

Abstract
Much of the focus in muscle regeneration has been placed on the identification and delivery of stem cells to promote regenerative capacity. As those efforts have advanced, we have learned that complex features of the microenvironment in which regeneration occurs can determine success or failure. The immune system is an important contributor to that complexity and can determine the extent to which muscle regeneration succeeds. Immune cells of the myeloid lineage play major regulatory roles in tissue regeneration through two general, inductive mechanisms: instructive mechanisms that act directly on muscle cells; and permissive mechanisms that act indirectly to influence regeneration by modulating angiogenesis and fibrosis. In this article, recent discoveries that identify inductive actions of specific populations of myeloid cells on muscle regeneration are presented, with an emphasis on how processes in muscle and myeloid cells are co-regulated.
Keywords: Muscle regeneration, Macrophage phenotype, Signaling systems
Introduction
Acute trauma, chronic disease or disruption of vascularization cause rapid death of skeletal muscle. Functional recovery of the damaged tissue is determined by interacting cellular responses, including stem cell activation, proliferation and differentiation, vascular repair, and tissue fibrosis during healing. Experimental approaches to improve muscle regeneration have focused on identifying stem cell populations that promote regeneration and refining their delivery, while using genetic or pharmacological manipulations to influence angiogenesis and fibrosis. However, those approaches are sensitive to the presence of myeloid cells in the injured muscle, suggesting that manipulations of myeloid cells can provide novel strategies to potentiate muscle regeneration. As knowledge of myeloid cell involvement in muscle regeneration has grown, we have come to appreciate that multiple subpopulations interact in complex and delicately balanced ways to influence regeneration. Perturbations in myeloid cell number, phenotype or the stage of regeneration at which they are present can yield vastly different outcomes in the regenerative process. As these complexities become better understood, there is hope that knowledge can be exploited to improve muscle regeneration therapy.
In this Review, inductive processes through which myeloid cells influence muscle regeneration are presented, including instructive processes that directly influence developmental gene expression in muscle and permissive processes that modify the environment in which regeneration occurs. Here, muscle ‘regeneration’ refers to processes in damaged muscle tissue that lead to the restoration of normal structure and homeostasis, especially those events that influence the differentiation and growth of muscle cells that replace damaged tissue (Box 1). In addition, we emphasize discoveries that have contributed to understanding the mechanisms that coordinate phenotypic switches in myeloid cell populations with progressive stages of muscle regeneration.
Box 1. Indices of muscle regeneration
Muscle regeneration can be assessed based on the extent or rate at which damaged muscle recovers its normal structure and function. Routinely used indices of muscle regeneration include the following.
Growth of muscle cells. The most commonly used morphological indicator is the change in size of nascent, multinucleated muscle cells as they differentiate and grow following injury. In mice, cross-sectional areas are typically ∼100 μm2 at the onset of regeneration and ∼3000 μm2 when regeneration is complete.
Gene and protein expression. Assays for relative levels and timecourse of expression of muscle-specific transcription factors also provide an index of myogenesis following injury.
Translational activity. Increased binding of basophilic dyes in individual muscle fibers is associated with high levels of cytosolic mRNA, providing a surrogate readout for translational activity.
Position of nuclei. During regeneration of damaged muscle, the nuclei of nascent muscle fibers are located at the center of the cells, moving to a location subjacent to the cell membrane at the end of myogenesis. Thus, measuring the percentage of muscle fibers that have centrally located nuclei can serve as an index of regeneration. However, this is limited as an index of regeneration because other variables can influence the proportion of fibers that show central nucleation.
Functional assays. Force production in regenerating muscle in response to a single action potential (peak twitch tension) or maximally activated by a rapid series of stimuli (tetanic tension) is a particularly valuable assay to measure muscle regeneration.
Muscle structure and regeneration following injury
Fully differentiated skeletal muscle is composed of large, multinucleated cells called muscle fibers, which can be centimeters in length and contain a cytosolic volume that is tens of thousands of times greater than that of most cells. Because muscle fibers are superficially located in the body and routinely subjected to direct trauma, their capacity for repair is essential for normal, life-long function. However, muscle fibers are post-mitotic cells, incapable of replacing lost or damaged tissue by proliferation of the muscle fibers themselves. Instead, most of the regenerative capacity of injured or diseased muscle is attributable to mononucleated myogenic cells called satellite cells (Mauro, 1961), which normally reside in a quiescent state on the surface of muscle fibers.
