

Abstract
Autoimmune hemolytic anemia (AIHA) is not an uncommon clinical disorder and requires advanced, efficient immunohematological and transfusion support. Many AIHA patients have underlying disorder and therefore, it is incumbent upon the clinician to investigate these patients in detail, as the underlying condition can be of a serious nature such as lymphoproliferative disorder or connective tissue disorder. Despite advances in transfusion medicine, simple immunohematological test such as direct antiglobulin test (DAT) still remains the diagnostic hallmark of AIHA. The sensitive gel technology has enabled the immunohematologist not only to diagnose serologically such patients, but also to characterize red cell bound autoantibodies with regard to their class, subclass and titer in a rapid and simplified way. Detailed characterization of autoantibodies is important, as there is a relationship between in vivo hemolysis and strength of DAT; red cell bound multiple immunoglobulins, immunoglobulin G subclass and titer. Transfusing AIHA patient is a challenge to the immunohematologist as it is encountered with difficulties in ABO grouping and cross matching requiring specialized serological tests such as alloadsorption or autoadsorption. At times, it may be almost impossible to find a fully matched unit to transfuse these patients. However, transfusion should not be withheld in a critically ill patient even in the absence of compatible blood. The “best match” or “least incompatible units” can be transfused to such patients under close supervision without any serious side-effects. All blood banks should have the facilities to perform the necessary investigations required to issue “best match” packed red blood cells in AIHA. Specialized techniques such as elution and adsorption, which at times are helpful in enhancing blood safety in AIHA should be established in all transfusion services.
Keywords: Alloadsorption, alloantibody, autoadsorption, autoantibody, autoimmune hemolytic anemia, best match blood, flow cytometry, gel technology
Introduction
Immune hemolytic anemia (IHA) is the clinical condition in which antibodies of immunoglobulin G (IgG) and/or immunoglobulin M (IgM) bind to red cell surface antigens and initiate red cell destruction via the complement system and the reticulo-endothelial system. IHA is classified as either autoimmune, alloimmune or drug induced based on the antigenic stimulus responsible for the immune response.[1] Autoimmune Hemolytic Anemia (AIHA) is characterized by the production of autoantibodies directed against red blood cells (RBC). Usually these autoantibodies are directed against high incidence antigens. But, often they exhibit reactivity against allogenic red cells.[1] Though uncommon, but the disease is not rare. The overall incidence being 1 in 80,000 to 100,000 of a given population/year in the Caucasians.[2] More than 70% of new cases are seen annually in patients above 40 years of age. The peak incidence being between 60 and 70 years of age and the frequency of the disorder is usually more in females than in males. The male to female ratio is 40:60.[3]
In contrast, alloimmune hemolytic anemia requires exposure to allogenic red cells, the sources being pregnancy, blood product transfusion and transplantation. The resulting alloantibodies show no reactivity towards autologous red cells. Drug-induced antibodies can recognize either intrinsic red cell antigens or red cell-bound drugs. Antibodies that react with intrinsic red cell antigens are serologically indistinguishable from autoantibodies, whereas antibodies that react against red cell-bound drug require the drug for hemolysis.[1]
The pathogenesis of IHA ultimately overlaps for these three classifications. The degree of hemolysis depends on characteristic of the bound antibody as well as the target antigen. IgG antibodies are relatively poor activators of the classical complement pathway, but are easily recognized by the phagocytic cells. On the other hand, IgM antibodies readily activate the classical complement pathway and produce cytolysis.[4,5,6]
First described by Coombs et al. in 1945, the anti-human globulin test uses antibody to human globulin and in vivo coating of red cells with antibody or complement.[7] Generally, direct antiglobulin test (DAT) is used to determine whether the red cells have been coated in vivo with IgG or complement or both. However, manual DAT can only detect a level of 100-500 molecules of IgG/red cell and 400-1100 molecules of C3d/red cell.[7] The detection of small amounts of red cell bound IgG is becoming increasingly important in investigating and monitoring the clinical progress in AIHA. It has been seen that in so called “DAT negative AIHA”, more sensitive techniques such as enzyme linked DAT, flow cytometry (FC) and gel cards can detect IgG or C3d molecules coating the red cells.[8,9]
Serological characterization of autoantibody helps to differentiate various types of AIHA and gives a better assessment to the clinician regarding the likely course of disease and the form of treatment to be given. IgG subclass determination will depict more on the prognosis of the disease.[10] Determination of the presence or absence of autoantibodies in the serum by indirect antiglobulin test and titration of the particular Ig relates to the speed of response to therapy. Determination of the specificity of the autoantibody correlates the serum antibody with the antibody eluted from patient's red cells. The determination of thermal amplitude of the causative autoantibody correlates with the severity of the episodes of hemolysis in patients with AIHA following their exposure to warm or cold.[3]
Etio-Pathogenesis
It was Issit in 1985 who first described the series of events that led to the development of AIHA.[3] Firstly, an autoantibody is made and secondly this autoantibody has the capability of bringing about accelerated clearance of red cells thus reducing the in vivo life span of patient's own red cells. Thirdly, when the rate of in vivo red cell destruction is greater than the rate of marrow compensation anemia develops.[3] The basic cause of autoantibody production is the individual's immune system not able to recognize the host or self-antigens and this has been attributed to the failure of T cell regulation of B cells and less likely the subtle alteration in structure of the antigens on the patient's red cells.[3] Genetic factors, infection, inflammatory disorders, drugs, lymphoproliferative disorders etc., often serve as the trigger to initiate the emergence of autoantibodies.[11,12,13,14]
