

Abstract
The protein tyrosine phosphatase Shp2 (PTPN11) is crucial for normal brain development and has been implicated in dorsal telencephalic neuronal and astroglia cell fate decisions. However, its roles in the ventral telencephalon and during oligodendrogenesis in the telencephalon remain largely unknown. Shp2 gain-of-function (GOF) mutations are observed in Noonan syndrome, a type of RASopathy associated with multiple phenotypes, including cardiovascular, craniofacial, and neurocognitive abnormalities. To gain insight into requirements for Shp2 (LOF) and the impact of abnormal Shp2 GOF mutations, we used a Shp2 conditional mutant allele (LOF) and a cre inducible Shp2-Q79R GOF transgenic mouse in combination with Olig2cre/+ mice to target embryonic ventral telencephalic progenitors and the oligodendrocyte lineage. In the absence of Shp2 (LOF), neuronal cell types originating from progenitors in the ventral telencephalon were generated, but oligodendrocyte progenitor cell (OPC) generation was severely impaired. Late embryonic and postnatal Shp2 cKOs showed defects in the generation of OPCs throughout the telencephalon and subsequent reductions in white matter myelination. Conversely, transgenic expression of the Shp2 GOF Noonan syndrome mutation resulted in elevated OPC numbers in the embryo and postnatal brain. Interestingly, expression of this mutation negatively influenced myelination as mice displayed abnormal myelination and fewer myelinated axons in the white matter despite elevated OPC numbers. Increased proliferating OPCs and elevated MAPK activity were also observed during oligodendrogenesis after expression of Shp2 GOF mutation. These results support the notion that appropriate Shp2 activity levels control the number as well as the differentiation of oligodendrocytes during development.
Keywords: oligodendrocyte, Shp2
Introduction
The telencephalon exhibits immense cellular diversity, producing numerous neuronal subtypes and glia (oligodendrocytes and astrocytes) (for review, see Marín and Rubenstein, 2001; Rowitch and Kriegstein, 2010). Oligodendrocytes are generated from ventricular zone (VZ) progenitors that give rise to oligodendrocyte progenitor cells (OPCs), which subsequently proliferate, migrate, and differentiate into mature oligodendrocytes (OLs) that myelinate axons. A major question that remains in the field is to identify key regulators of signaling pathways that are responsible for or contribute to the generation and differentiation of OPCs from telencephalic progenitor cells.
Shp2 (PTPN11 in humans) encodes a nonreceptor protein tyrosine phosphatase that has a stimulatory role on the RAS/MAPK pathway, likely acting on negative regulator(s) of RAS (for review, see Neel et al., 2003; Dance et al., 2008). Germline gain of function (GOF) and dominant-negative mutations of PTPN11 have been identified in Noonan syndrome and LEOPARD syndrome patients, respectively (Gelb and Tartaglia, 2006). These developmental syndromes are referred to as neuro-cardio-facio-cutaneous syndromes as a result of overlapping phenotypes or “RASopathies” due to the abnormal regulation of the RAS/MAPK pathway (Gelb and Tartaglia, 2006; Bentires-Alj et al., 2006; Tidyman and Rauen, 2009). In addition to growth, cardiovascular, and craniofacial defects, various studies have reported that nearly 50% of Noonan syndrome patients exhibit some form of neurocognitive impairment, suggesting an impact of the Shp2 GOF mutations in human brain development (Lee et al., 2005; Pierpont et al., 2009; Alfieri et al., 2011).
Previous studies have shown a role for Shp2 in cell fate decisions by promoting neurogenesis and repressing astrogliogenesis in the developing hippocampus and cortex of the telencephalon (Gauthier et al., 2007; Ke et al., 2007). Moreover, Gauthier et al. (2007) showed that germline Noonan syndrome mice (PTPN11D61G/+) show more neurons and fewer astrocytes in the hippocampus and dorsal cortex at postnatal day 2, suggesting that Noonan syndrome mutations cause cellular brain abnormalities. Notably, white matter regions and oligodendrocytes were not analyzed in these mice. In vitro studies have suggested different roles for Shp2 during OL proliferation and maturation (Kuo et al., 2010; Liu et al., 2011). Yet, the precise in vivo role for Shp2 or consequence of Noonan syndrome Shp2 GOF mutations during telencephalic oligodendrogenesis remains unknown.
In this study, we explore the in vivo role of Shp2 during oligodendrogenesis in the telencephalon. We generated Shp2 conditional knock-out (cKOs) and conditional Shp2 GOF Noonan syndrome mice using Olig2cre/+ mice to target ventral telencephalic progenitors and the entire oligodendrocyte lineage. Our results show that Shp2 cKOs exhibit reduced OPC numbers at embryonic and postnatal stages and severe hypomyelination phenotypes at P21. Moreover, expressing a Shp2 GOF Noonan syndrome mutation elevated OPC numbers in the embryonic and postnatal brain but yielded abnormal myelination and fewer myelinated axons in the white matter. The phenotypes observed in both LOF and GOF mice were associated with changes in OPC proliferation and MAPK activity. Our results provide evidence that appropriate control of Shp2 activity levels is essential for proper development of oligodendrocytes in the telencephalon.
Materials and Methods
Animals.
Animal protocols were approved by the Cincinnati Children's Hospital Medical Center Institutional Animal Care and Use Committee in accordance with National Institutes of Health guidelines. Shp2 conditional mutant and Q79R-Shp2-tg mice were previously described (Krenz et al., 2008; Nakamura et al., 2009). Olig2cre/+ mice were described in Dessaud et al. (2007) and provided by Y. Yoshida. CC-eGFP cre reporter mice were described by Nakamura et al. (2006) and used as described by Waclaw et al. (2010). Shp2 conditional mutants were obtained by crossing double heterozygous males (Shp2loxP/+;Olig2cre/+) with Shp2 homozygous flox (Shp2loxP/loxP) females. Shp2 GOF mice (Q79R-Shp2-tg;Olig2cre/+) were obtained by crossing Olig2cre/+ males with Q79R-Shp2-tg females. All mice were maintained on mixed background that contains outbred CD-1 strain. For timed pregnancies, vaginal plug indicates embryonic day 0.5. At least 3 embryos or adult brains were analyzed for each genotype at each stage. Embryos and adult brains were fixed, processed for histology, and sectioned as previously described (Waclaw et al., 2006, 2010).
Immunohistochemistry/fluorescence.
Slides were treated with 0.3% hydrogen peroxide for 10 min and washed in KPBS. Primary antibodies were used at the following concentrations: guinea pig anti-Ascl1 (1:10,000, provided by J. Johnson, University of Texas Southwestern Medical Center, Dallas), rabbit anti-βIII-tubulin (1:1000, Covance), rabbit anti-CNPase (1:500, Cell Signaling Technology), rabbit anti-GFP (1:1000, Invitrogen), rabbit anti-Ki67 (1:1000, Abcam), chicken anti-MBP (1:500, AVES), mouse anti-neurofilament (1:100, 2H3, Developmental Studies Hybridoma Bank), rabbit anti-Nkx2.1 (1:1000, Seven Hills Bioscience), rabbit anti-Olig2 (1:2000, Millipore) (Mizuguchi et al., 2001), rabbit anti-Pdgfrα (1:200, Santa Cruz Biotechnology), rabbit anti-phosho-histone3 (1:200, Millipore), rabbit anti-Shp2 (1:200, Santa Cruz Biotechnology), and goat anti-Sox10 (1:200, Santa Cruz Biotechnology). Slides were incubated overnight with primary antibody at room temperature. The following day slides were washed in KPBS and incubated with biotinylated swine anti-rabbit antibodies (1:200, DAKO) or biotinylated horse anti-goat antibodies (1:200, Vector Laboratories) for 2 h. Slides were then washed in KPBS and incubated in ABC reagent (Vector Laboratories) for 1 h. DAB (Sigma) or VIP (Vector Laboratories) were used as the final chromagen to visualize the reaction. For immunofluorescence stains, secondary antibodies used were as follows: donkey anti-chicken antibodies conjugated to 488 (1:200, Jackson ImmunoResearch Laboratories), donkey anti-mouse antibodies conjugated to Cy3 (1:200, Jackson ImmunoResearch Laboratories), and donkey anti-rabbit antibodies conjugated to 488 or Cy3 (1:200, Jackson ImmunoResearch Laboratories). All bright-field pictures were taken on a Leica DM2500 microscope equipped with a Leica DFC500 camera. Fluorescent images were taken on a Nikon AZ-100 multizoom microscope equipped with a Nikon DS-Ri1 camera. Confocal images were taken on Nikon C2 confocal microscope.
Quantification.
