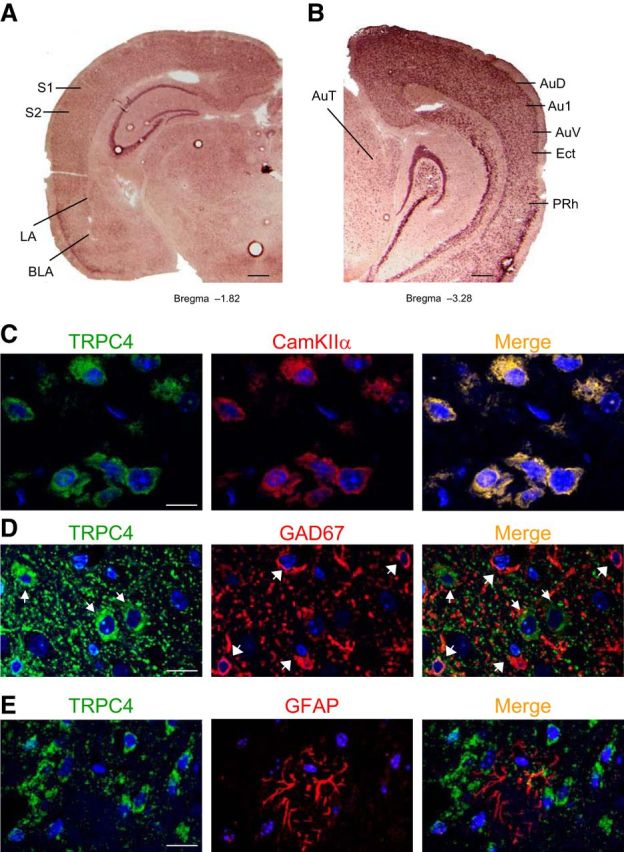
TRPC4 expression in adult mouse brain. A, B, In situ hybridization of TRPC4-mRNA in amygdala, hippocampus, somatosensory cortex, auditory thalamus, and auditory cortex. BLA, amygdala basolateral nucleus; S1, primary somatosensory cortex; S2, secondary somatosensory cortex; AuT, auditory thalamus; AuD, secondary auditory cortex, dorsal; Au1, primary auditory cortex; AuV, secondary auditory cortex, ventral; Ect, ectorhinal cortex; PRh, perirhinal cortex. Scale bar, 1 mm. C, TRPC4 (left) and CaMKIIα (middle; a marker of pyramidal neurons) colocalize in the LA (right). D, Cells expressing TRPC4 (red) and GFAP (green; a marker of glial cells) do not colocalize in the LA. E, Cells expressing TRPC5 (red) and GAD67 (green; a marker of interneurons) do not colocalize in the LA. Scale bars: C–E, 10 μm.
In immunohistochemical studies, we found that TRPC4 is colocalized with the neuron-specific marker CaMKIIα in the LA (Fig. 1C). It did not colocalize, however, with either the interneuron-specific marker, glutamic acid decarboxylase (GAD67), or the glial marker, GFAP (Fig. 1D,E). These findings show, that similar to TRPC5 (Riccio et al., 2009), TRPC4 expression is restricted to principal neurons.
Generation of TRPC4-null mice
Genetic ablation of the TRPC4 gene was achieved by removing the exon 4 genomic region encoding the amino acids 299–412 within the putative third transmembrane domain (Fig. 2A). This genetic modification introduced both a frame shift and premature stop codons after the deleted segment. The targeting construct was made using homologous recombination (Liu et al., 2002, 2003) and successful targeting was verified by Southern blotting (data not shown). The deletion of the exon 4 region, catalyzed by Cre-recombinase, was confirmed by PCR (Fig. 2B). The deletion of exon 4 was also confirmed by RT-PCR analysis and sequencing of the amplicon that spanned the deleted region of transcripts from brain microsomes of control and mutant mice (Fig. 2C). TRPC4 protein was not detected by Western blot in immunoprecipitation assays in brain microsomes from TRPC4−/− mice (Fig. 2D). Also, specific TRPC4 immunoreactivity was detected in brain sections from control mice but not in matched samples from TRPC4−/− mice (Fig. 2E). Matings between heterozygous animals generated offspring with normal Mendelian distribution of gender and genotype.
Figure 2.
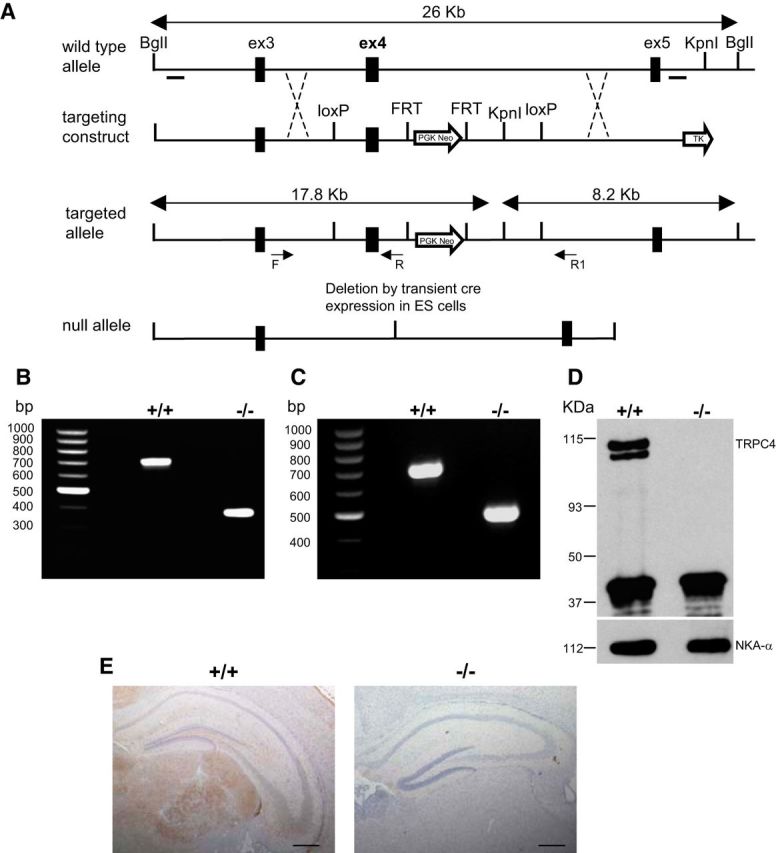
Generation and confirmation of TRPC4−/− mouse. A, Targeting strategy for the disruption of the TRPC4 gene. After homologous recombination, deletion of exon 4 region was catalyzed by Cre-recombinase in ES cells. B, Targeting of the TRPC4 locus is confirmed by PCR analysis of tail genomic DNA. C, RT-PCR analysis of whole-brain mRNA from control and TRPC4−/− littermates confirms the absence of exon 4. D, IP of TRPC4 protein in brain microsomes from control and TRPC4−/− mice reveals loss of TRPC4 protein in TRPC4−/− mice (top). Western blotting of NKA-α confirms equal protein loading in control and TRPC4−/− mice (bottom). E, Immunohistochemical staining of brain sections from control and TRPC4−/− littermates reveals selective loss of TRPC4 expression in mutant mice. Scale bar, 1 mm.