Cell destruction in AIHA
Immune hemolysis in vivo begins with opsonization of red cells by autoantibody. Abramson et al. in 1970 described a number of characteristic factors that determine the degree of hemolysis.[4] The antibody related factors include quantity of the antibody; its specificity, thermal amplitude, ability to fix complement and ability to bind tissue macrophages as well as characteristics of the target antigen which include the antigen density, its expression and patient age. It was observed by Sokol et al. in 1981 that in 80% of patients with AIHA red cell destruction is extravascular and involves red cells coated with antibody or complement or both, reacting with mononuclear phagocytes via specific receptors.[15] Less frequently hemolysis is intravascular and results from complement activation or coated red cells interacting with receptors on lymphoid cells or granulocytes. A varying affinity of IgG subclass for Fc receptor has been noticed by Sokol et al. in 1992 with IgG3 antibodies having a higher affinity for mononuclear phagocyte receptors than IgG1 and IgG2, IgG4 even much less efficient.[16] Moreover, monocyte reactions with IgG3 are much more rapid, greater rosette formation and fewer molecules are required to initiate erythrophagocytosis. Multiple IgG coating red cells are one of the major causes of hemolysis.[16] IgG antibodies are relatively poor activators of the classical complement pathway, but they in particular IgG1 and IgG3 antibodies are recognized readily by the Fc receptors on various reticulo-endothelial cells.[5,6] An optimum number of molecules of IgG/red cell are required to stimulate the phagocytosis. Duffy in 2002 observed that about 2000 molecules of IgG1 + IgG2 or IgG4/red cell and 230 molecules of IgG3/red cell are required to stimulate the phagocytosis and red cells coated with both IgG and complement are found to undergo further exaggerated destruction. The more the molecules of IgG, the more the activation of complement, the more is the red cell destruction.[2]
Classification of AIHA
Cases of AIHA generally are classified according to the characteristic temperature reactivity of the red cell autoantibody [Table 1]. Warm autoantibodies react more strongly near 37°C and exhibit decreased affinity at a lower temperature. Cold autoantibodies on the other hand, bind to red cells more strongly near 0-4°C and generally show little affinity at physiologic temperature. Occasionally patients have a combination of warm and cold autoantibodies.[1]
Table 1.
Classification of AIHA
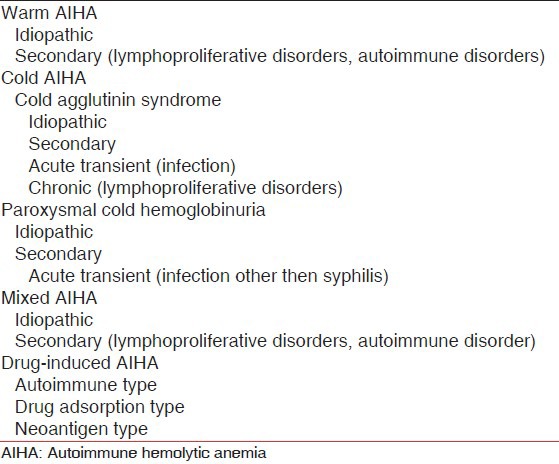
Warm AIHA
It was observed by Petz and Garratty in 1980 and Sokol et al. in 1981 that warm autoantibodies are responsible for 48-70% of AIHA cases. Lymphoproliferative disorders such as chronic lymphocytic leukemia, Hodgkin's disease, non-Hodgkin's lymphoma and Waldenstorm's macroglobulinemia are the leading causes of secondary cases.[15,17]
Cold agglutinin syndrome (CAS)
Cold-reactive autoantibodies cause two distinct clinical entities: CAS, (cold hemagglutinin disease) and paroxysmal cold hemoglobinuria (PCH). CAS represents approximately 16-32% of AIHA cases.[15,17] Primary CAS generally affects older adults, with a peak incidence at approximately 70 years of age with a slight female preponderance.[18] Infection and lymphoproliferative disorders are the predominant causes of secondary cases. The typical case of an infectious etiology involves mycoplasmal pneumonia or infectious mononucleosis in an adolescent or young adult.
PCH is a relatively uncommon form of AIHA, the incidence being 2-10% cases of hemolytic anemia.[17,19,20] Both idiopathic and secondary forms of PCH exist.
Mixed-type AIHA
Some of the patients with warm AIHA also possess a cold agglutinin. Whereas the majority of these cold agglutinins are not clinically significant, occasionally they have a sufficient thermal amplitude (>30°C) or high titer (>1:1000 at 0-4°C) to indicate CAS. Mixed type AIHA can be either idiopathic or secondary to lymphoproliferative disorders or SLE.[21]
Drug-induced AIHA
Drugs can produce hemolysis by both immune and non-immune mechanisms. Historically, alpha methyldopa and high dose penicillin were responsible for the majority of cases of drug-induced IHA. Sokol et al. in 1981published that these two drugs were responsible for 12-18% of the cases of drug-induced AIHA.[15] While the incidence of drug induced AIHA has likely decreased since then, however second and third generation cephalosporins particularly cefotetan and ceftriaxone have been associated increasingly with drug-induced AIHA that at times can be fatal.[22,23]
Clinical Features
As described by Pirofsky in 1976 warm AIHA has a highly variable clinical presentation. Typically patients insidiously develop anemic symptoms such as weakness, dizziness, fatigue and dyspnea on exertion; other less specific symptoms include fever, bleeding, coughing, abdominal pain and weight loss.[24]
Patients with primary or secondary cold AIHA have a mild, chronic hemolytic anemia producing pallor and fatigue, however there is exacerbation of the condition in a cold environment. Episodes of acute hemolysis with hemoglobinemia and hemoglobinuria are more common in the winter months. Patients also present with acrocyanosis during the exacerbations. Some patients experience Raynaud's phenomenon and rarely the red cell agglutination becomes significant enough to produce vascular occlusions with resulting necrosis.[25,26,27]
Patients with mixed-type AIHA have a chronic course interrupted by severe exacerbations, which can result in severe anemia at times. These exacerbations do not appear to be associated with cold exposure and do not result in acrocyanosis or Raynaud's phenomenon.[21]
In an Indian study, Das et al. observed that 66% of the patients were below 40 years of age with a female preponderance. In more than 60% of patients the presentation was insidious (>6 months). All patients presented with symptoms of anemia; however, in secondary AIHA the symptoms of underlying disorders were predominant. Three of the 43 patients complained of passing dark urine [Table 2].[28]
Table 2.