Olig2-, Pdgfrα-, and Ki67-positive cells in the parenchyma (outside of VZ) at E15.5 were counted in three serial sections at LGE/MGE levels at 100× magnification. Phospho-histone3 and phospho(p)-ERK1/2 expression at E15.5 was counted in VZ progenitors in three consecutive sections at LGE levels at 400× magnification. Olig2- and Pdgfrα-positive cells were counted in ventral forebrain (VF) and medial/lateral corpus callosum (CC), areas at E18.5 in sections beginning at anterior commissure (AC) levels at 200× for each level. Appropriate areas (VF or CC) were defined in ImageJ and numbers presented as cells/mm2. Ki67/Pdgfrα double stains were quantified at E15.5 in the parenchyma region at 400× in two separate regions of three consecutive sections. Ki67/Pdgfrα double stains were quantified at P21 in the medial CC at 200×. For all quantification, at least three different controls or experimental samples (cKO or GOF) were used. Statistics were performed between control and Shp2 cKO or Shp2 GOF using a Student's unpaired t test.
Western analysis.
White matter and cortex were dissected from control and Shp2 GOF mice. Western blot analysis was performed following standard methods (Dasgupta and Milbrandt, 2009). Briefly, tissues were homogenized in a tissue homogenizer with RIPA lysis buffer (20 mm Tris, 10 mm EGTA, 40 mm β-glycerophosphate, 1% NP40, 2.5 mm MgCl2, 2 mm orthovanadate 1 mm PMSF, 1 mm DTT, and protease inhibitor mixture). Protein was estimated by BCA method; 20 μg of protein was resolved by SDS-PAGE. The following antibodies were used: rabbit anti-pErk1/2, rabbit anti-Erk1/2, rabbit anti-pS6, rabbit anti-actin (all from Cell Signaling Technology).
Electron microscopy and g-ratio analysis.
For tissue processing, animals were anesthetized and perfused transcardially for 10–15 min with ice-cold mixture of 4% paraformaldehyde and 2.5% glutaraldehyde for electron microscopy (EM). Brains were removed after perfusion and postfixed in the same fixative overnight. The CC was microdissected and transferred to 0.175 m cacodylate buffer, osmicated, dehydrated, and embedded in Embed 812 (Ladd Research Industries) before processing for semithin and ultrathin sectioning. Sections were stained in uranyl acetate and lead citrate and viewed on a Hitachi Model H-7600 EM microscope. Plastic semithin sections were cut, stained with toluidine blue, and viewed under the scope to identify the medial area of the CC. Areas cut in cross section with respect to the axon fibers were retrimmed and ultrathin sections cut for analysis by electron microscopy. Axon diameters and g-ratios were measured from each animal by a naive, blinded, observer. The g-ratio was determined by dividing the diameter of each axon (without myelin) to the diameter of the same axon together with its myelin sheath. Regression line analysis of g-ratio data was performed in Excel.
Results
Conditional deletion of Shp2 using Olig2cre/+ results in severe myelination defects in the telencephalon
Shp2 has been characterized as a ubiquitously expressed signaling molecule (Neel et al., 2003; Lauriol and Kontaridis, 2011), and we confirmed that Shp2 protein is robustly expressed in telencephalic progenitor cells in the embryonic VZ (data not shown) and white matter cells at P21 (Fig. 1A). It has previously been shown that germline deletion of Shp2 results in defects in trophoblast stem cell survival, preventing the analysis of mature CNS structures (Yang et al., 2006). To overcome this issue, we used mice containing a Shp2 conditional mutant allele that contains loxP sites flanking the N-SH2 domain and hinge region of exon 3 and 4 (Nakamura et al., 2009). Cre-mediated deletion results in absence of Shp2 protein in targeted cell populations (Nakamura et al., 2009). To determine an in vivo role for Shp2 in ventral telencephalic progenitor cells and the entire oligodendrocyte lineage, we used Olig2cre/+ mice. The Olig2 lineage in the telencephalon gives rise to oligodendrocytes and neuronal populations (Lu et al., 2000; Cai et al., 2007; Miyoshi et al., 2007; Dimou et al., 2008). The Olig2cre/+ mice used in this study were initially characterized during spinal cord development (Dessaud et al., 2007) and recently used in the ventral telencephalon (Chapman et al., 2013). We confirmed that the Olig2cre/+ mice are a robust Cre driver because the fate-map showed strong EGFP expression derived from a Cre reporter allele in 62% of DAPI-positive white-matter cells at P21 (Fig. 1C).
Figure 1.
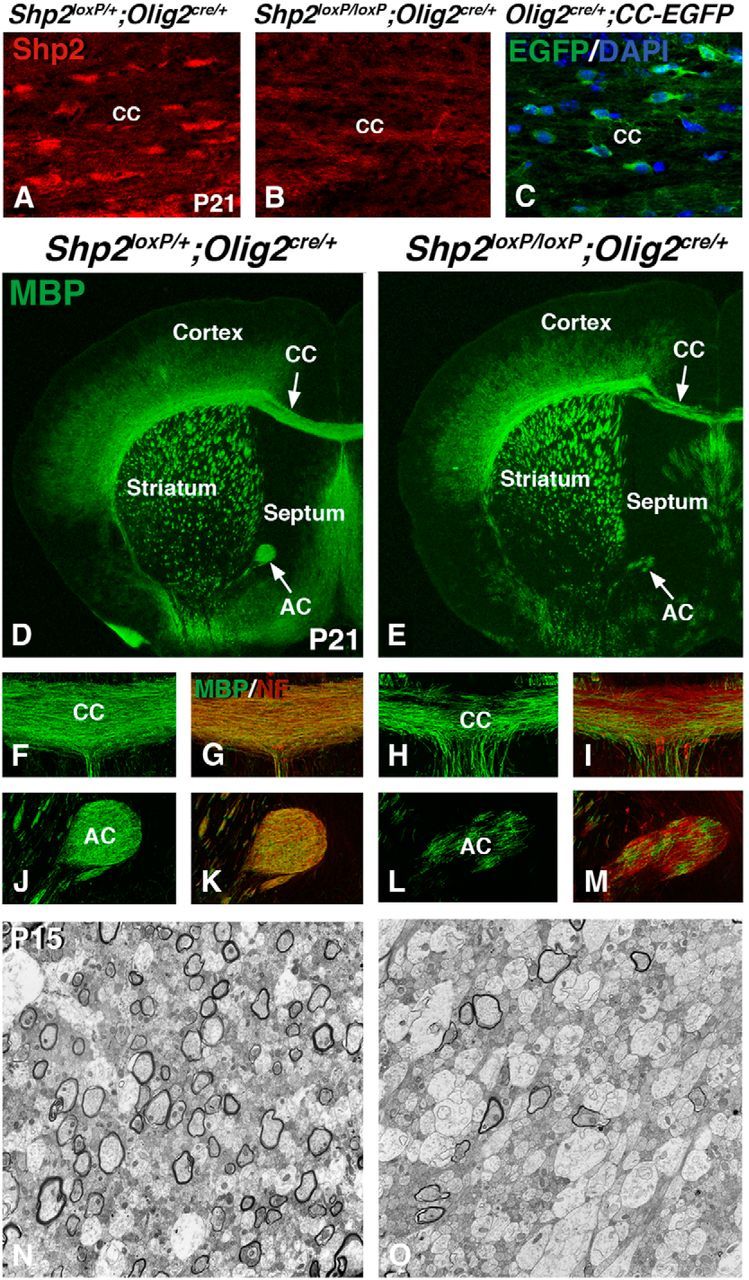
Conditional deletion of Shp2 using Olig2cre/+ results in myelination defects. Shp2 cKOs (Shp2loxP/loxP;Olig2cre/+) show reduced Shp2 protein expression in the postnatal CC (B) compared with controls (Shp2loxP/+;Olig2cre/+) at P21 (A). Olig2 fate-map (Olig2cre/+;CC-EGFP) shows high level EGFP expression in many DAPI-positive cells in the CC at P21 (C). Immunofluorescence for MBP expression at P21 reveals myelination defects in the CC and AC (compare arrows in E, D) in Shp2 cKOs (E, H, L) compared with controls (D, F, J). Overlay images of MBP and neurofilament (NF) expression show the presence of axons in the white matter of Shp2 cKOs (I, M) compared with controls (G, K). Transmission electron micrographs of CC regions show the reduction in myelinated large axons in the Shp2 cKOs (O) compared with controls (N). EM analysis was done during developmental myelination (P15), so many axons are still unmyelinated, even in the controls at this stage. CC, Corpus callosum; AC, anterior commissure.
Shp2 conditional mutants were generated using Olig2cre/+ mice. Shp2 conditional mutants (Shp2loxP/loxP;Olig2cre/+, hereafter, Shp2 cKOs) showed markedly reduced Shp2 expression in the postnatal white matter tracts (compare Fig. 1B with Fig. 1A). Postnatal Shp2 cKOs displayed severe shivering between 2 and 3 weeks of age but failed to survive beyond 3 weeks of age. The shivering phenotype is typical of myelin deficient mice (Molineaux et al., 1986). Immunofluorescence of 3-week-old Shp2 cKO telencephalon showed dramatic reductions of myelin as shown by MBP expression (compare Fig. 1E with Fig. 1D) in the white matter (CC and AC) regions (compare Fig. 1H,L with 1F,J). Although MBP expression was reduced, neuronal axons in the white matter tracts remained detectable with anti-neurofilament (2H3) staining (compare red stain in Fig. 1I,M with Fig. 1G,K). The most severe changes in MBP expression in Shp2 cKOs were observed in the medial CC and AC compared with the lateral CC, and also in the septum and other VF areas. We observed these defects at both rostral and caudal telencephalic regions (data not shown). We examined ultrastructure in the medial CC using transmission electron microscopy during developmental myelination at P15. At this stage, postnatal myelination is not complete in control animals; however, many axons show mature myelin sheaths. Shp2 cKOs showed a clear increase in unmyelinated axons compared with controls (compare Fig. 1O with Fig. 1N; Shp2 cKOs 83.5 ± 5.3% unmyelinated axons and controls 30.3 ± 7.7% unmyelinated axons, p < 0.002, n = 3). These findings suggest that Shp2 plays a crucial role in development of oligodendrocytes.
