

Abstract
Rare, familial, early-onset autosomal dominant forms of familial Alzheimer's disease (FAD) are caused by mutations in genes encoding β-amyloid (Aβ) precursor protein (APP), presenilin-1 (PS1), and presenilin-2. Each of these genes is expressed ubiquitously throughout the CNS, but a widely held view is that excitatory neurons are the primary (or sole) source of the Aβ peptides that promote synaptic dysfunction and neurodegeneration. These efforts notwithstanding, APP and the enzymes required for Aβ production are synthesized by many additional cell types, and the degree to which those cells contribute to the production of Aβ that drives deposition in the CNS has not been tested. We generated transgenic mice in which expression of an ubiquitously expressed, FAD-linked mutant PSEN1 gene was selectively inactivated within postnatal forebrain excitatory neurons, with continued synthesis in all other cells in the CNS. When combined with an additional transgene encoding an FAD-linked APP “Swedish” variant that is synthesized broadly within the CNS, cerebral Aβ deposition during aging was found to be unaffected relative to mice with continued mutant PS1 synthesis in excitatory neurons. Thus, Aβ accumulation is non-cell autonomous, with the primary age-dependent contribution to cerebral Aβ deposition arising from mutant PS1-dependent cleavage of APP within cells other than excitatory neurons.
Keywords: amyloid deposition, APP, dementia, mouse model, neurodegeneration, presenilin
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by impairments in cognition and memory, neuronal death, and deposition of 40–42 amino acid β-amyloid (Aβ) peptides that are derived from larger amyloid precursor proteins (APPs). Familial, early-onset, autosomal dominant forms of AD (FAD) are caused by inheritance of mutant genes encoding APP, presenilin-1 (PS1), and presenilin-2 variant polypeptides that are expressed ubiquitously in all CNS cell types and peripheral organs. Presenilins are the catalytic subunits of the γ-secretase complex that promotes intramembranous proteolysis of APP and a number of type I membrane proteins (De Strooper, 2003). Importantly, FAD-linked mutant forms of presenilins cause disease by elevating the ratio of Aβ42/Aβ40 peptides, ultimately leading to early and selective cerebral deposition of Aβ42 peptides (Price and Sisodia, 1998).
Neuronal-specific overexpression of FAD-linked APP or PSEN1 transgenes leads to cerebral Aβ deposition in transgenic mice but attempts to evaluate the effect on Aβ metabolism or deposition of expressing these mutant genes in other CNS cell types has not been forthcoming. To explore the effect on Aβ levels and deposition, we now report the outcome of selective excision of a widely expressed mutant PSEN1ΔE9 transgene within excitatory neurons, because these cells are known to produce high levels of Aβ peptides in an activity-dependent manner (Kamenetz et al., 2003). Despite >80% reduction in steady-state PS1ΔE9 polypeptide levels in the brain by the age of 7 months, age-dependent accumulation of Aβ and amyloid deposition (at 12 months of age) was unaffected, and the levels of soluble and insoluble Aβ peptides in younger mice (8–9 months) were only modestly affected. However, loss of mutant presenilin expression by excitatory neurons did markedly suppress Aβ deposition in early adult life during aging (between 5 and 7 months of age). We further demonstrate that cultured astrocytes and microglia secrete high levels of Aβ peptides. Thus, during aging of this mouse model, cells other than excitatory neurons provide a sustained source of secreted Aβ peptides that may accrete onto “seeds” that form independent of Aβ production by excitatory neurons and/or serve as templates for seeding and aggregation at later stages of amyloidogenesis.
Materials and Methods
Animals.
Male mice expressing prion promoter (PrP)-driven PS1 transgenes [PrP.APPswe (Borchelt et al., 1997), PrP.PS1ΔE9flox (line 13; Veeraraghavalu and Sisodia, 2013), and αCaMKIICre (line T29-2; Tsien et al., 1996)] were maintained heterozygous for the transgene in (C3H/HeJ × C57BL/6J F3) × C57BL/6J n1 background. Mice homozygous for PS1ΔE9flox and αCaMKIICre transgenes were crossed to PrP.APPswe mice to generate APPswe/PS1ΔE9flox/CaMKIICre mice carrying all three transgenes. Animal experiments were conducted in accordance with institutional approval and National Institutes of Health guidelines.