Clinical details of AIHA patients (N = 43)
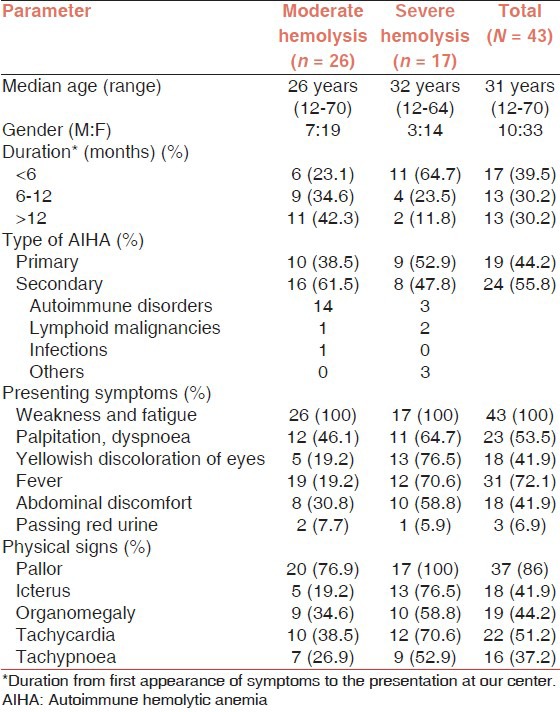
Idiopathic/primary AIHA was seen in 44.2% of patients while remaining were secondary to some underlying diseases, amongst which autoimmune disorders were the main.[28] Another study from India reported 34.2% of secondary AIHA in their series of 79 patients.[29] Das et al. observed a significant association (P < 0.05) between laboratory parameters and severity of in vivo hemolysis [Figure 1].[28]
Figure 1.
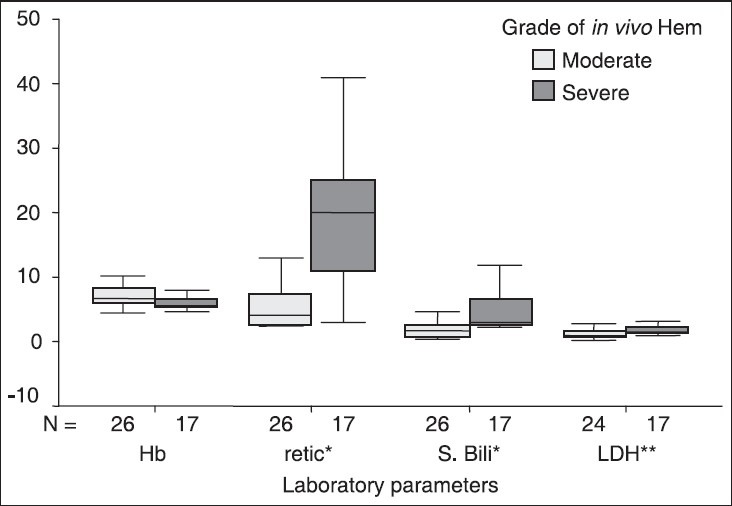
Hematological and biochemical parameters of autoimmune hemolytic anemia patients with different grades of in vivo hemolysis *P = 0.000, **P = 0.007: Mann-Whitney U-test. Hb: Hemoglobin concentration (g/dl), Retic: Reticulocyte count (%), S. Bili: Serum Bilirubin (mg/dl), LDH: Lactate dehydrogenase (×103 IU/ml)
Serological evaluation of warm AIHA
As defined by Gehrs et al in 2002, warm autoantibodies react more strongly at 37°C than at a lower temperature and are generally polyclonal.[1] Sokol et al. in 1981,[15] Petz et al in 1980[17] and Chaplin in 1973[30] have shown that over 95% of warm AIHA cases have a positive DAT and is consistent with the high prevalence of IgG. Among the DAT positive cases, 20-66% have only IgG detected on the red cell surface, 24-63% have both IgG and C3 on the surface and 7-14% have only C3 on the surface. The vast majority of the IgG autoantibodies are in the IgG1 subclass; the IgG3 is the next most common, but it is found alone in <7% of warm AIHA patients.[31,32]
Serological evaluation of CAS
Patients with CAS have more homogenous DAT results than with warm AIHA. Since the pathophysiology of CAS typically involves IgM autoantibodies and complement, patients almost exclusively have positive DAT with anti-C3 and polyspecific reagents and a negative result with anti-IgG. The IgM autoantibodies dissociate from the red cells subsequent to C3 binding and hence are generally not detected in vitro. Cold autoantibodies react more strongly at 0-4°C than at higher temperatures. Pathological cold autoantibodies are characterized by large thermal amplitude or a high titer, with thermal amplitude as the better predictor of hemolysis.[33] Activity at 37°C always correspond to clinically significant cold autoantibodies. In addition pathologic cold autoantobodies generally have a titer of greater than 1:1000 at 0-4°C. Primary CAS and CAS secondary to lymphoproliferative disorders usually exhibit higher titer than CAS secondary to infection.[1]
It was Issitt in 1967 who first observed that cold autoantibodies commonly show specificity against the Ii blood group system, with approximately 90% directed against the I antigen and most of the remaining ones directed against the i antigen.[34] Other reported specificities include Pr, Gd, Sa, Lud, Fl, Vo, M, N, D and P.[35]
Serological evaluation of PCH
PCH is caused by a biphasic IgG autoantibody (Donath-Landsteiner antibody) that fixes complement at low temperature but ultimately dissociates at a higher temperature. As a result, the DAT is positive with anti-C3, but it is generally negative with anti IgG unless performed at colder temperatures. Biphasic IgG autoantibodies bind red cells efficiently at 0-4°C and subsequently fix complement C1 at that temperature.[1] However, the other complement components bind more efficiently and cause lysis at a temperature nearer normal body temperature.
