

Abstract
Objectives
This study sought to examine the effect of oral metformin (Mf) therapy on endothelialization in the setting of drug-eluting stents (DES).
Background
Mf is a commonly used therapy in diabetic patients receiving DES. Mf and locally eluted mammalian target of rapamycin (mTOR) inhibitors used in DES have convergent molecular signaling; however, the impact of this drug interaction on stent endothelialization is unknown.
Methods
We examined human endothelial aortic cells (HAECs) and a rabbit model of stenting to determine points on molecular convergence between these 2 agents and their impact on stent endothelialization.
Results
Western blotting of HAECs treated with Mf and the mTOR inhibitor sirolimus and 14-day rabbit iliacs treated with the combination of zotarolimus-eluting stents (ZES) and oral Mf demonstrated greater inhibition of S6 kinase (S6K), a downstream effector of mTOR complex 1, than either treatment alone. HAEC proliferation was significantly inhibited by Mf or sirolimus treatments alone and further reduced when they were combined. Knockdown of S6K via short interfering RNA in HAECs impaired cell proliferation via a cyclin D1–dependent mechanism, whereas its overexpression rescued the antiproliferative effects of both agents. Last, endothelialization and endothelial cell proliferation at 14 days were assessed in rabbits receiving ZES or bare-metal stents and Mf or placebo by scanning electron microscopy and bromodeoxyuridine/CD31 labeling, respectively. Both endpoints were inhibited by ZES treatment alone and were further reduced by the combination of Mf and ZES.
Conclusions
Significant convergence of signaling occurs between Mf and locally delivered mTOR inhibitors at S6K. This further impairs endothelial recovery/proliferation via an S6K-dependent mechanism. Patients receiving Mf in combination with stents that elute mTOR inhibitors are potentially at increased risk of delayed endothelial healing and stent thrombosis.
Keywords: cell proliferation, drug-eluting stents, endothelium, metformin, S6K
Coronary artery disease is a leading cause of death and disability in patients with diabetes. Treatment strategies aimed at reducing events in diabetic patients have included both optimal medical therapy and catheter-based percutaneous coronary intervention with drug-eluting stents (DES) (1).
Although DES have dramatically reduced restenosis rates, their use has been associated with an increased risk of late stent thrombosis, especially in diabetic patients (2-5). Mechanisms behind this phenomenon remain unknown; however, the primary substrate underlying these cases is lack of endothelialization (6,7). The majority of DES in clinical use elute inhibitors of the mammalian target of rapamycin (mTOR), a phosphatidylinositol kinase-related family of serine/threonine kinase, which include sirolimus [SRL], everolimus [EVL] and zotarolimus (8,9). mTOR exists in 2 distinct protein complexes with specific binding partners, including raptor in mTOR complex 1 (mTORC1) and rictor in mTOR complex 2. The best known mTORC1 substrates are S6 kinase (S6K) and eIF4E-binding proteins (4E-BP), whereas mTOR complex 2 phosphorylates the hydrophobic motif of Akt. The delay in endothelialization seen in stents that elute mTOR inhibitors suggests a key role for mTOR and its downstream effectors in distinct processes critical for endothelial recovery after arterial injury. Of these processes, cell proliferation may be the most essential and likely the exclusive domain of S6K (10).
Because mTOR also integrates signals from upstream pathways such as insulin and senses cellular nutrient status and energy levels (11), it is affected by antidiabetic medications that alter insulin content and nutrient/energy levels (12). We previously showed the interaction of the oral peroxisome proliferator–activated receptor γ agonist rosiglitazone with locally eluted SRL further delays stent healing due to convergence of molecular signaling (13).
Metformin (Mf), a biguinide, is the most widely used oral diabetic agent and inhibits mitochondrial respiratory chain complex I, altering the adenosine monophosphate–to–adenosine triphosphate ratio, thus resulting in the activation of 5′-adenosine monophosphate–activated protein kinase (AMPK) (14,15). AMPK activation by Mf leads to the inhibition of mTORC1 (16) and its downstream effectors (i.e., S6K). Despite its clinical relevance, it remains uncertain how this potential convergence in molecular signaling between locally eluting mTOR inhibitors and systemic Mf could affect vascular endothelial recovery after stent placement.