Shp2 cKOs show defects in the generation of OPCs during development
To determine whether the myelination phenotype in Shp2 cKOs was a result of an inability of OLs to myelinate and/or a defect in the generation of OLs/OPCs, we analyzed OPCs and OLs in Shp2 cKOs at P21 using CNPase to label OLs, Olig2 to label the entire OL lineage, Pdgfrα to label OPCs, and Sox10 to label OPCs and OLs (for review, see Nicolay et al., 2007). Shp2 cKOs displayed a similar reduction in CNPase expression (compare Fig. 2B with Fig. 2A) as was observed with MBP expression (Fig. 1). There are clear reductions in the CC and AC of the white matter in Shp2 cKOs (Fig. 2B, arrows) compared with controls (Fig. 2A). Although some Olig2-positive cells were detected in Shp2 cKOs, especially in the lateral CC between the cerebral cortex and striatum, their overall density was markedly reduced in both the gray and white matter throughout the telencephalon (Fig. 2C,D; and data not shown). To determine whether these cells were recombined and lost Shp2 expression, we stained for Olig2 and Shp2 (Fig. 2C′). We did not detect any Shp2 staining in the cytoplasm (red) of Olig2+ cells (green) remaining in Shp2 cKOs, suggesting that these cells were recombined with Cre (compare Fig. 2D′ with Fig. 2C′). The most severe reduction of Olig2+ cells was observed in the medial CC (compare Fig. 2F with Fig. 2E) where severe myelination defects were detected (Fig. 1). Quantification of Olig2+ cells in the medial CC revealed a 79% decrease (control 54.9 ± 4.0 and Shp2 cKO 11.4 ± 2.8 cells per 400× field in the CC, p < 0.005, n = 3). Both Pdgfrα+ (compare Fig. 2H with Fig. 2G) and Sox10+ (compare Fig. 2J with Fig. 2I) cells were also severely reduced in the medial CC. Quantification of Pdgfrα+ cells revealed a 65% decrease (control 13.6 ± 1.1 and Shp2 cKO 4.8 ± 1.5 cells per 400× field in the CC, p < 0.005, n = 3), whereas Sox10+ cells were decreased by 75% (control 37.1 ± 1.8 and Shp2 cKO 9.2 ± 4.1 cells per 400× field in the CC, p < 0.005, n = 3). These results demonstrate that Shp2 cKOs display reduced OPCs and OLs in the CC at P21, which is likely to be a main cause for the myelination defect (Fig. 1).
Figure 2.
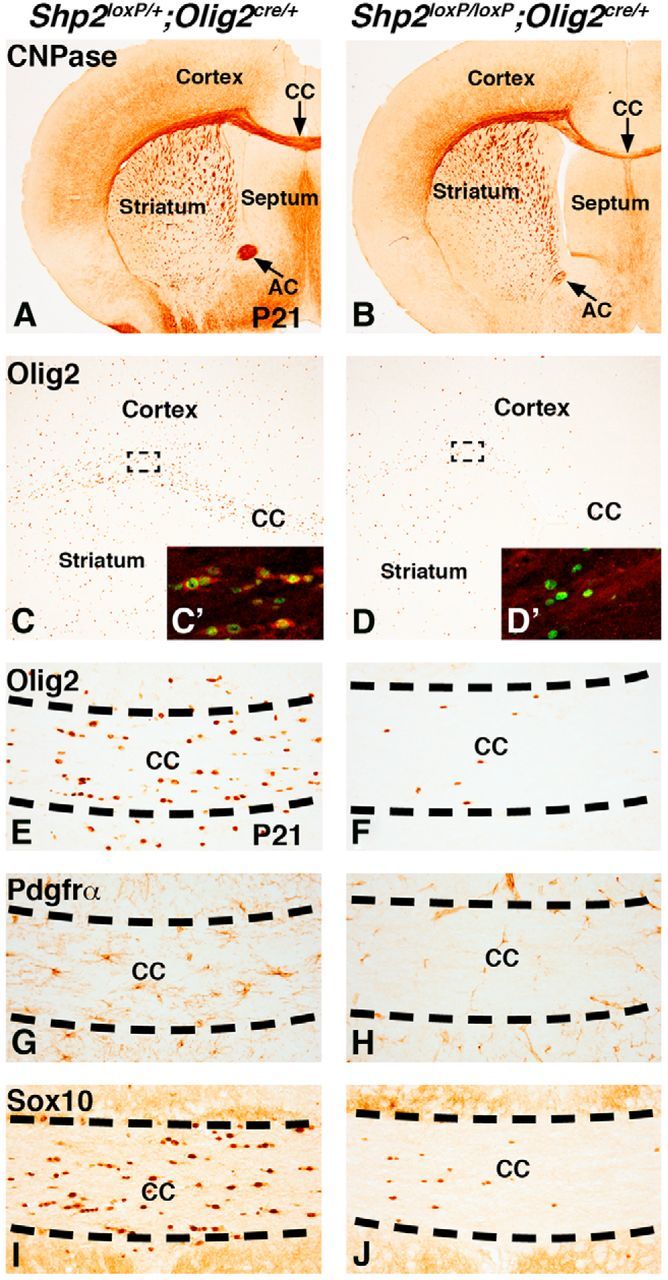
Reduced OLs and OPCs in Shp2 cKOs at P21. CNPase expression in OLs is reduced in the postnatal white matter regions (CC and AC) in Shp2 cKOs (Shp2loxP/loxP;Olig2cre/+) (B) compared with controls (Shp2loxP/+;Olig2cre/+) (A). Olig2 expression labels both OLs and OPCs at P21 (C) and is generally reduced in Shp2 cKOs (D). Dashed lines indicate representative areas for C and D insets that show Shp2 (red) and Olig2 (green) double stains. Shp2 staining in the cytoplasm is detected in Olig2-positive cells (C′) in controls. However, the few remaining Olig2 cells in Shp2 cKOs do not show any Shp2 staining in the cytoplasm (D′). Olig2-, Pdgfrα-, and Sox10-positive cells are severely reduced in the medial CC of Shp2 cKOs (F, H, and J, respectively) compared with controls (E, G, and I, respectively).
Next, we analyzed the expression of Olig2 and Pdgfrα, which label telencephalic OPCs during development (for review, see Rowitch and Kriegstein, 2010). We analyzed multiple telencephalic regions at E18.5 to define OPC populations affected by Shp2 cKO (Fig. 3A–J). First, we analyzed the CC at midline levels to determine whether the postnatal phenotypes (Figs. 1 and 2) are a result of a defect in embryonic OPC generation. Indeed, both the numbers of Olig2+ and Pdgfrα+ cells were reduced in the medial CC compared with controls (Fig. 3A,B,F,G). Shp2 cKOs showed a 52% reduction in Olig2 (control 626.0 ± 88.1 cells/mm2 and Shp2 cKOs 300.1 ± 99.1 cells/mm2, p < 0.05, n = 3; Fig. 3E) and a 79% reduction in Pdgfrα (control 282.9 ± 14.4 cells/mm2 and Shp2 cKOs 59.0 ± 37.8 cells/mm2, p < 0.005, n = 3; Fig. 3J). In other telencephalic regions, Olig2+ cells were differently affected by loss of Shp2. Olig2+ cells were dramatically reduced in VF regions (compare red asterisk in Fig. 3D with Fig. 3C) showing a 73% reduction (control 290.2 ± 23.2 cells/mm2 and Shp2 cKOs 77.2 ± 17.1 cells/mm2, p < 0.001, n = 3), whereas their density was not significantly affected in the lateral CC (compare dashed lines in Fig. 3D with Fig. 3C; control 1126.1 ± 88.1 cells/mm2 and Shp2 cKOs 960.3 ± 157.2 cells/mm2, p = 0.204, n = 3). Pdgfrα+ cells were also reduced at VF regions with Shp2 cKOs (compare red asterisk in Fig. 3I with Fig. 3H) showing a 73% reduction compared with controls (control 198.4 ± 2.5 cells/mm2 and Shp2 cKOs 53.1 ± 20.4 cells/mm2, p < 0.005, n = 3). However, unlike Olig2+ cells, Shp2 cKOs showed a small reduction (34%) in Pdgfrα+ cells in the lateral CC (controls 699.4 ± 39.3 cells/mm2 and Shp2 cKOs 460.4 ± 70.2 cells/mm2, p < 0.05, n = 3) (compare dashed lines in Fig. 3I with Fig. 3K). These results suggest that Shp2 is required for the generation of OPCs during embryonic development and that such a requirement varies among OPCs in different regions. Moreover, the analysis of the entire telencephalon revealed that Pdgfrα+ OPCs seem to be more affected by Shp2 loss compared with Olig2+ cells, suggesting different roles for Shp2 in these two cell populations. A significant fraction of Olig2+ cells remaining in Shp2 cKOs may be glial progenitors that eventually differentiate into astrocytes and OLs (Cai et al., 2007; Zhu et al., 2012), which would suggest that we observe an OL lineage-specific phenotype. In line with this, we did not notice an obvious change in GFAP protein expression in midline at E18.5 (data not shown).
