

Abstract
S aureus internalized by bone cells and shielded from the immune system provides a reservoir of bacteria in recurring osteomyelitis. Its targeting by the antibiotic therapy may thus be more relevant for treating chronic bone infection than eliminating only the pathogens colonizing the bone matrix. Assessed was the combined osteogenic and antibacterial effect of clindamycin-loaded calcium phosphate nanoparticles of different monophasic compositions on co-cultures comprising osteoblasts infected with S aureus. Antibiotic-carrying particles were internalized by osteoblasts and minimized the concentration of intracellular bacteria. In vitro treatments of the infected cells, however, could not prevent cell necrosis due to the formation of toxic byproducts of the degradation of the bacterium. Antibiotic-loaded particles had a positive morphological effect on osteoblasts per se, without reducing their viability, alongside stimulating upregulation of expression of different bone growth markers in infected osteoblasts to a higher degree than achieved during the treatment with antibiotic only.
Keywords: Calcium Phosphate, Controlled Drug Delivery, Nanoparticles, Osteogenesis, Osteomyelitis, Staphylococcus aureus
Introduction
The continuously aging population on Earth insinuates a rise in the number of bone diseases, including the infectious ones, such as osteomyelitis [1]. The incidence of osteomyelitis in the US is 1 – 2 %, but the disease is far more prevalent in the developing countries as well as among particular patient populations: approximately 1 in 5000 among children, 1 in 1000 among neonates, 1 in 250 among sickle cell patients, 1 in 7 among diabetics and 1 in 3 among patients with punctured foot [2 – 5]. Since bone tissues are relatively well shielded from the external pathogens that may cause the infection, osteomyelitis is particularly common among the orthopedically postoperative and posttraumatic patients [6]. Conversely, however, this means that, once infected, bone tissues are difficult to target by conventional antibiotic therapies. The traditional approach to the treatment of osteomyelitis involves (a) repetitive administration of antibiotics, typically by intravenous or oral means for up to six weeks, frequently followed by a 6-month course of oral antibiotics in the case of chronic infection, and (b) surgical removal of the osteonecrotic portion of bone malformed due to disease so as to prevent chronically recurrent infection due to sequestra inaccessible to immune cells or antibiotics [7]. The main downsides of this conventional type of treatment include: (a) systemic distribution of the antibiotic throughout the organism and the corresponding side effects [8]; (b) its lower concentration in the target area, potentially promoting bacterial resistance to the therapy; (c) the necessity of physical removal of the necrotic bone and the installment of lasting prostheses or implants; and (d) cost-ineffectiveness, as a daily dose of orally or intravenously administered antibiotics ranges in excess of $100 per patient. Incentives thus exist to develop an antibiotic delivery system for (a) sustained therapeutic concentration of the drug in the target zone and (b) the ability to enhance the remineralization of the tissue lost to disease.
Although osteomyelitis can be caused by a variety of pathogens, most of which reside in healthy oral flora, S aureus stands for the main causative agent thereof [9]. S aureus, which is found in healthy oral and nasal flora, has the ability to penetrate endothelial, epithelial and osteoblastic cells, and thrive in the intracellular environment where it could be less susceptible to antibiotic-related destruction [10 – 12]. S aureus internalized by the cells and shielded from the host immune system is thought to provide a reservoir of bacteria in recurring osteomyelitis and its targeting by the antibiotic may thus be more relevant for treating chronic bone infection than eliminating merely pathogens colonizing the bone matrix [13]. Another rationale behind the usage of particulate carriers for intracellular delivery of antibiotics comes from the fact that many colonies create an acidic milieu that can drastically reduce the potency of antibiotic in its pure form [14]. Our previous studies have shown that calcium phosphates (CAPs) of various stoichiometries could present a viable candidate for drug delivery carriers for tunable release of antibiotics in the treatment of osteomyelitis [15]. Our earlier report confirming the antibacterial effectiveness against S aureus in both suspended and surface-bound forms [16] is supplemented hereby with the assessment of the bactericidal performance of antibiotic-loaded CAP particles with respect to osteoblastic cells infected with the given bacterium as well as of their osteoconductive and osteogenic potentials.
The choice for drug delivery agents utilized in this work has been derived from the incentives to come up with a biocompatible, biodegradable and osteogenic material with tunable degradation and drug release kinetics. Local and sustained delivery of gentamicin has been clinically achieved since the mid-1970s by means of surgical implantation of millimetric polymethyl methacrylate beads [17], which still present the gold standard in local antibiotic therapy for the treatment of osteomyelitis [18]. Their non-osteogenic and non-biodegradable nature that requires secondary surgical procedure for their removal from the patient has, however, made it impossible to promote conditions for spontaneous regeneration of diseased bone [19]. The premise of this study stems from a belief that a compositional similarity between the ceramic component of bone and artificial calcium phosphates makes the latter an ideal candidate for the carriers of locally deliverable antibiotics in the treatment of osteomyelitis. In the past and with respect to the broad spectrum of phase compositions of CAPs, only two CAP phases were utilized for the sake of controlling the biodegradation rate of monophasic or biphasic mixtures thereof: hydroxyapatite (HAP) and tricalcium phosphate. In particular, the approach to bone regeneration by means of surgical implantation of synthetic CAPs has paradigmatically relied on HAP, the phase already present in bone. However, we believe that since osteoblasts eventually build new hard tissues from its ionic and organic ingredients, more resorbable phases, including dicalcium phosphate (DCPA), shown in our former work to boost gene expression that promotes in vitro bone growth more than other, less soluble CAP phases, should be paid more attention to. In this study, we have focused on three different phases of CAP that cover a broad range of solubilities, from pKsp = 7 (DCPA) to pKsp = 20–25 (amorphous CAP; ACP) to pKsp = 117 (HAP). Our previous work has shown that sustained release of drugs could be made directly relatable to the degradation rate of CAP carriers [20]. In theory, by combining these different CAP phases in various proportions, drug release profiles could be tailored to the therapeutic occasion. By varying the phase composition of the particles, an optimal balance between bioactivity and biodegradability could be achieved, so as to provide an osteoconductive surface for osteoblasts to adhere onto and match the rather slow rate of new bone formation, while at the same time ensuring sufficient degradation and drug release rates to eradicate the pathological source of infection and provide ionic species that will serve as ingredients of the newly forming bony tissues.
Finally, it should be noted that methodologically wedded in this study are molecular biology, classical bacteriology and materials science, all under one umbrella. It is a necessity today, when research at the frontier is, as a rule, of interdisciplinary nature, to combine methodologies that have traditionally belonged to separate fields. To the same degree as they are challenging for the interpreters and the inquirers alike, these phenomena lying at the boundary between different fields carry a potential for the discovery of new and more sophisticated technological and therapeutic tools.
Materials and methods
Synthesis and characterization of clindamycin-containing CAP nanopowders
Synthesis of different monophasic nanoparticulate powders of CAP and their loading with clindamycin phosphate (CL), a common antibiotic of choice in the treatment of acute ostemyelitis [21], was described in a previous report [16]. Briefly, an aqueous solution of NH4H2PO4 was admixed to an identical volume of an aqueous solution of Ca(NO3)2 at different pHs, ionic concentrations and rates, depending on the intended phase composition to be obtained. Table 1 summarizes these parameters and includes other physical properties of these powders. For the formation of all powders except ACP, the precipitate was additionally brought to the boiling point, then immediately cooled down at room temperature, aged for 24 h, washed with distilled water, centrifuged, and dried in vacuum (p = −20 mm Hg) at room temperature. In this method, CL dissolved in water was introduced to CAP suspensions immediately after their cooling down to room temperature. To ensure that CL was stably confined in the porous pockets of the powder and released at a sustained rate, CAP particles with CL physisorbed on them were dried in vacuum. Compact blocks were formed thereby, protecting the bulk of the drug from immediate contact with the solvent and premature release. Cobalt-doped HAP as the negative control for osteoblastic compatibility tests was prepared with 5 wt% of Co according to a previously reported procedure [22]. The phase composition of the particles was analyzed on a Bruker AXS D4 Endeavour X-ray diffractometer using CuKα = 1.5418 Å as the wavelength of the radiation source. The step size was 0.02 °, with 2 s of the sample irradiation time per step. The average size of the crystallites was determined using a structural refinement approach (Topas 2.0) based on the previously identified crystal structure (PCPDFWIN & Eva). The size distribution profiles and morphology of the powders were analyzed on a Hitachi S-4300SE/N scanning electron microscope (SEM). Mac Biophotonics ImageJ software was used to measure the average particle size from the obtained electron micrographs. High-resolution transmission electron microscopy (HR-TEM) analysis of CAP nanopowders was performed on a FEI monochromated F20 UT Tecnai HR-TEM under the electron acceleration voltage of 200 kV. Raman spectra were taken on a Renishaw inVia Reflex Raman microscope, in the high throughput mode, using low laser power of 2 – 5 mW, λ = 532 nm, the groove grating of 2400, and a 50x objective.