To test our hypothesis that Mf in combination with locally eluted mTOR inhibitors results in a significant delay in endothelial recovery due to further modulation of mTOR signaling cascades, we examined points of molecular convergence between these 2 agents in cultured endothelial cells and explored the consequences of this interaction on endothelial cell proliferation, an essential cellular function needed for re-endothelialization. We then modeled the effects of this interaction on stent endothelialization and endothelial proliferation in vivo in rabbits receiving oral Mf or placebo in combination with zotarolimus-eluting stents (ZES) or bare-metal stents (BMS).
Methods
Cell culture, immunoblotting, quantification of cell proliferation/viability and apoptosis, quantitative polymerase chain reaction, plasmid and short interfering RNA transfection, and lentiviral transduction
Human aortic endothelial cells (HAECs) (Cell Applications, San Diego, California) were maintained in endothelial cell growth medium, and passages 2 and 8 were used for all experiments unless otherwise specified. Short interfering RNA target sequences are provided (Online Table 1). Further experimental details are available in the Online Appendix.
Rabbit model of iliac artery stenting, assessment of endothelialization, and endothelial cell proliferation
New Zealand white male rabbits were given Mf (100 mg/kg/day orally), the dose based on body surface area calculations of therapeutic human dosing (2 g/day), stents were placed and removed 14 days post-procedure as previously described (17). En face scanning electron microscopy was used to assess stent endothelialization. Bromodeoxyuridine was given 18 and 12 h before removal, and immunostaining of bromodeoxyuridine was used to assess proliferation on stent surfaces. See the Online Appendix for further details.
Statistical analysis
Statistical analysis was performed using JMP Pro version 10 (SAS Institute, Cary, North Carolina). All data were expressed as mean ± SD. Differences were evaluated using an unpaired Student t test between 2 groups. For multiple group comparisons a 1- or 2-way analysis of variance was used. If the variance ratio test (F test) was significant, a more detailed post hoc analysis of differences between groups was made using a Tukey-Kramer honest significance difference test. A p value <0.05 was considered statistically significant. (See the Online Appendix for further details.)
Results
To determine how Mf interacts with mTOR inhibitors at the vascular endothelium, we examined their effects, alone and in combination with HAECs, on the activity of downstream effectors of mTOR signaling. First, to confirm Mf-activated AMPK in HAECs, we demonstrated that the Mf-treated HAECs increased AMPK phosphorylation compared with controls, whereas no effect was seen with SRL alone (Fig. 1A). SRL groups (i.e., control and Mf) showed inhibition of all downstream targets of mTOR (i.e., S6K, 4E-BP, and Akt); however, S6K phosphorylation was differentially inhibited with SRL-Mf compared with SRL-control (Fig. 1B). Mf alone inhibited S6K but did not significantly inhibit Akt or 4E-BP activity (Figs. 1C and 1D). Collectively, the significant observed interaction that occurs between Mf and SRL was at S6K.
Figure 1. Downstream Products of Mammalian Target of Rapamycin Complex 1, But Not Complex 2, Are Inhibited by Mf in Human Aortic Endothelial Cells.

Western blotting was conducted for phospho-5′-adenosine monophosphate–activated protein kinase (AMPK) (A), phospho-S6 kinase (S6K) (B), phospho-eIF4E binding protein (4E-BP) (C), and phospho-Akt (D) and densitometry values calculated. Densitometry was normalized to the total protein product (n = 4 per group). Cont = control; Mf = metformin; SRL = sirolimus.
Previous studies suggested a dominant role for mTORC1 and S6K in cell-cycle progression and proliferation (18,19). Given the significant overlap in molecular signaling observed between Mf and SRL at S6K, we examined their effects on processes likely involved in endothelialization. HAEC proliferation was observed to decrease in a dose-dependent manner with Mf, SRL, and the SRL analog EVL, respectively (Figs. 2A and 2B). The concentration of Mf (30 mmol/l) that inhibited 50% of proliferation matched serum levels in patients receiving Mf therapy (20). We then compared the agents, in combination, both in normoglycemic (5 mmol/l) and hyperglycemic (30 mmol/l) conditions, respectively. The combination of SRL with Mf (SRL-Mf) resulted in the greatest inhibition of cell proliferation while there was also a significant inhibition with the combination of EVL with Mf (EVL-Mf) compared with either agent alone, regardless of the glycemic condition (Figs. 2C and 2D).
Figure 2. Endothelial Cell Proliferation is Differentially Inhibited by Mf in Combination With Mammalian Target of Rapamycin Inhibition Regardless of Glycemic Environment.