Serological evaluation of mixed-type AIHA
In mixed-AIHA the serological work-up shows that the DAT is positive for both IgG and C3. Mixed type AIHA produce difficulties with the antibody screening and cross matching due to its association with a number of underlying causes. The red cell eluate typically indicates panreactive warm IgG autoantibody. The cold autoantibody usually exhibits specificity against I antigen, but reactivity against i has also been reported.[21,36,37] Donor units have to be released as cross- match least-incompatible due to the presence of autoantibodies.
Serological evaluation of drug-induced AIHA
Drug-induced AIHA is serologically indistinguishable from warm AIHA; a presumptive diagnosis can be made only if the patient responds to withdrawal of the drug. Alpha-methyldopa is the prototypical drug operating by the induction of autoantibodies, producing a positive DAT in 11-36% of patients (dose-dependent) within 3-6 months of initiation.[38,39]
Das et al. observed that 81.4% of their patients were of warm type where IgG was the only autoantibody bound to red cells in 69.8% of total patients; subclass IgG1 or IgG3 or both were found in 46.5% of patients [Table 3].[28] The authors also observed that red cells coated with multiple autoantibodies undergo severe in vivo hemolysis compared to red cells coated with single autoantibody (P = 0.000) [Figure 2].[28]
Table 3.
Immunoglobulin class, subclass and complement fraction in AIHA (N = 43)
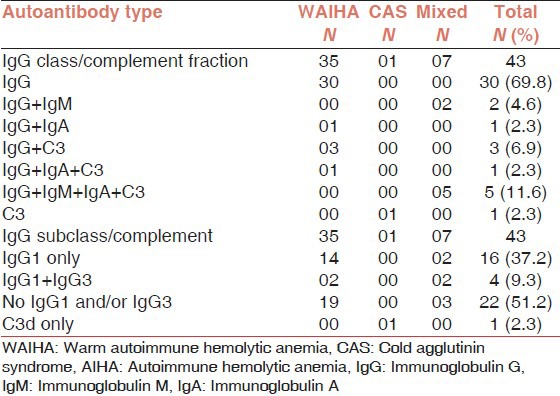
Figure 2.
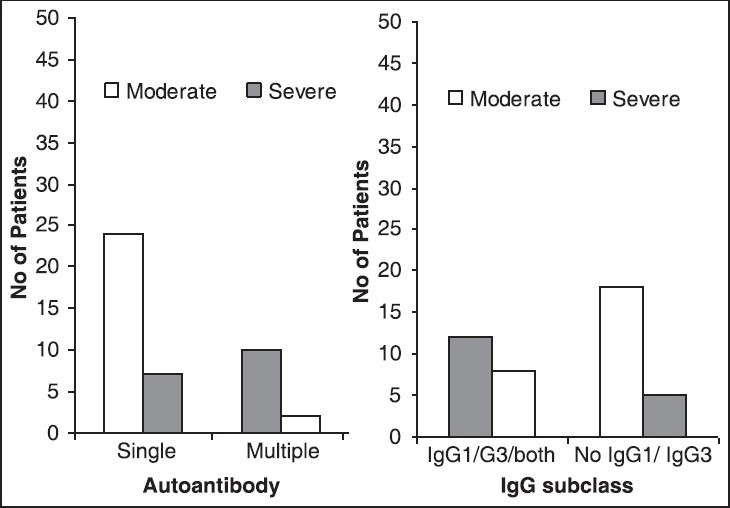
Multiple autoantibodies, immunoglobulin G (IgG) subclass and severity of in vivo hemolysis in Autoimmune hemolytic anemia(a) Compares single versus multiple autoantibodies in relation to the severity of hemolysis (P = 0.000), (b) subclass of IgG and its correlation with severity of in vivo hemolysis (P = 0.035). Chi-square test
Treatment of AIHA
Warm AIHA
The treatment of warm AIHA generally depends upon the severity of the hemolysis, though folic acid supplementation is recommended for all. If the bone marrow can compensate, then the patient can continue to be monitored. However, once anemia develops, glucocorticoids are the first-line treatment.[1] If the patient has no initial response to steroids, then the next line of therapy includes splenectomy and cytotoxic drugs. Other therapies such as plasmapheresis, IVIG, danazol have been tried with variable success.[40,41]
Cold AIHA
Treatment for CAS is dependent on its etiology and severity. With primary CAS, most patients only have mild anemia. Therefore, avoidance of cold exposure is the primary therapy which necessitates moving to a warmer climate in some patients.[42] In cases with severe hemolysis, immunosuppression with chlorambucil or cyclophosphamide may be beneficial.[42] Significant responses have also been seen with alpha-interferon.[43] Steroids are found effective only in those patients having either low titer or high thermal amplitude IgM cold agglutinins or IgG cold agglutinins. Generally extravascular hemolysis in CAS occurs in the liver, so splenectomy has only benefited those patients with IgG cold agglutinins.[44] Plasmapheresis can provide a temporary improvement in cases of severe hemolysis and it may be used prophylactically for surgeries requiring cold exposure. For secondary CAS, treating the underlying disease is the main stay of treatment. Most cases of PCH are self-limited. Treatment is usually symptomatic and also includes keeping the patient warm.
