

ABSTRACT
In veterinary medicine, hyperferritinemia is often observed in dogs with various diseases (e.g., histiocytic sarcoma and immune-mediated hemolytic anemia) without evidence of iron overload. The mechanism underlying hyperferritinemia development is not well understood. Anemia caused by inflammation is termed as anemia of chronic disease (ACD), and experimentally induced ACD is known to cause slight hyperferritinemia. However, almost all these studies were based on short-term acute inflammation. Hepcidin, a protein mainly produced by hepatocytes, is thought to be a key regulator in iron release from reticuloendothelial cells (RECs), and its expression is related to ACD. We hypothesized that in the case of long-term ACD, iron deposition in RECs increases through hepcidin, causing a diachronic increase in serum ferritin levels. In the present study, we used a canine model with repeated subcutaneous administration of turpentine oil every 3 days over a period of 42 days (15 injections) and induced long-term inflammatory conditions; furthermore, we evaluated the change in serum ferritin concentration. Hypoproliferative anemia, bone marrow iron deposition and hypoferremia, which are characteristic of ACD, were observed on administering the turpentine injections. Hepatic iron content, hepatic hepcidin mRNA expression and serum ferritin concentration increased during the early period after turpentine injection, but returned to normal levels later. These results show that experimentally induced long-term ACD caused hypoproliferative anemia without sustained increase in hepcidin expression and did not cause systemic iron overload. Thus, chronic inflammation may not contribute greatly to increase in hyperferritinemia.
Keywords: ACD, canine, chronic inflammation, ferritin, hepcidin
Ferritin is an iron storage protein that is ubiquitous in all mammals. It has an iron core within a 24-mer globular protein complex, consisting of heavy (H) and light (L) subunits with molecular masses of 21 and 19 kDa, respectively [17, 46]. Ferritin can accommodate 3,000–4,500 iron molecules and protects cells from reactive oxygen species [18]. While ferritin is found in all cells present in tissues in mammals, it is most abundant in the liver, spleen and bone marrow [19].
Ferritin synthesis is tightly regulated by iron at the translational level [41]. In mammals, including dogs, ferritin normally circulates in the serum at a relatively low concentration (<1 µg/ml), and the serum ferritin concentration is positively correlated with the level of iron stored in the body [19, 32]. The serum ferritin concentration increases in various disease states, such as iron overload, inflammatory diseases, liver damage and malignancies [19, 46]. In veterinary medicine, it has been reported that canine serum ferritin levels increase in histiocytic sarcoma, immune-mediated hemolytic anemia (IMHA) and lymphoma [12, 21, 29]. In addition, serum ferritin concentration is reported to be a useful serological marker of histiocytic sarcoma and IMHA in dogs [12]. It is likely that high concentrations of serum ferritin are due to increases in the level of storage iron, damage to the liver (which contains a high level of ferritin), inflammation (ferritin is an acute-phase protein) and direct release from tumors [46]. Despite much research interest in this phenomenon, the precise mechanism underlying hyperferritinemia is not well understood.
Inflammation associated with infections, neoplasia and immune-mediated diseases decreases the serum iron concentration and reduces erythropoiesis, thereby leading to anemia [44]. This condition is known as anemia of chronic disease (ACD). ACD, also known as anemia of inflammation, is commonly found in veterinary and human cases [4]. The mechanism underlying ACD has not yet been completely understood, but recently, hepcidin, an iron-regulated acute-phase protein that is composed of 25 amino acids, has been found to play an important role in ACD development. Hepcidin, a peptide that has antibacterial activity and is present in the liver, was discovered in 2000 [23]. Pigeon et al. reported that hepcidin mRNA expression increased when mice were given iron orally and that a similar change was generated by lipopolysaccharide (LPS) administration [34]. These findings strongly suggest that hepcidin is related with iron metabolism and is upregulated under inflammatory conditions. Reticuloendothelial cells (RECs) phagocytize aging red blood cells (RBC) and recycle iron from heme; RECs release iron into the blood through the iron exporter ferroportin on the cell surface [9, 10]. It has been reported that hepcidin regulates the functions of ferroportin, which is the only iron exporter in cells, and regulates the absorption of iron from the gastrointestinal tract and iron release from RECs. Hepcidin regulates the function of ferroportin and decreases iron release from and promotes iron deposition in RECs [16, 28]. Therefore, hepcidin is thought to play an important role in iron metabolism and ACD development.
We hypothesize that in the case of ACD induced by long-term inflammation, iron is continuously deposited in RECs by hepcidin and that ferritin synthesis increases in the cell, causing a diachronic increase in the serum ferritin level. Many previous studies have investigated experimentally induced ACD in animal models, and it is known that serum ferritin levels increase slightly with inflammation [36]. However, most of these experiments involved short-term observation and, to the authors’ knowledge, no study has evaluated changes in serum ferritin levels in the case of long-term inflammation. The purpose of this study was to determine whether abnormalities in iron metabolism due to chronic inflammation are involved in hyperferritinemia. Therefore, we established a canine ACD model by using turpentine oil and examined changes in the serum ferritin concentration.