(A) Human aortic endothelial cells (HAECs) were incubated in the presence of increasing doses of Mf (0 to 50 mmol/l) for 24 h, and cell proliferation was measured by bromodeoxyuridine (BrdU) cell proliferation assay (n = 4 per group). (B) HAECs were incubated in the presence of increasing doses of SRL (0 to 500 nmol/l) and everolimus (EVL) (0 to 500 nmol/l) for 24 h, and proliferation was measured (n = 4 per group). (C, D) HAECs were incubated in normoglycemic (NG) (5 mmol/l glucose) and hyperglycemic (HG) (30 mmol/l) environments, respectively, for 48 h followed by exposure to Mf (30 mmol/l) and SRL (500 nmol/l)/EVL (500 nmol/l) alone or in combination for 24 h, and proliferation was measured (n = 4 per group). Abbreviations as in Figure 1.
Cell viability under each treatment was also assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and was decreased in a dose-dependent manner by Mf and SRL/EVL, in addition to being differentially decreased with the combination of Mf and SRL/EVL compared with either agent alone (Online Figs. 1A and 1B).
Apoptosis was assessed using a combination of annexin V and propidium iodide staining and were seen to increase in a dose-dependent manner with all agents; however, although the combination of EVL and Mf did significantly increase apoptosis, the combination of SRL and Mf did not (Online Figs. 1C and 1D). This suggests that the interaction of Mf with SRL/EVL acts predominantly on proliferation/viability rather than on apoptosis.
Effect of Mf on endothelial cell proliferation is AMPK dependent
To observe the effect of AMPK activation, selective AMPK activators 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranosyl 3′,5′ cyclic monophosphate (AICAR) and A-769662, were used (21,22). Dose-dependent inhibition of proliferation with AICAR or A-769662 was observed with each agent alone in addition to further suppression of proliferation when combined with SRL (Figs. 3A and 3B). Transfection with short interfering RNA (siRNA) for AMPK (α1/2), rescued HAEC proliferative ability despite treatment with increasing doses of Mf, suggesting that Mf requires AMPK for its antiproliferative effect (Fig. 3C, Online Fig. 2). Transfection with short interfering for AMPK (a1/2) rescued HAEC proliferative ability despite treatment with increasing doses of Mf, suggesting that Mf requires AMPK for its antiproliferative effect.
Figure 3. Inhibition of Cell Proliferation by Mf Is AMPK Dependent.

(A, B) Increasing doses of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranosyl 3′,5′ cyclic monophosphate (AICAR) (0 to 10 mmol/l), an AMP mimetic, and A-769662 (0 to 1,000 μmol/l), a selective AMPK activator, were incubated with HAECs for 24 h, and cell proliferation was measured (n = 4 per group). (C) HAECs were incubated with AICAR (2 mmol/l), A-769662 (100 μmol/l) and SRL (500 nmol/l) alone or in combination for 24 h, and proliferation was measured (n = 4 per group). (D) HAECs were transfected with short interfering RNA (siRNA) for AMPK α1/2 or nontargeting, scrambled siRNA (Scr) and were subsequently treated with increasing doses of Mf (0 to 30 mmol/l) for 24 h. Cell proliferation was measured at 24 h with a BrdU assay (n = 4 per group). Abbreviations as in Figures 1 and 2.
Effect of Mf on endothelial recovery and proliferation is dependent on the mTORC1/S6K axis
Given the inhibition of S6K and cell proliferation observed with the combination of Mf and ZES, we decided to examine whether suppression of mTORC1 alone could affect endothelial stent-strut coverage using a novel in vitro stent strut assay. HAECs were transduced with short hairpin RNA (shRNA) for raptor, a vital protein component of mTORC1, or scrambled shRNA, and allowed to grow on an in vitro stent-strut coverage model for 2 weeks. Expression of platelet endothelial cell adhesion molecule 1 (PECAM1)/CD31, a commonly used endothelial marker, was measured from the stent surface. The raptor shRNA group demonstrated a significant decrease in PECAM1/CD31 expression compared with scrambled shRNA-treated cells (Fig. 4A, Online Fig. 2). Stents were also immunostained for PECAM1/CD31 with representative examples from each group shown (Fig. 4A).
Figure 4. Genetic Knockdown of Mammalian Target of Rapamycin Complex 1 and S6K, Its Downstream Effector, Inhibits Stent Endothelialization and Endothelial Cell Proliferation, Respectively.