Table 1.
Initial ionic concentrations and pHs of aqueous precursors for the synthesis of CAP powders of different monophasic compositions by precipitation, along with their solubility products (pKsp), admixing time, empirically derived solubilities [23], particle sizes (D) and crystallite sizes (d).
| Phase | Chemical formula | [Ca(NO3)2] (M) | pH of Ca(NO3)2 (aq) | [NH4H2PO4] (M) | pH of NH4H2PO4 (aq) | Time (min) | pKsp | Solubility (g/dm3) | D (nm) | d (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| HAP | Ca10(PO4)6(OH)2 | 0.1 | 11.5 | 0.06 | 10.5 | 30 | 117 | 3 · 10−4 | 83.8 +− 7.3 | 12 |
| ACP | Ca3(PO4)2·nH2O | 0.5 | 6.8 | 0.2 | 10.5 | 1.6· 10−2 | 25 | 2.5 · 10−3 | 61.8 +− 9.5 | 0.4 |
| DCPA | CaHPO4 | 0.33 | 6.8 | 0.25 | 7.6 | 30 | 7 | 4.8 · 10−2 | 74.3 +− 6.2 | 33 |
Cell culture
Mouse calvarial preosteoblastic cell line, MC3T3-E1 subclone 4, was purchased from American Tissue Culture Collection (ATCC, Rockville, MD) and cultured in Alpha Minimum Essential Medium (α-MEM; Gibco) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and no ascorbic acid (AA). The medium was replaced every 48 h, and the cultures were incubated at 37 °C in a humidified atmosphere containing 5% CO2. Every 7 days, the cells were detached from the surface of the 75 cm2 cell culture flask (Greiner Bio-One) using 0.25 wt% trypsin, washed, centrifuged (1000 rpm x 3 min), resuspended in 10 ml α-MEM and subcultured in 1:7 volume ratio. Cell passages 8 – 16 were used for the experiments reported hereby. The cultures were regularly examined under an optical microscope to monitor growth and possible contamination.
Differentiation and invasion assay
Cells were seeded on glass cover slips placed in 24 well plates and 500 μl of α-MEM supplemented with 10% fetal bovine serum (FBS, Invitrogen) and no AA at the density of 6 × 104 cells per well. After 5 days of incubation, nearly confluent cells were treated with α-MEM containing 50 μg/ml AA as the mineralization inductor. After 2 days, the media were aspirated and the cells were washed with PBS and had antibiotic-free RPMI-1640 medium buffered with 25 mM HEPES and containing sodium bicarbonate added to them. Previously, a single colony of Staphylococcus aureus (ATCC 25923) cultured on a blood agar plate over 48 hours was stabbed with a pipette tip and added to 5 ml of 37 mg/ml brain heart infusion (BHI) broth and kept on an incubator shaker (Innova 44) overnight at 37 °C and 225 rpm. To fluorescently label the bacteria, fluorescein isothiocyanate (FITC) was added to the bacterial broth 4 h into the inoculation to the concentration of 100 μg/ml, following a modified previously described protocol [24]. Five microliters of this freshly inoculated broth were added to 500 μl of 25 mM of NaCO3-containing HEPES media previously added to the cells. The given bacterial amount corresponded to circa 6.25 × 107 colony forming units, according to the results of comparison of optical transparency with McFarland standards prepared by mixing 1 wt% BaCl2 solution and 1 wt% H2SO4 in the volume ratio of 1:200, respectively. To induce intracellular infection, cells were incubated for 2 h at 37 °C, as in accordance with the previously reported invasion time scale [25 – 27], in a CO2-free incubator, after which they were washed once with PBS, replenished with fresh antibiotic-free RPMI-1640 media and had 2 mg/cm2 of CAP particles loaded with CL (Tokyo Chemical Industry) added to them. The amount of the particles added to each well was normalized to the surface area rather than to cell culture medium volume because particles initially sediment to the bottom of the wells where cells are exclusively found. Normalized to the cell culture medium volume, the amount of CL-loaded particles used to treat the infected cells with equaled 4 mg/ml. Negative controls (C−) were uninfected and positive controls (C+) were infected but not treated with either CAP particles or CL. When treated with CL only, the infected cells were given the amount of antibiotic that equaled the amount of CL-loaded CAP particles that other experimental groups were treated with. Subsequent incubation in a CO2-free environment took place over different periods of time, ranging from 1 to 48 h, after which a portion of the samples was analyzed for the amount of mitochondrial dehydrogenases, indicative of the metabolic activity of the cells, using MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide) in vitro toxicological assay (Sigma). Another portion of the samples was stained for osteoblastic cell nucleus and cytoskeletal f-actin using a previously described protocol [16]. All the experiments were done in triplicates and the fluorescence of the FITC-tagged bacterium was measured under identical excitations from four randomly selected images in each sample. The subsequent average pixel intensity analysis was performed using ImageJ and NIS Elements software. Both single-plane and volume-rendered z-stack images (12 – 15 of them) spaced by 1 μm were also collected. The third portion of the samples was lysed with 0.1 wt% Tween-20 for 5–10 min. To minimize lysis of intracellular and extracellular bacterial cell walls during permeabilization, the effect known to occur when Triton-X, the standard permeabilization agent, is used [28], the latter was substituted with a milder, nonionic surfactant, Tween-20 known to be non-injurious to fastidious microbial pathogens [29]. One part of the lysed samples was mixed with BHI broths in 1:10 ratio and incubated for 4 – 24 h at 37 °C and 225 rpm. After the given period of time, the broths were analyzed for optical density at 600 nm on a UV/Vis spectrophotometric microplate reader (Molecular Devices: Spectra Max 190). Two microliters of the lysates were also spread on top of sheep blood agar plates, incubated overnight at 37 °C and counted for the number of colonies the following day. Invasion assays were also carried out using mutants of S aureus potentially resistant to the antibiotic. For that purpose, differentiated MC3T3-E1 cells were co-cultured with the bacterium and 2 mg/cm2 CL for 48 h using the aforementioned procedure, lysed, streaked on an agar plate and incubated at 37 °C overnight. A single colony was picked off the plate and added to a fresh BHI broth. The latter was inoculated overnight and a portion of it was used to infect differentiated MC3T3-E1 cells via the aforementioned procedure.
For the purpose of qPCR analysis, MC3T3-E1 cells were seeded in 96 well plates and 100 μl of α-MEM supplemented with 10% fetal bovine serum (FBS, Invitrogen) and no AA at the density of 2 × 104 cells per well. After 3 days of incubation, the medium was replaced with α-MEM containing 50 μg/ml AA as the mineralization inductor. After a 24 h incubation, the cells had 100 μl of antibiotic-free RPMI-1640 medium buffered with 25 mM HEPES and containing sodium bicarbonate added to them, altogether with 2 μl of S aureus in a freshly inoculated BHI broth (equivalent to circa 3 × 107 colony forming units). After 2 h of the invasion time, the medium was refreshed and 1 mg of HAP, with and without the antibiotic, including antibiotic alone (1 mg), were added to the co-culture and incubated for 24 h. After the given time, cell lysis, reverse transcription (Bio-Rad) and qPCR (Applied Biosystems, StepONEPlus) were performed using the Fast SYBR Green Cells-to-CT kit (Ambion) in accordance with the manufacturer’s instructions. Each experiment was done in triplicates and each experimental replica was analyzed for mRNA expression in triplicates too (n = 3 × 3). The expressions of one housekeeping gene, β-actin (ACTB), and five osteogenic markers, mouse type I procollagen (Col I), osteocalcin (BGLAP), alkaline phosphatase (ALP), osteopontin (BSP-1) and Runx2, were analyzed. Table 2 shows the primer pair sequences used [30 – 32]. The real-time PCR results were analyzed using the ΔΔCt method [33] and all the data were normalized to ACTB expression levels. Comparison in the mRNA transcript levels of ACTB itself was carried out by normalizing the cycle number (Ct) for each sample to fit the Ct range between that for the uninfected, negative control (~ 21) and the maximal cycle number on the given device (~ 40), approximated to correspond to zero cell viability.
Table 2.