Figure 3.

Reduced OPCs in Shp2 cKOs are observed at late embryonic stages. Olig2 expression in OPCs at E18.5 is reduced in the Shp2 cKO (Shp2loxP/loxP;Olig2cre/+) in the medial CC (B) and VF regions (D, red asterisk) compared with controls (Shp2loxP/+;Olig2cre/+) (compare CC in B with A and red asterisk in D with C). Olig2 expression in the lateral CC between the striatum and cortex (dashed lines) was largely unaffected in Shp2 cKOs (D, dashed lines) compared with controls (C, dashed lines). Pdgfrα expression was reduced at all telencephalic levels in Shp2 cKOs (I) compared with controls (H), including medial CC (compare G with F), VF regions (compare red asterisk in I with H), and lateral CC (compare dashed lines in I with H). Graphical representation of Olig2 cell counts in E and Pdgfrα cell counts in J. **p < 0.005 (Student's t test). *p < 0.05 (Student's t test).
To further determine whether Shp2 is required for early telencephalic OPCs that originate in the ventral regions at E15.5 (Kessaris et al., 2006; for review, see Rowitch and Kriegstein, 2010), we analyzed Olig2 and Pdgfrα in the parenchyma region away from the MGE and LGE at E15.5. Shp2 cKOs displayed a clear reduction in the density of Olig2+ cells (compare red asterisk in Fig. 4B and Fig. 4A) and nearly complete depletion of Pdgfrα OPCs (compare red asterisk in Fig. 4D and Fig. 4C). Specifically, Shp2 cKOs displayed a 53% reduction in Olig2+ cells (control 193.2 ± 4.1 and Shp2 cKO 90.7 ± 3.3 cells per 100× field, p < 0.001, n = 3) and an 83% reduction in Pdgfrα+ cells (control 141.0 ± 12.4 and Shp2 cKOs 23.7 ± 3.3 cells per 100× field, p < 0.001, n = 3) compared with controls. Interestingly, Olig2 expression (compare MGE in Fig. 4B with Fig. 4A) and Ascl1 expression (compare arrows in Fig. 4F and Fig. 4E) in VZ progenitor cells of the MGE and LGE were not overtly different in Shp2 cKOs compared with controls, suggesting that the initial specification of progenitors in the LGE and MGE by E15.5 is established correctly in the VZ, but the subsequent generation of the Olig2+ cells and Pdgfrα+ OPCs away from the progenitor regions was impaired. Together, our analysis at E18.5 and E15.5 demonstrate that Shp2 is required for proper generation of OPCs during embryogenesis, which is likely a major reason for a global reduction of OPCs and hypomyelination of the white matter at P21.
Figure 4.
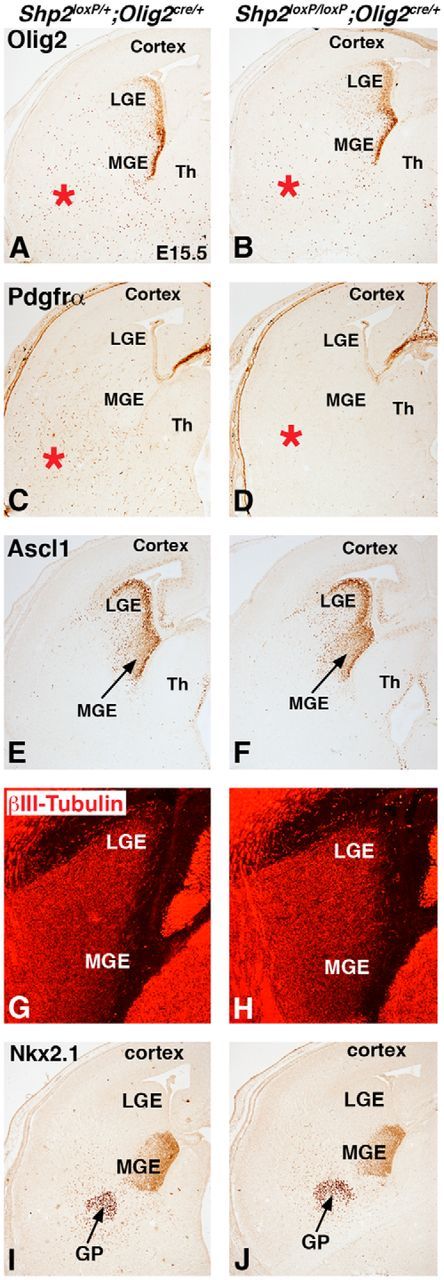
Loss of Shp2 has different effects on the generation of OPCs and neurons from the Olig2 lineage. Olig2 expression labels progenitor cells in the VZ of the MGE and LGE and in OPCs in the parenchyma (A, red asterisk) of controls (Shp2loxP/+;Olig2cre/+). Shp2 cKOs (Shp2loxP/loxP;Olig2cre/+) showed no change in the MGE/LGE expression of Olig2, but fewer Olig2 cells were observed in the parenchyma (B, red asterisk). Pdgfrα expression is nearly absent in the parenchyma of the ventral telencephalon in Shp2 cKO embryos (D, red asterisks) compared with controls (C, red asterisks). Ascl1 expression in the progenitors of the MGE and LGE (E) was not changed in Shp2 cKOs (F). βIII-Tubulin expression in newborn neurons is not changed in the MGE and LGE of Shp2 cKOs (H) compared with controls (G). Nkx2.1 expression in MGE progenitors and the developing GP neurons is unchanged in Shp2 cKOs (J) compared with controls (I).
Subcortical neuronal populations that originate from the Olig2 lineage are unaffected in Shp2 cKOs
The developmental expression of Olig2 is not limited to the OL lineage. Olig2 fate maps at embryonic stages show robust recombination in VZ progenitors cells of the LGE and MGE and many neuronal subtypes in major subcortical structures, namely, striatum and globus pallidus (data not shown; Chapman et al., 2013). Unlike the OPC and OL phenotype in the Shp2 cKOs, these ventrally derived neuronal populations were not as severely affected. We found no overt differences in βIII-tubulin expression in newborn neurons in the LGE and MGE of the ventral telencephalon (compare Fig. 4H and Fig. 4G). We also analyzed Nkx2.1 expression, which labels the MGE progenitors and differentiating globus pallidus neurons. Nkx2.1 expression was not changed in Shp2 cKO compared with controls (compare Fig. 4J and Fig. 4I). Moreover, at postnatal stages when severe OPC/OL phenotypes were observed in Shp2 cKOs (Figs. 1 and 2), there was no obvious difference in postnatal parvalbumin+ neurons in the globus pallidus or Darpp32+ striatal projection neurons (data not shown). Thus, although previous reports indicate that Shp2 gene function is crucial for neurogenesis in the cortex and hippocampus, the areas derived from dorsal telencephalic progenitors (Gauthier et al., 2007; Ke et al., 2007), we found that subcortical neurons (i.e., striatum and globus pallidus) are generated in its absence. This suggests that, unlike dorsal telencephalic progenitors, the loss of Shp2 in progenitors of the ventral telencephalon is crucial for the generation of OPCs/OLs but not neuronal populations.