The mRNA expression levels of TRPC1, 3, 5, 6, and 7 remained unchanged in the whole brain of TRPC4−/− mice compared with control littermates (Fig. 3A–E; unpaired t test, A, t(10) = 1.9, p = 0.10; B, t(10) = 0.80, p = 0.4; C, t(10) = 0.88, p = 0.39; D, t(10) = 1.44, p = 0.17; E, t(10) = 1.0, p = 0.34). This finding provides evidence that, similar to the TRPC5 KO, TRPC4 ablation was not associated with detectable compensatory changes in the expression of related genes.
Figure 3.

qRT-PCR determination of TRPC mRNA levels in the brain. A–E, Whole-brain mRNA levels of TRPC1, TRPC3, TRPC5, TRPC6, and TRPC7 were not different between control and TRPC4−/− littermates. Data are expressed as arbitrary units normalized to β-actin to correct for RNA quantity and integrity and presented as mean ± SEM for triplicate reverse transcription reactions from two RNA pools (triplicate data from each of 2 control and 2 KO mice).
Innate fear responses are diminished in TRPC4−/− mice
Our preliminary tests indicated that TRPC4−/− mice had no abnormalities of spontaneous behaviors (body position, tremor, touch escape, transfer arousal, tail elevation), neurological reflexes (righting reflex, postural reflex, ear twitch reflex, grip strength, and whisker orientation reflex) or body weight.
We found previously that TRPC5−/− mice were less anxious than their control littermates (Riccio et al., 2009). As shown above, TRPC4 is also expressed in brain areas implicated in control of fear-related behaviors, prompting us to probe the level of fear and anxiety in TRPC4−/− mice. TRPC4, apparently, has no role in the mechanisms of learned fear, as we did not observe differences between groups in conditioned fear memory after single-trial fear conditioning (Fig. 4A; unpaired t test, t(18) = 0.06, p = 0.95) or in contextual freezing at 24 h post training (Fig. 4B; two-way ANOVA, F(1,144) = 0.04, p = 0.99). In contrast, control and mutant mice differed in their responses to innately aversive stimuli. The elevated plus-maze paradigm is an ethological test for anxiety-related behavior built on the natural conflict between exploration of a novel environment and the tendency to avoid elevated, exposed areas (Pellow et al., 1985; Lister, 1987). In our experiments, TRPC4−/− mice entered the open arms of the maze more frequently (Fig. 4C; unpaired t test, t(18) = 3.1, p < 0.01) and spent more time there than control mice (Fig. 4D; unpaired t test, t(18) = 4.4, p < 0.005), indicating that TRPC4-null mice were less anxious than their control littermates. The groups of mice did not differ in the closed-arm entries (Fig. 4E; t(18) = 0.7, p = 0.46) or total entries (Fig. 4E; t(18) = 1.71, p = 0.16). These findings are consistent with the notion that TRPC4 may be specifically implicated in control of innate fear responses.
Figure 4.

TRPC4−/− mice exhibit an anxiolytic-like phenotype. A, B, No differences in the percentage of conditioned freezing (A) or contextual freezing (B) were observed between control and null mice 24 h post training (n = 10 mice per each group). C–E, Elevated plus maze experiments (n = 10 mice per each group). TRPC4−/− mice entered the open arms more commonly (C) and spent more time in the open arms (D), but did not differ in the closed-arm entries (E, left) or total entries (E, right). F–H, Open field test (100 lux). TRPC4−/− mice entered the center more frequently (F), spent significantly more time in the center of the arena (G), and were more active (H). Data from 10 control and 9 null mice. I–K, In open field tests under red light (nonanxiogenic) conditions, there were no differences between control (n = 10) and null (n = 10) mice in exploration of the center of the arena for entries into the center (I), time spent in the center (J), or general exploratory activity (K). Results are shown as mean ± SEM.
To explore further the role of TRPC4 in unconditioned fear, we tested the performance of control and mutant mice in the novel open field test when the open field was either brightly lit (100 lux) or illuminated by red light (<1 lux). The open field test is an ethologically based measure of anxiety built on the conflict between the desire of mice to explore novel environments but to actively avoid brightly lit, open areas (Britton and Britton, 1981; File, 1985). When placed in a well lit, novel open field, TRPC4−/− mice spent significantly more time in the anxiogenic center of the arena (Fig. 4F; unpaired t test, t(17) = 3.2, p < 0.01) throughout the course (30 min) of the test (data not shown). Moreover, TRPC4 KO mice entered the center more frequently (Fig. 4G; unpaired t test, t(17) = 3.54, p < 0.01) and were more active than control littermates as measured by the total distance traveled (Fig. 4H; unpaired t test, t(17) = 3.57, p < 0.01). There were no overall differences between the genotypes in exploration of the center of the arena or in the distance traveled in the same novel open field under less anxiogenic, red light illumination (Fig. 4I–K; unpaired t test, I, t(18) = 0.06, p = 0.95; J, t(18) = 0.69, p = 0.49; K, t(18) = 0.15, p = 0.88). These findings demonstrate that anxiolytic-like behavioral modifications in mice lacking TRPC4 are more readily revealed under anxiety-provoking conditions.
TRPC4−/− mice did not exhibit general behavioral deficits reflecting anxiety levels unrelated to changes in behavioral responses. Thus, mutant mice did not exhibit impairment in the acoustic startle reflex (tested at 90, 95, 100; and 105 dB; Fig. 5A; two-way ANOVA, F(1,72) = 0.08, p = 0.97). We also did not observe differences between control and TRPC4−/− mice in the tail suspension test that is used as a measure of behavioral despair. In this behavioral paradigm, mice subjected to the short-term stress of being suspended by their tail acquire an immobile posture (behavioral despair) after a period of agitation (escape-oriented behavior). In our experiments, mice in both groups rapidly attained an immobile posture (Fig. 5B; unpaired t test, t(18) = 0.61, p = 0.55) and remained immobile for the same amount of time (Fig. 5C; unpaired t test, t(18) = 0.21, p = 0.83). As motor coordination was impaired in TRPC5−/− mice (Puram et al., 2011), we tested the performance of control and TRPC4−/− mice in balance beam assays. There were no group differences in the occurrence of foot-slip errors (Fig. 5D; unpaired t test, t(18) = 0.43, p = 0.67), number of steps (Fig. 5E; unpaired t test, t(18) = 0.52, p = 0.61), and mean crossing time (Fig. 5F; unpaired t test, t(18) = 0.39, p = 0.69). The gait analysis also did not reveal differences between genotypes in any of the parameters assayed, including paw angle variability (Fig. 5G; unpaired t test, t(18) = 0.42, p = 0.67), ataxia coefficient (deviation of the minimum and maximum stride length from the mean stride length; Fig. 5H; unpaired t test t(18) = 0.51, p = 0.61), and stride length variability (Fig. 5I; t(18) = 1.04, p = 0.31). The relative duration of each phase of gait remained unaltered in TRPC4−/− mice (data not shown). These findings show that motor coordination is unaffected by the ablation of TRPC4. Together, these results indicate that TRPC4 may serve an essential function in the regulation of anxiety-related behaviors.