MATERIALS AND METHODS
Animals and inflammatory treatment: Healthy male (n=2) and female (n=3) beagle dogs (mean body weight, 9.0 kg) aged 1–4 years were used in this study (Nihon Crea, Tokyo, Japan). The dogs were maintained in a temperature- and light-controlled environment and were given standard laboratory food, CD-5M (Nihon Crea), once a day, with free access to water. Before the experiment, the dogs were acclimatized to the research facility for 2 weeks. Chronic inflammation was produced by subcutaneous injection of 0.5 ml turpentine oil (Wako Pure Chemical Industries, Ltd., Osaka, Japan) between the scapulas, every 3 days over a 42-days period (15 injections), by using a 22-gauge needle and a 2.5-ml syringe. Physical examination of the dogs was performed every day. Before performing this study, the procedures used were approved by the Kitasato University Animal Committee.
Anesthesia and operative treatment: The dogs were administered a subcutaneous injection of atropine sulfate (0.025 mg/kg; Mitsubishi Tanabe Pharma, Osaka, Japan), butorphanol (0.2 mg/kg; Bristol-Myers, Tokyo, Japan) and acepromazine maleate (0.1 mg/kg; Boehringer Ingelheim Japan, Tokyo, Japan) as preanesthetic medication. Anesthesia was induced by administering 5 mg/kg propofol solution (Mylan, Tokyo, Japan) intravenously and was maintained by isoflurane (Dainippon Sumitomo Pharma, Osaka, Japan) in 100% oxygen. Liver samples were collected from the right lobe of the anesthetized dogs, which were held in the dorsal position, using a 16-gauge Tru-cut biopsy needle (Super Core; Medical Device Technologies, Gainesville, FL, U.S.A.) with ultrasound guidance. Bone marrow core samples were obtained from the humerus by using an 8-gauge Jamshidi needle (Medical Device Technologies).
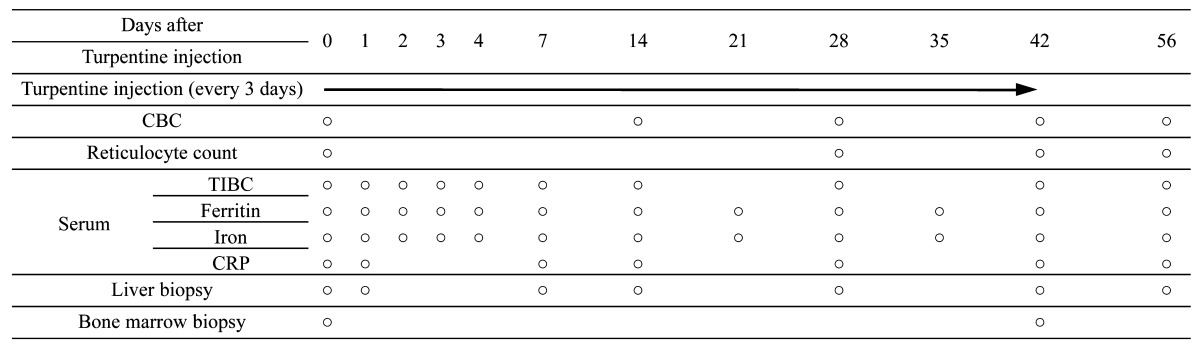
Samples: Blood, liver tissue and bone marrow were extracted according to the time course mentioned in Table 1 . Two blood samples (5 ml each) were obtained from the jugular vein of each animal. One sample was mixed with ethylenediaminetetraacetic acid (EDTA), as an anticoagulant, for determining complete blood cell count (CBC). The other sample was left at room temperature for 30 min and then centrifuged (1,560 × g, 5 min) to separate the serum for determining the C-reactive protein (CRP) level, iron level, total iron-binding capacity (TIBC) and ferritin level. The liver sample was divided into 2 portions. One portion (0.2 g wet weight) was immersed in RNAlater (Life Technologies, Carlsbad, CA, U.S.A.) and frozen at −80°C until use for quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) analysis. The second portion (0.3 g wet weight) was prepared as a 10% homogenate by using a previously described method [20, 43] and was stored at −80°C until use for determining hepatic iron content. Bone marrow tissue was fixed in 10% formalin solution, and 4-µm paraffin-embedded sections were cut and stained with Prussian blue stain to detect hemosiderin.
Table 1. Time course of turpentine administration and sample extraction.