(A) HAECs were transduced, with raptor/mammalian target of rapamycin complex 1 short hairpin RNA (shRNA) or nontargeting scrambled shRNA (Scr), and allowed to grow on an in vitro stent strut coverage model for 2 weeks. Stents were immunostained with anti-CD31/platelet endothelial cell adhesion molecule 1 antibody, and endothelial coverage was assessed with confocal microscopy and on-cell enzyme-linked immunosorbent assay, normalized to Scr (n = 4 per group). Representative confocal images are shown at ×20 magnification. HAECs were transfected with siRNA for S6K1, 4E-BP3, or nontargeting, scramble siRNA (Scr). Cell proliferation was measured with a BrdU cell-proliferation assay at 24 h (n = 4 per group, inset). (B to D) HAECs overexpressing wild-type (WT) or a sirolimus-resistant mutant (MT) S6K were treated with Mf (0 to 30 mmol/l) or SRL (0 to 10 nmol/l) or EVL (0 to 10 nmol/l), respectively, for 24 h and cell proliferation measured with a BrdU cell-proliferation assay (n = 4 per group). Abbreviations as in Figures 1 through 3.
To assess which downstream effector of mTORC1 (i.e., S6K or 4E-BP) is critical for proliferation, HAECs were transfected with siRNA for S6K1 and 4E-BP3 to target their major catalytic isoforms (23), respectively, or nontargeting, scrambled siRNA (Scr). Proliferation was significantly decreased in HAECs transfected with siRNA for S6K1 compared with 4E-BP3 (Fig. 4A). To determine whether the antiproliferative effect of Mf is mediated via S6K, we overexpressed either a constitutively active wild-type S6K or an SRL-resistant mutant via plasmid transfection in HAECs and then treated cells with increasing doses of Mf (Fig. 4B, Online Fig. 2). Overexpression of either form of S6K mitigated the antiproliferative effect of Mf. Overexpression of S6K was repeated with increasing doses of SRL/EVL confirming that their inhibition of cell proliferation occurs via S6K (Figs. 4C and 4D) (18).
Endothelial cell proliferation by S6K is mediated via cyclin D1 regulation
Control of endothelial cell proliferation by S6K is thought to be mediated via transcriptional control of cyclin D1, a member of the cyclin protein family that is involved in cell-cycle G1/S phase progression (10). HAECs treated with Mf and SRL, alone and in combination, demonstrated decreased protein expression of cyclin D1, although the SRL-Mf group inhibited cyclin D1 to the greatest extent (Fig. 5A). To demonstrate a direct connection between S6K inhibition and cyclin D1 mRNA regulation, we used an siRNA against S6K1. Knockdown of S6K1 significantly decreased cyclin D1 mRNA expression (Fig. 5B). Last, overexpression of cyclin D1 via plasmid transfection mitigated the effects of increasing doses of both Mf and SRL on cell proliferation (Figs. 5C and 5D).
Figure 5. S6K Controls Proliferation by Regulating Cyclin D1 Expression.

(A) HAECs were incubated with SRL (500 nmol/l) or Mf (30 mmol/l), alone and in combination, and Western blots performed for cyclin D1. Relative expression normalized to beta-actin levels were measured by densitometry (n = 4 per group). (B) HAECs were transfected with siRNA for S6K1 and HAECs were transfected with siRNA for S6K1 and nontargeting, scrambled siRNA (Scr). Cyclin D1 mRNA expression measured by quantitative polymerase chain reaction (n = 4 per group). Cyclin D1 mRNA expression measured by quantitative polymerase chain reaction nontargeting, scrambled siRNA (Scr) (n = 4 per group). (C, D) Cyclin D1 was overexpressed in HAECs via plasmid transfection and exposed to increasing doses of Mf (0 to 30 mmol/l) or SRL (0 to 10 nmol/l), respectively, with cell proliferation measured at 24 h (n = 4 per group). Abbreviations as in Figures 1 through 3.
Effect of Mf on the mTOR signaling pathway in rabbits receiving DES and oral Mf
Given the effects of Mf on the mTOR signaling pathway seen in HAECs in vitro, we examined whether these same interactions occur in vivo in rabbits receiving BMS or DES (ZES, ENDEAVOR stent, Medtronic, Minneapolis, Minnesota) in the presence of oral Mf (100 mg/kg/day) or placebo for 14 days (Online Fig. 3). Zotarolimus, like EVL, is a semisynthetic analogue of sirolimus designed for increased lipophilicity but preserving its specificity for mTOR inhibition (9).