Shp2 cKO embryos show a reduction in MAPK signaling and reduced proliferation in the ventral telencephalon
Genetic disruption of the MAPK pathway results in abnormalities in glial cell development (Galabova-Kovacs et al., 2008; Fyffe-Maricich et al., 2011; and Li et al., 2012). In the developing telencephalon, we observed active MAPK signaling detected by phospho-ERK1/2 (p-ERK1/2) in VZ progenitors cells in the ventral telencephalon and ventral/lateral pallium at E15.5 (Fig. 5A,B). We found that Shp2 cKOs showed a 39% reduction of p-ERK1/2+ cells in the ventral regions of the LGE at E15.5 (Fig. 5D) compared with controls (Fig. 5B) at E15.5 (controls 74.8 ± 1.9 cells per 400× field and Shp2 cKOs 45.7 ± 2.5 cells per 400× field, p < 0.001, n = 3). Importantly, however, we did not detect changes in proliferation observed by pH3 immunostaining in VZ progenitors (Fig. 5E–H; controls 19.7 ± 1.5 cells in VZ 400× field and Shp2 cKOs 17.0 ± 1.3 cells in VZ 400× field, p < 0.5, n = 3). By contrast, we did notice a 60% reduction in Ki67+ cells in the parenchyma of Shp2 cKOs (Fig. 5L) compared with controls (Fig. 5J; controls 112.1 ± 6.8 cells per 100× field and Shp2 cKOs 44.4 ± 6.0 cells per 100× field, p < 0.001, n = 3). Indeed, we found that 26% of Ki67+ cells in the ventral parenchyma (outside of the VZ) were Pdgfrα+ OPCs (Fig. 5J′). Similar to the reduction in total Pdgfrα+ cells, Shp2 cKOs displayed a dramatic reduction in Pdgfrα+ Ki67+ double-positive cells (Fig. 5L′). Quantification revealed a 70% reduction in the parenchyma (controls 26.1 ± 2.6% Ki67 cells that express Pdgfrα per 400× field and Shp2 cKOs 7.8 ± 1.6% Ki67 cells that express Pdgfrα per 400× field, p < 0.005, n = 3). The vast majority of Ki67-positive cells remaining in Shp2 cKOs were negative for Pdgfrα (Fig. 5L′, green arrow). However, the few remaining Pdgfrα+ cells in the Shp2 cKO did express Ki67. These data suggest that the small numbers of Pdgfrα+ cells that are generated in Shp2 cKO are proliferative, but it remains unknown whether a defect in proliferation rate exists (i.e., expansion). Combined, these results suggests that loss of Shp2 in the ventral telencephalon reduces MAPK signaling in the VZ and alters the number of proliferating OPCs in the ventral embryonic parenchyma, where early telencephalic OPCs emerge (Kessaris et al., 2006). Therefore, we conclude that Shp2 cKOs have a deficiency in either the generation of OPCs and/or OPC expansion by proliferation.
Figure 5.

Shp2 cKOs show reduced MAPK signaling in the VZ and reduced proliferation in the parenchyma. P-ERK1/2 staining primarily labels VZ progenitors in the LGE (A, dashed box represents high power in B) and is reduced in Shp2 cKO VZ (C, dashed box represents high power in D). Phospho-histone3 expression in the LGE is unchanged in Shp2 cKOs (G, dashed box represents high power in H) compared with controls (E, dashed box represents high power in F). Ki67 expression is robust in the progenitors of the LGE and MGE (I) and also in scattered cells of the parenchyma (I, dashed box represents image in J). J, Inset, Double-positive cells for Ki67 and Pdgfrα in the parenchyma (J′, red arrows). Shp2 cKOs show no change in Ki67 expression in the progenitor regions (K) but exhibit fewer cells in the parenchyma (K, dashed box represents image in L). Many remaining Ki67-positive cells in the parenchyma are Pdgfrα-negative (L′, green arrows).
Consequence of the Q79R-Shp2 GOF mutation on OPC generation and myelination
The studies described above suggest that Shp2 is required for OPC generation and/or expansion in the telencephalon and myelination in the postnatal brain. We next wanted to determine whether Shp2 GOF mutations that are observed in Noonan syndrome patients impact oligodendrogenesis. We used a Cre-inducible transgenic mouse that expresses a Q79R mutation in the regulatory N-SH2 domain of Shp2 that results in elevated Shp2 phosphatase activity (Krenz et al., 2008; Nakamura et al., 2009). Because the impact of Shp2 GOF mutations has not been addressed in the ventral telencephalon progenitors and OL lineage, we used the Olig2cre/+ mice in combination with Q79R-Shp2-tg mice.
Shp2 cKOs showed reductions in OPC markers at E18.5 (Fig. 3). Therefore, we analyzed Q79R-Shp2-tg;Olig2cre/+ double transgenic embryos in the same regions (the medial and lateral CC, and VF) at the same stage (Fig. 6A–N). We found an increase in Olig2+ and Pdgfrα+ cells in all of these regions at E18.5 in Q79R-Shp2-tg;Olig2cre/+ embryos (Fig. 6B,E,F,I,L,M) compared with controls (Fig. 6A,C,D,H,J,K). Quantification of Olig2+ cells (Fig. 6G) revealed a 32% increase in the medial CC (controls 619.8 ± 36.7 cells/mm2 and Q79R-Shp2-tg;Olig2cre/+ 820.6 ± 74.4 cells/mm2, p < 0.05, n = 3), 46% increase in Olig2+ cells in the lateral CC (controls 1351.9 ± 46.3 cells/mm2 and Q79R-Shp2-tg;Olig2cre/+ 1974.3 ± 242.2 cells/mm2, p < 0.05, n = 3), and 61% increase in Olig2+ cells in the VF (controls 377.7 ± 20.4 cells/mm2 and Q79R-Shp2-tg;Olig2cre/+ 608.4 ± 3.2 cells/mm2, p < 0.001, n = 3). Quantification of Pdgfrα+ cells (Fig. 6N) showed 29% increase in in the medial CC (controls 306.5 ± 8.9 cells/mm2 and Q79R-Shp2-tg;Olig2cre/+ 394.7 ± 28.6 cells/mm2, p < 0.05, n = 3), 65% increase in Pdgfrα cells in the lateral CC (controls 623.4 ± 0.7 cells/mm2 and Q79R-Shp2-tg;Olig2cre/+-1028.9 ± 72.1 cells/mm2, p < 0.005, n = 3), and 73% increase in Pdgfrα cells in the VF (controls 197.4 ± 15.1 cells/mm2 and Q79R-Shp2-tg;Olig2cre/+ 342.4 ± 25.5 cells/mm2, p < 0.005, n = 3). Thus, the impact of the Shp2 GOF mutation is more profound on Pdgfrα+ cells than Olig2+ cells. Because Shp2 GOF mutations have been reported to promote neurogenesis in the dorsomedial cortex (Gauthier et al., 2007), we next examined neural markers from the MGE (Nkx2.1) and septum (Zic1) and did not find any overt difference in expression after Q79R-Shp2 expression (data not shown). Q79R-Shp2-tg;Olig2cre/+ embryos showed an increase of OPC markers in all regions analyzed, suggesting that the Shp2 GOF mutation is sufficient to increase both Olig2 and Pdgfrα expression in the telencephalic parenchyma, affecting either initial OPC specification and/or expansion. Given the opposite effects of the LOF mutation on OPC numbers, our results demonstrate that proper levels of Shp2 activity are crucial in determining the normal number of OPCs in the telencephalon during development.
Figure 6.

Increased embryonic OPCs after expression of Q79R-Shp2 GOF mutation in Olig2 lineage. Embryonic OPCs labeled by Olig2 (A–F) and Pdgfrα (H–M) are increased in Q79R-Shp2-tg;Olig2cre/+ embryos (Olig2, B, E, F; Pdgfrα, I, L, M) compared with Q79R-Shp2-tg controls (Olig2, A, C, D; Pdgfrα, H, J, K). Images show the increased Olig2 (compare B, E, F with A, C, D) and Pdgfrα (compare I, L, M with H, J, K) in Q79R-Shp2-tg;Olig2cre/+ embryos compared with controls in the forming white matter tracts: medial/lateral CC and AC and VF regions. Graphical representation of Olig2 cell counts in G and Pdgfrα cell counts in N. **p < 0.005 (Student's t test). *p < 0.05 (Student's t test).
Unlike Shp2 cKOs, which were not viable beyond 3 weeks of age, Q79R-Shp2-tg;Olig2cre/+ animals survived to adulthood. Interestingly, we did not detect obvious changes in MBP expression in Shp2 GOF mice at these stages (see Fig. 8A,B). We noticed, however, two myelination phenotypes upon analysis of individual axons at P60 using electron microscopy. Q79R-Shp2-tg;Olig2cre/+ animals showed more unmyelinated axons (Fig. 7B,D,E; 21.5 ± 3.3% unmyelinated axons) compared with controls (Fig. 7A,C,E; 8.4 ± 2.3% unmyelinated axons, p < 0.001, n = 4). In addition, we noticed abnormally wrapped myelinated axons (Fig. 7D, red arrows) and in rare cases what appeared to be a thicker myelin sheath (Fig. 7D, purple arrows). Indeed, g-ratio analysis (Fig. 7F) revealed a slightly lower g-ratio in Q79R-Shp2-tg;Olig2cre/+ animals compared with controls (Q79R-Shp2-tg;Olig2cre/+ 0.693, control 0.698), indicating abnormal myelin and/or increased myelin thickness. These data suggest that, despite increasing OPC numbers, expression of the Shp2 GOF mutation results in increased unmyelinated axons and abnormal myelin in axons of the CC.
Figure 8.