Figure 5.

Lack of generalized behavioral deficits in TRPC4−/− mice. A, No significant differences were observed between control (empty bars) and null mice (filled bars) in acoustic startle responses to auditory stimuli at 90, 95, 100, and 105 dB (n = 10 mice per group). Responses were calculated as the maximum force in Newtons (N) based on the highest absolute value (during extension or retraction of the legs). B, C, Tail suspension test. When suspended by the tail for a 6 min test session, mice in both groups (n = 10 mice per each group) assumed an immobile posture within 40–60 s (B) and remained immobile for the times shown in C. There were no significant differences between genotypes. D–F, Beam walking test. Control and null mice (n = 10 mice per each group) did not show significant differences in percentage of foot slips (errors) on a narrow (4 mm wide) balance beam (D), number of steps (E), or mean crossing times (F). G–I, Gait analyses. No significant differences between groups were observed in paw angle variability (G), ataxia coefficient (H), and stride length variability (I) parameters. Results are shown as mean ± SEM.
Synaptic transmission and LTP in inputs to LA neurons are normal in TRPC4−/− mice
The amygdala is a key structural component of brain circuits underlying fear-related behavioral processes (LeDoux, 2000; Maren and Quirk, 2004). We, therefore, tested the possibility that behavioral deficits in TRPC4−/− mice might be associated with functional impairments in the LA (implicated in both learned and innate fear (Shumyatsky et al., 2005). We first explored the effect of TRPC4 ablation on the firing properties of LA neurons, determining the number of spikes generated in response to prolonged depolarizing current injections of gradually increasing intensity (from 50 to 450 pA) under current-clamp conditions (Fig. 6A,B). We found no differences between control and mutant mice in spike generation by injected current (Fig. 6C; n = 26 neurons from four control mice, n = 29 neurons from four TRPC4−/− mice; two-way ANOVA, F(1,371) = 0.64, p = 0.8 between groups), indicating that membrane excitability of neurons in the LA was unaffected by loss of TRPC4.
Figure 6.

Basal synaptic transmission and LTP are normal at the cortico-amygdala and thalamo-amygdala synapses in TRPC4−/− mice. A, Schematic representation of a brain slice preparation containing the amygdala that shows the position of the stimulation electrode (Sthalamic, Scortical) and recording (R) pipette. EC, external capsule. B, Responses of LA neurons in slices from control and TRPC4−/− mice to a prolonged current injection (200 pA, 500 ms) recorded in current-clamp mode. C, Number of APs in LA neurons evoked by current injections of increasing intensity in slices from control (open symbols) and TRPC4−/− (filled symbols) mice, recorded as in B. D, Synaptic input–output curves for the EPSCs recorded at the cortico-LA synapses. The EPSCs were recorded under voltage-clamp conditions at a holding potential of –70 mV. E, Spike timing-dependent LTP of the cortico-amygdala EPSPs recorded in LA neurons in slices from control and mutant mice. Insets are averages of 10 EPSPs recorded before and 35 min after induction. F, Summary of LTP experiments in cortical input to the LA. G, Synaptic input–output curves for the EPSCs recorded at the thalamo-LA synapses. The EPSCs were recorded under voltage-clamp conditions at a holding potential of –70 mV. H, LTP in thalamic input to the LA. Insets show the average of 10 EPSPs recorded before and 35 min after induction. I, Summary of LTP experiments in thalamic input to the LA. Results are shown as mean ± SEM.
Auditory CS information during fear conditioning and fear memory retrieval is transmitted to the LA through projections from the auditory thalamus, specifically from the medial division of the medial geniculate nucleus and the posterior intralaminar nucleus, and the auditory cortex (Romanski and LeDoux, 1992; Campeau and Davis, 1995; LeDoux, 2000). To investigate the role of TRPC4 in synaptic function in cortical and thalamic projections to the LA, we stimulated fibers in the external capsule (cortical input) or internal capsule (thalamic input; Shin et al., 2006, 2010; Cho et al., 2012), and recorded glutamatergic EPSCs from LA neurons in slices from TRPC4−/− and control mice. We previously demonstrated that these two inputs, converging on the same neurons in the LA, could be activated independently under our experimental conditions (Tsvetkov et al., 2004). Recorded cells were classified as principal neurons if they showed significant spike frequency adaptation in response to depolarizing current injection under current-clamp recording conditions (Tsvetkov et al., 2002). We found that TRPC4 ablation had no effect on synaptic strength at both auditory inputs to the LA, as input–output curves for AMPA receptor-mediated EPSCs did not differ between control and TRPC−/− mice (Fig. 6D, cortical input: n = 20 neurons from five control mice, n = 19 neurons from five TRPC4−/− mice, two-way ANOVA, F(1,333) = 0.21, p = 0.99 between groups; Fig. 6G, thalamic input: n = 19 neurons from five control mice, n = 21 neurons from six TRPC4−/− mice, two-way ANOVA, F(1,342) = 0.13, p = 1.0 between groups). To further assay possible contributions of TRPC4 in regulation of synaptic functions in fear circuits, we recorded spontaneous mEPSCs in LA neurons in the presence of TTX (1 μm), a sodium channel blocker. Neither the frequency of mEPSCs (an index of presynaptic function) nor their amplitude (indicative of sensitivity of postsynaptic AMPA receptors to glutamate), were affected by the TRPC4 ablation (Fig. 7A–E; n = 18 neurons from four control mice, n = 17 neurons from four TRPC4−/− mice;C, frequency: unpaired t test, t(33) = 1.44, p = 0.16 between groups; E, amplitude: unpaired t test, t(33) = 0.78, p = 0.44 between groups). These findings indicate that basal glutamatergic synaptic transmission in the LA was unaffected by deletion of the TRPC4 subunit.
Figure 7.