Laboratory evaluation: The RBC count, hematocrit (HCT) value, hemoglobin (Hb) concentration, mean corpuscular volume (MCV) and mean corpuscular hemoglobin concentration (MCHC) were determined using an automatic analyzer (Celltac αMEK-6358; Nihon Kohden, Tokyo, Japan). Peripheral blood smears were prepared and stained by new methylene blue staining for evaluating the reticulocyte response. An absolute reticulocyte count was performed in which 1,000 erythrocytes were counted and categorized as either aggregate reticulocytes or normal cells [25], and the corrected reticulocyte percent for HCT was calculated using the following formula [7]:
| Corrected reticulocyte percent (%)=Observed reticulocyte (%) × HCT (%)/45 |
The serum CRP concentration was measured using a nephelometric immunoassay performed with a canine CRP detection kit (Arrows, Osaka, Japan) [31]. The serum iron level and hepatic iron content were measured using a colorimetric method involving 3-(2-pyridyl)-5,6-bis (4-phenylsulfonicacid)-1,2,4-triazine (Ferrozine; Sigma-Aldrich, St. Louis, MO, U.S.A.) [35, 38]. The serum ferritin concentration was measured using a sandwich ELISA, performed as reported in a previous study [6]. The TIBC was measured by the 2-nitroso-5-(N-propyl-N-sulfopropylamino) phenol (nitroso-PSAP) method at SRL (Tokyo, Japan).
RNA extraction and complementary DNA (cDNA) synthesis: Liver tissue samples were cut into 5-mm pieces and homogenized in 1 ml Trizol Reagent (Life Technologies) by using a Polytron PT3100 homogenizer (Kinematica, Lucerne, Switzerland). Total RNA was extracted, and RNA purity was confirmed with a BioSpec-nano system (Shimadzu, Kyoto, Japan). The RNA extracted was reverse transcribed using a Primescript II cDNA synthesis kit (TAKARA, Otsu, Japan) for cDNA synthesis.
qRT-PCR analysis: Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal reference gene for analysis of hepcidin mRNA expression [33]. Primers were designed using Primer3 (http://primer3.sourceforge.net/) with the canine hepcidin cDNA sequences [13]. For hepcidin qRT-PCR analysis, the primer sequences were 5′-CCA GAC AGG ACA GCT CAC AG-3′ and 5′-GGT GTT TTA CAG CAG CCA CA-3′, and the amplicon size was 147 bp. For the GAPDH internal reference gene, the primer sequences were 5′-TGT CCC CAC CCC CAA TGT ATC-3′ and 5′-CTC CGA TGC CTG CTT CAC TAC CTT-3′, and the amplicon size was 100 bp. The reactions mixtures comprised primers (200 nM each), Power SYBR Green Master Mix (Life Technologies) and diluted cDNA solution and were added to the wells of a 96-well plate (MicroAmp Fast 96-Well Reaction Plate; Life Technologies). PCR was performed using a Step One Plus system (Life Technologies) with the following protocol: 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min. All the samples were analyzed in triplicate, and the specificity of the amplified products was confirmed using a melting curve, which showed a single peak. Hepcidin mRNA expression was standardized to GAPDH expression and was expressed as the relative change from Day 0.
Statistical analysis: The values have been expressed in terms of mean ± standard deviation (SD). The data at each time point were compared with those for day 0 (before turpentine administration) by using repeated measures analysis of variance (ANOVA) and Dunnett’s test. Values were considered statistically significant when the P-value was less than 0.05.
RESULTS
We measured the serum CRP concentration as a nonspecific inflammatory marker for confirming inflammation following turpentine injection [5]. The serum CRP concentration after turpentine injection was significantly higher than the value on Day 0, and the increased value exceeded the standard upper limit (1.0 mg/dl) over the course of the turpentine injection period (Fig. 1). We performed hematological analysis to examine the existence and type of anemia. The data for the CBC, absolute reticulocyte count and corrected reticulocyte percent have been provided in Table 2. The RBC count, HCT value and Hb concentration significantly decreased from Day 14 onwards until the end of the turpentine injection period, compared with the values on Day 0. The MCV decreased from Day 14 until the end of the turpentine injection period, and the MCHC decreased from Day 42 to Day 56, compared with the values for Day 0. To evaluate the reticulocyte response to anemia, we calculated the reticulocyte count. Both the absolute reticulocyte count and corrected reticulocyte percent did not change significantly over the course of the turpentine injection period, compared with those on Day 0; however, they significantly increased on Day 56.
Fig. 1.

Changes in serum C-reactive protein (CRP) concentration during the course of experimental inflammation. The dotted line shows 1 mg/dl, which is the standard upper limit. *P<0.05 versus Day 0.
Table 2. Changes in the RBC, Hematocrit, Hb level, MCV, MCHC and reticulocyte response, over the course of the experiment.
| Day 0 | Day 14 | Day 28 | Day 42 | Day 56 | |
|---|---|---|---|---|---|
| RBC (×104cells /µl) | 662 ± 37 | 585 ± 23* | 465 ± 30* | 468 ± 22* | 552 ± 30* |
| Hematocrit (%) | 46.1 ± 3.1 | 38.7 ± 2.4* | 30.8 ± 1.5* | 31.4 ± 1.4* | 38.0 ± 1.5* |
| Hemoglobin (g/dl) | 15.8 ± 1.0 | 13.1 ± 0.6* | 10.5 ± 0.8* | 10.1 ± 1.0* | 12.4 ± 0.9* |
| MCV (fg) | 69.6 ± 1.0 | 66.0 ± 1.0* | 66.2 ± 1.7* | 67.1 ± 0.6* | 68.8 ± 1.2 |
| MCHC (%) | 34.3 ± 0.9 | 33.9 ± 0.5 | 34.1 ± 0.4 | 32.2 ± 1.2* | 32.6 ± 1.1* |
| Absolute reticulocyte count (×104cells /µl) | 2.3 ± 1.1 | NDa) | 4.8 ± 2.1 | 2.1 ± 3.5 | 17.0 ± 5.0* |
| Corrected reticulocyte percent (%) | 0.36 ± 0.13 | ND | 0.71 ± 0.34 | 0.31 ± 0.16 | 2.6 ± 1.1* |
*: P<0.05 versus Day 0, a) ND: Not detected.