Increased OPCs and elevated MAPK after expression of Q79R-Shp2 GOF mutation in Olig2 lineage. No change in MBP expression was observed in Q79R-Shp2;Olig2cre/+ brains (B) compared with Q79R-Shp2-tg controls (A) at P21. Q79R-Shp2;Olig2cre/+ brains show increased expression of the OPC markers Pdgfrα (D) and Sox10 (F) compared with controls (C, E) at P21. Increased dividing cells labeled with Ki67 were detected in the CC near the SVZ in Q79R-Shp2;Olig2cre/+ brains (H) compared with Q79R-Shp2-tg controls (G) at P21. Black arrows indicate Ki67 cells near SVZ. Red arrow indicates Ki67 cells at the midline of Q79R-Shp2;Olig2cre/+ brains, which are not observed in controls. H, Inset (H′), Ectopic Ki67 cells in the white matter express Pdgfrα. Graphical representation of Pdgfrα and Sox10 cell counts in 400× field of postnatal CC (I). Significance was determined using Student's t test. **p < 0.01. *p < 0.05. J, Western blots of dissected CC/cortex from control and Q79R-Shp2;Olig2cre/+ at P21 show elevated p-ERK1/2 (MAPK) but no change in p-S6 (mTORC1) expression. Graph of the density ratio from each well for p-ERK1/2/ERK1/2 (K).
Figure 7.
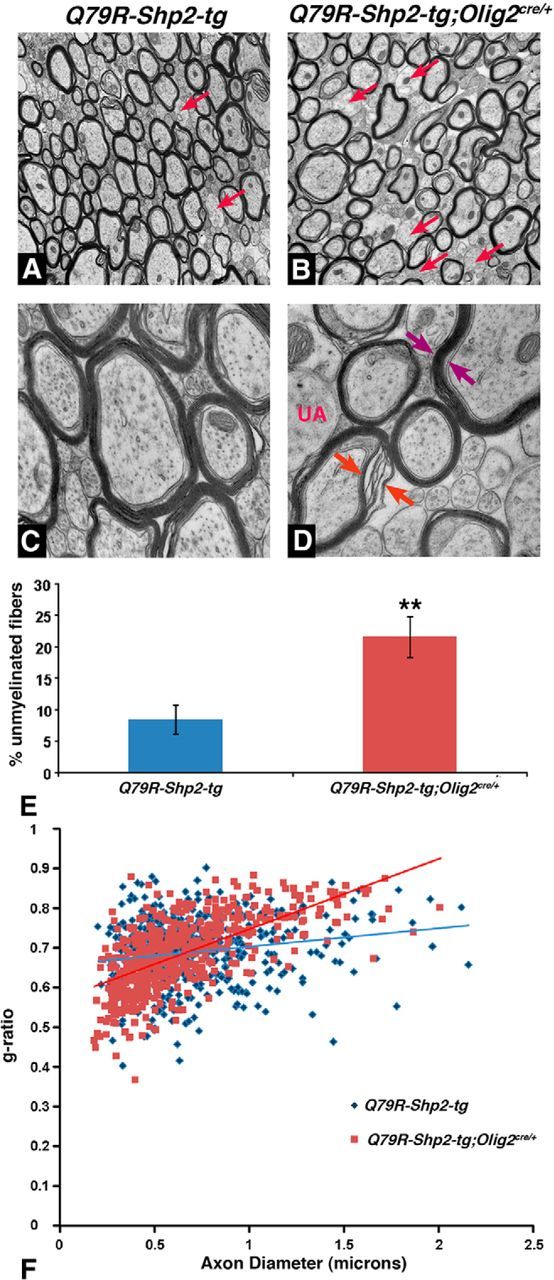
Abnormal myelination in postnatal forebrain after expression of Q79R-Shp2 GOF mutation in Olig2 lineage. Electron micrographs at P60 show fewer myelinated axons (B, red arrows) in Q79R-Shp2-tg;Olig2cre/+ animals compared with Q79R-Shp2-tg controls (A). Q79R-Shp2-tg;Olig2cre/+ animals displayed abnormal myelination (D, red arrows) and in rare cases what appeared to be a thicker myelin sheath (D, purple arrows) compared to controls (C). E, Graphical representation of unmeylinated axons shows that Q79R-Shp2;Olig2cre/+ mice exhibited increased unmyelinated fibers in the CC (N = 3) compared with controls. Significance was determined using a Student's t test. **p < 0.005. F, Graphical representation of g-ratio, which is the ratio of the diameter of the axon to the diameter of the axon and myelin, shows Q79R-Shp2;Olig2cre/+ with a g-ratio of 0.693 (N = 3) compared with controls, which show a g-ratio of 0.698 (N = 3 for each genotype).
Similar to the embryonic GOF phenotype (Fig. 6), postnatal Q79R-Shp2-tg;Olig2cre/+ displayed more Pdgfrα+ (compare Fig. 8D with Fig. 8C) and Olig2+ cells (data not shown) at P21. We also analyzed Sox10 expression and found an increase Sox10+ OPCs and OLs in the CC of Q79R-Shp2-tg;Olig2cre/+ brains (Fig. 8F) compared with controls at P21 (Fig. 8E). Quantification of these cells revealed a 110% increase of Pdgfrα+ cells (controls 9.4 ± 0.9 cells per 400× field; and Q79R-Shp2-tg;Olig2cre/+ 19.7 ± 2.2 cells per 400× field, p < 0.01, n = 3) and a 32% increase of Sox10+ cells (controls 48.3 ± 4.9 cells per 400× field; and Q79R-Shp2-tg;Olig2cre/+ 64.0 ± 5.2 cells per 400× field, p < 0.05, n = 3) in the CC (Fig. 8I). In the late postnatal and adult brain, the majority of proliferating cells are observed in the subventricular zone and the subgranular zone of the hippocampus (for review, see Alvarez-Buylla and Lim, 2004), and only a very few scattered Ki67+ cells are detectable in the CC. We found, however, more Ki67+ cells in the white matter (CC) of Q79R-Shp2-tg;Olig2cre/+ animals (Fig. 8H) compared with controls at P21 (Fig. 8G). It appeared that these cells were streaming out from SVZ regions into more medial areas of the CC (compare arrows in Fig. 8H with Fig. 8G). Furthermore, these Ki67+ cells expressed the Pdgfrα (Fig. 8H, inset). Indeed, we found a 111% increase in Ki67 Pdgfrα double-positive cells in the medial CC at P21 in Shp2 GOF mutants (controls 2.7 ± 0.9 average cells per 200× field in medial CC and Q79R-Shp2-tg;Olig2cre/+ 5.7 ± 0.4 cells per 200× field in medial CC, p < 0.05, n = 3). Together, these data suggest that expression of a Shp2 GOF mutation maintains elevated numbers of OPCs in the postnatal brain and also interferes with normal myelination. Moreover, the fact that fewer axons are myelinated despite the presence of elevated numbers of OPCs in Shp2 GOF mice suggests that proper levels of Shp2 activity are important for the formation of normal myelin sheath, a crucial aspect in differentiation of oligodendrocytes.
Shp2 GOF animals show increased MAPK activity in the white matter
GOF mutations of the Shp2 human gene (PTPN11) observed in Noonan syndrome result in enhanced phosphatase activity, thereby inhibiting negative regulators of the RAS/MAPK pathway and yielding elevated RAS/MAPK signaling (Araki et al., 2004). Consistent with this, isolated CC/cortex (Fig. 8J) and VF (data not shown) regions from Q79R-Shp2-tg;Olig2cre/+ animals showed elevated p-ERK1/2 protein (Fig. 8J). Interestingly, we also detected more total ERK1/2 protein, suggesting a positive feedback on the MAPK pathway. However, the ratio of p-ERK/1/2 to total ERK1/2 was clearly increased in Q79R-Shp2-tg;Olig2cre/+ tissue (Fig. 8J,K), suggesting activation of this pathway in Q79R-Shp2-tg;Olig2cre/+. Thus, contrary to the Shp2 cKOs that exhibit reduced MAPK signaling in progenitor cells, expression of a Shp2 GOF mutation yielded hyperactivation of MAPK signaling. The mTORC1 pathway can be activated by MAPK signaling and/or AKT signaling and is also crucial for oligodendrocyte differentiation (Tyler et al., 2009). Interestingly, we did not observe changes in phospho-S6 ribosomal protein (Fig. 8J), which is a major downstream effector of mTORC1. This suggests that increased MAPK signaling via Shp2 GOF mutation does not lead to elevated mTORC1 signaling, potentially signaling through an alternate pathway.
Discussion
Using an in vivo conditional mouse genetics approach, we have identified a crucial role for appropriately regulated Shp2 function in telencephalic oligodendrocyte development. Our results indicate that loss of Shp2 in ventral telencephalon progenitors and also in the entire oligodendrocyte lineage results in severe hypomyelination phenotypes in the white matter that are most likely to be a consequence of a role for this gene in the generation and/or expansion of OPCs during embryonic and early postnatal stages. Previous reports have shown that Shp2 regulates neurogenesis in the cortex of dorsal telencephalon in vivo (Gauthier et al., 2007; Ke et al., 2007). Our data, however, demonstrate that OPCs but not neuronal populations are most dependent upon Shp2 function among the cell types that emerge from the ventral telencephalon. Moreover, during embryogenesis Shp2 influences normal MAPK signaling in VZ progenitors of the telencephalon. We also found that inappropriate Shp2 function triggered by transgenic expression of a Shp2 GOF mutation (Noonan syndrome-Q79R) increased embryonic and postnatal OPCs, elevated MAPK signaling, and resulted in abnormal myelination in the white matter. Thus, our LOF and GOF analyses support a critical role for Shp2 in the regulation of OPC numbers. Together with work on the role of other key RAS/MAPK signaling molecules (Bennett et al., 2003; Zhu et al., 2005; Li et al., 2012; Wang et al., 2012), these findings add support to the impact of RASopathy mutations on glial cell development and implicate inappropriate Shp2 function (GOF mutation) during OPC generation and normal myelination in the telencephalon.