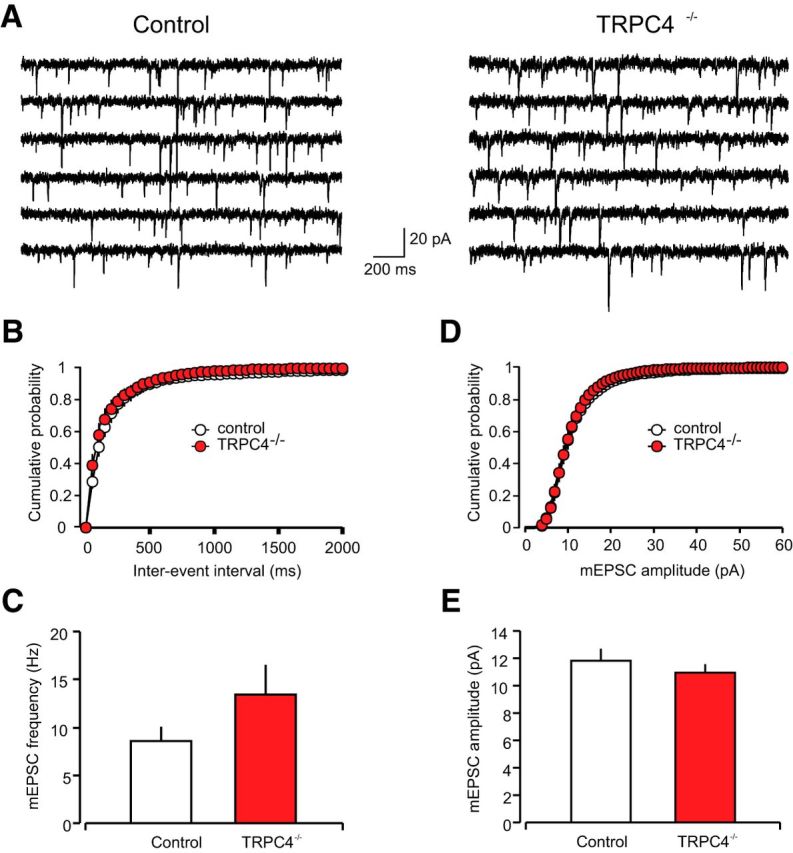
TRPC4 ablation had no effect on parameters of glutamatergic mEPSCs. A, mEPSCs recorded in LA neurons in slices from control (left) and TRPC4−/− mice (right). B, Cumulative interevent interval histograms of mEPSCs recorded in LA neurons in slices from control and TRPC4−/− mice. C, Summary plot showing averaged mEPSC frequency data. D, Cumulative amplitude histograms of mEPSCs recorded in LA neurons in slices from control and TRPC4−/− mice. E, Summary plot showing mean peak amplitude data from same recordings as in D. Results are shown as mean ± SEM.
We also assayed the effects of TRPC4 ablation on GABAA receptor-mediated inhibition in the LA. In these experiments, we recorded IPSCs in LA neurons in the presence of the AMPA receptor antagonist (CNQX, 20 μm) at –70 mV with a chloride-based intrapipette solution (Shumyatsky et al., 2005; Fig. 8A,B). The stimulation electrode was placed within the LA, thus allowing direct activation of local circuit interneurons. We found that the input–output relations for the GABA-mediated IPSCs were not different between control and TRPC4−/− mice (Fig. 8C; n = 12 neurons from four control mice, n = 8 neurons from four null mice; two-way ANOVA, F(1,108) = 0.55, p = 0.74). Moreover, there were no differences between control and mutant mice in the frequency or amplitude of sIPSCs (Fig. 8D–F; n = 15 neurons from four control mice, n = 12 neurons from four null mice; frequency: unpaired t test, t(25) = 0.30, p = 0.77 between groups; amplitude: unpaired t test, t(25) = 0.43, p = 0.67 between groups). Thus, GABAergic inhibitory neurotransmission in the LA was also normal in TRPC4−/−-null mice.
Figure 8.

GABAA receptor-mediated synaptic responses in LA neurons are normal in TRPC4−/− mice. A, Schematic representation of the neuronal circuit for fast GABAergic inhibition in the LA and the experimental design. LA/PN, principal neuron in the LA; LA/IN, interneurons in the LA. B, IPSCs (average of 10 traces), which were evoked in LA neurons by stimulation pulses of two different intensities in slices from control and TRPC4−/− mice. The stimulation electrode was placed within the LA, and the external solution contained CNQX (20 μm). The IPSCs were recorded with a chloride-based intrapipette solution (Shumyatsky et al., 2005). C, Input–output relations for evoked GABAAR IPSCs in slices from control and TRPC4−/− mice. D, Representative recordings of sIPSCs in LA neurons at –70 mV in slices from control and TRPC4−/− mice. E, Cumulative interevent interval (left) and amplitude (right) histograms of sIPSCs recorded in slices from control and TRPC4−/− mice. F, Summary of sIPSCs parameters for data in E. The graph shows averaged sIPSC frequency (left) and mean peak amplitude (right). Results are shown as mean ± SEM.
It has been repeatedly demonstrated that LTP-like synaptic enhancement in the CS pathways are implicated in fear learning and retention of fear memory during auditory fear conditioning (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997; Tsvetkov et al., 2002; Rumpel et al., 2005; Cho et al., 2012; Li et al., 2013). In our earlier experiments, we did not observe detectable differences in the magnitude of spike timing-dependent LTP, induced either in cortical or thalamic inputs to the LA, between control and TRPC5−/− mice (Riccio et al., 2009). However, TRPC5 and TRPC4 exhibit different expression patterns in the brain, with only TRPC4 being expressed in both the auditory thalamus and auditory cortex. This suggested to us that these two TRPC subunits could differ in their contribution to the inducibility of LTP. Therefore, we assayed the effect of TRPC4 subunit ablation on this form of synaptic plasticity at the cortico-amygdala or thalamo-amygdala synapses, stimulating the external capsule or internal capsule, respectively. LTP of glutamatergic EPSPs, recorded in current-clamp mode, was induced by pairing low-frequency presynaptic stimulation (2 Hz) for 40 s with APs evoked in a postsynaptic neuron with 4–8 ms delay from the onset of each EPSP (Shin et al., 2006; Riccio et al., 2009; Fig. 6E,H). The recordings were performed in the presence of the GABAA receptor antagonist picrotoxin (PTX; 50 μm). We found that, similar to TRPC5−/− mice, there was no difference in the magnitude of LTP in either of two studied inputs between control and TRPC4−/− mice (Fig. 6F, cortical input: n = 23 neurons from seven control mice, n = 13 neurons from five TRPC4−/− mice, unpaired t test, t(34) = 0.35, p = 0.73 between groups; I, thalamic input: n = 16 neurons from eight control mice, n = 17 neurons from six TRPC4−/− mice, unpaired t test, t(31) = 0.31, p = 0.76 between groups), indicating that TRPC4 does not play a role in the induction of spike timing-dependent LTP in CS pathways.
mGluR-EPSCs and CCK2 receptor-mediated membrane currents in LA neurons are diminished in mice lacking TRPC4
We next investigated whether the mGluR-mediated and CCK2 receptor-mediated responses in LA neurons are affected by ablation of the TRPC4 subunit for two reasons. We previously found that Group I mGluR-EPSCs and CCK2 receptor-mediated currents in LA neurons were reduced in TRPC5−/− mice. There is also significant experimental evidence indicating that both aforementioned Gq/11 protein-coupled receptor systems regulate fear-related behaviors (Frankland et al., 1997; Rodrigues et al., 2002; Wang et al., 2005; Pérez de la Mora et al., 2006). To evoke mGluR-EPSCs, we delivered short trains (10 pulses) of stimuli at a high frequency (100 Hz) to either cortical or thalamic projections in the presence of selective antagonists of AMPA receptors (CNQX, 20 μm), NMDA receptors (d-APV, 50 μm; and MK-801, 10 μm), GABAA receptors (PTX, 50 μm) and the GABABR blocker (CGP35348, 300 μm; Fig. 9A,B). We found that the magnitude of a residual component of the EPSP recorded in the presence of the blockers, which reflects synaptic activation of Group I mGluRs (Faber et al., 2006; Riccio et al., 2009), was diminished significantly in LA neurons from TRPC4−/− mice both in cortico-amygdala and thalamo-amygdala pathways (Fig. 9C; cortical input: n = 7 neurons from four control mice, n = 8 neurons from four TRPC4−/− mice, unpaired t test, t(13) = 2.17, p = 0.049 between groups; thalamic input: n = 9 neurons from six control mice, n = 6 neurons from four TRPC4−/− mice, unpaired t test, t(13) = 2.71, p = 0.018 between groups). Under our present recording conditions, these slow synaptic responses were sensitive to the mGluR5 and mGluR1 antagonists, MPEP (10 μm) and CPCCOEt (40 μm), respectively (cortical input: n = 4 neurons, paired t test, t(3) = 4.46, p = 0.02 vs baseline; thalamic input: n = 4 neurons, paired t test, t(3) = 3.78, p = 0.03 vs baseline), thus confirming that they were mediated by Group I mGluRs (Riccio et al., 2009).