Primer pair sequences used for the qPCR analysis.
| Gene | Forward 5′–3′ primer | Reverse 5′–3′ primer |
|---|---|---|
| ACTB | GGCCCAGAGCAAGAGAGGTATCC | ACGCACGATTTCCCTCTCAGC |
| BSP-1 | AGGAGGAGGCAGAGCACA | CTGGTATGGCACAGGTGATG |
| BGLAP | CTCACAGATGCCAAGCCCA | CCAAGGTAGCGCCGGAGTCT |
| Col I | GCGAAGGCAACAGTCGCT | CTTGGTGGTTTTGTATTCGATGAC |
| Runx2 | AAATGCCTCCGCTGTTATGAA | GCTCCGGCCCACAAATCT |
Results and discussion
SEM images of CL-loaded HAP powders shown in Fig. 1a–b demonstrate their nanoparticulate nature, with particles being spherical in shape and with sizes in the nanoparticle range (< 100 nm). The same nanosized and spherical nature was observed for the other two CAP phases: DCPA (Fig. 1d) and ACP (Fig. 1e). XRD patterns of three different CAP phases with different solubilities utilized in this study - DCPA, ACP and HAP – are displayed in Fig. 2a. Naturally, the average particle sizes were bigger than the average crystallite sizes for each of the powders: 84 vs. 12 nm for HAP, 74 vs. 33 nm for DCPA, and 62 vs. less than 5 Å for ACP (Table 1). The amount of CL captured by each powder per unit weight was inversely proportional to the average particle size, being highest, in excess of 5 wt%, for ACP and smallest, less than 1 wt%, for DCPA [15]. As could be seen from the portion of Raman spectrum of HAP shown in Fig. 2b, the characteristic non-degenerate symmetric stretch of the phosphate tetrahedron was detected at 961 cm−1, while the corresponding stretch of the carbonate ion was detected at 1075 cm−1, suggesting a particle substitution of PO43− ions of HAP with CO32−, the product of which is the so-called B-type carbonated HAP. Unlike the A-type which typically forms upon annealing at elevated temperatures [34] and wherein CO32− occupy the place of OH− groups in the unit cell of HAP, exhibiting the blue shift of ν1(CO3) to ~ 1100 cm−1, the B-type is the typical product of precipitation of HAP from aqueous solutions under atmospheric conditions. Carbonate ions are intrinsic to the crystal lattice of biological apatite too [35], where their role is to decrease the crystallite size [36] and increase the lattice strain [37] and solubility [38], thus promoting the bone resorption process and speeding up its turnover and regeneration. The content of carbonate ions in HAP used in this study is not supposed to be higher than 1 wt% [39].
Fig. 1.

Scanning electron micrographs of spherical and monodisperse HAP nanoparticles (a), forming compact microscopic blocks and capturing the adsorbed drug upon desiccation (b). (c) A transmission electron micrograph of a single nanoscopic particle of HAP with denoted defect-laden crystalline particle core and the largely amorphous, disorder surface atomic layers thereof. DCPA (d) and ACP (e) powders display similar nanoparticle size and shape characteristics.
Fig. 2.
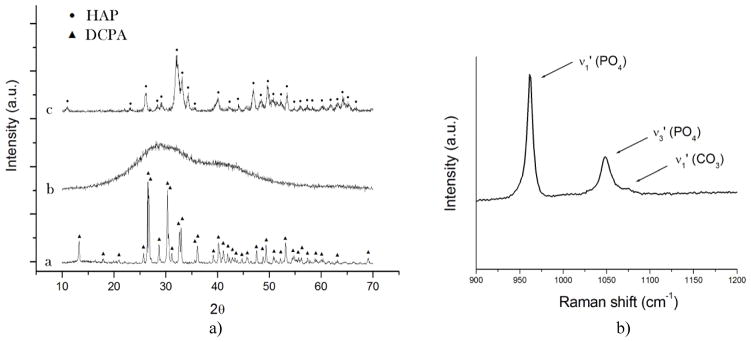
(a) XRD patterns of three different monophasic CAP nanopowders with different solubility products (Ksp) utilized in this study: (a) DCPA (pKsp = 7); (b) ACP (pKsp = 20 – 25); (c) HAP (pKsp = 114). (b) Raman spectrum of HAP in 900 – 1200 cm−1 region, showing three bands originating from the symmetrical (ν1) and asymmetrical (ν3) stretches of the phosphate ion and from the symmetrical stretch of the carbonate ion, at 961, 1048 and 1075 cm−1, respectively.
Largely accepted is both in vitro and in vivo growth of micro- and nano-particles of CAP via aggregation of 1–2 nm sized growth units, a.k.a. Posner’s clusters [40 – 42]. One such basic unit of growth was discerned in the course of the HR-TEM analysis of precipitated HAP nanoparticles, possessing ~ 2 nm sized crystalline core abundant with twinned interfaces and 3 – 4 atomic layers thick and largely disordered surface (Fig. 1c). Aggregation of the nanoparticles formed by coalescence of Posner’s singlets during the powder desiccation process in vacuum into bigger, microsized blocks, two of which are shown in Fig. 1b, presented the mechanism for the loading of the powders with the drug. Premature release of the drug merely adsorbed on the nanoparticle surface is thus avoided and its retention within the powder is enabled despite the immersion of the powder in the solvent for prolonged periods of time [15]. Although developed independently from other groups, similar methods for drug capture have been used earlier to ensure the extended release of vancomycin [43] and aminoglycoside antibiotic, isepamacin sulfate [44] at a sustained rate from compacted calcium phosphate powders. Although the loading method is different, using soaking rather than desiccation-induced particle aggregation, PerOssal, a commercial mixture HAP and calcium sulfate also relies on one such compaction of nanoparticles to ensure sustained release of antibiotics [45]. Other methods for the fabrication of compact and mesoporous blocks of HAP, without sintering at elevated pressures or temperatures at which the degradation of encapsulated organics would have occurred, have also been reported [46 – 47].
Our previous studies on monocultures have confirmed the ability of CL-containing CAP powders to inhibit the bacterial growth both on solid surfaces and in broths, regardless of the CAP phase loaded with CL, with the minimal inhibitory concentrations ranging from 1 – 15 mg/ml of the particles. In this work, we have looked at the ability of the given antibiotic powders to eradicate the same bacterium, S aureus, from infected osteoblastic cells, while preserving their osteogenic potency.
As shown in Fig. 3a, following 2 h of co-culture, individual bacteria become visible inside of the infected osteoblasts, while after 48 h of subsequent incubation of infected osteoblasts with CL-containing HAP (Fig. 3d), ACP (Fig. 3e) and DCPA (Fig. 3f), the number of bacteria visible both inside and outside of the cells becomes drastically reduced. In contrast, the bacterial population in the untreated co-culture increased during the same time (Fig. 3c). The cytoskeletal f-actin pattern in the untreated sample (Fig. 3c), however, revealed a healthier, more striated and elongated appearance in comparison with the co-cultures treated with the CL-containing nanoparticles (Fig. 3d–f). The release of cell debris, necrotic factors and proteases following the antibiotic suppression of the pathogen thus obviously has a negative effect on cell viability, whereas a state more akin to symbiotic homeostasis is reached in untreated co-cultures, at least throughout the first 48 h following the infection. A larger number of apoptotic cells, distinguishable by their clumped cytoskeletal microfilaments (Fig. 3f) and extensive cell aggregation (Fig. 3e), is consequently found in the treated samples after 48 of co-culture time.
Fig. 3.

Z-stacked confocal optical images of fluorescently stained osteoblastic cell nuclei (blue) and cytoskeletal f-actin (red), invaded with S aureus (green): (a) positive control stained immediately following the 2 h infection, showing intracellular presence of the bacterium; (b) negative control at 48 h of incubation time; (c) positive control at 48 h of incubation time; co-cultured samples incubated with 2 mg/cm2 of HAP/CL (d), ACP/CL (e), and DCPA/CL (f) at 48 h of incubation time. The sizes of images are 270 × 270 μm (a, b), 450 × 450 μm (c, d, f), and 350 × 350 μm (e).
Streaking of the osteoblastic cell lysates on agar plates has resulted in concordantly decreased number of colonies or even reduced down to zero for HAP and ACP after 24 and 48 h of incubation, respectively (Fig. 4a–b). The fact that no colonies were visible on agars despite the presence of a markedly lower amount of intracellular pathogens is explained by the remnant antibiotic in the lysates, able to prevent the surface growth of the sufficiently low concentration of the bacterium. BHI broths incubated for 4 h with fresh cell lysates obtained after a 4 h incubation with different particles also exhibited markedly lower optical densities measured at λ = 600 nm, directly proportional to the bacterial quantity, in comparison with the positive control, invaded with the bacterium (Fig. 4c). This suggests a significantly higher concentration of colony forming units in untreated co-cultured samples compared to those treated with any of the antibiotic-containing nanoparticles or the pure antibiotic. The same insight was derived from the measurements of fluorescence intensity of the FITC-tagged bacterium per surface area of co-cultured samples, the results of which are shown in Fig. 4d. As this intensity was significantly higher in the untreated samples, a moderately effective inhibition of the bacterial growth by means of the antibiotic-containing nanoparticles can be inferred, despite the negative effects on the osteoblastic cell viability that the bacterial degradation process entailed. Interestingly, the concentration of the fluorescent bacteria detected was higher in co-cultures treated with pure CL than in those treated with CL-loaded CAPs (Fig. 4d), indicating greater antibiotic efficiency of CL delivered by means of nanoparticulate CAP carriers compared to CL per se.