To evaluate changes in the iron status, we measured the serum iron concentration and TIBC (Fig. 2). After the first turpentine injection (on Day 2), the serum iron level decreased approximately threefold and then returned to a normal level on Day 4. Then, the serum iron level significantly decreased from Day 14 to Day 42, compared with the level on Day 0. The TIBC significantly decreased from Days 2 to 56, compared with the level on Day 0. The serum ferritin concentration significantly decreased on Day 2 and was significantly elevated from Days 7 to 21, compared with the values for Day 0; subsequently, it decreased to a normal level (Fig. 3).
Fig. 2.

Changes in serum iron concentration and total iron-binding capacity (TIBC). *P<0.05 versus Day 0.
Fig. 3.

Changes in serum ferritin concentration. *P<0.05 versus Day 0.
To evaluate the total amount of stored iron in the body, we measured hepatic iron content and bone marrow iron staining. The hepatic iron concentration in liver biopsy specimens and estimation of stainable iron in bone marrow biopsy specimens are reported to be acceptable methods for assessing body iron stores [2, 24]. The hepatic iron content was significantly elevated from Days 14 to 28, compared with the levels on Day 0. Subsequently, it decreased to a normal level (Fig. 4). Bone marrow specimens were stained with Prussian blue for iron staining and observed with an optical microscope. The number of Prussian blue-positive cells was clearly lesser on Day 42 than on Day 0 (Fig. 5).
Fig. 4.

Changes in hepatic iron content. Values have been provided for the iron content of 10% liver homogenate solution, which was prepared as described in Materials and Methods. *P<0.05 versus Day 0.
Fig. 5.
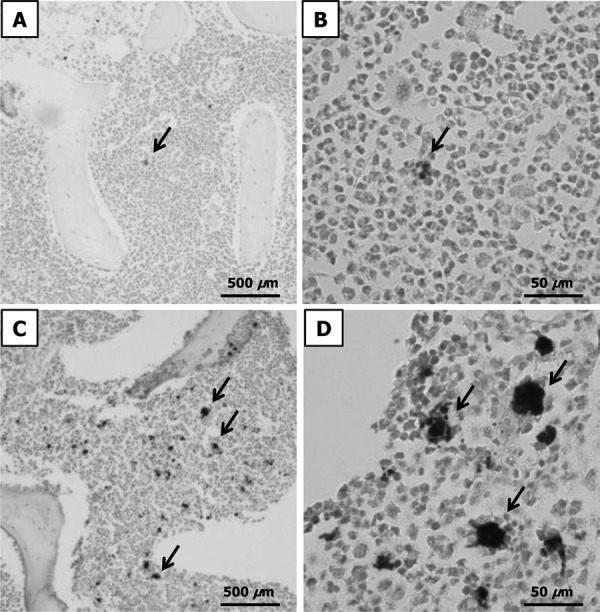
Histopathological changes in bone marrow tissue sections, analyzed using Prussian blue staining. The arrows show hemosiderin. A, B: low- and high-power light microscopy images for Day 0. C, D: low- and high-power light microscopy images for Day 42.
We measured hepcidin mRNA expression in the liver. Hepatic hepcidin mRNA expression increased on Day 1 (approximately 15-fold increase; P<0.01 versus Day 0) and Day 7 (approximately 6-fold increase; P<0.05 versus Day 0) after turpentine injection and returned to a normal level by the end of the study period (Fig. 6).
Fig. 6.

qRT-PCR analysis for hepcidin mRNA expression. Values were standardized to GAPDH mRNA expression. *P<0.05, **P<0.01 versus Day 0.
DISCUSSION
Turpentine induces local inflammation, caused by tissue injury at the injection site, and leads to increased expression of positive acute-phase proteins, such as CRP, fibrinogen, haptoglobin and ceruloplasmin and decreased expression of negative acute-phase proteins, such as transferrin and albumin [5]. Additionally, the inflammatory response continues for several days and is believed to peak at 24–48 hr after turpentine injection [5, 36].