Tissue preparation, Western blot analyses, immunocytochemistry, and sandwich ELISA assays.
Animals were deeply anesthetized and perfused transcardially with cold 0.9% saline solution, and brains were harvested. One hemisphere was snap frozen for biochemical analysis, and the other half was fixed in 4% paraformaldehyde and transferred into 30% sucrose; 40-μm-thick coronal sections were stored at −20°C in cryoprotective buffer before histology. For Western blot analysis, brain tissue was homogenized in buffer containing 1% SDS and protein concentration in the soluble fraction determined using BCA protein assay (Pierce). For immunofluorescence studies, every 6th or 12th coronal section spanning the proximal to distal end of the hippocampus including cortical regions was used. Sections were incubated with Aβ-specific 3D6 antibody and bound antibody visualized using a fluorescently labeled secondary antibody. Amyloid area fraction and plaque numbers were quantified using NIH Image J, as described previously (Veeraraghavalu et al., 2013). Data are reported as mean ± SEM. Student's t test (unpaired) was performed for comparisons of quantitative data. Values of p < 0.05 were used as the criterion for statistical significance. The levels of TBS-soluble and 70% formic acid-soluble Aβ40 and Aβ42 peptides were determined using ELISA protocols as described previously (Veeraraghavalu et al., 2013).
Results
Postnatal deletion of mutant PSEN1 in forebrain excitatory neurons
In previous studies, we characterized a transgenic mouse line harboring a PrP.PS1ΔE9lox/lox.cGFP transgene (PS1ΔE9flox mice), wherein the ubiquitously expressed murine PrP drives expression of a mutant PS1ΔE9 cDNA flanked by loxP sites and includes a downstream cDNA encoding the copepod green fluorescent protein (cGFP; Veeraraghavalu and Sisodia, 2013); in PS1ΔE9flox mice, expression of the PS1ΔE9 transgene occurs in all neuronal and non-neuronal cell types, leaving expression of the cGFP reporter silent. Cell-type-specific expression of Cre recombinase results in excision of the PS1ΔE9 cDNA cassette, thus activating expression of the cGFP reporter in cells wherein recombination has occurred (Veeraraghavalu and Sisodia, 2013).
To examine the roles of excitatory neurons on amyloid deposition and Aβ production, we crossed the PS1ΔE9flox mice with αCaMKIICre transgenic mice (line T29-1; (Tsien et al., 1996), wherein Cre recombinase is expressed postnatally and is restricted to excitatory neurons of the forebrain. Western blot analysis of detergent solubilized brain lysates prepared from either 1-, 2-, 3-, 6-, or 20-week-old PS1ΔE9flox mice or PS1ΔE9flox/CaMKIICre mice were performed with a rat monoclonal antibody specific for human PS1 (Lah et al., 1997) or cGFP (Fig. 1A). Compared with 1- or 3-week-old nontransgenic (NTg) mice, expression of the PS1ΔE9 polypeptide was observed in PS1ΔE9flox mice at all ages (Fig. 1A, compare lanes 1, 6 with 2, 4, 7, 9, 11). However, compared with PS1ΔE9flox mice (Fig. 1A, lanes 2–5), a prominent reduction in PS1ΔE9 polypeptide levels was evident in PS1ΔE9flox/CaMKIICre mice beginning at 6 weeks of age (Fig. 1A, compare lanes 9 with 10 and 11 with 12). In line with these observations, cGFP reporter expression was observed only in lysates prepared from PS1ΔE9flox/CaMKIICre mice beginning at 6 weeks of age (Fig. 1A, lanes 10, 12, cGFP panel).
Figure 1.