As seen in vitro, increased AMPK activation in the arterial wall of all Mf groups was observed compared with controls (Fig. 6A). Next, we examined S6K/4E-BP and Akt phosphorylation, downstream effectors of mTORC1 and mTOR complex 2, respectively. S6K was inhibited to the greatest extent in the ZES-Mf group compared with all other groups (Fig. 6B). Interestingly, the BMS-Mf group demonstrated reduced S6K phosphorylation; however, no effect on 4E-BP or Akt was seen (Figs. 6C and 6D). All ZES groups (i.e., control and Mf) showed inhibition of Akt and S6K phosphorylation but not 4E-BP. The latter finding is consistent with the known milder and transient effects of mTOR inhibitors on 4E-BP (24). These data confirm that the significant overlap in mTOR signaling at S6K in the arterial wall in vivo in animals receiving oral Mf and ZES was seen in vitro with SRL and Mf.
Figure 6. Downstream Effectors of Mammalian Target of Rapamycin Complex 1, But Not Complex 2, Are Differentially Inhibited in Animals Receiving ZES and Oral Mf.

Representative Western blots and densitometry of 14-day rabbit iliac arteries stented with a bare-metal stent (BMS) or zotarolimus-eluting stent (ZES) in the presence or absence of Mf (100 mg/kg/day) for phospho-AMPK (A), an upstream inhibitor of mammalian target of rapamycin complex 1, phospho-S6K (B) and phospho-4EBP (C), downstream effectors of mammalian target of rapamycin complex 1, and phospho-Akt (D), a downstream effector of mammalian target of rapamycin complex 2. Densitometry was normalized to the total protein product (n = 3 per group). Abbreviations as in Figure 1.
Oral Mf further impairs endothelial cell proliferation and coverage and in rabbits receiving ZES.
A combination of PECAM1/CD31 for endothelial staining and bromodeoxyuridine labeling of proliferating cells was used to localize proliferating endothelial cells on the stent surface in our animal model by confocal microscopy (Fig. 7). The ZES-control group had significantly less proliferation compared with BMS groups, consistent with the antiproliferative effects of mTOR inhibition. Similar to the in vitro findings, there was significantly less proliferation in the combination ZES-Mf group compared with BMS groups (BMS-control and BMS-Mf) and numerically less than the ZES-control group (Table 1).
Figure 7. Stent Surface Endothelial Cell Proliferation Is Impaired to the Greatest Extent in Rabbits Treated With ZES-Mf.
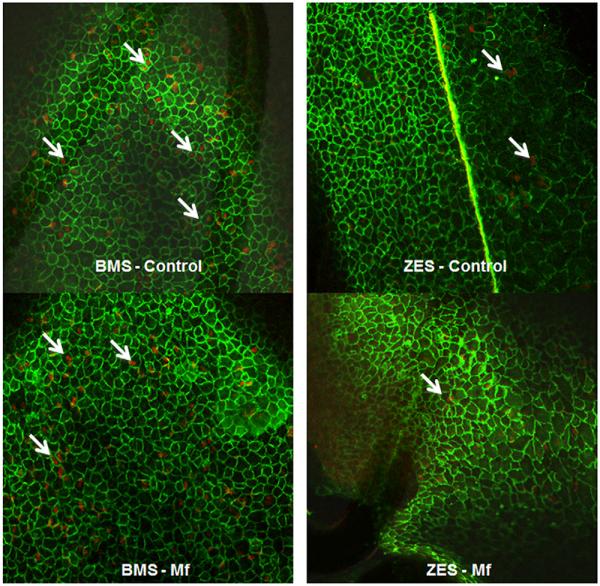
Confocal microscopy was conducted on 14-day whole-mount stented (BMS or ZES) arterial segments in the presence or absence of Mf (100 mg/kg/day) using dual immunofluorescence for platelet endothelial cell adhesion molecule 1/CD31 (green) and BrdU (red). Representative figures are shown at ×100 magnification. White arrows indicate examples of proliferating endothelial cells. Abbreviations as in Figures 1 and 6.
Table 1.