Mixed-type AIHA
Mixed-type AIHA appears to respond to treatment in a similar manner as warm AIHA. Patients generally respond to steroids and immunosuppressive agents and splenectomy has been successfully employed. To optimize the recovery, the underlying diseases have to be treated as well.[1]
Drug-induced AIHA
Despite the high incidence of a positive DAT associated with the uses of a-methyldopa, the responsible autoantibodies result in hemolytic anemia in <1% of patients.[45] The offending drug should be discontinued if the hemolysis is clinically evident. Usually within several days of discontinuing the drug, this type of AIHA gets resolved, occasionally months may be required for complete resolution.[46] In cases of severe hemolysis, steroids may aid in the recovery. RBC transfusion if needed should be given, however, these red cells will be hemolyzed at a similar rate as the endogenous red cells particularly when donor unit is cross-match incompatible.[1]
Transfusion support in AIHA
Although RBC transfusion is not a contraindication in AIHA, however its use should be limited to cases of life-threatening anemia or a high risk of cardiac or cerebrovascular ischemic events. The serologic work-up is made complicated by the panagglutinating warm autoantibodies that often mask the existing alloantibodies thus rendering cross-match incompatible. If the patient has not been recently transfused and does not have a high titer autoantibody, autoadsorption techniques can eliminate the confounding autoantibody and reduce the risk. When transfusion is needed then the least incompatible unit should be issued and the infusion should be slow and carefully monitored. Donor RBCs are destroyed at the same rate as autologous RBCs unless the autoantibodies exhibit specificity, in which case antigen-negative units should be transfused if available.[47] Transfusion has always a tendency to induce further autoantibody production.[48]
In CAS, red cell transfusions are only indicated when there is a life-threatening anemia causing crisis. Least incompatible units may have to be transfused to manage such patients. Most of the cold autoantibodies are directed against I antigen and I antigen negative donor units are extremely rare, so red cell transfusion may potentiate hemolysis. Hemolysis can also be accelerated by complement present in the exogenous donor plasma; so washed red cell transfusion may reduce such risk. The risk of additional, transfusion-related hemolysis can be reduced by using an in-line blood warmer at 37°C and by keeping the patient warm.[49] In PCH, blood units negative for P antigens are ideal, but rarity of these units precludes their use. P antigen positive blood can be beneficial when a blood warmer is employed.[50]
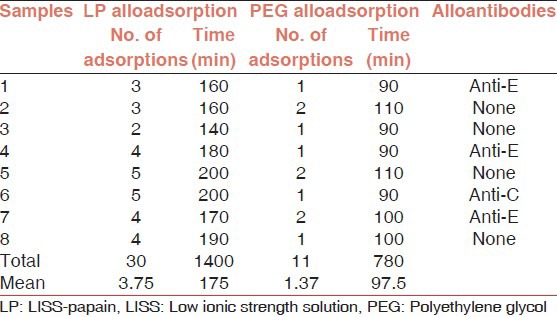
In context to Indian scenario Das et al opined that decision to transfuse in AIHA should be based on the clinical condition of the patient. No critical patient should be denied blood transfusion due to serological incompatibility. All transfusion services should follow the definite protocol and perform the minimum test required to issue safe and “best match” packed red blood cells in AIHA [Figure 3]. Twenty four of their 59 patients received blood transfusion with mean 2.9 units/patient. The main determinants of blood transfusion were the deranged hematological and biochemical hemolytic markers and all transfusions were uneventful.[51] Underlying alloantibodies could be detected in 7 of the 23 patients (30.4%) and all these were specific to Rhesus antigens. Mean number of alloadsorptions for complete autoantibody removal using PEG was 1.43 and that using low ionic strength solution (LISS)-papain method was 3.9 (P < 0.05). The mean time required by PEG alloadsorption and LISS-papain alloadsorption for autoantibody removal was 93.6 min and 177.7 min respectively (P < 0.05) [Table 4].[52]
Figure 3.
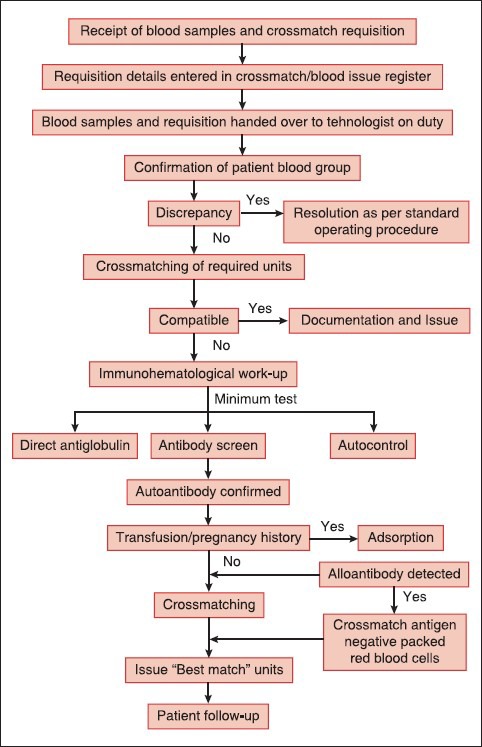
Process flow of immunohematological work-up for issuing “best match” blood in autoimmune hemolytic anemia
Table 4.