Postnatal deletion of PS1ΔE9 transgene expression in PS1ΔE9flox/CaMKIICre mice. A, Total brain protein lysates from 1-, 2-, 3-, 6-, or 20-week-old mice were immunoblotted (20 μg/lane) with antibodies specific to human PS1 (hPS1), cGFP, or βIII-tubulin (Tuj1). One- or 3-week-old NTg brain lysate was loaded as controls. B, Total protein lysates from NTg mice (50 μg), PS1ΔE9flox mice (50, 25, 10, 5, 2.5, 1 μg), or PS1ΔE9flox/CaMKIICre mice (25, 10, 5 μg) at 2, 3, 5, or 7 months immunoblotted with antibodies specific to human PS1 (hPS1) or cGFP. Equal protein content of 50 μg/lane was loaded by supplementing the samples from different dilutions with NTg protein lysate. C, Total brain protein lysates prepared from 5, 7, 8, or 12 month age-matched PS1ΔE9flox mice or PS1ΔE9flox/CaMKIICre mice were immunoblotted (20 μg/lane) with PS1-NT antibody (hPS1NT). Brain lysate from 12-month-old APPswe mice was loaded as control. D, Images on the right reveal cGFP expression (green), turned on by Cre-mediated excision of PS1ΔE9 cassette. Gradual increase in endogenous green fluorescence signal was observed within the cortex of PS1ΔE9flox/CaMKIICre mice brain sections at 1.5, 2, 5, and 7 months of age. No cGFP expression was detected in cortex or hippocampus of 5-month-old PS1ΔE9flox mice brain section (top row). Images on the left are the respective DAPI-stained images. Scale bar, 250 μm. E, Confocal image reveals that neurons located in the layers III, V, and VI expressed cGFP in PS1ΔE9flox/CaMKIICre mice by 3 months of age (Ei), and the overlay with DAPI counterstain is shown below (Eii). Scale bar, 100 μm. F, Confocal image of dentate gyrus in PS1ΔE9flox/CaMKIICre mice brain sections at 1.5, 2, 3, 5, and 7 months of age. Progressive expression of cGFP was observed in the cell bodies and neurites of granule cell neurons in the granule cell layer (GCL) from the outermost layers to innermost layers of the dentate gyrus. HL, Hilus. Scale bar, 10 μm. G, Images of dentate gyrus from 4-month-old PS1ΔE9flox/CaMKIICre mice, double stained with antibodies specific to neuronal markers Prox1 (red) or NeuN (red). cGFP-expressing dentate granule cells were colabeled with Prox1 and NeuN. Scale bar, 25 μm. M, Month; W, week.
To semiquantitatively determine the extent of reduction in PS1ΔE9 levels, human PS1-specific antibody was used in Western blot analysis of serial diluted total protein lysates prepared from the hippocampi of age-matched PS1ΔE9flox or PS1ΔE9flox/CaMKIICre mice (Fig. 1B). These studies reveal that, in 2-, 3-, 5-, or 7-month-old PS1ΔE9flox/CaMKIICre mice, PS1ΔE9 polypeptide levels are reduced by 48, 58.3, 69.7, and 78.5%, respectively, relative to the levels seen in PS1ΔE9flox mice. We speculate that the residual PS1ΔE9 signal in the PS1ΔE9flox/CaMKIICre mice samples represents mutant PS1 expression in cells that do not express Cre. Western blot analysis of protein lysates prepared from the brains of 5-, 7-, 8-, or 12-month-old PS1ΔE9flox using polyclonal PS1-amino terminal (NT) antibody that recognizes both human and mouse PS1 (Thinakaran et al., 1997) revealed that the endogenous mouse N-terminal derivative (NTF) is “replaced” (Fig. 1C, lanes 1, 3, 5, 7). Conversely, in brains of 5-, 7-, 8-, or 9-month-old PS1ΔE9flox/CaMKIICre mice, excision of the PS1ΔE9flox cassette led to the accumulation of mouse NTF (Fig. 1C, lanes 2, 4, 6, 8, respectively), as was expected if endogenous PS1 can compete with the limiting components of the γ-secretase complex (Thinakaran et al., 1997).
Consistent with the Western blot analyses, cGFP fluorescence was observed in the cortex and hippocampus of PS1ΔE9flox/CaMKIICre mice beginning at the age of 1.5 months that increased in intensity in an age-dependent manner (Fig. 1D–F); the cGFP-positive cells express the neuronal antigens Prox1 and/or NeuN but not antigens specific for astrocytes (GFAP and S100β) or microglia (Iba1) (Veeraraghavalu and Sisodia, 2013).