Quantification of (14)-Day Endothelial Cell Proliferation (%)
| p Value |
||||
|---|---|---|---|---|
| Group | %, Mean ± SD | vs. BMS Control |
vs. BMS Mf |
vs. ZES Control |
| BMS-control (n = 3) | 48.3 ± 6.8 | |||
| BMS-Mf (n = 3) | 43.0 ± 7.9 | 0.6629 | ||
| ZES-control (n = 3) | 20.1 ± 0.9 | 0.0011* | 0.0043* | |
| ZES-Mf (n = 3) | 11.6 ± 3.9 | 0.0002* | 0.0006* | 0.3112 |
Statistically significant (p < 0.05).
BMS = bare-metal stent(s); Mf = metformin; ZES = zotarolimus-eluting stent(s).
Last, we explored the effects of this interaction on endothelial coverage after stent placement by scanning electron microscopy (Fig. 8). ZES-control significantly impaired endothelial coverage compared with BMS-control, as previously reported (17). Oral Mf decreased endothelial coverage in BMS (BMS-Mf) compared with BMS-control; however, this was not statistically significant. Conversely, there was a significant decrease in coverage with ZES and systemic Mf treatment (ZESMf) compared with ZES placement alone (ZES-control) as well as BMS (BMS-control and BMS-Mf) (Table 2).
Figure 8. Endothelial Stent Strut Coverage Is Significantly Impaired in ZES in Combination With Oral Mf in the Rabbit Iliac Model.

Representative scanning electron micrographs (×15 magnification) of 14-day BMS and ZES in the presence and absence of oral Mf (100 mg/kg/day) treatment. Note the significantly decreased endothelial strut coverage in the ZES-Mf group versus all others.
Table 2.
Quantification of (14)-Day Endothelial Coverage by Scanning Electron Microscopy (%)
| p Value |
||||
|---|---|---|---|---|
| Group | %, Mean ± SD | vs. BMS Control |
vs. BMS Mf |
vs. ZES Control |
| BMS-control (n = 6) | 79.7 ± 18.3 | |||
| BMS-Mf (n = 5) | 59.0 ± 18.2 | 0.1979 | ||
| ZES-control (n = 5) | 43.2 ± 20.5 | 0.0087* | 0.4468 | |
| ZES-Mf (n = 6) | 13.3 ± 6.4 | <0.0001* | 0.0012* | 0.0359* |
Statistically significant (p < 0.05).
Abbreviations as in Table 1.
Discussion
Although DES using mTOR inhibitors prevent restenosis, they also result in delayed stent endothelialization, a finding that implicates mTOR and its downstream effectors as critical in this process. Although local drug delivery minimizes systemic toxicity, it does not eliminate the potential for drug–drug interactions with systemic medications. Clinical studies of sirolimus and newer generation everolimus-eluting stents suggest differential effects in nondiabetic and diabetic patients, with the latter having significantly higher rates of late stent thrombosis (2-5), a phenomenon related to delayed stent endothelialization (6,7). This study indicates that Mf interacts with mTOR signaling and when combined with locally eluted mTOR inhibitors resulted in further suppression of endothelial proliferation both in our in vivo and in vitro models. A convergence in molecular signaling between these 2 agents was found at S6K via an AMPK-dependent mechanism, which our data demonstrate is critical for endothelial proliferation via control of cyclin D1. These findings suggest that patients receiving oral Mf and DES that elute mTOR inhibitors may be at increased risk of delayed stent healing.
Previous studies showed important roles for Akt and 4E-BPs in cell proliferation (23,25). We found that the effect of knockdown of 4E-BP and Akt (data not shown) on HAEC proliferation was relatively little compared with S6K. The antiproliferative effect of Mf has been reported in multiple cell types, especially in response to injury or malignancy, with the suppressed downstream mediators appearing to be cell type and context specific (10,26-29). Our data, however, suggest that S6K plays a dominant role in endothelial proliferation.
Although AMPK activation and mTOR inhibition may affect apoptosis individually, their combined effect was variable, and both were a function of their action on Akt activity and environmental context (30-33). Although SRL/EVL proapoptotic action is consistent with its suppression of Akt, Metformin, via activation of LKB1/AMPK, may also promote proapoptosis-mediated pathways (34). Eventual feedback activation of Akt, via combined mTORC1 suppression by Mf and SRL/EVL may occur, leading to variable effects on apoptosis when agents are combined (Online Fig. 4) (35).