Fig. 4.
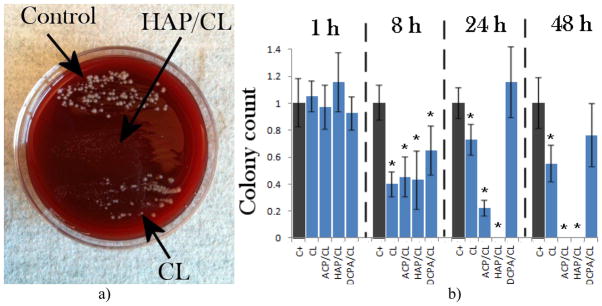
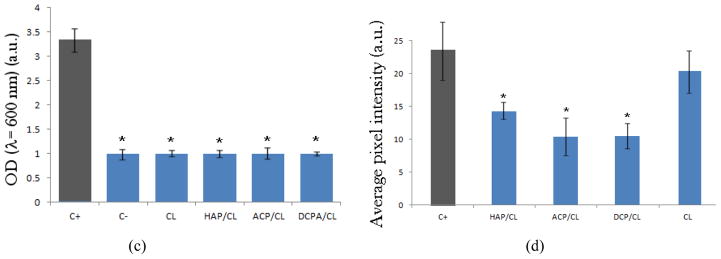
(a) Bacterial colonies forming on sheep blood agar plates following streaking of lysates of cells invaded by S aureus and incubated with either no particles (control), or with CL-loaded HAP (HAP), or pure CL (CL); (b) the number of bacterial colonies counted following overnight incubation of streaked lysates of cells invaded by S aureus and incubated for different times (1, 8, 24 h, and 5 days) with either no particles (C+), or with CL-loaded HAP (HAP), CL-loaded ACP (ACP), CL-loaded DCPA (DCPA), or pure CL (CL); (c) optical densities at λ = 600 nm for BHI broths incubated for 4 h with fresh cell lysates obtained after a 4 h incubation with different particles; (d) average pixel intensity, directly relatable to the bacterial number in co-cultured samples, measured at the excitation wavelength of 488 nm and the detection wavelength range of 520 – 550 nm. Data are shown as arithmetic means with error bars representing standard deviation (* => p < 0.05 with respect to the C+ control group).
Previous studies have shown that S aureus harvested from the colonized osteoblasts that had been treated with an antibiotic tends to become more resistant to the given therapy during subsequent intracellular colonization [48]. Co-culture experiments carried out using S aureus made potentially resistant to CL therapy showed similar intracellular invasion efficiency after 2 h of infection (Fig. 5a). Their ability to eradicate the host cells was, however, greater than for their regular counterparts. Namely, a few osteoblastic cells that remained in the invaded culture uninhibited by the antibiotic-containing nanoparticles after 48 h were fully pervaded by the bacterium and in the final stage of necrosis (Fig. 5c). In contrast, the concentration of S aureus colonies drastically dropped in co-cultures supplemented with CAP/CL particles (Fig. 5d–f). A large portion of the cells, ~ 60 – 80 %, however, underwent apoptosis during the treatment period. In view of the fact that actin microfilaments play a role in the intracellular invasion of osteoblasts by S aureus [49], the interrupted structure of f-actin, along with the formation of focal aggregates, visible in invaded cells treated with HAP/CL (Fig. 5d) and ACP/CL (Fig. 5e) particles, is the natural consequence of the infection and the subsequent bactericidal action. Due to its comparatively low drug loading capacity and a greater acidity, DCPA has shown to be nowhere as equally antibacterially effective as ACP and HAP (Fig. 5f).
Fig. 5.

Z-stacked confocal optical images of fluorescently stained osteoblastic cell nuclei (blue) and cytoskeletal f-actin (red), invaded with S aureus (green) previously made potentially resistant to CL therapy: (a) positive control stained immediately following the infection, showing intracellular presence of the bacterium; (b) negative control at 48 h of incubation time; (c) positive control at 48 h of incubation time; co-cultured samples incubated with 2 mg/cm2 of HAP/CL (d), ACP/CL (e), and DCPA/CL (f). The size of each image is 450 × 450 μm.
Results of the tests used to determine the mitochondrial activity of differentiated MC3T3-E1 cells following the infection are shown in Fig. 6, indicating a higher metabolic activity of cells in the presence of CAP/CL powders compared to those incubated either without any antibiotics or with CL per se, including the negative, uninfected control, 24 h after the infection. An insignificant decrease in viability of osteoblasts following infection and antibacterial treatment suggests that despite the intracellular infection, no cell death initially occurs as the bacteria, following a short reproductive period, enter a symbiotic balance with the host cells and protect them from premature apoptosis. This finding is in agreement with the previously observed protective effect on macrophages infected by S aureus against cell death induced by the apoptotic initiator, staurosporine [50]. Byproducts of bacterial degradation in the presence of the antibiotic include necrotic factors and proteases, and even though S aureus does not release endotoxins, as gram-negative bacteria do, this release may explain why CL alone, uncoupled with the osteoconductive CAP drug carriers, leads to lower metabolic activity compared to negative and positive controls after 24 h of incubation. Seventy two hours after the infection, the mitochondrial activity of positive controls, infected but not treated, becomes markedly higher compared to samples with reduced bacterial population due to the antimicrobial effect of CL-containing HAP and ACP powders. Increased respiration activity of cells has been reported earlier to result from their infection [51 – 52]. This effect can be seen as a natural response of the cell directed to cope with the deleterious effects caused by the pathogen [53]. Increased mitochondrial ATPase activity is also observed to mark an early event during the infection-induced cell death [54]. Indeed, 24 h after the noticed increase in mitochondrial activity, the bacterial population wiped out the osteoblasts. Decreasing the amount of the initially added colony forming units fourfold delayed the peak in mitochondrial activity to 4–5 days after the infection and the onset of full necrosis to 5–7 days following the invasion (data not shown). Osteoblasts treated with the CL-containing nanoparticles likewise underwent necrosis after a sufficient period of time.
Fig. 6.
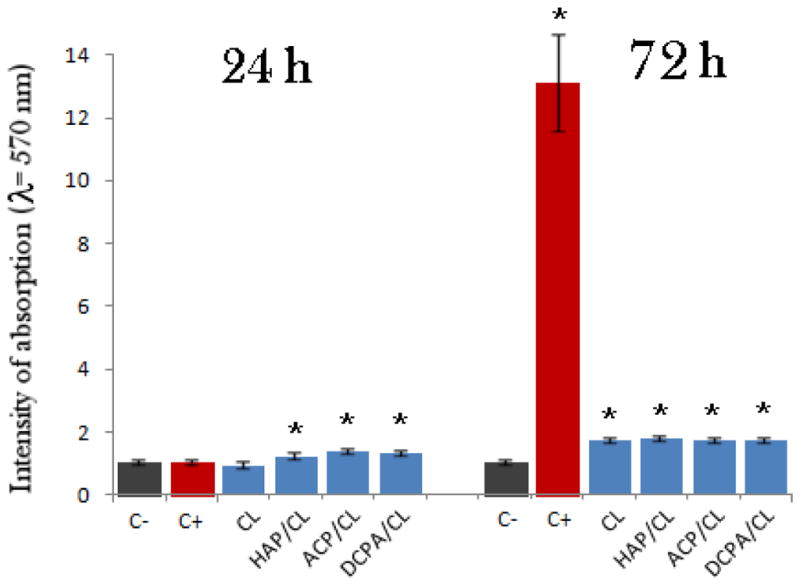
Mitochondrial activity indicative of cell viability after different treatment intervals (24 and 72 h), normalized to the negative control (C−) and determined by the MTT assay for different types of CL-containing particles (HAP, ACP, DCPA), including CL alone and positive control, infected but not subjected to the treatment with the CL-containing particles (C+). Data normalized to the optical density at λ = 570 nm of the negative control are shown as arithmetic means with error bars representing standard deviation (* => p < 0.05 with respect to the C− control group).