Effect of αCaMKIICre-mediated excision of the PS1ΔE9 cassette in excitatory neurons on Aβ deposition and steady-state Aβ levels
We first bred PS1ΔE9flox mice to PrP promoter-driven APPswe transgenic mice that express APPswe at ∼2.5-fold over endogenous APP and only exhibit sparse amyloid deposits beginning at ∼18–20 months of age (Borchelt et al., 1997). The resulting APPswe/PS1ΔE9flox mice were then crossed with CaMKIICre mice to generate APPswe/PS1ΔE9flox/CaMKIICre mice. Cohorts of male APPswe, APPswe/PS1ΔE9flox, or APPswe/PS1ΔE9flox/CaMKIICre mice were aged to 4, 5–7, 8–9, or 10–12 months, and the extent of Aβ deposition was revealed by immunostaining with an Aβ-specific 3D6 monoclonal antibody. As expected, we did not observe 3D6-positive Aβ deposits in the brains of APPswe mice aged to 4, 5–7, 8–9, or 10–12 months (Fig. 2Ai,Aiv,Avii,Ax, respectively; quantified in B) or in the brains of 4-month-old APPswe/PS1ΔE9flox and APPswe/PS1ΔE9flox/CaMKIICre mice (Fig. 2Aii,Aiii; quantified in B).
Figure 2.
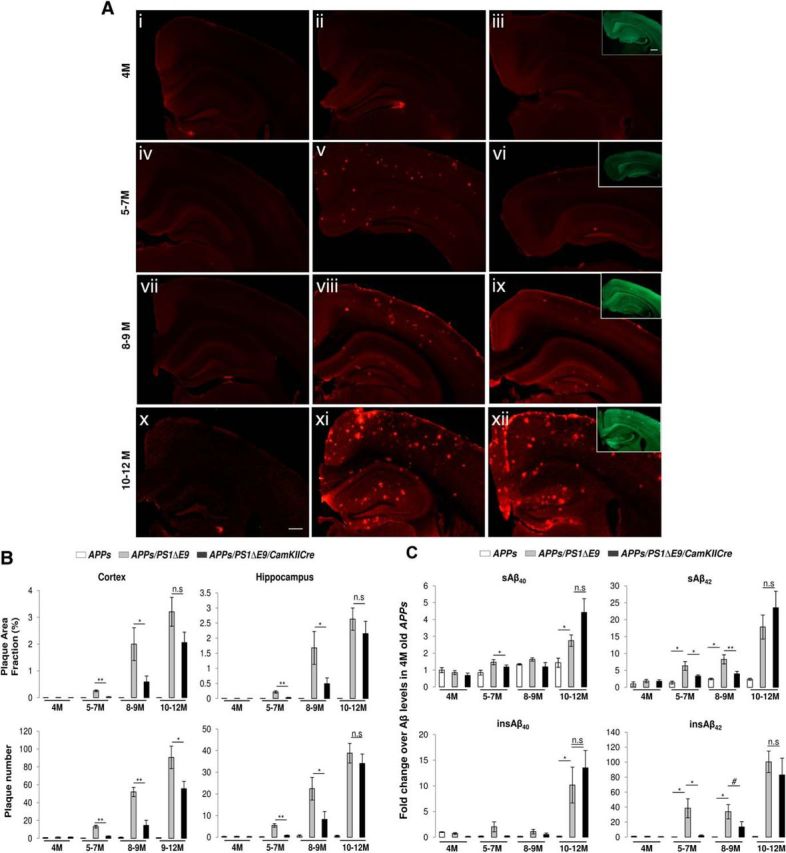
Amyloid deposition and Aβ levels as a function of age in brains of APPswe, APPswe/PS1ΔE9flox, or APPswe/PS1ΔE9flox/CaMKIICre mice. A, Extent of amyloid deposition after postnatal ablation of FAD-linked mutant PS1 expression in mature excitatory neurons. Images of hemibrain sections from APPswe (Ai, Aiv, Avii, Ax), APPswe/PS1ΔE9flox (Aii, Av, Aviii, Axi), or APPswe/PS1ΔE9flox/CaMKIICre (Aiii, Avi, Aix, Axii) mice aged to 4, 5–7, 8–9, or 10–12 months immunostained with 3D6 (red). cGFP expression as observed by cGFP fluorescence (green) in APPswe/PS1ΔE9flox/CaMKIICre mice are shown within the insets in Aiii, Avi, Aix, and Axii. Scale bar, 500 μm. B, Quantification of amyloid deposition after postnatal ablation of FAD-linked mutant PS1 expression in mature excitatory neurons. Histograms show the amyloid plaque area fraction (percentage) and plaque numbers in the cortex or hippocampus of APPswe, APPswe/PS1ΔE9flox, or APPswe/PS1ΔE9flox/CaMKIICre mice aged to 4, 5–7, 8–9, or 10–12 months. n = 6 (4 months, APPswe), 7 (4 months, APPswe/PS1ΔE9flox), 6 (4 months, APPswe/PS1ΔE9flox/CaMKIICre), 5 (5–7 months, APPswe), 8 (5–7 months, APPswe/PS1ΔE9flox; plaque area fraction in the cortex and hippocampus was 0.255 ± 0.033 and 0.218 ± 0.039%, respectively, and the plaque number was 13.15 ± 1.49 and 5.47 ± 0.79, respectively), 8 (5–7 months, APPswe/PS1ΔE9flox/CaMKIICre), 3 (8–9 months, APPswe), 6 (8–9 