Figure 9.
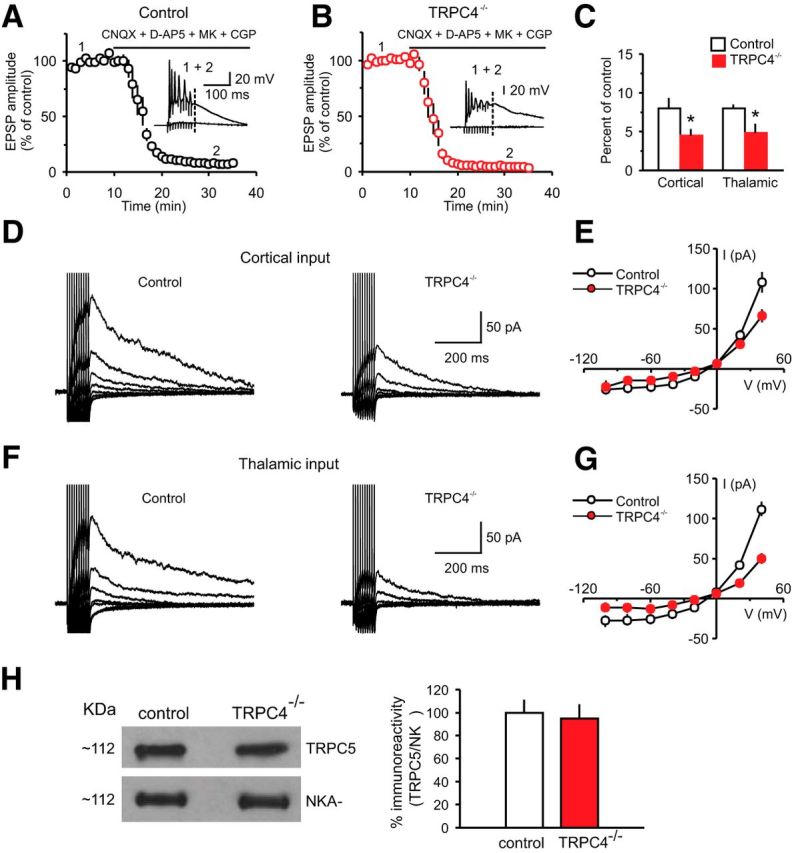
mGluR-EPSCs in LA neurons are suppressed in TRPC4−/− mice. A, Synaptic responses in cortical input to the LA neuron in a slice from a control mouse evoked by trains of high-frequency stimulation before and during addition of CNQX (AMPA receptor antagonist; 20 μm), NMDA receptor antagonists, d-APV (50 μm), and MK-801 (10 μm) and GABABR antagonist CGP 35348 (300 μm) recorded as described previously (Riccio et al., 2009). Stimulation trains consisted of 10 pulses at 100 Hz which were delivered once every 30 s. Inset shows synaptic responses recorded in current-clamp mode before (1) and after (2) the addition of antagonists to the external medium. The dashed line indicates the time point where the EPSP amplitude was measured. B, The experiment was identical to A, but the recording was obtained from a TRPC4−/− mouse. C, Summary data for the experiments as in A and B, performed in both cortical and thalamic inputs to the LA. The amplitude of the residual component of the EPSP in the presence of antagonists was smaller in both pathways in slices from TRPC4−/− mice compared with control littermates. D, EPSCs in cortical input recorded in voltage-clamp mode at holding potentials ranging from –100 mV to +40 mV in slices from control (left) and TRPC4−/− (right) mice in the presence of the antagonists (as in A). E, Current–voltage (I–V) plots of the peak current in cortical input (as in D) in slices from control and TRPC4−/− mice. F, EPSCs in thalamic input recorded in voltage-clamp mode at holding potentials ranging from –100 mV to +40 mV in slices from control (left) and TRPC4−/− (right) mice in the presence of the antagonists (as in A). G, Current–voltage (I–V) plots of the peak current in thalamic input (as in F) in slices from control and TRPC4−/− mice. H, Left, Representative immunoblot shows similar amounts of TRPC5 protein in brain microsomes extracted from control or TRPC4−/− littermates. Western blotting of Na+-K+-ATPase indicates equal protein loading (bottom). Right, Quantification of three different blots. Results are shown as mean ± SEM.
We then recorded isolated mGluR-EPSCs at cortical and thalamic inputs over a range of holding potentials (from –100 mV to +40 mV) under whole-cell conditions to compare the current–voltage (I–V) relations of the peak EPSC amplitude between control and mutant mice (Fig. 9D,F). Whereas there were no differences in the shape of I–Vs for mGluR-EPSCs between the groups, the EPSC amplitude in slices from TRPC4−/− mice was smaller than in slices from control mice at more positive potentials (+40 mV), where the amplitude of EPSCs was relatively large, and, therefore the differences were more readily detectable (Fig. 9E, cortical input: n = 10 neurons from six control mice, n = 10 neurons from four TRPC4−/− mice, unpaired t test, t(18) = 2.79, p = 0.012 for peak amplitudes at Vh = +40 mV between groups; G, thalamic input: n = 10 neurons from six control mice, n = 10 neurons from four TRPC4−/− mice, unpaired t test, t(18) = 5.83, p = 0.001 for peak amplitudes at Vh = +40 mV). Levels of TRPC5 protein in the IP assay remained unchanged in brain microsomes from the whole-brain and LA lysates obtained from control or TRPC4−/− mice (Fig. 9H, right; unpaired t test, t(4) = 0.26, p = 0.8). These results confirm that the observed effects on mGluR-EPSCs in TRPC4−/− mice were not due to a compensatory downregulation of TRPC5 expression (Fig. 3).