In vitro approaches using rat cortical cultures and a combination of Shp2/phosphatase inhibitors or siRNAs have suggested Shp2's role in either OPC proliferation or maturation (Kuo et al., 2010; Liu et al., 2011). Deleting Shp2 with Olig2cre/+ mice allowed us to conduct a cell type-specific in vivo analysis that could explore the role of Shp2 in ventral telencephalic VZ progenitor cells (LGE and MGE) and the entire oligodendrocyte lineage. Shp2 cKOs displayed a severe reduction in OPCs that normally emerge from the embryonic ventral telencephalon. In line with this result, previous work has shown OPC defects in the developing spinal cords from Shp2 cKOs generated with Olig1cre/+ (Zhu et al., 2010), suggesting crucial roles for Shp2 in OPC generation in multiple CNS regions. We also observed a more severe effect on the ventral/medial Olig2-positive OPC cells compared with lateral CC Olig2-positive OPCs, suggesting that Shp2 is most important for the generation of embryonic ventral/medial OPCs. It is possible that a small number of regional OLs are generated independent of Shp2. However, this regional difference may be an indirect effect of the timing of Shp2 deletion because ventral cells are targeted in the VZ, whereas dorsal cells are targeted as Olig2 begins to be expressed in OPCs while leaving the VZ in Olig2cre/+ mice. Shp2 protein is robustly expressed in the VZ of the cortex and a previous report deleting Shp2 in all VZ progenitors using Nestin-cre resulted in severe microcephaly, including defects in cortical neurons and reduced NG2+ cells at birth (Ke et al., 2007). Our approach of deleting Shp2 using Olig2cre/+ has revealed that Shp2 plays a key role for proper generation of OPCs from the ventral VZ progenitors and/or their subsequent expansion during embryogenesis. Future experiments will be need to be performed to isolate Shp2 loss to distinct OL stages using NG2-cre in OPCs or PLP-creER OLs to determine OPC/OL stage-specific effects.
One major role that has been proposed for Shp2 in the developing brain is to promote neurogenesis in the cortex of the dorsal telencephalon (Gauthier et al., 2007; Ke et al., 2007). The present results suggest a different role for Shp2 in the embryonic ventral telencephalon where Shp2 is more important for the generation of OPCs compared with the generation of subcortical neurons. Olig2cre/+ robustly targets the VZ progenitors that generate subcortical neurons as well as OPCs (Chapman et al., 2013). However, the most severe phenotypes observed in our Shp2 cKOs are in the OPC populations, whereas development of subcortical neurons remains largely unaffected, supporting the notion that in ventral progenitors Shp2 is vital for glial cells (ventral OPCs) and not ventral-derived neurons. Mechanisms underlying this regional difference in the requirement for Shp2 in the production of different neuronal and glial cell types are currently unknown and an important subject in future studies.
Shp2 is known to be a positive regulator of the RAS/MAPK signaling pathway (for review, see Neel et al., 2003; Dance et al., 2008). For example, Shp2 can dephosphorylate and subsequently inhibit Sprouty, which is an inhibitor of RAS/MAPK signaling, resulting in sustained signaling (Hanafusa et al., 2004). Therefore, in the case of loss of Shp2 protein (and phosphatase activity), less regulation of inhibitors of RAS activity is expected to occur causing a downstream negative effect on MAPK signaling. During development, p-ERK1/2 staining is at high levels in the VZ progenitors cells of the ventral and lateral telencephalon but not in the parenchyma region (Fig. 5). Moreover, we found that Shp2 cKOs display a reduction but not a complete loss of p-ERK1/2 staining in LGE VZ progenitors. This is quite different in other cell types as conditional loss of Shp2 in developing neural crest and lens tissue results in a near complete loss of p-ERK1/2 (Nakamura et al., 2009; Pan et al., 2010). Hence, depending on the tissue type, Shp2 may be either essential for MAPK signaling or required for appropriate levels MAPK signaling. It will be interesting to determine whether activated MAPK signaling in Shp2-deficient cells rescues the OPC phenotype because it is known that conditional loss of B-raf, Mek1/2, and Erk1/2 in the telencephalon exhibits some aspect of OL phenotype (Galabova-Kovacs et al., 2008; Fyffe-Maricich et al., 2011; Li et al., 2012; Ishii et al., 2012).
In this study, we also used an inducible transgenic mouse to gain new insight into the consequence of a Shp2 GOF Noonan syndrome mutation during brain development. Although the D61G GOF mutation has been reported to promote neurogenesis and repress astrocyte generation in the cortex (Gauthier et al., 2007), we found that expression of the Q79R mutation in the ventral telencephalon increased OPC numbers, indicating that Noonan syndrome mutations also influences the oligodendrocyte lineage. Moreover, Shp2 GOF mice display abnormal myelination suggesting that the increased OPCs expressing the Q79R Noonan syndrome mutation exhibit defects in oligodendrocyte differentiation and myelination.
It has been suggested that the impact of Shp2 GOF mutations is cell context specific because MEFs do not show enhanced ERK activation after D61G expression (Araki et al., 2004). On the other hand, Shp2 GOF Noonan syndrome mutations (D61G or Q79R) enhance p-ERK1/2 expression in craniofacial mesenchyme and in progenitor cells of the heart valves, contributing to pathogenic phenotypes observed in the cardiovascular and craniofacial systems (Araki et la., 2004; Krenz et al., 2008; Nakamura et al., 2009). We detected elevated p-ERK1/2 expression and increased OPC proliferation in Q79R-Shp2-tg;Olig2cre/+ white matter regions. In line with this result, recent reports have suggested that in vivo MEK GOF or NF1 LOF in glial progenitors and/or mature OLs result in elevated MAPK signaling (Hegedus et al., 2007; Li et al., 2012; Wang et al., 2012; Mayes et al., 2013). Because it is known that some patients with various RASopathies (Noonan syndrome-Shp2/PTPN11, neurofibromatosis type 1-NF1, or cardiofaciocutaneous syndrome-MEK1) exhibit neurocognitive abnormalities (Tidyman and Rauen, 2009; Acosta et al., 2012), it will be interesting to explore further whether glial phenotypes are the underlying cause of some behavioral deficits observed in these patients.
It will also be interesting to determine whether the extent of these glial phenotypes are entirely from misregulation of the MAPK pathway. Recently, it was suggested that Shp2 acts via the AKT pathway to promote OL maturation in OPC cultures (Liu et al., 2011). Interestingly, in vivo upregulation of AKT signaling via transgenic expression of active AKT or Pten conditional deletion results in hypermyelination from increased myelin production and not an increase in OPCs (Flores et al., 2008; Harrington et al., 2010). The Shp2 GOF phenotype described here is quite different, showing increased OPCs and fewer myelinated axons. Although in some cases we also detected abnormal and/or thicker myelin, this was minor compared with the global hypermyelination observed by AKT activation (Flores et al., 2008; Harrington et al., 2010). Moreover, we did not detect increased phospho-S6 ribosomal protein expression, which is a readout of the activity of the PI3/AKT/mTOR pathway and indicative of AKT activation in the oligodendrocyte lineage (Narayanan et al., 2009; Harrington et al., 2010). Therefore, in our Shp2 GOF mice, AKT is not likely a major contributor to the phenotype.
In conclusion, Shp2 is a crucial regulator for normal OPC generation and myelination in the telencephalon. MAPK signaling, embryonic/postnatal OPC generation, and white matter myelination are impacted by complete loss of Shp2 as well as abnormal Shp2 function caused by Shp2 GOF Noonan syndrome mutation. Thus, appropriate Shp2 function likely regulates levels of MAPK signaling and maintains normal numbers of OPCs that differentiate and myelinate correctly.
Footnotes
This work was supported by start-up funds provided by Cincinnati Children's Hospital Medical Center Trustee Grant to R.R.W., National Institutes of Health Grant 5P30CA-149239 to N.R., Grant 1R01NS075291-01A1 to B.D., and Grant R01HL116525 to M.K. We thank Drs. Tom Jessell and Ben Novitch for the Olig2cre/+ mice; Dr. Jane Johnson for the Ascl1 antibody; and especially Dr. Kenny Campbell for critical reading and discussion of the manuscript.
The authors declare no competing financial interests.