Clinical relevance
Mf is the most widely used antidiabetes medication in the world and is associated with improved endothelial function in diabetic patients (36). We suggest, however, that Mf may have a direct deleterious effect on endothelial recovery after stent-induced injury because of its effects on proliferation in the setting of DES that elute mTOR inhibitors. Because poor stent endothelialization has been shown as an underlying substrate for late stent thrombosis in both pathological and clinical studies, this study suggests that long-term Mf therapy may augment the potential risk of late in-stent thrombosis. In a pooled analysis from several randomized clinical trials of EVLeluting stents, Stone et al. (3) recently reported a differential clinical response to EVL-eluting stents in patients with and without diabetes with significantly increased stent thrombosis and myocardial infarction rates in diabetic patients not treated with insulin compared with those without diabetes at 2 years. Prospective studies are needed to determine whether the impairment of endothelial coverage occurs in patients treated with the combination of systemic Mf and local mTOR inhibition, corresponding to an increase risk of late in-stent thrombosis.
Study limitations
The use of a nonatherosclerotic, nondiabetic animal model of stenting may have underestimated the effect of Mf in combination with ZES on endothelial healing.
Conclusions
This is the first study to elucidate the effects of oral Mf on endothelial recovery in the setting of stent placement. Patients receiving Mf in combination with newer generation DES that use mTOR inhibitors are potentially at increased risk of delayed stent endothelialization and stent thrombosis.
Supplementary Material
Acknowledgment
This study was supported by the Carlyle Fraser Heart Center, CVPath Inc., American Heart Association and US NIH grant RO1 HL096970-01A. CVPath Institute has research grants from Medtronic CardioVascular, Abbott Vascular, Terumo Corporation, Atrium Medical, Boston Scientific, Cordis/Johnson & Johnson, OrbusNeich Medical, and Biosensors International. Dr. Narula is a consultant for N-terminus Research Laboratory, en-N-Tech and St. Jude Medical, Inc. and is on the advisory board for Philips Healthcare, GE HealthCare and Atkins Nutritional Inc. Dr. Virmani is a consultant for Medtronic CardioVascular, Abbott Vascular, Terumo Corporation, Atrium Medical, W.L. Gore, and Lutonix. Dr. Finn has sponsored research agreements with Medtronic CardioVascular and Boston Scientific and is on the advisory board of Medtronic CardioVascular. Dr. Habib is supported by an American Heart Association Postdoctoral Fellowship grant (Greater Southeast Affiliate). All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Neil Kleinman, MD, has served as Guest Editor for this paper.
The authors would like to thank the Emory University Core Laboratories for their assistance with flow cytometry.
Abbreviations and Acronyms
- 4E-BP
eIF4E binding protein
- AICAR
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranosyl 3′,5′ cyclic monophosphate
- AMPK
5′-adenosine monophosphate–activated protein kinase
- BMS
bare-metal stent(s)
- DES
drug-eluting stent(s)
- EVL
everolimus
- HAEC
human aortic endothelial cell
- Mf
metformin
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- PECAM1
platelet endothelial cell adhesion molecule 1
- S6K
S6 kinase
- shRNA
short hairpin RNA
- siRNA
short interfering RNA
- SRL
sirolimus
- ZES
zotarolimus-eluting stent(s)
Footnotes
REFERENCES
- 1.Frye RL, August P, Brooks MM, et al. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360:2503–15. doi: 10.1056/NEJMoa0805796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daemen J, Wenaweser P, Tsuchida K, et al. Early and late coronary stent thrombosis of sirolimus-eluting and paclitaxel-eluting stents in routine clinical practice: data from a large two-institutional cohort study. Lancet. 2007;369:667–78. doi: 10.1016/S0140-6736(07)60314-6. [DOI] [PubMed] [Google Scholar]
- 3.Stone GW, Kedhi E, Kereiakes DJ, et al. Differential clinical responses to everolimus-eluting and paclitaxel-eluting coronary stents in patients with and without diabetes mellitus. Circulation. 2011;124:893–900. doi: 10.1161/CIRCULATIONAHA.111.031070. [DOI] [PubMed] [Google Scholar]
- 4.Caixeta A, Leon MB, Lansky AJ, et al. 5-year clinical outcomes after sirolimus-eluting stent implantation insights from a patient-level pooled analysis of 4 randomized trials comparing sirolimuseluting stents with bare-metal stents. J Am Coll Cardiol. 2009;54:894–902. doi: 10.1016/j.jacc.2009.04.077. [DOI] [PubMed] [Google Scholar]
- 5.Kimura T, Morimoto T, Nakagawa Y, et al. Very late stent thrombosis and late target lesion revascularization after sirolimus-eluting stent implantation: five-year outcome of the j-Cypher Registry. Circulation. 2012;125:584–91. doi: 10.1161/CIRCULATIONAHA.111.046599. [DOI] [PubMed] [Google Scholar]