We also found that membrane depolarization in LA neurons, induced by bath application of the specific agonist of CCK2 receptors, CCK4 (3 μm), was significantly reduced in slices from TRPC4−/− mice compared with control littermates (Fig. 10B; n = 15 neurons from six control mice, n = 16 neurons from seven TRPC4−/− mice; unpaired t test, t(29) = 1.95, p = 0.031 between groups). The observed decrease in membrane depolarization was associated with a diminished ability of exogenously applied CCK4 to increase spike firing in response to depolarizing current injections in slices from mice lacking the TRPC4 subunit (Fig. 10A,C,D; n = 15 neurons from six control mice, n = 16 neurons from seven TRPC4−/− mice; t test, t(29) = 3.01, p = 0.003). The relative densities of LA innervation by CCK-positive fibers did not differ between control and TRPC4−/− mice, indicating that TRPC4 ablation had no effect on peptidergic innervation of the LA (Fig. 10E, right, unpaired t test, t(10) = 0.25, p = 0.81). Thus, similar to TRPC5−/− mice, the observed behavioral impairments could partially be due to the lack of activation of TRPC4-containing channels via mGluRs and CCK2 receptors in the brain's anxiety circuits.
Figure 10.

CCK-mediated increase in spike firing in LA neurons is diminished in TRPC4−/− mice. A, Spikes evoked in LA neurons by current injection (150 pA) recorded in current-clamp mode under baseline conditions and in the presence of 3 μm CCK4 in a slice from a control mouse. B, Summary plot showing CCK4-induced depolarization in LA neurons in slices from control and mutant mice. C, Spikes evoked in LA neuron under baseline conditions and in the presence of 3 μm CCK4 in a slice from a TRPC4−/− mouse. D, The percentage increase in spike frequency in the presence of CCK4 relative to the baseline frequency (taken as 100%) in slices from control and null mice. E, Left, Fluorescence double labeling for CCKergic fibers (green) and CaMKIIα (red) reveals no differences in innervation of the LA by CCK-containing fibers in brain sections from control and TRPC4−/− littermates. Right, Mean CCK fluorescence after normalization to CaMKIIα fluorescence from six images per group taken from different stainings (two images from each of 3 control and 3 KO mice). Scale bar, 10 μm. Results are shown as mean ± SEM.
RNAi-induced knockdown of TRPC4 in the LA recapitulates behavioral phenotype of TRPC4−/− mice
To strengthen a link between decreased anxiety and altered function of the amygdala in TRPC4−/− mice, we explored whether the targeted inactivation of TRPC4 in the LA of control mice would, in fact, reproduce the behavioral phenotype observed in constitutive mutant animals. In these experiments, we used an in vivo RNA interference (RNAi) approach, as there are no specific TRPC4 blockers currently available. We previously generated shRNAs that target the TRPC4 gene and confirmed the induced knockdown of TRPC4 protein in neuronal cultures (Puram et al., 2011). Here, we knocked down TRPC4 by using a lentivirus vector expressing both anti-TRPC4 shRNA and GFP under different promoters. Ten-week-old control mice received bilateral stereotaxic intra-LA injections of LV-GFP-shTRPC4 or scrambled control LV-GFP-shSCRM and were killed after 4 weeks to verify knockdown. Sustained expression of GFP was observed in brain sections in the LA, whereas surrounding regions did not exhibit significant infection (Fig. 11A). Western blot analysis confirmed significant decreases in TRPC4 protein expression in LA homogenates obtained from mice injected with LV-GFP-shTRPC4 relative to LV-GFP-shSCRM controls, indicating that TRPC4 shRNA was effective at depleting TRPC4 protein in vivo (Fig. 11B, right; unpaired t test, t(8) = 4.4, p < 0.01 vs LV-SCRM-GFP controls). We did not detect significant changes in the TRPC5 expression, indicating a lack of compensatory effects (Fig. 11B; unpaired t test, t(8) = 0.38, p = 0.72). Mice were subjected to behavioral tests 4 weeks after microinjections. We found that TRPC4 knockdown in the LA resulted in diminished innate fear responses, assayed with both the elevated plus maze (Fig. 11C–E; unpaired t test, C, t(18) = 6.36, p < 0.001; D, t(18) = 4.64, p < 0.005; E, t(18) = 0.58, p = 0.51) and open field (Fig. 11F–H; unpaired t test, F, t(18) = 6.5, p < 0.005; G, t(18) = 5.77, p < 0.005; H, t(18) = 1.1, p = 0.31) tests. Conversely, TRPC4 knockdown had no effect on auditory (Fig. 11I; unpaired t test, t(18) = 0.68, p = 0.42) or contextual fear conditioning (Fig. 11J; unpaired t test, t(18) = 0.32, p = 0.74). Thus, the behavioral consequences of TRPC4 knockdown in the LA were very similar to the anxiolytic behavioral phenotype observed in genetically modified mice lacking TRPC4. These findings, providing direct evidence for the role of TRPC4 in the LA in control of innate fear, justify the focus of our electrophysiological recordings in the LA of control and TRPC4−/− mice.
Figure 11.
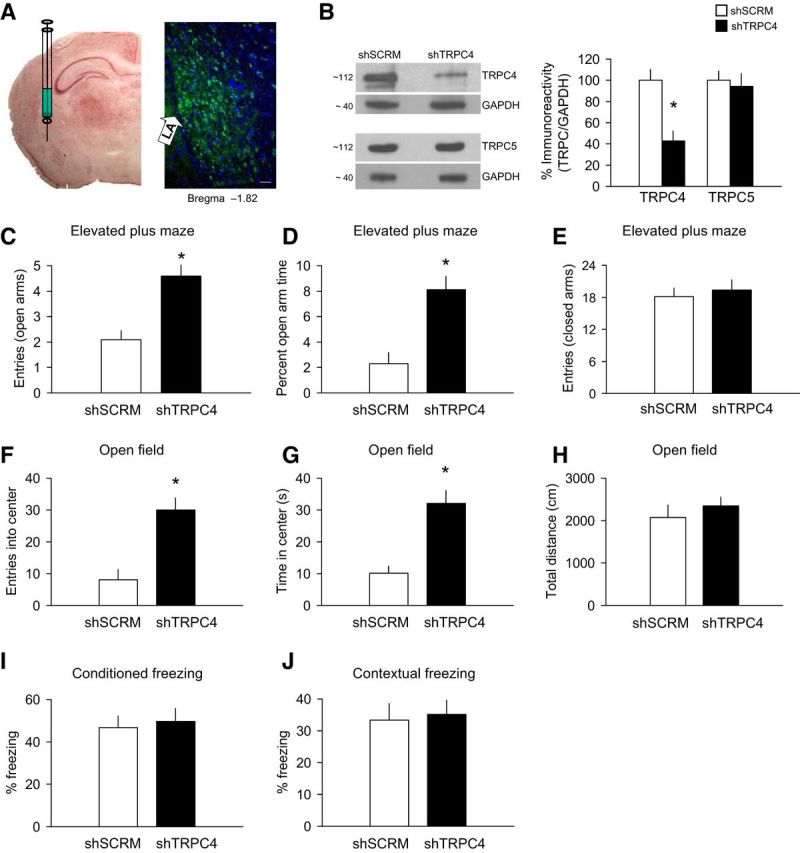
Knockdown of TRPC4 in the LA. A, Left, An image illustrating the injection experiments. Right, a microscopic image showing expression of LV-shTRPC4-GFP in the LA (green). Scale bar, 50 μm. B, Left, Representative Western blots showing TRPC4 knockdown and TRPC5 expression in the LA. Right, Analysis of TRPC4 and TRPC5 in LA homogenates taken 4 weeks following intra-LA infusion of LV-shTRPC4-GFP (n = 5) or LV-SCRM-GFP (n = 5). Protein levels were normalized to GAPDH). C–E, Elevated plus maze experiments (n = 10 mice per a group). Mice with TRPC4 knockdown exhibited an increased number of entries into the open arms (C), spent more time in the open arms (D), but showed no differences in closed-arm entries (E). F–H, Open field test (100 lux; n = 10 mice per group). TRPC4 knockdown mice entered the center of the arena more frequently (F) and spent more time in it (G), and travel the same distance as control mice (H). I, J, No differences in freezing responses (n = 10 mice per a group) were observed in fear conditioning (I) or contextual fear tests (J). Results are shown as mean ± SEM.