Although necrotic and apoptotic effects of internalization of S aureus have been reported for different cell types [55 – 59], observed was also homeostatic persistence of the bacterium within vacuolar compartments inside of phagocytes [60] and adipocytes [61] for up to 5 days, the same time scale as that observed in our study, before the cells finally succumbed to infection and died. Low levels of apoptosis leading to bacterial persistence in the course of 120 h after the infection were also observed in the case of epithelial HT-29 enterocytes [62]. This intracellular dormancy that S aureus is capable of presents its important virulence strategy that assists in protecting the bacterium from antibiotics and host defense system and provides a reservoir for chronic or systemic colonization of the organism. Treatment of the infected cells even with a relatively high concentration of pure CL – 1 mg/ml – managed to inhibit the bacterial growth, but not eliminate the source of infection. The large population of intracellular bacteria still remained after 48 h, as evident from the number of colonies formed upon plating them on agars (Fig. 4a–b). Frequent replenishment of the cell culture media, however, led to weakening of the cell response to antibiotic therapy by means of CL, which, like penicillin, has bacteriostatic rather than bactericidal potency, and after 3–5 days of post-infection incubation with CL-containing particles, the bacteria began to multiply and induce osteoblastic cell death. Research is under way as to how to extend the antibiotic release within the therapeutic window that suppresses the growth of bacteria for longer periods of time. The minimization of the bacterial presence inside of the infected cells with the use of the antibiotic-loaded CAP particles is still promising in view of the fact that the uncompromised immune system can eliminate a vast majority of even severe bacterial infections for as long as the bacterial growth is somewhat suppressed by the therapeutic means.
Internalized S aureus flocci after 2 h of infection were shown in Figs. 3a, while cells displaying a markedly lesser concentration of the intracellular bacterial contaminants after 2 days of incubation with the antibiotic-containing CAP powders were shown in Fig. 3d–f. To explain a higher antimicrobial potency of CL coupled to CAP rather than CL alone, optical confocal imaging was performed on MC3T3-E1 fibroblasts incubated with the antibiotic-loaded CAP powders. After 48 h of the incubation time, a large amount of CAP particles was found within the cells (Fig. 7a–c), as indicated by the overlap of fluorescence signals originating from CL-loaded CAP particles (green) and from intracellular structures, in this case nucleus (blue) and cytoskeletal microfilaments (red). All the monophasic CAP particles, from the most soluble DCPA to the least soluble HAP, irrespective of whether they were loaded with CL or not, were able to penetrate the cell membrane. The hazier fluorescence derived from CAP particles in the case of DCPA suggests smaller and more dispersed particles present inside of the cell, as opposed to HAP and ACP, which maintained the form of larger aggregates. Nanoparticles of CAP and various other compositions are known to be able to facilely permeate the cell membrane and localize themselves in the intracellular environment [63]. Comparatively high transfection efficiency of CAP powders is, in fact, the reason why CAPs have been one of the most affordable candidates for non-viral gene delivery carriers [64 – 67] ever since they were first utilized for this purpose in 1973 [68]. The drug delivery efficacy is known to greatly depend on the physicochemical nature of the carrier and reported was a 70-fold increase in the transfection efficacy of DNA plasmids bound to polyethyleneimine-functionalized nanoscopic diamond particles able to internalize themselves intracellularly [69]. A greater bioactivity of an anticancer therapeutic was also found out to result from its delivery in the nanoparticle-bound form, along with a reduced myelosuppression and, thus, toxicity of the treatment compared to therapies based on applying the pure drug [70]. Longer intracellular retention times of the drug delivered by means of a nanocarrier were also observed in the case of delivery of doxorubicin to cancer cells using liposomes, leading to a 5000-fold enhancement of the therapeutic efficiency in vitro [71]. It can be speculated that while the drug per se tends to be partially ejected from the cell following its uptake, this effect is prone to happen to a much lesser extent when the permeability of the cell becomes compromised by the penetration of the nanoparticles that, in addition, prevent the drug excretion from occurring by keeping it bound to their surface. The nanostructured nature of the particles is a vital factor in ensuring their excellent cell permeation capabilities [72] as much as their aggregation is vital in preventing an overly fast release of the adsorbed drug molecules. The bactericidal effect of CL is tied to its ability to inhibit the bacterial protein synthesis by binding to ribosomal subunits [73]. As such, the effectiveness of the antibiotic therapy by means of CL is conditioned by the cytoplasmic penetration of CL released from the CAP particle surface as well as by the cellular internalization of CL-containing nanosized CAP particles. The latter process does not take place instantaneously and only after a few hours of incubation do the first intracellular particles begin to be observed, as displayed in Fig. 7d.
Fig. 7.

Single plane confocal optical images of fluorescently stained osteoblastic cell nucleus (blue) and cytoskeletal f-actin (red), and CL-containing CAP particles (green) of different monophasic compositions – HAP (a, d), ACP (b), and DCPA (c) – following 48 h (a) and 4 h (d) of incubation. The sizes of images are 270 × 270 μm.
While comparatively effectively eliminating both the extracellular and intracellular source of bacterial infection, CAP particles loaded with CL have demonstrated high levels of osteoconductivity, as evidenced by the morphology of osteoblastic cells adhering onto the given particles. Their excellent spreading and intimate contact with the particles (Fig. 8a–c), altogether with the healthy appearance of f-actin pattern (Fig. 8d), known to play a crucial role in the internalization of S aureus by osteoblasts [74], serve as an evidence of this. Aside from maximizing the specific surface area available for adsorption of the drug, the nanoparticulate nature of CAP carriers interacting with the cells may be an important factor in ensuring the high levels of observed osteoconductivity. Namely, increased osteoblast adhesion has already been detected on compacts composed of nanoparticulate HAP compared to those comprising microsized particles of the same chemical composition [75]. Furthermore, an inverse proportionality was observed between (a) the level of adherence and proliferation of osteoblasts, and (b) the size of the nanoparticles (> 20 nm) comprising the surface that the osteoblasts adhered to [76, 77].
Fig. 8.
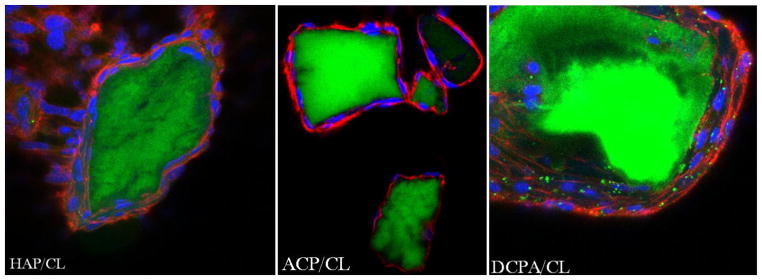
Single-plane confocal optical images of fluorescently stained HAP/CL, ACP/CL and DCPA/CL particles (green) and osteoblastic MC3T3-E1 cells (f-actin - red; nucleus - blue) following 21 days of incubation in the differentiation medium. The sizes of the images are 675 × 675 μm.
CAP particles have been shown to exhibit toxic effects on cells at high concentrations [78] due to release of Ca2+ ions that interfere with the ionic channel transport [79]. In our case, this effect has not been observed at concentrations up to 20 mg/cm2. The example of toxic effects of a carrier incorporated in CAP particles is shown in Fig. 9 in terms of the disrupted f-actin network (Fig. 9a) and morphology (Fig. 9b) of osteoblastic cells in contact with cobalt-doped (5 – 12 wt%) HAP particles. Whereas the osteoconductive HAP surface promotes an evident spreading of osteoblasts on it, the release of cobalt ions incorporated in the crystal lattice of HAP exerts a somewhat necrotic effect on the cells, which was also evidenced by a markedly lower mitochondrial activity: only 15 – 20 % of the control, as compared to the approximately twice higher activity for CL-containing HAP particles with respect to the control (Fig. 6), following an identical incubation time. Overall, favorable material-cell interaction is the foundation of facile bioresorption and repair of the diseased and demineralized hard tissue. Viable osteoblastic cells with healthy morphologies observed in close contact with the antibiotic-loaded particles unequivocally suggest their moderate-to-high levels of osteogenic potency.
Fig. 9.
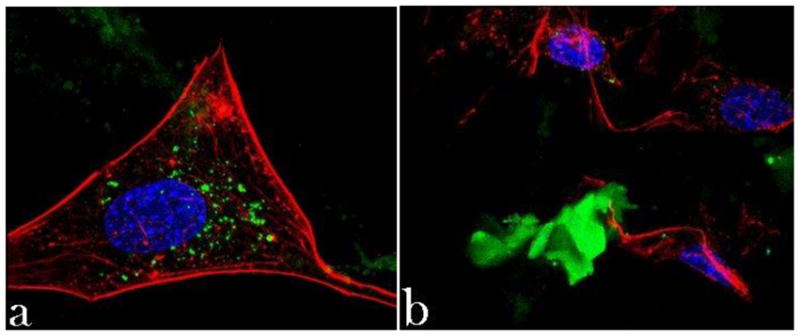
Confocal optical micrographs of osteoblastic cells exhibiting disrupted cytoskeletal f-actin pattern (red) following incubation with Co-doped HAP particles (green) (a) as well as becoming morphologically deformed in contact with them (b). The sizes of the images are 270 × 270 μm.