In the present study, when the dogs were administered repeated subcutaneous turpentine injections, sterile turpentine-induced abscesses were formed, the serum CRP levels continuously increased and the TIBC, commonly assessed by the total transferrin concentration, continuously decreased. We also evaluated the levels of serum albumin, which showed a decrease similar to that seen for TIBC in this study (data not shown). Therefore, we thought that chronic inflammation is induced through repeat turpentine injections. However, in the present study, mild anemia was observed in dogs, and the reticulocyte response did not change during the turpentine-injection period, thereby indicating the presence of hypoproliferative anemia. Morphological analysis showed that both MCV and MCHC decreased significantly in this experiment, compared with the results obtained before turpentine injection. These changes were similar to those of a previous study wherein it was found that ACD is hypoproliferative and typically both normocytic and normochromic, but may progress to microcytic and hypochromic anemia [3]. In addition, the serum iron concentration decreased, and hypoferremia was observed. These results suggest that ACD developed in dogs with long-term turpentine induced inflammation. Acute inflammation has been reported to be induced in a canine ACD model by a single dose of Freund’s complete adjuvant; furthermore, hypoferremia, decreased TIBC and increased hepatic non-heme iron content were observed [11], and these changes were similar to the observations obtained during the early period after turpentine-induced inflammation in this study.
In the early period of this experiment, the serum iron concentration decreased, and hepatic hepcidin gene expression increased after the first turpentine injection. This change was rapid and is in accordance with previous studies where the serum iron level decreased at 16 hr after turpentine-induced inflammation developed in mice and at 22 hr after LPS administration in humans, and it was thought that these findings could be related to rapid hepcidin synthesis [22, 27]. In addition, it has been reported that hepcidin gene expression is induced as soon as 6 hr after turpentine injection in mice [30]. Therefore, we thought that the dogs as well as other species have an inherent mechanism, that rapidly regulates iron metabolism under inflammatory conditions. Hepcidin is a key regulator of iron metabolism, decreases iron absorption from the gastrointestinal tract and inhibits its release from RECs [10, 16]. Hepcidin interacts with ferroportin, the only known cellular iron exporter, and causes its internalization and degradation [28]. Hepcidin is reported to downregulate ferroportin, decrease iron release from RECs into the blood circulation, induce hypoferremia and suppress erythropoiesis [16]. Interestingly, in the later period of this study, although increased serum CRP levels, hypoproliferative anemia, decreased TIBC and hypoferremia were observed, the hepatic iron content and hepatic hepcidin gene expression gradually decreased to the level seen before turpentine injection. These changes are similar to those seen in the turpentine-induced mouse chronic inflammation model, in which hepatic hepcidin mRNA expression increased during the early period and then returned to a normal level [39]. In addition, ACD mice with experimentally transplanted tumors and an in vivo model using human melanoma cells have also been reported to show increase in hepatic hepcidin mRNA expression only in the early period [42].
We thought that the regulation of hepatic hepcidin expression in long-term inflammation would involve complex mechanisms in connection with prevention of severe anemia. It is known that canine ACD does not develop into severe anemia (HCT value of less than 20%) [3]. Hepcidin expression is upregulated by LPS and IL-6 and downregulated by tumor necrosis factor-α (TNF-α) [27]. In addition, it has been reported that the anti-inflammatory cytokine IL-10 controls the expression of IL-6 and IL-1β in human monocytes [26]. The change in hepcidin expression in this study may have been caused by changes in the cytokine level with continuation of chronic inflammation. However, we did not investigate this issue. The involvement with erythropoietin (EPO) was also thought to be the other reason for the decrease in hepcidin gene expression. EPO centrally regulates erythroid cell proliferation, and it has been reported that EPO injection downregulates hepatic hepcidin mRNA expression under normal, acute and chronic inflammatory conditions in mice [39]. However, we did not evaluate EPO in this study, and the reticulocyte response did not change during the inflammation period. It has been reported that EPO decreases hepcidin expression only in the case of functional erythropoiesis [42]. In addition, a study reported that the serum EPO level did not increase in human patients with ACD [40]. Therefore, we thought that it is not highly related to serum EPO level for change of iron metabolism in this study. Iron deficiency was also considered to be the cause of decrease in hepatic non-heme iron content and hepcidin gene expression during the later period in this experiment. It has been reported that hepcidin gene expression markedly decreases in dogs with experimentally induced nutritional iron deficiency [14]. However, the hepatic non-heme iron content and hepcidin gene expression returned to the levels seen before turpentine injection, and the intensity of bone marrow iron staining did not decrease in the later period in this experiment. Therefore, we thought that the dogs did not develop iron deficiency. Interestingly, no link was detected between the change in bone marrow iron storage and hepatic hepcidin gene expression. Some factors other than hepcidin may participate in bone marrow iron storage. In chronic inflammation, part of the acquisition of iron by macrophages takes place through transmembrane import of ferrous iron by the protein divalent metal transporter 1 (DMT1) [1]. Interferon-γ and TNF-α upregulate the expression of DMT-1, accompanied by increased uptake of iron into activated macrophages. In addition, these proinflammatory cytokines also induce the deposition of iron in macrophages by downregulating the expression of ferroportin [26]. Therefore, because hepatocytes and bone marrow macrophages, the main iron-storage regions, have different reactions for iron storage through hepcidin and inflammatory cytokines, the iron composition in these regions may differ.