Discussion
The results of the present study provide evidence that normal expression of the TRPC4 subunit in brain circuits might be required for behavioral responses triggered by exposure of experimental subjects to anxiety-inducing stimuli, since TRPC4−/− mice exhibited an anxiolytic-like behavioral phenotype. Consistent with its role in regulation of fear-related behaviors, we found enriched TRPC4 levels in the LA, where CS and US information converge during formation of fear memory (LeDoux, 2000; Davis and Whalen, 2001). Brain regions providing CS and US signals, including the auditory cortex and auditory thalamus (Romanski and LeDoux, 1992; Campeau and Davis, 1995; Maren and Quirk, 2004) and somatosensory cortical areas (Shi and Davis, 1999; Lanuza et al., 2004; Shumyatsky et al., 2005), also express TRPC4. Since the LA is an essential component of the neural network underlying both innate and learned fear, we assessed whether knockdown of TRPC4 in the LA affected fear-related behaviors. In mice, TRPC4 protein expression was knocked down via viral-mediated expression of TRPC4-shRNAi. The behavioral studies on these mice revealed that innate fear responses were impaired, similar to the phenotype detected in constitutive TRPC4-null mice. These observations demonstrate that TRPC4 in the LA is required for normal fear responses. The decreased anxiety levels in TRPC4−/− mice could result from diminished Group I mGluR-mediated and CCK2 receptor-mediated responses. Notably, mGluR1 receptor-mediated synaptic responses in cerebellar Purkinje cells evoked by stimulation of parallel fibers were normal in TRPC1–TRPC4 double KO mice (Hartmann et al., 2008). This suggests that the extent of neurotransmitter-evoked recruitment of specific Gq/11-linked signaling mechanisms might differ between different regions of the brain.
The observed anxiety deficits in TRPC4−/− mice were not due to changes in motor coordination. In contrast to TRPC5-null mice in previous work (Puram et al., 2011), motor coordination was unaffected in mice lacking TRPC4. Interestingly, TRPC4 and TRPC5 are both expressed in the cerebellum (Allen Atlas, http://www.brain-map.org/), implicated in coordinated motor control. The observed behavioral differences could be due to differential expression of these two channels in distinct subpopulations of granule neurons and/or differences in TRPC4 and TRPC5 interactions with TRPC1 subunits. Therefore, it might be interesting to explore TRPC1-null mice in future behavioral and electrophysiological experiments.
By analogy with TRPC5−/− mice (Riccio et al., 2009), the observed ability of cortico-amygdala and thalamo-amygdala synapses in TRPC4-null mice to undergo normal associative LTP is in agreement with the lack of impairment in conditioned fear memory in the absence of TRPC4. Given that baseline excitatory synaptic transmission was unaffected by TRPC4 removal, afferent signals, requiring formation of the CS–US association, could be normally transmitted to the LA during fear conditioning. The resulting synaptic enhancements in the CS pathways (due to the convergence of CS and US signals), implicating LTP mechanisms (McKernan and Shinnick-Gallagher, 1997; Tsvetkov et al., 2002; Rumpel et al., 2005; Cho et al., 2012), could then be translated into the enhanced firing output of LA neurons, relaying the CS information to other components of fear-conditioning circuitry, and, eventually producing normal learned fear responses. Conversely, Gq/11-mediated signaling, triggered by activation of Group I mGluRs and/or CCK2 receptors, was suppressed in TRPC4-deficient mice, thus potentially decreasing the spike output of neurons in the LA, and, subsequently, transmission of signals to downstream areas involved in fear reactions. These findings could explain the observed decreases in innate fear in the absence of TRPC4, as both receptor systems (Group I mGluRs and CCK2 receptors) were implicated previously in regulation of anxiety mechanisms (Frankland et al., 1997; Rodrigues et al., 2002; Wang et al., 2005).
Whereas behavioral modifications and changes in Gq/11 protein-mediated signaling in the present work were similar to previously observed behavioral and functional consequences of TRPC5 ablation (Riccio et al., 2009), the expression profiles of TRPC4 and TRPC5 did not ideally overlap. TRPC4 was expressed in both auditory cortex and auditory thalamus, while TRPC5 expression was not observed in the auditory thalamus. The difference in expression patterns suggests the interesting possibility that these two members of the TRPC1/4/5 subfamily might differentially contribute to certain aspects of fear related behaviors (e.g., CS discrimination during fear conditioning; Armony et al., 1997), warranting future comparative investigation. However, the differences in expression profiles between TRC5 and TRPC4 subunits in normal mouse brain did not result in mutation-specific changes in baseline synaptic transmission or in the inducibility and maintenance of LTP in the amygdala, as synaptic transmission and plasticity remained normal in slices from both mutant mice. Apparently, both TRPC4 and TRPC5 are not directly implicated in control of synaptic function, either presynaptically or postsynaptically, but rather serving modulatory roles when activated or potentiated by Gq/11-coupled neurotransmitter receptors.
TRPC4's expression profile was very similar to that of a phosphoprotein stathmin, a negative regulator of microtubule formation, which is present in both the US and CS neural circuits, controlling learned and innate fear responses (Shumyatsky et al., 2005). Despite similarities in expression profiles of TRPC4 and stathmin, behavioral consequences of TRPC4 deletion were limited to the impairments in innate fear. Although these findings do not challenge the notion that behavioral mechanisms could be controlled by the neural circuitry-specific gene expression, they suggest that the behavioral outcome may eventually depend on a complex interplay between multiple cellular and molecular events governed by the products of these genes.
Footnotes
This work was supported by grants from the National Institutes of Health (NIH; to V.Y.B., grant MH090464), and (to D.E.C., grant MH090293), and the Neurodevelopmental Behavioral Core and the Gene Manipulation Facility of the Boston Children's Hospital Mental Retardation and Developmental Disabilities Research Center (NIH grant P30-HD18655).