References
- Acosta MT, Bearden CE, Castellanos FX, Cutting L, Elgersma Y, Gioia G, Gutmann DH, Lee YS, Legius E, Muenke M, North K, Parada LF, Ratner N, Hunter-Schaedle K, Silva AJ. The Learning Disabilities Network (LeaDNet): using neurofibromatosis type 1 (NF1) as a paradigm for translational research. Am J Med Genet A. 2012;158A:2225–2232. doi: 10.1002/ajmg.a.35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfieri P, Cesarini L, Mallardi M, Piccini G, Caciolo C, Leoni C, Mirante N, Pantaleoni F, Digilio MC, Gambardella ML, Tartaglia M, Vicari S, Mercuri E, Zampino G. Long term memory profile of disorders associated with dysregulation of the RAS-MAPK signaling cascade. Behav Genet. 2011;41:423–429. doi: 10.1007/s10519-011-9446-5. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron. 2004;41:683–686. doi: 10.1016/S0896-6273(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–857. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Rizvi TA, Karyala S, McKinnon RD, Ratner N. Aberrant growth and differentiation of oligodendrocyte progenitors in neurofibromatosis type 1 mutants. J Neurosci. 2003;23:7207–7217. doi: 10.1523/JNEUROSCI.23-18-07207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentires-Alj M, Kontaridis MI, Neel BG. Stops along the RAS pathway in human genetic disease. Nat Med. 2006;12:283–285. doi: 10.1038/nm0306-283. [DOI] [PubMed] [Google Scholar]
- Cai J, Chen Y, Cai WH, Hurlock EC, Wu H, Kernie SG, Parada LF, Lu QR. A crucial role for Olig2 in white matter astrocyte development. Development. 2007;134:1887–1899. doi: 10.1242/dev.02847. [DOI] [PubMed] [Google Scholar]
- Chapman H, Waclaw RR, Pei Z, Nakafuku M, Campbell K. The homeobox gene Gsx2 controls the timing of oligodendroglial fate specification in mouse lateral ganglionic eminence progenitors. Development. 2013;140:2289–2298. doi: 10.1242/dev.091090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 2008;20:453–459. doi: 10.1016/j.cellsig.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. 2009;16:256–270. doi: 10.1016/j.devcel.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, Briscoe J. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature. 2007;450:717–720. doi: 10.1038/nature06347. [DOI] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Götz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28:10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe-Maricich SL, Karlo JC, Landreth GE, Miller RH. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci. 2011;31:843–850. doi: 10.1523/JNEUROSCI.3239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galabova-Kovacs G, Catalanotti F, Matzen D, Reyes GX, Zezula J, Herbst R, Silva A, Walter I, Baccarini M. Essential role of B-Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol. 2008;180:947–955. doi: 10.1083/jcb.200709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier AS, Furstoss O, Araki T, Chan R, Neel BG, Kaplan DR, Miller FD. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron. 2007;54:245–262. doi: 10.1016/j.neuron.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb BD, Tartaglia M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet. 2006;15:R220–R226. doi: 10.1093/hmg/ddl197. [DOI] [PubMed] [Google Scholar]
- Hanafusa H, Torii S, Yasunaga T, Matsumoto K, Nishida E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J Biol Chem. 2004;279:22992–22995. doi: 10.1074/jbc.M312498200. [DOI] [PubMed] [Google Scholar]
- Harrington EP, Zhao C, Fancy SP, Kaing S, Franklin RJ, Rowitch DH. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol. 2010;68:703–716. doi: 10.1002/ana.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus B, Dasgupta B, Shin JE, Emnett RJ, Hart-Mahon EK, Elghazi L, Bernal-Mizrachi E, Gutmann DH. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell. 2007;1:443–457. doi: 10.1016/j.stem.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci. 2012;32:8855–8864. doi: 10.1523/JNEUROSCI.0137-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Zhang EE, Hagihara K, Wu D, Pang Y, Klein R, Curran T, Ranscht B, Feng GS. Deletion of Shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol. 2007;27:6706–6717. doi: 10.1128/MCB.01225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz M, Gulick J, Osinska HE, Colbert MC, Molkentin JD, Robbins J. Role of ERK1/2 signaling in congenital valve malformations in Noonan syndrome. Proc Natl Acad Sci U S A. 2008;105:18930–18935. doi: 10.1073/pnas.0806556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo E, Park DK, Tzvetanova ID, Leiton CV, Cho BS, Colognato H. Tyrosine phosphatases Shp1 and Shp2 have unique and opposing roles in oligodendrocyte development. J Neurochem. 2010;113:200–212. doi: 10.1111/j.1471-4159.2010.06596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauriol J, Kontaridis MI. PTPN11-associated mutations in the heart: has LEOPARD changed Its RASpots? Trends Cardiovasc Med. 2011;21:97–104. doi: 10.1016/j.tcm.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DA, Portnoy S, Hill P, Gillberg C, Patton MA. Psychological profile of children with Noonan syndrome. Dev Med Child Neurol. 2005;47:35–38. doi: 10.1111/j.1469-8749.2005.tb01037.x. [DOI] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD. MEK is a key regulator of gliogenesis in the developing brain. Neuron. 2012;75:1035–1050. doi: 10.1016/j.neuron.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li Y, Zhang Y, Lu Y, Guo W, Liu P, Zhou J, Xiang Z, He C. SHP-2 promotes the maturation of oligodendrocyte precursor cells through Akt and ERK1/2 signaling in vitro. PLoS One. 2011;6:e21058. doi: 10.1371/journal.pone.0021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, McMahon AP, Stiles CD, Rowitch DH. Sonic hedgehog–regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron. 2000;25:317–329. doi: 10.1016/S0896-6273(00)80897-1. [DOI] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Mayes DA, Rizvi TA, Titus-Mitchell H, Oberst R, Ciraolo GM, Vorhees CV, Robinson AP, Miller SD, Cancelas JA, Stemmer-Rachamimov AO, Ratner N. Nf1 loss and Ras hyperactivation in oligodendrocytes induce NOS-driven defects in myelin and vasculature. Cell Rep. 2013;4:1197–1212. doi: 10.1016/j.celrep.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Butt SJ, Takebayashi H, Fishell G. Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J Neurosci. 2007;27:7786–7798. doi: 10.1523/JNEUROSCI.1807-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi R, Sugimori M, Takebayashi H, Kosako H, Nagao M, Yoshida S, Nabeshima Y, Shimamura K, Nakafuku M. Combinatorial roles of olig2 and neurogenin2 in the coordinated induction of pan-neuronal and subtype-specific properties of motoneurons. Neuron. 2001;31:757–771. doi: 10.1016/s0896-6273(01)00413-5. [DOI] [PubMed] [Google Scholar]
- Molineaux SM, Engh H, de Ferra F, Hudson L, Lazzarini RA. Recombination within the myelin basic protein gene created the dysmyelinating shiverer mouse mutation. Proc Natl Acad Sci U S A. 1986;83:7542–7546. doi: 10.1073/pnas.83.19.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Gulick J, Colbert MC, Robbins J. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc Natl Acad Sci U S A. 2009;106:11270–11275. doi: 10.1073/pnas.0902230106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- Nicolay DJ, Doucette JR, Nazarali AJ. Transcriptional control of oligodendrogenesis. Glia. 2007;55:1287–1299. doi: 10.1002/glia.20540. [DOI] [PubMed] [Google Scholar]
- Pan Y, Carbe C, Powers A, Feng GS, Zhang X. Sprouty2-modulated Kras signaling rescues Shp2 deficiency during lens and lacrimal gland development. Development. 2010;137:1085–1093. doi: 10.1242/dev.042820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E, Seidenberg MS. Genotype differences in cognitive functioning in Noonan syndrome. Genes Brain Behav. 2009;8:275–282. doi: 10.1111/j.1601-183X.2008.00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, Wood TL. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29:6367–6378. doi: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waclaw RR, Allen ZJ, 2nd, Bell SM, Erdélyi F, Szabó G, Potter SS, Campbell K. The zinc finger transcription factor Sp8 regulates the generation and diversity of olfactory bulb interneurons. Neuron. 2006;49:503–516. doi: 10.1016/j.neuron.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Waclaw RR, Ehrman LA, Pierani A, Campbell K. Developmental origin of the neuronal subtypes that comprise the amygdalar fear circuit in the mouse. J Neurosci. 2010;30:6944–6953. doi: 10.1523/JNEUROSCI.5772-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y. ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell. 2012;150:816–830. doi: 10.1016/j.cell.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, George EL, Neel BG. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell. 2006;10:317–327. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zuo H, Maher BJ, Serwanski DR, LoTurco JJ, Lu QR, Nishiyama A. Olig2-dependent developmental fate switch of NG2 cells. Development. 2012;139:2299–2307. doi: 10.1242/dev.078873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, Burns DK, Bajenaru ML, Gutmann DH, Parada LF. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132:5577–5588. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Park J, Hu X, Zheng K, Li H, Cao Q, Feng GS, Qiu M. Control of oligodendrocyte generation and proliferation by Shp2 protein tyrosine phosphatase. Glia. 2010;58:1407–1414. doi: 10.1002/glia.21016. [DOI] [PMC free article] [PubMed] [Google Scholar]