Classically, ACD has been defined as normocytic or microcytic anemia that develops in the setting of a chronic inflammatory condition without another known cause [36]. On the basis of the results of this study, we thought that the anemia observed was ACD. The serum ferritin concentration decreased significantly on Day 2, compared to the values on Day 0; subsequently, it returned to a normal level and decreased significantly again. Throughout the experiment, the changes in serum ferritin concentration were similar to those in hepatic iron content. This trend may reflect that of body iron storage, because hepatic iron content and serum ferritin level have been reported to be indicators of iron storage in the body, including the bone marrow [2, 46]. However, unlike the results for the hepatic iron content, the finding that the serum ferritin concentration was significantly lower on Day 2 as compared to the values obtained before turpentine injection was unexpected. We think that the change in serum ferritin concentration on Day 2 was not related to total body iron storage, because the hepatic iron content did not decrease. No previous studies have reported similar results, and the mechanisms underlying the decrease in serum ferritin concentration are unclear. In the present study, similar to past reports, increase in serum ferritin concentration was observed during the early period of turpentine-induced inflammation. However, in the later period of this experiment, serum ferritin concentration returned to the level seen before turpentine injection. We found that turpentine-induced long-term inflammation does not cause hyperferritinemia and does not sustain hepatic mRNA expression of hepcidin.
This study has a limitation in that the experimentally induced inflammation does not completely reflect ACD caused by various diseases in clinical cases. It is thought that a complex mechanism underlies the involvement of hepcidin and inflammatory cytokines in ACD, and it is difficult to reproduce them experimentally. Some treatments for experimentally induced ACD have been reported, such as implantation of a Staphylococcus epidermidis-coated catheter in the peritoneum in a mouse infection model [15], collagen-induced arthritis in a rheumatoid arthritis mouse model [45] and oral feeding of dextran sulfate sodium in an inflammatory bowel disease mouse model [37]. Because ACD is not a simple condition, but is a complex response to inflammation, it is thought that many kinds of animal models would be required to study ACD.
In conclusion, when long-term ACD is induced by repeated subcutaneous injection of turpentine oil in dogs, body iron storage increases only during the early period of inflammation and does not induce remarkable hyperferritinemia. Therefore, it is suggested that the persistence of local inflammation does not greatly contribute to increase in the serum ferritin concentration. To our knowledge, this is the first study to evaluate serum ferritin levels in dogs with chronic inflammation-induced ACD.
REFERENCES
- 1.Andrews N. C. 1999. The iron transporter DMT1. Int. J. Biochem. Cell Biol. 31: 991–994. doi: 10.1016/S1357-2725(99)00065-5 [DOI] [PubMed] [Google Scholar]
- 2.Angelucci E., Brittenham G. M., Mclaren D. E., Ripalta M., Baronciani D., Giardino C., Galimberti M., Polichi P., Lucarelli G. 2000. Hepatic iron concentration and total body iron stores in thalassemia major. N. Engl. J. Med. 343: 327–331. doi: 10.1056/NEJM200008033430503 [DOI] [PubMed] [Google Scholar]
- 3.Barger A. M. 2003. The complete blood cell count: a powerful diagnostic tool. Vet. Clin. North Am. Small Anim. Pract. 33: 1207–1222. doi: 10.1016/S0195-5616(03)00100-1 [DOI] [PubMed] [Google Scholar]
- 4.Bergman P. J. 2007. Paraneoplastic syndromes. pp. 77–94. In: Small Animal Clinical Oncology, 4th ed. (Withrow, S. J. and Vail, D. M. eds.), Saunders, Philadelphia. [Google Scholar]
- 5.Cerón J. J., Eckersall P. D., Subiela S. M. 2005. Acute phase proteins in dogs and cats: current knowledge and future perspectives. Vet. Clin. Pathol. 34: 85–99. doi: 10.1111/j.1939-165X.2005.tb00019.x [DOI] [PubMed] [Google Scholar]
- 6.Chikazawa S., Hori Y., Hoshi F., Kanai K., Ito N., Sato J., Orino K., Watanabe K., Higuchi S. 2013. Development of a sandwich enzyme-linked immunosorbent assay to detect and measure serum levels of canine ferritin. J. Vet. Med. Sci. 75: 515–517. doi: 10.1292/jvms.12-0324 [DOI] [PubMed] [Google Scholar]