months, APPswe/PS1ΔE9flox; plaque area fraction was 1.99 ± 0.61 and 1.67 ± 0.54%, respectively, and plaque numbers were 51.96 ± 5.18 and 22.4 ± 5.24, respectively), 7 (8–9 months, APPswe/PS1ΔE9flox/CaMKIICre; plaque area fraction in the cortex and hippocampus was 0.589 ± 0.21 and 0.49 ± 0.19%, respectively, and plaque numbers were 14.57 ± 5.64 and 8.22 ± 3.64, respectively), 3 (10–12 months, APPswe), 4 (10–12 months, APPswe/PS1ΔE9flox; plaque area fraction and plaque numbers in cortex were 3.2 ± 0.53% and 90.7 ± 12.6, respectively, and the plaque area fraction and plaque numbers in hippocampus were 2.62 ± 0.36% and 38.85 ± 4.55, respectively), and 5 (10–12 months, APPswe/PS1ΔE9flox/CaMKIICre; plaque area fraction and plaque number in the cortex were 2.05 ± 0.39% and 55.6 ± 8.36, respectively, and the plaque area fraction and plaque numbers in the hippocampus were 2.15 ± 0.41% and 34.08 ± 4.35, respectively). Plaque area fraction in the cortex: *p = 0.021 for8–9 months and **p = 0.00013 for 5–7 months for APPswe/PS1ΔE9flox versus APPswe/PS1 ΔE9flox/CaMKIICre groups. Plaque numbers in the cortex: *p = 0.023 for 10–12 months,**p < 0.0001 for 5–7 months, and **p = 0.008 for 8–9 months for APPswe/PS1ΔE9flox versus APPswe/PS1ΔE9flox/CaMKIICre groups. Plaque area fraction in the hippocampus: *p = 0.025 for 8–9 months and **p = 0.001 for 5–7 months for APPswe/PS1ΔE9flox versus APPswe/PS1ΔE9flox/CaMKIICre groups. Plaque numbers in the hippocampus: *p = 0.022 for 8–9 months and **p = 0.0004 for 5–7 months for APPswe/PS1ΔE9flox versus APPswe/PS1ΔE9flox/CaMKIICre groups. n.s., Not significant. C, Changes in steady-state levels of Aβ40 and Aβ42 peptides after postnatal ablation of FAD-linked mutant PS1 expression in mature excitatory neurons. Histograms show fold changes in sAβ40, sAβ42, insAβ40, or insAβ42 levels in hemibrains of APPswe, APPswe/PS1ΔE9flox, or APPswe/PS1ΔE9flox/CaMKIICre mice aged to 4, 5–7, 8–9, or 10–12 months over the levels observed in age-matched APPswe mice group. n = 6 (4 months, APPswe), 7 (4 months, APPswe/PS1ΔE9flox), 7 (4 months, APPswe/PS1ΔE9flox/CaMKIICre), 5 (5–7 months, APPswe), 8 (5–7 months, APPswe/PS1ΔE9flox), 8 (5–7 months, APPswe/PS1ΔE9flox/CaMKIICre), 2 (8–9 months, APPswe), 5 (8–9 months, APPswe/PS1ΔE9flox), 6 (8–9 months, APPswe/PS1ΔE9flox/CaMKIICre), 3 (10–12 months, APPswe), 5 (10–12 months, APPswe/PS1ΔE9flox), and 5 (10–12 months, APPswe/PS1ΔE9flox/CaMKIICre). * p < 0.05, **p < 0.01, #p = 0.058. n.s., Not significant.