LP versus PEG alloadsorptions on same samples
DAT Negative AIHA
Although positive DAT is considered the hallmark of AIHA, the incidence of a negative DAT in patients with AIHA is reported to be between 2% and 4% respectively.[53] A possible explanation given by Issitt et al for these findings was that the number of IgG molecules/red cell necessary for accelerated in vivo destruction is sometimes lower than the number necessary to yield a positive DAT.[54] In other patients with negative DAT, but with clinical and hematological features typical of AIHA, IgA autoantibodies or monomeric IgM may be involved.[55] Sometimes low affinity antibodies may be involved in the causation of AIHA and these antibodies can easily dissociate from the RBC surface on repeated washing. Garratty in 1988 also found that washing the patient's red cells with ice cold saline, preferably in a refrigerated centrifuge, helps to keep low-affinity IgG bound to the RBCs.[56] This cold wash DAT resulted strongly positive agglutination with anti-IgG serum.[53] In recent years more sensitive tests are available, which helps in the diagnosis of conventional DAT negative warm AIHA. These tests include column agglutination technology (CAT), enzyme linked antiglobulin test (ELAT) and FC.[8,57,58] FC is the most sensitive of all these techniques and can detect antibodies as low as 35 IgG molecules/red cell.[57]
Methods of DAT
There are a number of methods available for DAT right from conventional test tube (CTT) method, CAT, ELAT, radiolabelled DAT and FC. Each has its own advantages and disadvantages. The manual method has the disadvantage that it is least sensitive for detection of red cell bound Ig. Moreover, it requires meticulous washing of red cells, which can be cumbersome. The CAT is easy to perform as it avoids washing phase and is more sensitive (93.5%). Sokol et al. in 1985 developed ELAT, which can detect very small amounts of IgG, IgA and IgM, which are normally present on red cells. It is helpful in the diagnosis of DAT negative AIHA.[8] Similarly, radiolabelled DAT is also very sensitive but has the major disadvantage that radioactive reagents are involved in addition to being cumbersome. Recently, FC is being increasingly used for DAT and other immunohematological investigations.[58]
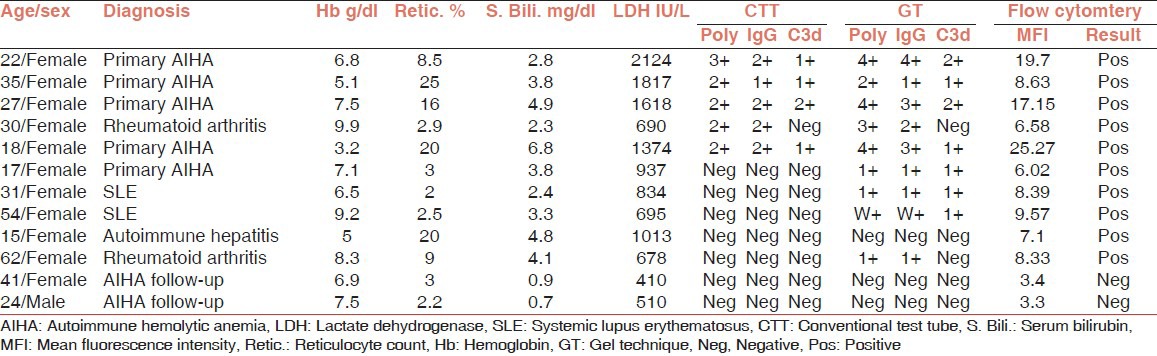
In an elaborate study conducted by Das et al. in 2006 the CAT was found to be advantageous over the CTT in the evaluation of red cell bound immunoglobulin and complement.[59] In another Indian study, Chaudhary et al. concluded that FC is a very useful tool in assessing Coomb's negative AIHA and should be employed when CTT or CAT give discordant results and there is a strong clinical suspicion of AIHA. All the five samples negative for CT were found to be positive by FC for monospecific IgG (mean fluorescence intensity = 7.88) [Table 5].[60]
Table 5.
Flow cytometric analysis of AIHA patients
Therefore, it can be concluded that AIHA is not uncommon and requires advanced, efficient immunohematological and transfusion support. Detailed characterization of autoantibodies is important, as there is a relationship between in vivo hemolysis and antibody characteristics. Though transfusing these patients is a challenge to the immunohematologist; however, transfusion should not be withheld in a critically ill-patient. All blood banks should have the facilities to perform the necessary investigations required to issue the safest blood in AIHA.
Footnotes
Source of Support: Nil
Conflicting Interest: None declared.
References
- 1.Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258–71. doi: 10.1002/ajh.10062. [DOI] [PubMed] [Google Scholar]
- 2.Duffy TP. Autoimmune hemolytic anemia and paroxysmal nocturnal hemoglobinuria. In: Simon TL, Dzik WH, Synder EL, Stowell CP, Strauss RG, editors. Rossi's Principles of Transfusion Medicine. 3rd ed. Philadelphia, USA: Lippincott Williams and Wilkins Publication; 2002. [Google Scholar]
- 3.Issit P. Serological diagnosis and characterization of causative antibody. In: Chaplin H Jr, editor. Methods in Hematology – Immune Hemolytic Anemia. USA: Churchill Livingston; 1985. [Google Scholar]
- 4.Abramson N, Gelfand EW, Jandl JH, Rosen FS. The interaction between human monocytes and red cells. Specificity for IgG subclasses and IgG fragments. J Exp Med. 1970;132:1207–15. doi: 10.1084/jem.132.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson CL, Looney RJ. Human leukocyte IgG Fc receptors. Immunol Today. 1986;7:264. doi: 10.1016/0167-5699(86)90007-1. [DOI] [PubMed] [Google Scholar]
- 6.Ravetch JV, Kinet JP. Fc receptors. Annu Rev Immunol. 1991;9:457–92. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- 7.Coombs RRA, Mourant AE, Race RR. A new test for the detection of weak and incomplete Rh agglutinins. Br J Exp Pathol. 1945;26:255–66. [PMC free article] [PubMed] [Google Scholar]