- 7.Cowgill E. S., Neel J. A., Grindem C. B. 2003. Clinical application of reticulocyte counts in dogs and cats. Vet. Clin. North Am. Small Anim. Pract. 33: 1223–1244. doi: 10.1016/S0195-5616(03)00099-8 [DOI] [PubMed] [Google Scholar]
- 8.de Waal Malefyt R., Abrams J., Bennett B., Figdor C. G., Vries J. E. 1991. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174: 1209–1220. doi: 10.1084/jem.174.5.1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delaby C., Pilard N., Gonçalves A. S., Beaumont C., Hergaux F. C. 2005. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 106: 3979–3984. doi: 10.1182/blood-2005-06-2398 [DOI] [PubMed] [Google Scholar]
- 10.Delaby C., Pilard N., Hetet G., Driss F., Grandchamp B., Beaumont C., Canonne-Hergaux C. 2005. A physiological model to study iron recycling in macrophages. Exp. Cell Res. 310: 43–53. doi: 10.1016/j.yexcr.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 11.Feldman B. F., Kaneko J. J., Farver T. B. 1981. Anemia of inflammatory disease in the dog: clinical characterization. Am. J. Vet. Res. 42: 1109–1113 [PubMed] [Google Scholar]
- 12.Friedrichs K. R., Thomas C., Piler M., Andrews G. A., Chavey P. S., Young K. M. 2010. Evaluation of serum ferritin as a marker for canine histiocytic sarcoma. J. Vet. Intern. Med. 24: 904–911. doi: 10.1111/j.1939-1676.2010.0543.x [DOI] [PubMed] [Google Scholar]
- 13.Fry M. M., Liggett J. L., Beak S. J. 2004. Molecular cloning and expression of canine hepcidin. Vet. Clin. Pathol. 33: 223–227. doi: 10.1111/j.1939-165X.2004.tb00377.x [DOI] [PubMed] [Google Scholar]
- 14.Fry M. M., Kirk C. A., Liggett J. L., Daniel G. B., Baek S. J., Gouffon J. S., Chimakurthy P. M., Rekapalli B. 2009. Change in hepatic gene expression in dogs with experimentally induced nutritional iron deficiency. Vet. Clin. Pathol. 38: 13–19. doi: 10.1111/j.1939-165X.2008.00081.x [DOI] [PubMed] [Google Scholar]
- 15.Gallimore B., Gagnon R. F., Subang R., Richards G. K. 1991. Natural history of chronic Staphylococcus epidermidis foreign body infection in a mouse model. J. Infect. Dis. 164: 1220–1223. doi: 10.1093/infdis/164.6.1220 [DOI] [PubMed] [Google Scholar]
- 16.Ganz T. 2011. Hepcidin and iron regulation, 10 years later. Blood 117: 4425–4433. doi: 10.1182/blood-2011-01-258467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison P. M., Arosio P. 1996. The ferritins: molecular properties, iron storage function and cellular regulation. Biochem. Biophys. Acta 1275: 161–203. doi: 10.1016/0005-2728(96)00022-9 [DOI] [PubMed] [Google Scholar]
- 18.Hintze K. J., Theli E. C. 2006. Cellular regulation and molecular interactions of the ferritins. Cell Mol. Life Sci. 63: 591–600. doi: 10.1007/s00018-005-5285-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs A., Worwood M. 1975. Ferritin in serum: Clinical and biochemical implications. N. Engl. J. Med. 292: 951–956. doi: 10.1056/NEJM197505012921805 [DOI] [PubMed] [Google Scholar]
- 20.Kakuta K., Orino K., Yamamoto S., Watanabe K. 1997. High levels of ferritin and its iron in fetal bovine serum. Comp. Biochem. Physiol. A Physiol. 118: 165–169. doi: 10.1016/S0300-9629(96)00403-3 [DOI] [PubMed] [Google Scholar]
- 21.Kazmierski K. J., Ogilvie G. K., Fettman M. J., Lana S. E., Walton J. A., Hansen R. A., Richardson K. L., Harmar D. W., Bedwell C. L., Andrews G., Chavey S. 2001. Serum zinc, chromium, and iron concentrations in dogs with lymphoma and osteosarcoma. J. Vet. Intern. Med. 15: 585–588. doi: 10.1111/j.1939-1676.2001.tb01595.x [DOI] [PubMed] [Google Scholar]
- 22.Kemna E., Pickkers P., Nemeth E., Hoeven H., Swinkels D. 2005. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in human injected with LPS. Blood 106: 1864–1866. doi: 10.1182/blood-2005-03-1159 [DOI] [PubMed] [Google Scholar]
- 23.Krause A., Neitz S., Mägert H. J., Schultz A., Forssmann W. G., Knappe P. S., Adermann K. 2000. LEAP-1 a novel highly disulfide-bounded human peptide, exhibits antimicrobial activity. FEBS. Lett. 480: 147–150. doi: 10.1016/S0014-5793(00)01920-7 [DOI] [PubMed] [Google Scholar]
- 24.Krause J. R., Stolc V. 1979. Serum ferritin and bone marrow iron stores: I. Correlation with absence of iron in biopsy specimens. Am. J. Clin. Pathol. 72: 817–820 [DOI] [PubMed] [Google Scholar]
- 25.Lassen E. D., Weiser G. 2004. Laboratory technology for veterinary medicine. pp. 18–20. In: Veterinary Hematology and Clinical Chemistry (Thrall, M. A., Baker, D. C., Campbell, T. W., DeNicola, D., Fettman, M. J., Lassen, E. D., Rebar, A. and Weiser, G. eds.), Lippincott Williams and Wilkins, Philadelphia. [Google Scholar]