Our gene expression analysis, the results of which are displayed in Fig. 10, has shown that not only are the antibiotic-containing HAP particles able to partially reverse this trend and minimize the presence of the pathogen, but they are also able to boost the expression of mineralization markers. In our previous study, we showed that while CL per se induced significant downregulation of osteogenic markers BGLAP, Col I and Runx2 in osteoblastic cells, the gene expression induced by HAP loaded with CL was at the same level as in the presence of pure HAP, significantly upregulated with respect to the control [20]. Here we show similar downregulation of BGLAP and Runx2 in osteoblasts invaded with S aureus and incubated with CL only. In contrast, cells incubated with HAP/CL demonstrate significant upregulation of the given genes with respect to both the control and the cells incubated with HAP or CL alone. Apparently, HAP/CL particles are able to compensate for the negative effect of the pure drug on the expression of osteogenic markers and provide a favorable synergy between antibacterial and osteogenic potencies of the composite biomaterial designed for the treatment of bone infection.
Fig. 10.
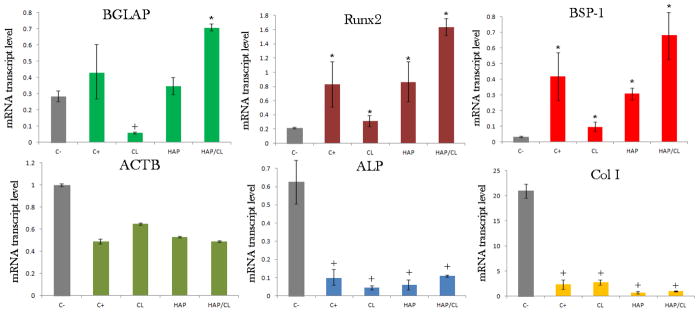
The comparative effect of pure HAP, pure CL and HAP loaded with CL (HAP/CL) on the mRNA expression of the housekeeping gene ACTB and different osteogenic markers - BGLAP, Runx2, BSP-1, ALP and Col I - in osteoblastic MC3T3-E1 cells infected with S aureus for 2 h and then incubated for 24 h with HAP, CL or HAP/CL. mRNA expression was detected by quantitative RT-polymerase chain reaction relative to the housekeeping gene ACTB. Data normalized to the expression of ACTB are shown as arithmetic means with error bars representing standard deviation. Genes significantly (p < 0.05) upregulated with respect to the control group are marked with *. Genes significantly (p < 0.05) downregulated with respect to the negative control group (C−) are marked with +.
The majority of mature bone extracellular matrix, ~ 90 % of it, is composed of collagenous proteins, the most pervasive of which is collagen type I, while the rest of it is comprised of noncollagenous proteins, the most abundant of which is osteocalcin (BGLAP), directly involved in nucleation of apatite crystals [80]. Protocollagen type I (Col I), a metabolic precursor for collagen type I, appeared to be downregulated for both the positive control, infected but untreated, as well as for infected cells treated with HAP, CL, and HAP/CL (Fig. 10). The reason for this effect relates to the fact that, unlike BGLAP and Runx2 that are specific for the differentiated MC3T3-E1 cells, Col I is expressed by non-differentiated fibroblasts too and the infection process obviously reduces the ability of cells to produce this vital ingredient of the extracellular matrix. Moreover, the two osteogenic markers BGLAP and Runx2 were upregulated for the positive control, suggesting that the infection process boosts the metabolic and osteogenic activity of cells to counteract the necrotic effects of the bacterial invasion. Despite the fact that it has been shown that the infection of cultured osteoblasts with S aureus leads to diminished proliferation, increased apoptosis and inhibited mineralization [81], our results indicate that osteoblasts can coexist with the opportunistic intracellular pathogen for days as well as that the infection entails an increase in the expression of proteins involved in the mineralization process, a completely opposite effect from that induced by the pure antibiotic. Still, however, the mRNA transcript level corresponding to the expression of the housekeeping gene β-actin (ACTB) becomes significantly diminished among the cell population that underwent invasion, even though the antibiotic therapies, with and without the CAP particles as the drug carrier, lead to a partial recovery of this unfavorable effect that indicates the ongoing loss of integrity of cytoskelatal microfilaments due to the infection. Two more genes that encode for proteins playing roles in bone formation, ALP and BSP-1, were also analyzed and whereas markedly lower expressions of ALP were observed for co-cultured samples in comparison with the uninfected control, the opposite effect was observed for BSP-1. Aside from expressing osteopontin, an acidic phosphoprotein associated with (a) pathological and inhibited mineralization [82], (b) acute and chronic inflammation [83], and (c) tumor invasiveness, progression and metastasis [84, 85], BSP-1 is a negative regulator of proliferation and differentiation in MC3T3-E1 cells [86], and its upregulation coincides with the gradual reduction in cell viability caused by the infection. Due to an array of mutually antagonistic effects that ALP downregulation can suggest, the causes of its diminished expression are more difficult to speculate about, except for the readily hypothesized ongoing reduction in the cell viability that the infection process induces.
Summary
Staphylococcus aureus is the main causative agent of osteomyelitis, an infectious disease that often entails postoperative bone tissue treatments and is on the rise due to the continuously aging population. Earlier we have shown that calcium phosphates of various stoichiometries could present a viable candidate for drug delivery carriers for tunable release of antibiotics in the treatment of osteomyelitis [15, 20]. In this work we have extended our assessment of the simultaneous osteogenic and antibacterial performance of antibiotic-loaded calcium phosphate nanoparticles to co-cultures comprising osteoblastic cells infected with Staphylococcus aureus. To that end, we have tested clindamycin-loaded calcium phosphate nanoparticles of different monophasic compositions, including monetite, amorphous calcium phosphate and hydroxyapatite. Internalization of the bacterium by the osteoblasts has been evidenced to initially hinder their antibody- or neutrophil-mediated destruction and forestall an effective antibiotic therapy by means of pure bacteriostatics. Our results have demonstrated that clindamycin-loaded hydroxyapatite and amorphous calcium phosphate particles appear to be more effective in decreasing the intracellular bacterial population and slowing down the growth of the pathogen than the pure antibiotic. Simultaneously, the osteoblastic cells were shown to engage in an intimate interface with the microscopic compacts composed of nanosized calcium phosphate particles loaded with the antibiotic, which is a strong sign of the osteoconductive nature of the material. Moreover, the bone growth markers were upregulated in the presence of clindamycin-loaded hydroxyapatite, illustrating the crucial effect of the osteogenic nature of the drug carrier in facilitating revitalization of bone tissues subjected to necrosis due to infection. More specifically, the upregulation of the expression of different bone growth markers, indicative of the increased osteogenic activity of the osteoblastic cells, was larger in the presence of calcium phosphate particles loaded with the antibiotic than in the case of (i) untreated infection, (ii) infection treated with the antibiotic only, or (iii) infection treated with antibiotic-free CAP particles. As in agreement with our previous studies, here we have confirmed the ability of clindamycin-loaded calcium phosphate particles to compensate for the negative effect of the pure drug on the expression of osteogenic markers and provide a synergy between antibacterial and osteogenic potencies promising in the consideration of the material as a clinical candidate. Namely, the simultaneous osteogenic and antimicrobial performance of the material developed in this study, altogether with its ability to exhibit sustained drug release profiles, may favor it as an alternative to prolonged antibiotic administration and surgical debridement typically prescribed in the treatment of osteomyelitis.
Highlights.
Calcium phosphate nanoparticles were loaded with clindamycin phosphate.
Efficacy of the particles was evaluated against osteoblasts infected with S aureus.
Particles were internalized and minimized the amount of intracellular bacterium.
Cell necrosis was not prevented due to formation of toxic degradation byproducts.
Particles upreguled the expression of osteogenic markers in infected osteoblasts.
Acknowledgments
Presented were the results of a study supported by the NIH/NIDCR grant K99-DE021416. The authors would like to thank Zoran Stojanović of the Institute of Technical Sciences of the Serbian Academy of Sciences and Arts for the synthesis of cobalt-substituted hydroxyapatite and Nikon Imaging Center at UCSF at which confocal microscopy data for this study were acquired.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Del Pozo JL, Patel R. N Engl J Med. 2009;361:787–794. doi: 10.1056/NEJMcp0905029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bisland SK, Chien C, Wilson BC, Burch S. Photochem Photobio Sci. 2006;5:31–38. doi: 10.1039/b507082a. [DOI] [PubMed] [Google Scholar]
- 3.Bone Health and Osteoporosis: A Report of the Surgeon General. US Department of Health and Human Services, Office of the Surgeon General; Rockville, MD: 2004. [PubMed] [Google Scholar]
- 4.Paluska SA. Clin Fam Practice. 2004;6:127–156. [Google Scholar]
- 5.King RW, Johnson DL. Stearns DA, Talavera F, Weiss EL, Halamka JD, Kulkarni R, editors. Osteomyelitis: eMedicine Medscape Reference. 2011 retrieved from http://emedicine.medscape.com/article/785020-overview#a0101.