- 8.Sokol RJ, Hewitt S, Booker DJ, Stamps R. Enzyme linked direct antiglobulin tests in patients with autoimmune haemolysis. J Clin Pathol. 1985;38:912–4. doi: 10.1136/jcp.38.8.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynen R, Krone O, Legler TJ, Köhler M, Mayr WR. A newly developed gel centrifugation test for quantification of RBC-bound IgG antibodies and their subclasses IgG1 and IgG3: Comparison with flow cytometry. Transfusion. 2002;42:612–8. doi: 10.1046/j.1537-2995.2002.00076.x. [DOI] [PubMed] [Google Scholar]
- 10.Dacie SJ. The immune haemolytic anaemias: A century of exciting progress in understanding. Br J Haematol. 2001;114:770–85. doi: 10.1046/j.1365-2141.2001.02945.x. [DOI] [PubMed] [Google Scholar]
- 11.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 12.Meite M, Léonard S, Idrissi ME, Izui S, Masson PL, Coutelier JP. Exacerbation of autoantibody-mediated hemolytic anemia by viral infection. J Virol. 2000;74:6045–9. doi: 10.1128/jvi.74.13.6045-6049.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pirofsky B. Hereditary aspects of autoimmune hemolytic anemia; a retrospective analysis. Vox Sang. 1968;14:334–47. doi: 10.1111/j.1423-0410.1968.tb01723.x. [DOI] [PubMed] [Google Scholar]
- 14.Cobo F, Pereira A, Nomdedeu B, Gallart T, Ordi J, Torne A, et al. Ovarian dermoid cyst-associated autoimmune hemolytic anemia: A case report with emphasis on pathogenic mechanisms. Am J Clin Pathol. 1996;105:567–71. doi: 10.1093/ajcp/105.5.567. [DOI] [PubMed] [Google Scholar]
- 15.Sokol RJ, Hewitt S, Stamps BK. Autoimmune haemolysis: An 18-year study of 865 cases referred to a regional transfusion centre. Br Med J (Clin Res Ed) 1981;282:2023–7. doi: 10.1136/bmj.282.6281.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sokol RJ, Booker DJ, Stamps R. The pathology of autoimmune haemolytic anaemia. J Clin Pathol. 1992;45:1047–52. doi: 10.1136/jcp.45.12.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petz LD, Garratty G. New York: Churchill Livingstone; 1980. Acquired Immune Hemolytic Anemias. [Google Scholar]
- 18.Schubothe H. The cold hemagglutinin disease. Semin Hematol. 1966;3:27–47. [PubMed] [Google Scholar]
- 19.Van Loghem JJ, Hart MV, Dorfmeier H. New York: Grune and Stratton; 1958. Serological study in acquired hemolytic anemia. Sixth International Congress of the International Society of Hematology. [Google Scholar]
- 20.Heddle NM. Acute paroxysmal cold hemoglobinuria. Transfus Med Rev. 1989;3:219–29. doi: 10.1016/s0887-7963(89)70082-1. [DOI] [PubMed] [Google Scholar]
- 21.Shulman IA, Branch DR, Nelson JM, Thompson JC, Saxena S, Petz LD. Autoimmune hemolytic anemia with both cold and warm autoantibodies. JAMA. 1985;253:1746–8. [PubMed] [Google Scholar]
- 22.Shammo JM, Calhoun B, Mauer AM, Hoffman PC, Baron JM, Baron BW. First two cases of immune hemolytic anemia associated with ceftizoxime. Transfusion. 1999;39:838–44. doi: 10.1046/j.1537-2995.1999.39080838.x. [DOI] [PubMed] [Google Scholar]
- 23.Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239–46. doi: 10.1046/j.1537-2995.1999.39111239.x. [DOI] [PubMed] [Google Scholar]
- 24.Pirofsky B. Clinical aspects of autoimmune hemolytic anemia. Semin Hematol. 1976;13:251–65. [PubMed] [Google Scholar]
- 25.Mitchell AB, Pergrum GD, Gill AM. Cold agglutinin disease with Raynaud's phenomenon. Proc R Soc Med. 1974;67:113–5. [PMC free article] [PubMed] [Google Scholar]
- 26.Shelley WB, Shelley ED. Acrocyanosis of cold agglutinin disease successfully treated with antibiotics. Cutis. 1984;33:556–7. [PubMed] [Google Scholar]
- 27.Roelcke D. Cold agglutination. Transfus Med Rev. 1989;3:140–66. doi: 10.1016/s0887-7963(89)70075-4. [DOI] [PubMed] [Google Scholar]
- 28.Das SS, Nityanand S, Chaudhary R. Clinical and serological characterization of autoimmune hemolytic anemia in a tertiary care hospital in North India. Ann Hematol. 2009;88:727–32. doi: 10.1007/s00277-008-0674-6. [DOI] [PubMed] [Google Scholar]
- 29.Naithani R, Agrawal N, Mahapatra M, Pati H, Kumar R, Choudhary VP. Autoimmune hemolytic anemia in India: Clinico-hematological spectrum of 79 cases. Hematology. 2006;11:73–6. doi: 10.1080/10245330500345587. [DOI] [PubMed] [Google Scholar]
- 30.Chaplin H., Jr Clinical usefulness of specific antiglobulin reagents in autoimmune hemolytic anemias. Prog Hematol. 1973;8:25–49. [PubMed] [Google Scholar]
- 31.Engelfriet CP, Overbeeke MA, von dem Borne AE. Autoimmune hemolytic anemia. Semin Hematol. 1992;29:3–12. [PubMed] [Google Scholar]
- 32.Garratty G. Factors affecting the pathogenicity of red cell auto and alloantibodies. In: Nance SJ, editor. Immune Destruction of Red Blood Cells. Arlington, VA: American Association of Blood Banks; 1989. p. 109. [Google Scholar]
- 33.Rosse WF, Adams JP. The variability of hemolysis in the cold agglutinin syndrome. Blood. 1980;56:409–16. [PubMed] [Google Scholar]
- 34.Issitt PD. I blood group system and its relation to other blood group systems. J Med Lab Technol. 1967;24:90–7. [PubMed] [Google Scholar]
- 35.von dem Borne AE, Mol JJ, Joustra-Maas N, Pegels JG, Langenhuijsen MM, Engelfriet CP. Autoimmune haemolytic anaemia with monoclonal IgM (kappa) anti-P cold autohaemolysins. Br J Haematol. 1982;50:345–50. doi: 10.1111/j.1365-2141.1982.tb01925.x. [DOI] [PubMed] [Google Scholar]