- 26.Ludwiczek S., Aigner E., Theurl I., Weiss G. 2003. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood 101: 4148–4154. doi: 10.1182/blood-2002-08-2459 [DOI] [PubMed] [Google Scholar]
- 27.Nemeth E., Rivera S., Gabayan V., Keller C., Taudorf S., Pedersen B. K., Ganz T. 2004. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 113: 1271–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nemeth E., Rivera S., Gabayan V., Keller C., Gueux E., Ruivard M., Coudray C., Rayssiguier Y. 2004. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090–2093. doi: 10.1126/science.1104742 [DOI] [PubMed] [Google Scholar]
- 29.Newlands C. E., Houston D. M., Vasconcelos D. Y. 1994. Hyperferritinemia associated with malignant histiocytosis in a dog. J. Am. Vet. Med. Assoc. 205: 849–851 [PubMed] [Google Scholar]
- 30.Nicolas G., Chauvet C., Viatte L., Danan J. L., Bigard X., Devaux I., Beaumont C., Kahn A., Vaulont S. 2002. The gene expression the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 110: 1037–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Onishi T., Inokuma H., Ohno H., Soeda K., Noguchi S., Sasaki K. 2000. C-reactive protein concentrations in normal and diseased dogs − measured by laser nephelometric immunoassay. J. Jpn. Vet. Med. Assoc. 53: 595–601 [Google Scholar]
- 32.Orino K., Watanabe K. 2008. Molecular, physiological and clinical aspects of the iron storage protein ferritin. Vet. J. 178: 191–201. doi: 10.1016/j.tvjl.2007.07.006 [DOI] [PubMed] [Google Scholar]
- 33.Piek C. J., Brinkhof B., Rothuizen J., Dekker A., Penning L. C. 2011. Leukocyte count affects expression of reference genes in canine whole blood samples. BMC. Res. Notes 4: 36. doi: 10.1186/1756-0500-4-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pigeon C., Ilyin G., Courselaud B., Leroyer P., Turlin B., Brissot P., Loreal O. 2001. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276: 7811–7819. doi: 10.1074/jbc.M008923200 [DOI] [PubMed] [Google Scholar]
- 35.Rebouche C. J., Wilcox C. L., Widness J. A. 2004. Microanalysis of non-heme iron in animal tissues. J. Biochem. Biophys. Methods 58: 239–251. doi: 10.1016/j.jbbm.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 36.Rivera S., Ganz T. 2009. Animal models of anemia of inflammation. Semin. Hematol. 46: 351–357. doi: 10.1053/j.seminhematol.2009.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schubert T. E., Obermaier F., Ugocsai P., Männel D. N., Echtenacher B., Hofstädter F., Haerle P. 2008. Murine models of anaemia of inflammation: extramedullary haematopoiesis represents a species specific difference to human anaemia of inflammation that can be eliminated by splenectomy. Int. J. Immunopathol. Pharmacol. 21: 577–584 [DOI] [PubMed] [Google Scholar]
- 38.Stookey L. L. 1970. Ferrozine − a new spectrophotometric reagent for iron. Anal. Chem. 42: 779–781. doi: 10.1021/ac60289a016 [DOI] [Google Scholar]
- 39.Sun X. F. 2006. An iron regulator hepcidin is affected by EPO. Zhonqquo Shi Yan Xue Ye Xue Za Zhi 14: 778–782 [PubMed] [Google Scholar]
- 40.Theurl I., Mattle V., Seifert M., Mariani M., Marth C., Weiss G. 2006. Dysregulated monocyte iron homeostasis and erythropoietin formation in patients with anemia of chronic disease. Blood 107: 4142–4148. doi: 10.1182/blood-2005-08-3364 [DOI] [PubMed] [Google Scholar]
- 41.Torti F. M., Torti S. V. 2002. Regulation of ferritin genes and protein. Blood 99: 3505–3516. doi: 10.1182/blood.V99.10.3505 [DOI] [PubMed] [Google Scholar]
- 42.Vokurka M. 2011. Hepcidin expression in the liver of mice with implanted tumour reacts to iron deficiency, inflammation and erythropoietin administration. Folia Biologica 57: 248–254 [PubMed] [Google Scholar]
- 43.Watanabe K., Muranishi N., Murata Y., Orino K., Okano S., Yamamoto S. 2000. Biochemical properties of canine serum ferritin: iron content and nonbinding to concanavalin A. BioMetals 13: 319–324. doi: 10.1023/A:1009276323707 [DOI] [PubMed] [Google Scholar]
- 44.Weiss G. 1999. Iron and anemia of chronic disease. Kidney Int. Suppl. 69: S12–S17. doi: 10.1046/j.1523-1755.1999.055Suppl.69012.x [DOI] [PubMed] [Google Scholar]
- 45.Williams R. O. 2004. Collagen-induced arthritis as a model for rheumatoid arthritis. Methods Mol. Med. 98: 207–216 [DOI] [PubMed] [Google Scholar]
- 46.Worwood M. 1990. Ferritin. Blood Rev. 4: 259–269. doi: 10.1016/0268-960X(90)90006-E [DOI] [PubMed] [Google Scholar]