- 6.Qureshi AT, Terrell L, Monroe WT, Dasa V, Janes ME, Gimble JM, Hayes DJ. J Tissue Eng Regen Med. 2012 doi: 10.1002/term.1532. in press. [DOI] [PubMed] [Google Scholar]
- 7.Cunha BA. Clin Infect Dis. 2002;35:287–193. doi: 10.1086/341417. [DOI] [PubMed] [Google Scholar]
- 8.Mouzopoulos G, Kanakaris NK, Kontakis G, Obakponovwe O, Townsend R, Giannoudis PV. Injury. 2001;42:S18–23. doi: 10.1016/S0020-1383(11)70128-0. [DOI] [PubMed] [Google Scholar]
- 9.Mayo Foundation for Medical Education and Research. Osteomyelitis. 2010 retrieved from http://www.mayoclinic.com/print/osteomyelitis/DS00759/DSECTION=all&METHOD=print.
- 10.Almeida RA, Matthews KR, Cifrian E, Guidry AJ, Oliver SP. J Dairy Sci. 1996;79:1021–1026. doi: 10.3168/jds.S0022-0302(96)76454-8. [DOI] [PubMed] [Google Scholar]
- 11.Bayles KW, Wesson CA, Liou LW, Fox LK, Bohach GA, Trumble WR. Infect Immun. 1998;66:336–342. doi: 10.1128/iai.66.1.336-342.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellington JK, Harris M, Webb L, Smith B, Smith T, Tan K, Hudson M. J Bone Joint Surg. 2003;85B:918–921. [PubMed] [Google Scholar]
- 13.Bost KL, Ramp WK, Nicholson NC, Bento JL, Marriott I, Hudson MC. J Infect Dis. 1999;180:1912–1920. doi: 10.1086/315138. [DOI] [PubMed] [Google Scholar]
- 14.Radovic-Moreno AF, Lu TK, Puscasu VA, Yoon CJ, Langer R, Farokhzad OC. ACS Nano. 2012;6:4279–4287. doi: 10.1021/nn3008383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uskoković V, Desai TA. J Biomed Mat Res A. 2013;101:1427–1436. doi: 10.1002/jbm.a.34437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uskoković V, Hoover C, Vukomanović M, Uskoković DP, Desai TA. Mat Sci Eng C: Mat Biol Appl. 2013;33:3362–3373. doi: 10.1016/j.msec.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dingeldein E, Wahlig H. In: Gentamycin-Konzentration in Korperflussigkeit von Patienten nach Implantation von Gentamycin-PMMA-Kugeln, Unfallchirurgie, Symposium Munchen. Contzen H, editor. Vol. 1. VLE Verlag; Erlangen: 1976. p. 8. [Google Scholar]
- 18.Kraus R, Schiefer U, Schnettler R. Local Surgical Treatment of Osteomyelitis with a Resorbable, Osteoconductive Antibiotic Carrier. In: Schnettler Reinhard, Steinau Hans-Ulrich., editors. Septic Bone and Joint Surgery. Thieme; Stuttgart: 2010. pp. 126–132. [Google Scholar]
- 19.Klemm K. Septopal – A New Way of Local Antibiotic Therapy. In: van Rens Th JG, Kayser FH., editors. Local Antibiotic Treatment in Osteomyelitis and Soft-Tissue Infections. Excerpta Medica; Amsterdam, NL: 1981. [Google Scholar]
- 20.Uskoković V, Desai TA. J Biomed Mat Res A. 2013;101:1416–1426. doi: 10.1002/jbm.a.34426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breithaupt H. Oral Antibiotic Therapy. In: Schnettler Reinhard, Steinau Hans-Ulrich., editors. Septic Bone and Joint Surgery. Thieme; Stuttgart: 2010. pp. 72–78. [Google Scholar]
- 22.Ignjatović N, Ajduković Z, Savić V, Najman S, Mihailović D, Vasiljević P, Stojanović Z, Uskoković V, Uskoković D. J Mat Sci: Mat Med. 2013;24:343–354. doi: 10.1007/s10856-012-4793-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorozhkin S. J Funct Biomater. 2013;4:209–311. doi: 10.3390/jfb4040209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krut O, Utermöhlen O, Schlossherr X, Krönke M. Infect Immun. 2003;71:2716–2723. doi: 10.1128/IAI.71.5.2716-2723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudson MC, Ramp WK, Nicholson NC, Williams AS, Nouslainen MT. Microb Pathog. 1995;19:409–419. doi: 10.1006/mpat.1995.0075. [DOI] [PubMed] [Google Scholar]
- 26.Oelschlaeger TA, Guerry P, Kopecko DJ. Proc Natl Acad Sci USA. 1993;90:6884–6888. doi: 10.1073/pnas.90.14.6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schramm N, Wyrick PB. Infect Immun. 1995;63:324–332. doi: 10.1128/iai.63.1.324-332.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cornett JB, Shockman GD. J Bacteriol. 1978;135:153–160. doi: 10.1128/jb.135.1.153-160.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zierdt CH. J Clin Microbiol. 1982;15:172–174. doi: 10.1128/jcm.15.1.172-174.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SW, Her SJ, Park SJ, Kim D, Park KS, Lee HK, Han BH, Kim MS, Shin CS, Kim SY. Bone. 2005;37:359–369. doi: 10.1016/j.bone.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 31.Rousseau M, Boulzaguet H, Biagianti J, Duplat D, Milet C, Lopez E, Bedouet L. J Biomed Mat Res A. 2007;85A:487–497. doi: 10.1002/jbm.a.31553. [DOI] [PubMed] [Google Scholar]
- 32.Celetti A, Testa D, Staibano S, Merolla F, Guarino V, Castellone MD, Iovine R, Mansueto G, Somma P, De Rosa G, Galli V, Melillo RM, Santoro M. Clin Cancer Res. 2005;11:8019–1027. doi: 10.1158/1078-0432.CCR-05-0641. [DOI] [PubMed] [Google Scholar]
- 33.Pfaffl MW. Nucl Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Somasundaran P. Encyclopedia of Surface and Colloid Science. CRC Press; Boca Raton, FL: 2004. p. 567. [Google Scholar]
- 35.Fleet ME. Front Biosci (Elite Ed) 2013;5:643–652. doi: 10.2741/e645. [DOI] [PubMed] [Google Scholar]
- 36.LeGeros RZ, Tung MS. Caries Res. 1983;17:419–429. doi: 10.1159/000260696. [DOI] [PubMed] [Google Scholar]
- 37.Baig AA, Fox JL, Young RA, Wang Z, Hsu J, Higuchi WI, Chhettry A, Zhuang H, Otsuka M. Calcif Tissue Int. 1999;64:437–449. doi: 10.1007/pl00005826. [DOI] [PubMed] [Google Scholar]
- 38.Tang R, Henneman ZJ, Nancollas GH. J Crystal Growth. 2003;249:614–624. [Google Scholar]
- 39.Marković S, Veselinović Lj, Lukić M, Karanović Lj, Bračko I, Ignjatović N, Uskoković D. Biomed Mat. 2011;6:045005. doi: 10.1088/1748-6041/6/4/045005. [DOI] [PubMed] [Google Scholar]
- 40.Onuma K, Ito A. Chem Mat. 1998;10:3346–3351. [Google Scholar]
- 41.Dorozhkin SV. J Mat Sci: Mat Med. 2007;18:363–366. doi: 10.1007/s10856-006-0701-x. [DOI] [PubMed] [Google Scholar]
- 42.Li D, Nielsen MH, Lee JR, Frandsen C, Banfield JF, De Yoreo JJ. Science. 2012;336:1014–1018. doi: 10.1126/science.1219643. [DOI] [PubMed] [Google Scholar]
- 43.Petrone C, Hall G, Langman M, Filiaggi MJ. Acta Biomater. 2008;4:403–413. doi: 10.1016/j.actbio.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Itokazu M, Yang W, Aoki T, Ohara A, Kato N. Biomater. 1998;19:817–819. doi: 10.1016/s0142-9612(97)00237-8. [DOI] [PubMed] [Google Scholar]
- 45.Rauschmann MA, Wichelhaus TA, Stirnal V, Dingeldein E, Zichner L, Schnettler R, Alt V. Biomater. 2005;26:2677–2684. doi: 10.1016/j.biomaterials.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 46.Okada M, Furuzono T. J Colloid Interface Sci. 2011;360:457–462. doi: 10.1016/j.jcis.2011.04.068. [DOI] [PubMed] [Google Scholar]