- 6.Finn AV, Joner M, Nakazawa G, et al. Pathological correlates of late drug-eluting stent thrombosis: strut coverage as a marker of endothelialization. Circulation. 2007;115:2435–41. doi: 10.1161/CIRCULATIONAHA.107.693739. [DOI] [PubMed] [Google Scholar]
- 7.Guagliumi G, Sirbu V, Musumeci G, et al. Examination of the in vivo mechanisms of late drug-eluting stent thrombosis findings from optical coherence tomography and intravascular ultrasound imaging. J Am Cardiol Intv. 2012;5:12–20. doi: 10.1016/j.jcin.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 8.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Chen YW, Smith ML, Sheets M, et al. Zotarolimus, a novel sirolimus analogue with potent anti-proliferative activity on coronary smooth muscle cells and reduced potential for systemic immunosuppression. J Cardiovasc Pharmacol. 2007;49:228–35. doi: 10.1097/FJC.0b013e3180325b0a. [DOI] [PubMed] [Google Scholar]
- 10.Espeillac C, Mitchell C, Celton-Morizur S, et al. S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. J Clin Invest. 2011;121:2821–32. doi: 10.1172/JCI44203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 12.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finn AV, John M, Nakazawa G, et al. Differential healing after sirolimus, paclitaxel, and bare metal stent placement in combination with peroxisome proliferator-activator receptor gamma agonists: requirement for mTOR/Akt2 in PPARgamma activation. Circ Res. 2009;105:1003–12. doi: 10.1161/CIRCRESAHA.109.200519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasaki H, Asanuma H, Fujita M, et al. Metformin prevents progression of heart failure in dogs: role of AMP-activated protein kinase. Circulation. 2009;119:2568–77. doi: 10.1161/CIRCULATIONAHA.108.798561. [DOI] [PubMed] [Google Scholar]
- 15.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joner M, Nakazawa G, Finn AV, et al. Endothelial cell recovery between comparator polymer-based drug-eluting stents. J Am Coll Cardiol. 2008;52:333–42. doi: 10.1016/j.jacc.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 18.Vinals F, Chambard JC, Pouyssegur J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J Biol Chem. 1999;274:26776–82. doi: 10.1074/jbc.274.38.26776. [DOI] [PubMed] [Google Scholar]
- 19.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biology. 2004;24:200–16. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50:81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 21.Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–16. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Peyton KJ, Liu XM, Yu Y, Yates B, Durante W. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J Pharmacol Exp Ther. 2012;342:827–34. doi: 10.1124/jpet.112.194712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowling RJ, Topisirovic I, Alain T, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–6. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–9. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fosbrink M, Niculescu F, Rus V, Shin ML, Rus H. C5b-9-induced endothelial cell proliferation and migration are dependent on Akt inactivation of forkhead transcription factor FOXO1. J Biol Chem. 2006;281:19009–18. doi: 10.1074/jbc.M602055200. [DOI] [PubMed] [Google Scholar]
- 26.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–12. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 27.Ben Sahra I, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang Y, Miskimins WK. Cell cycle arrest in metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8:88–96. doi: 10.4161/cc.8.1.7499. [DOI] [PubMed] [Google Scholar]
- 30.Dormond O, Madsen JC, Briscoe DM. The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J Biol Chem. 2007;282:23679–86. doi: 10.1074/jbc.M700563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res. 2009;105:114–27. doi: 10.1161/CIRCRESAHA.109.201590. [DOI] [PubMed] [Google Scholar]
- 32.Guo D, Chien S, Shyy JY. Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ Res. 2007;100:564–71. doi: 10.1161/01.RES.0000259561.23876.c5. [DOI] [PubMed] [Google Scholar]
- 33.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong D, Liu X, Khuri FR, Sun SY, Vertino PM, Zhou W. LKB1 is necessary for Akt-mediated phosphorylation of proapoptotic proteins. Cancer Res. 2008;68:7270–7. doi: 10.1158/0008-5472.CAN-08-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Yue P, Kim YA, Fu H, Khuri FR, Sun SY. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008;68:7409–18. doi: 10.1158/0008-5472.CAN-08-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mather KJ, Verma S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol. 2001;37:1344–50. doi: 10.1016/s0735-1097(01)01129-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.