References
- Armony JL, Servan-Schreiber D, Romanski LM, Cohen JD, LeDoux JE. Stimulus generalization of fear responses: effects of auditory cortex lesions in a computational model and in rats. Cereb Cortex. 1997;7:157–165. doi: 10.1093/cercor/7.2.157. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- Britton DR, Britton KT. A sensitive open field measure of anxiolytic drug activity. Pharmacol Biochem Behav. 1981;15:577–582. doi: 10.1016/0091-3057(81)90212-4. [DOI] [PubMed] [Google Scholar]
- Campeau S, Davis M. Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2312–2327. doi: 10.1523/JNEUROSCI.15-03-02312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Bayazitov IT, Meloni EG, Myers KM, Carlezon WA, Jr, Zakharenko SS, Bolshakov VY. Coactivation of thalamic and cortical pathways induces input timing-dependent plasticity in amygdala. Nat Neurosci. 2012;15:113–122. doi: 10.1038/nn.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Clapham DE. SnapShot: mammalian TRP channels. Cell. 2007;129:220. doi: 10.1016/j.cell.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, Blüthmann H, Möhler H, Rudolph U. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Natl Acad Sci U S A. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Faber ES, Sedlak P, Vidovic M, Sah P. Synaptic activation of transient receptor potential channels by metabotropic glutamate receptors in the lateral amygdala. Neuroscience. 2006;137:781–794. doi: 10.1016/j.neuroscience.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- File SE. What can be learned from the effects of benzodiazepines on exploratory behavior? Neurosci Biobehav Rev. 1985;9:45–54. doi: 10.1016/0149-7634(85)90031-4. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Josselyn SA, Bradwejn J, Vaccarino FJ, Yeomans JS. Activation of amygdala cholecystokininB receptors potentiates the acoustic startle response in the rat. J Neurosci. 1997;17:1838–1847. doi: 10.1523/JNEUROSCI.17-05-01838.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. San Diego: Academic; 2007. [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanuza E, Nader K, Ledoux JE. Unconditioned stimulus pathways to the amygdala: effects of posterior thalamic and cortical lesions on fear conditioning. Neuroscience. 2004;125:305–315. doi: 10.1016/j.neuroscience.2003.12.034. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Li Y, Meloni EG, Carlezon WA, Jr, Milad MR, Pitman RK, Nader K, Bolshakov VY. Learning and reconsolidation implicate different synaptic mechanisms. Proc Natl Acad Sci U S A. 2013;110:4798–4803. doi: 10.1073/pnas.1217878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister RG. The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology. 1987;92:180–185. doi: 10.1007/BF00177912. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. Efficient Cre-loxP-induced mitotic recombination in mouse embryonic stem cells. Nat Genet. 2002;30:66–72. doi: 10.1038/ng788. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Meis S, Munsch T, Sosulina L, Pape HC. Postsynaptic mechanisms underlying responsiveness of amygdaloid neurons to cholecystokinin are mediated by a transient receptor potential-like current. Mol Cell Neurosci. 2007;35:356–367. doi: 10.1016/j.mcn.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky G, Kotton DN, Fabian AJ, Gray JT, Lee JS, Mulligan RC. Efficiency of transduction of highly purified murine hematopoietic stem cells by lentiviral and oncoretroviral vectors under conditions of minimal in vitro manipulation. Mol Ther. 2005;11:932–940. doi: 10.1016/j.ymthe.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12:218. doi: 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellow S, Chopin P, File SE, Briley M. Validation of open–closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods. 1985;14:149–167. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- Pérez de la Mora M, Lara-García D, Jacobsen KX, Vázquez-Garcia M, Crespo-Ramírez M, Flores-Gracia C, Escamilla-Marvan E, Fuxe K. Anxiolytic-like effects of the selective metabotropic glutamate receptor 5 antagonist MPEP after its intra-amygdaloid microinjection in three different nonconditioned rat models of anxiety. Eur J Neurosci. 2006;23:2749–2759. doi: 10.1111/j.1460-9568.2006.04798.x. [DOI] [PubMed] [Google Scholar]
- Pietraszek M, Sukhanov I, Maciejak P, Szyndler J, Gravius A, Wislowska A, Płaźnik A, Bespalov AY, Danysz W. Anxiolytic-like effects of mGlu1 and mGlu5 receptor antagonists in rats. Eur J Pharmacol. 2005;514:25–34. doi: 10.1016/j.ejphar.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Puram SV, Riccio A, Koirala S, Ikeuchi Y, Kim AH, Corfas G, Bonni A. A TRPC5-regulated calcium signaling pathway controls dendrite patterning in the mammalian brain. Genes Dev. 2011;25:2659–2673. doi: 10.1101/gad.174060.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Riccio A, Medhurst AD, Mattei C, Kelsell RE, Calver AR, Randall AD, Benham CD, Pangalos MN. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res Mol Brain Res. 2002;109:95–104. doi: 10.1016/S0169-328X(02)00527-2. [DOI] [PubMed] [Google Scholar]
- Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, Gapon S, Yao GL, Tsvetkov E, Rodig SJ, Van't Veer A, Meloni EG, Carlezon WA, Jr, Bolshakov VY, Clapham DE. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell. 2009;137:761–772. doi: 10.1016/j.cell.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Bauer EP, Farb CR, Schafe GE, LeDoux JE. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J Neurosci. 2002;22:5219–5229. doi: 10.1523/JNEUROSCI.22-12-05219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Romanski LM, LeDoux JE. Equipotentiality of thalamo-amygdala and thalamo-cortico-amygdala circuits in auditory fear conditioning. J Neurosci. 1992;12:4501–4509. doi: 10.1523/JNEUROSCI.12-11-04501.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- Shi C, Davis M. Pain pathways involved in fear conditioning measured with fear-potentiated startle: lesion studies. J Neurosci. 1999;19:420–430. doi: 10.1523/JNEUROSCI.19-01-00420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin RM, Tsvetkov E, Bolshakov VY. Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron. 2006;52:883–896. doi: 10.1016/j.neuron.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin RM, Tully K, Li Y, Cho JH, Higuchi M, Suhara T, Bolshakov VY. Hierarchical order of coexisting pre and postsynaptic forms of LTP at synapses in amygdala. Proc Natl Acad Sci U S A. 2010;107:19073–19078. doi: 10.1073/pnas.1009803107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumyatsky GP, Malleret G, Shin RM, Takizawa S, Tully K, Tsvetkov E, Zakharenko SS, Joseph J, Vronskaya S, Yin D, Schubart UK, Kandel ER, Bolshakov VY. stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell. 2005;123:697–709. doi: 10.1016/j.cell.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/S0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY. Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron. 2002;34:289–300. doi: 10.1016/S0896-6273(02)00645-1. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Shin RM, Bolshakov VY. Glutamate uptake determines pathway specificity of long-term potentiation in the neural circuitry of fear conditioning. Neuron. 2004;41:139–151. doi: 10.1016/S0896-6273(03)00800-6. [DOI] [PubMed] [Google Scholar]
- Vollenweider I, Smith KS, Keist R, Rudolph U. Antidepressant-like properties of a2-containing GABAA receptors. Behav Brain Res. 2011;217:77–80. doi: 10.1016/j.bbr.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wong PT, Spiess J, Zhu YZ. Cholecystokinin-2 (CCK2) receptor-mediated anxiety-like behaviors in rats. Neurosci Biobehav Rev. 2005;29:1361–1373. doi: 10.1016/j.neubiorev.2005.05.008. [DOI] [PubMed] [Google Scholar]