Sandwich ELISA analysis of TBS soluble (s) and formic acid-extractable insoluble (ins) Aβ peptides from hemibrains of the same cohorts of animals that were assessed by 3D6 immunocytochemistry (Fig. 2B) revealed no significant differences in the levels of sAβ40, sAβ42, insAβ40, or insAβ42 between APPswe, APPswe/PS1ΔE9flox, or APPswe/PS1ΔE9flox/CaMKIICre mice at 4 months of age (Fig. 2C).
Compared with the 5- to 7-month-old APPswe mice (Fig. 2Aiv), the APPswe/PS1ΔE9flox mice exhibited robust amyloid deposition (Fig. 2Av; quantified in B). Supporting these findings, we observed a significant increase in sAβ and insAβ species in APPswe/PS1ΔE9flox mice compared with APPswe mice (Fig. 2C). Interestingly, in 5- to 7-month-old APPswe/PS1ΔE9flox/CaMKIICre mice, we observed a dramatic reduction in 3D6 immunoreactivity compared with the APPswe/PS1ΔE9flox mice (Fig. 2, compare Avi with Av); the cortex and hippocampus of APPswe/PS1ΔE9flox/CaMKIICre mice exhibited significant 10.14- and 7.08-fold reductions in plaque area, respectively, and 5.59- and 6.63-fold reduction in plaque number, respectively (Fig. 2B). Indeed, biochemical analyses revealed a significant decrease in sAβ42 and insAβ42 levels in 5- to 7-month-old APPswe/PS1ΔE9flox/CaMKIICre cohorts compared with the age-matched APPswe/PS1ΔE9flox cohorts (Fig. 2C).
At 8–9 months of age, the APPswe/PS1ΔE9flox mice exhibited even higher levels of Aβ deposits compared with the 5- to 7-month-old APPswe/PS1ΔE9flox cohorts, as expected (Fig. 2, compare Aviii with Av; quantified in B). Moreover, we observed a modest reduction in Aβ deposition in the cortex and hippocampus of the 8- to 9-month-old APPswe/PS1ΔE9flox/CaMKIICre mice compared with their APPswe/PS1ΔE9flox cohorts (Fig. 2, compare Aix with Aviii). Compared with the 8- to 9-month-old APPswe/PS1ΔE9flox mice, the cortex and hippocampus of APPswe/PS1ΔE9flox/CaMKIICre mice exhibited a significant 3.38- and 3.39-fold reduction in plaque area, respectively, and a 3.56- and 2.72-fold reduction in plaque number, respectively (Fig. 2B). These morphological findings were confirmed by Aβ ELISAs (Fig. 2C). Thus, although there appears to be significant reductions in plaque area and number in the brains of 8- to 9-month-old APPswe/PS1ΔE9flox/CaMKIICre mice compared with their APPswe/PS1ΔE9flox cohorts of the same age, the extent of reduction is not as dramatic as that observed compared with their respective 5- to 7-month-old cohorts.
At 10–12 months of age, the APPswe/PS1ΔE9flox mice exhibited even higher levels of Aβ deposits compared with the 5- to 7-month-old or 8- to 9-month-old APPswe/PS1ΔE9flox cohorts (Fig. 2, compare Axi with Av, Aviii; quantified in B). However, compared with amyloid deposition in 10- to 12-month-old APPswe/PS1ΔE9flox mice (Fig. 2Axi), 10- to 12-month-old APPswe/PS1ΔE9flox/CaMKIICre mice (Fig. 2Axii) exhibited a very modest 1.56- and 1.63-fold reduction in plaque area and plaque number in the cortex, respectively, and a 1.22- and 1.13-fold reduction in plaque area and plaque number in the hippocampus, respectively (Fig. 2, compare Axii with Axi; quantified in B). Indeed, ELISAs failed to detect a statistically significant difference in the levels of sAβ or insAβ peptide species in APPswe/PS1ΔE9flox/CaMKIICre mice compared with APPswe/PS1ΔE9flox mice (Fig. 2C).