- 47.Uehira M, Okada M, Takeda S, Matsumoto N. Appl Surf Sci. 2013;287:195–202. [Google Scholar]
- 48.Ahmed S, Meghji S, Williams RJ, Henderson B, Brock JH, Nair SP. Infect Immun. 2001;69:2872–2877. doi: 10.1128/IAI.69.5.2872-2877.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellington JK, Reilly SS, Ramp WK, Smeltzer MS, Kellam JF, Hudson MC. Microb Pathog. 1999;26:317–323. doi: 10.1006/mpat.1999.0272. [DOI] [PubMed] [Google Scholar]
- 50.Koziel J, Maciag-Gudowska A, Mikolajczyk T, Bzowska M, Sturdevant DE, Whitney AR, Shaw LN, DeLeo FR, Potempa J. PLoS One. 2008;4:e5210. doi: 10.1371/journal.pone.0005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaarbo M, Ager-Wick E, Osenbroch PO, Kilander A, Skinnes R, Muller F, Eide L. Mitochondrion. 2011;11:935–945. doi: 10.1016/j.mito.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 52.Liebert G, Pinkert H, Fechner F, Gaunitz I, Schäfer P, Seibel C, Claus S, Chey S, Heinrich M, Reins B, Richardt S. J Virol. 2011;85:3881–3892. doi: 10.1128/JVI.02492-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tunholi-Alves VM, Tunholi VM, Pinheiro J, Thiengo SC. Exp Parasitol. 2012;131:143–147. doi: 10.1016/j.exppara.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Li C. J Cell Death. 2009;2:41–44. [PMC free article] [PubMed] [Google Scholar]
- 55.Alexander EH, Rivera FA, Marriott I, Anguita J, Bost KL. BMC Microbiol. 2003;2:3–5. doi: 10.1186/1471-2180-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mempel M, Schopp C, Hojka M, Fesq H, Weidinger S, Schaller M, Korting HC, Ring J, Abeck D. Br J Dermatol. 2002;146:943–951. doi: 10.1046/j.1365-2133.2002.04752.x. [DOI] [PubMed] [Google Scholar]
- 57.Kahl BC, Goulian M, Wamel Wv, Herrmann M, Simon SM, Kaplan G, Peters G, Cheung AL. Infect Immun. 2000;68:5385–5392. doi: 10.1128/iai.68.9.5385-5392.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Menzies BE, Kourteva I. Infect Immun. 1998;66:5994–5998. doi: 10.1128/iai.66.12.5994-5998.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murai M, Sakurada J, Seki K, Shinji H, Hirota Y, Masuda S. Microbiol Immunol. 1999;43:653–661. doi: 10.1111/j.1348-0421.1999.tb02453.x. [DOI] [PubMed] [Google Scholar]
- 60.Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, Golda A, Maciag-Gudowska A, Brix K, Shaw L, Foster T, Potempa J. PLoS One. 2008;3:e1409. doi: 10.1371/journal.pone.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanses F, Kopp A, Bala M, Buechler C, Falk W, Salzberger B, Schaggler A. Endocrinology. 2011;152:4148–4157. doi: 10.1210/en.2011-0103. [DOI] [PubMed] [Google Scholar]
- 62.Hess DJ, Henry-Stanley MJ, Erickson EA, Weiss CL. J Surg Res. 2003;114:42–49. doi: 10.1016/s0022-4804(03)00314-7. [DOI] [PubMed] [Google Scholar]
- 63.Uskoković V, Uskoković DP. J Biomed Mat Res B: Appl Biomat. 2011;96B:152–191. doi: 10.1002/jbm.b.31746. [DOI] [PubMed] [Google Scholar]
- 64.Bisht S, Bhakta G, Mitra S, Maitra A. Int J Pharm. 2005;288:157–168. doi: 10.1016/j.ijpharm.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 65.Zhang M, Kataoka K. Nano Today. 2009;4:508–517. [Google Scholar]
- 66.Epple M, Ganesan K, Heumann R, Klesing J, Kovtun A, Neumann S, Sokolova V. J Mat Chem. 2010;20:18–23. [Google Scholar]
- 67.Kakizawa Y, Miyata K, Furukawa S, Kataoka K. Adv Mat. 2004;16:699–702. [Google Scholar]
- 68.Graham FL, van der Eb AJ. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 69.Zhang XQ, Chen M, Lam R, Xu X, Osawa E, Ho D. ACS Nano. 2009;3:2609–2616. doi: 10.1021/nn900865g. [DOI] [PubMed] [Google Scholar]
- 70.Chow EK, Zhang XQ, Chen M, Lam R, Robinson E, Huang H, Schaffer D, Osawa E, Goga A, Ho D. Sci Transl Med. 2011;3:73ra21. doi: 10.1126/scitranslmed.3001713. [DOI] [PubMed] [Google Scholar]
- 71.Yan Y, Johnston AP, Dodds SJ, Kamphuis MM, Freguson C, Parton RG, Nice EC, Heath JK, Caruso F. ACS Nano. 2010;4:2928–2936. doi: 10.1021/nn100173h. [DOI] [PubMed] [Google Scholar]
- 72.Wang KW, Zhou LZ, Sun Y, Wu GJ, Gu HC, Duan YR, Chen F, Zhu YJ. J Mater Chem. 2010;20:1161–1166. [Google Scholar]
- 73.Spižek J, Novotna J, Rezanka T. Adv Appl Microbiol. 2004;56:121–154. doi: 10.1016/S0065-2164(04)56004-5. [DOI] [PubMed] [Google Scholar]
- 74.Jevon M, Guo C, Ma B, Mordan N, Nair SP, Harris M, Henderson B, Bentley G, Meghji S. Infect Immun. 1999;67:2677–2681. doi: 10.1128/iai.67.5.2677-2681.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Balasundaram G, Sato M, Webster TJ. Biomater. 2006;27:2798–2805. doi: 10.1016/j.biomaterials.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 76.Webster TJ, Ergun C, Doremus RH, Siegel RW, Bizios R. Biomater. 2000;21:1803–1810. doi: 10.1016/s0142-9612(00)00075-2. [DOI] [PubMed] [Google Scholar]
- 77.Webster TJ, Ergun C, Doremus RH, Siegel RW, Bizios R. J Biomed Mater Res. 2000;51:475–483. doi: 10.1002/1097-4636(20000905)51:3<475::aid-jbm23>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 78.Motskin M, Wright DM, Muller K, Kyle N, Gard TG, Porter AE, Skepper JN. Biomater. 2009;30:3307–3317. doi: 10.1016/j.biomaterials.2009.02.044. [DOI] [PubMed] [Google Scholar]
- 79.Yu SP, Canzoniero LMT, Choi DW. Curr Opin Cell Biol. 2011;13:405–411. doi: 10.1016/s0955-0674(00)00228-3. [DOI] [PubMed] [Google Scholar]
- 80.Hauschka P, Lian J, Cole D, Gundberg C. Physiol Rev. 1989;69:990–1047. doi: 10.1152/physrev.1989.69.3.990. [DOI] [PubMed] [Google Scholar]
- 81.Claro T, Widaa A, O’Seaghdha M, Miajlovic H, Foster TJ, O’Brien FJ, Kerrigan SW. PLoS ONE. 2011;6:e18748. doi: 10.1371/journal.pone.0018748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Bruyn JR, Goiko M, Mozaffari M, Bator D, Dauphinee RL, Liao Y, Flemming RL, Bramble MS, Hunter GK, Goldberg HA. PLoS One. 2013;8:e56764. doi: 10.1371/journal.pone.0056764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Regan A, Berman JS. Int J Exp Pathol. 2000;81:373–390. doi: 10.1046/j.1365-2613.2000.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chambers AF, Wilson SM, Kerkvliet N, O’Malley FP, Harris JF, Casson AG. Lung Cancer. 1996;15:311–323. doi: 10.1016/0169-5002(95)00595-1. [DOI] [PubMed] [Google Scholar]
- 85.Coppola D, Szabo M, Boulware D, Muraca P, Alsarraj M, Chambers AF, Yeatman TJ. Clin Cancer Res. 2004;10:184–190. doi: 10.1158/1078-0432.ccr-1405-2. [DOI] [PubMed] [Google Scholar]
- 86.Huang W, Carlsen B, Rudkin G, Berry M, Ishida K, Yamaguchi DT, Miller TA. Bone. 2004;34:799–808. doi: 10.1016/j.bone.2003.11.027. [DOI] [PubMed] [Google Scholar]