- 36.Sokol RJ, Hewitt S, Stamps BK. Autoimmune hemolysis: Mixed warm and cold antibody type. Acta Haematol. 1983;69:266–74. doi: 10.1159/000206903. [DOI] [PubMed] [Google Scholar]
- 37.Kajii E, Miura Y, Ikemoto S. Characterization of autoantibodies in mixed-type autoimmune hemolytic anemia. Vox Sang. 1991;60:45–52. doi: 10.1111/j.1423-0410.1991.tb00870.x. [DOI] [PubMed] [Google Scholar]
- 38.Carstairs KC, Breckenridge A, Dollery CT, Worlledge SM. Incidence of a positive direct coombs test in patients on alpha-methyldopa. Lancet. 1966;2:133–5. doi: 10.1016/s0140-6736(66)92422-6. [DOI] [PubMed] [Google Scholar]
- 39.Worlledge SM, Carstairs KC, Dacie JV. Autoimmune haemolytic anaemia associated with alpha-methyldopa therapy. Lancet. 1966;2:135–9. doi: 10.1016/s0140-6736(66)92423-8. [DOI] [PubMed] [Google Scholar]
- 40.Ahn YS. Efficacy of danazol in hematologic disorders. Acta Haematol. 1990;84:122–9. doi: 10.1159/000205048. [DOI] [PubMed] [Google Scholar]
- 41.Pignon JM, Poirson E, Rochant H. Danazol in autoimmune haemolytic anaemia. Br J Haematol. 1993;83:343–5. doi: 10.1111/j.1365-2141.1993.tb08293.x. [DOI] [PubMed] [Google Scholar]
- 42.Evans RS, Baxter E, Gilliland BC. Chronic hemolytic anemia due to cold agglutinins: A 20-year history of benign gammopathy with response to chlorambucil. Blood. 1973;42:463–70. [PubMed] [Google Scholar]
- 43.O’Connor BM, Clifford JS, Lawrence WD, Logue GL. Alpha-interferon for severe cold agglutinin disease. Ann Intern Med. 1989;111:255–6. doi: 10.7326/0003-4819-111-3-255. [DOI] [PubMed] [Google Scholar]
- 44.Silberstein LE, Berkman EM, Schreiber AD. Cold hemagglutinin disease associated with IgG cold-reactive antibody. Ann Intern Med. 1987;106:238–42. doi: 10.7326/0003-4819-106-2-238. [DOI] [PubMed] [Google Scholar]
- 45.Worlledge SM. Immune drug-induced haemolytic anemias. Semin Hematol. 1969;6:181–200. [PubMed] [Google Scholar]
- 46.Ewing DJ, Hughes CJ, Wardle DF. Methyldopa-induced auto-immune haemolytic anaemia-a report of two further cases. Guys Hosp Rep. 1968;117:111–8. [PubMed] [Google Scholar]
- 47.Plapp FV, Beck ML. Transfusion support in the management of immune haemolytic disorders. Clin Haematol. 1984;13:167–83. [PubMed] [Google Scholar]
- 48.Ness PM, Shirey RS, Thoman SK, Buck SA. The differentiation of delayed serologic and delayed hemolytic transfusion reactions: Incidence, long-term serologic findings, and clinical significance. Transfusion. 1990;30:688–93. doi: 10.1046/j.1537-2995.1990.30891020325.x. [DOI] [PubMed] [Google Scholar]
- 49.Rosenfield RE, Jagathambal Transfusion therapy for autoimmune hemolytic anemia. Semin Hematol. 1976;13:311–21. [PubMed] [Google Scholar]
- 50.Sokol RJ, Hewitt S, Stamps BK. Autoimmune haemolysis associated with Donath-Landsteiner antibodies. Acta Haematol. 1982;68:268–77. doi: 10.1159/000206992. [DOI] [PubMed] [Google Scholar]
- 51.Das SS, Chaudhary R. Transfusion support in autoimmune hemolytic anemia. Indian J Hematol Blood Transfus. 2006;1:9–13. [Google Scholar]
- 52.Das SS, Chaudhary R. Utility of adsorption techniques in serological evaluation of warm autoimmune haemolytic anaemia. Blood Transfus. 2009;7:300–4. doi: 10.2450/2009.0079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Issit PD. 3rd ed. Miami, Florida, USA: Montgomery Scientific Publications; 1985. Applied Blood Group Serology; pp. 536–8. [Google Scholar]
- 54.Issitt DP, Gutgsell NS. Clinically significant antibodies not detected by routine methods. In: Nance SJ, editor. Immune Destruction of Red Blood Cells. Arlington: American Association of Blood Banks; 1989. pp. 93–9. [Google Scholar]
- 55.Schreiber A, Gill FM, Manno CS. Autoimmune hemolytic anemia. In: Nathan D, Oski F, editors. Hematology of Infancy and Childhood. 4th ed. Philadelphia: WB Saunders; 1993. pp. 496–510. [Google Scholar]
- 56.Garratty G. The clinical significance (and insignificance) of red cell bound IgG and complement. In: Wallace ME, Levitt JS, editors. Current Application and Interpretation of the Direct Antiglobulin Test. Arlington: American Association of Blood Banks; 1988. pp. 1–16. [Google Scholar]
- 57.Nathalang O, Chuansumrit A, Prayoonwiwat W, Siripoonya P, Sriphaisal T. Comparison between the conventional tube technique and the gel technique in direct antiglobulin tests. Vox Sang. 1997;72:169–71. doi: 10.1046/j.1423-0410.1997.7230169.x. [DOI] [PubMed] [Google Scholar]
- 58.Roback JD, Barclay S, Hillyer CD. An automatable format for accurate immunohematology testing by flow cytometry. Transfusion. 2003;43:918–27. doi: 10.1046/j.1537-2995.2003.t01-1-00433.x. [DOI] [PubMed] [Google Scholar]
- 59.Das SS, Chaudhary R, Khetan D. A comparison of conventional tube test and gel technique in evaluation of direct antiglobulin test. Hematology. 2007;12:175–8. doi: 10.1080/10245330601111862. [DOI] [PubMed] [Google Scholar]
- 60.Chaudhary R, Das SS, Gupta R, Khetan D. Application of flow cytometry in detection of red-cell-bound IgG in Coombs-negative AIHA. Hematology. 2006;11:295–300. doi: 10.1080/10245330600915958. [DOI] [PubMed] [Google Scholar]