In summary, although postnatal deletion of the mutant PS1ΔE9 transgene in excitatory neurons results in a significant reduction in both Aβ deposition and steady-state Aβ levels in young animals, we failed to observe a reduction in either of these parameters in aged animals.
Generation of Aβ peptides in non-neuronal cells from transgenic mice
That the PrP promoter-driven transgene is ubiquitously expressed led us to investigate the possibility that the enzymatic machinery responsible for Aβ generation, namely β-secretase (BACE1) and presenilin/γ-secretase, are present in cells other than excitatory neurons that could contribute to the sustained elevation in steady-state levels of Aβ observed in older APPswe/PS1ΔE9flox/CaMKIICre mice. To address this issue, primary astrocytes, microglia, or adult neural progenitor cultures (NPCs) that are derived from radial glial-like precursors were established from the brains of newborn PS1ΔE9flox or APPswe mice or 8-week-old APPswe/PS1ΔE9flox mice. Conditioned medium (CM) collected from astrocyte (ACM), microglia (MCM), or NPC (NCM) cultures were fractionated by SDS-PAGE and subjected to Western blot analysis using the Aβ-specific monoclonal antibody 26D6. Secreted Aβ peptides were readily detected in the CM of all three cell types established from APPswe or APPswe/PS1ΔE9flox mice (Fig. 3A). Treatment of the cultures with a γ-secretase inhibitor (L-685,458 [1S-benzyl-4R-(1S-carbamoyl-2-phenylethylcarbamoyl-1S-3-methylbutylcarbamoyl)-2R-hydroxy-5-phenylpentyl] carbamic acid tert-butyl ester]) abolished Aβ secretion and resulted in the intracellular accumulation of APP C-terminal fragments, the penultimate substrate for Aβ generation (Fig. 3B). Moreover, both Aβ40 and Aβ42 peptides are present in the MCM of APPswe or APPswe/PS1ΔE9flox mice, with a notable elevation in Aβ42 peptide levels in APPswe/PS1ΔE9flox MCM (Fig. 3C). Collectively, these results reveal that the enzymes necessary for generating Aβ are present in multiple non-neuronal cell types, findings that would argue that these species contribute to amyloid deposition and steady-state levels of Aβ peptides in the adult CNS of our mouse model.
Figure 3.

Amyloidogenic processing in non-neuronal cells. A, Western blot analyses of ACM, MCM, or NCM cultures (150 μl; 3% of total volume) from mentioned transgenic genotypes, immunoblotted with 26D6 antibody. B, Western blot analyses of cell lysates (top 2 rows) or CM (third row) prepared from astrocyte, microglia, or NPC cultures established from APPswe/PS1ΔE9flox genotype after treatment with L-685,458 (1 μm) were immunoblotted with 369 antibody to detect full-length APP (FL APP) and C-terminal fragments (APP CTF) or 26D6 antibody. C, Western blot analyses of Aβ40 and Aβ42 peptides, immunoprecipitated with 6E10 from CM of APPswe or APPswe/PS1ΔE9flox microglia, were immunoblotted against 26D6. A total of 0.2 ng of pure Aβ40 and Aβ42 peptides were loaded as controls (lanes 1, 2).
Discussion
Although a series of preceding efforts established that neuronal overexpression of FAD-linked mutant APP and PSEN1 transgenes promotes CNS deposition of Aβ, it is clear that transcripts and encoded APP and PS1 are expressed in a wide variety of cell types in the adult CNS. For example, PSEN1 mRNA is highly expressed in neurons, astrocytes, and oligodendrocytes in white matter (Sherrington et al., 1995). Moreover, light and immuno-EM approaches revealed that PS1 is expressed in neurons, glial cells, and blood vessels in primate brain (Lah et al., 1997). Similarly, APP isoforms are expressed in non-neuronal cells of human brain white matter (Golde et al., 1990) and in cultured murine and human astrocytes, microglia, and endothelial cells (Haass et al., 1991; Forloni et al., 1992; Mönning et al., 1995). Thus, we sought to examine the effect of expressing FAD-linked mutant genes in CNS cell types other than excitatory neurons.
By selective inactivation of a widely expressed FAD-linked mutant presenilin gene in postnatal forebrain excitatory neurons (a principal source of secreted Aβ peptides that are released in an activity-dependent manner; Kamenetz et al., 2003; Cirrito et al., 2005), we have now demonstrated that the levels of insAβ peptides and amyloid burden in mice with or without excitatory neuron production of Aβ are indistinguishable by 10–12 months of age. Thus, production and accumulation of Aβ peptides during aging is independent of Aβ production by excitatory neurons, an observation that argues persuasively for alternative cellular source(s) and/or factor(s) that contribute to Aβ deposition in an age-dependent manner. Consistent with a non-neuron contribution, we demonstrate that cultured astrocytes and microglia from APPswe/PS1ΔE9flox mice secrete robust levels of Aβ peptides. Moreover, age-dependent, excitatory neuron-independent accumulation of Aβ occurs despite our evidence that excitatory neurons are a principal source of Aβ peptides deposited in younger animals.
Thus, we conclude that, during aging in this animal model, cells other than excitatory neurons provide the primary source of Aβ peptides that either accrete onto preexisting seeds or serve as templates for additional seeding, aggregation, and subsequent deposition. Additionally, expression of mutant PS1 in these non-excitatory neuronal cells may also effect conversion of sAβ monomers to “oligomers” that seed fibril formation and subsequent deposition. Indeed, we documented quantitative differences in the levels of selected cytokines and growth factors secreted by microglia isolated from adult transgenic mice expressing either wild-type human PS1 or mutant PS1 variants (Choi et al., 2008), and it is conceivable that these or other secreted factors may have a critical role in altering Aβ metabolism in a manner that influences seeding and/or deposition of these pathogenic peptides.
Footnotes
This work was supported by National Institutes of Health Grants AG021494 and AG027854 (S.S.S.), the Cure Alzheimer's Fund (S.S.S.), the Edward H. Levi Fund (K.V.), and the Adler Foundation (K.V., S.S.S.). We thank Dr. Vytas Bindokas, Microscopy Core Facility, for expert support with image analysis.
S.S.S. is a paid consultant of Eisai Research Labs, AZ Therapies, and Jannsen Pharmaceutica but is not a shareholder in any company that is a maker or owner of a Food and Drug Administration-regulated drug or device. The other authors declare no competing financial interests.
References
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/S0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, Sisodia SS. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. 2008;59:568–580. doi: 10.1016/j.neuron.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/S0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain Res Mol Brain Res. 1992;16:128–134. doi: 10.1016/0169-328X(92)90202-M. [DOI] [PubMed] [Google Scholar]
- Golde TE, Estus S, Usiak M, Younkin LH, Younkin SG. Expression of beta amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer's disease using PCR. Neuron. 1990;4:253–267. doi: 10.1016/0896-6273(90)90100-T. [DOI] [PubMed] [Google Scholar]
- Haass C, Hung AY, Selkoe DJ. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci. 1991;11:3783–3793. doi: 10.1523/JNEUROSCI.11-12-03783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mönning U, Sandbrink R, Weidemann A, Banati RB, Masters CL, Beyreuther K. Extracellular matrix influences the biogenesis of amyloid precursor protein in microglial cells. J Biol Chem. 1995;270:7104–7110. doi: 10.1074/jbc.270.13.7104. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/S0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Veeraraghavalu K, Sisodia SS. Mutant presenilin 1 expression in excitatory neurons impairs enrichment-mediated phenotypes of adult hippocampal progenitor cells. Proc Natl Acad Sci U S A. 2013;110:9148–9153. doi: 10.1073/pnas.1302106110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavalu K, Zhang C, Miller S, Hefendehl JK, Rajapaksha TW, Ulrich J, Jucker M, Holtzman DM, Tanzi RE, Vassar R, Sisodia SS. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models.”. Science. 2013;340:924-f. doi: 10.1126/science.1235505. [DOI] [PubMed] [Google Scholar]