Upon activation by injury or disease, satellite cells enter the cell cycle and divide. Daughter cells can then follow two developmental fates, either returning to quiescence to renew the satellite cell population or withdrawing from the cell cycle to begin terminal differentiation (Fig. 1) (Shafiq et al., 1968; Moss and Leblond, 1971). As differentiation proceeds, the cells fuse with neighboring myocytes to form multinucleated myotubes that undergo terminal differentiation to become muscle fibers (Füchtbauer and Westphal, 1992; Grounds et al., 1992; Yablonka-Reuveni and Rivera, 1994). This latter stage of development is characterized by extensive growth of individual muscle fibers, as proteins necessary for regulation of muscle contraction accumulate. Many stages of satellite cell activation and differentiation are functionally coupled with changes in the expression of transcription factors. Quiescent satellite cells express Pax7 but not MyoD, whereas following activation the cells express both Pax7 and MyoD (Grounds et al., 1992; Yablonka-Reuveni and Rivera, 1994; Seale et al., 2000). If they continue to differentiate, Pax7 expression ceases, while myogenin and then MRF4 (also known as Myf6) expression is elevated (Cornelison and Wold, 1997). Although the sequence of changes in gene expression in developing muscle is largely regulated by mechanisms intrinsic to muscle cells, extrinsic factors are also important in determining the duration of each developmental stage. Extrinsic factors can be derived from multiple sources, including endocrine and nervous tissues, but the presence of large numbers of inflammatory cells in injured muscle suggests that the immune system might also play a role in influencing the repair and growth of muscle following injury.
Fig. 1.
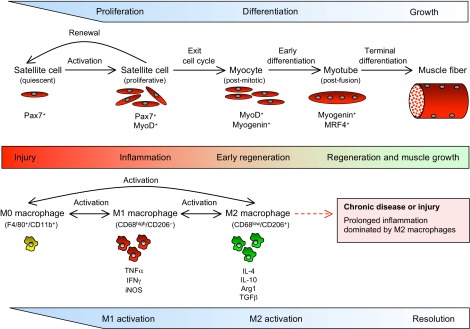
Changes in macrophage phenotype and stages of myogenesis in regenerative muscle following injury. During the initial stages of inflammation, Pax7+ satellite cells are activated, proliferate and begin expression of MyoD, initiating transcription of muscle-specific genes necessary for early differentiation. These events coincide with the activation of macrophages from the M0 phenotype to the CD68high/CD206- M1 phenotype, when they express elevated levels of TNFα, IFNγ and iNOS. As myogenesis proceeds, some activated satellite cells return to quiescence and renew the satellite cell reserve population, while others exit the cell cycle to undergo further differentiation. Those post-mitotic myocytes display a shift in gene expression that enables their fusion to form multinucleated myotubes that are able to undergo terminal differentiation. The progress of muscle cells into early regenerative stages coincides with a shift of macrophages to an M2 phenotype that expresses IL-10, IL-4, arginase 1 (Arg1) and TGFβ. As muscle regeneration and growth proceed to restore normal homeostasis, the inflammatory response is slowly resolved. However, in the case of chronic injury or repeated acute injuries, prolonged activity of M2 macrophages can exacerbate muscle dysfunction by driving the excessive accumulation of connective tissue, which is attributable to prolonged production of the pro-fibrotic cytokine TGFβ and to increased metabolism of arginine by Arg1.
Cells of the myeloid lineage dominate the inflammatory infiltrate in regenerating muscle
Early observations implicated inflammatory cells in forming new muscle by removing debris (Waldeyer, 1865) and possibly by promoting myogenesis (Volkmann, 1893). However, decades passed before the vast majority of invading cells were identified as belonging to the myeloid lineage (Kellner and Robertson, 1954) (Fig. 2). Following injury, these cells invade in large numbers with scant involvement of cells of the lymphoid lineage, regardless of whether muscle damage is caused by acute injury or chronic damage, such as in Duchenne muscular dystrophy (DMD) (Box 2). For example, in the mdx mouse model of DMD, myeloid cells can exceed 50,000 cells/mm3 of muscle (Wehling et al., 2001), meaning that a kilogram of muscle would contain more than 108 myeloid cells, the great majority of which are macrophages. However, other myeloid cells, such as neutrophils and eosinophils are also present in regenerative muscle, suggesting that they might also influence regeneration (Snow, 1977; Heredia et al., 2013), even though their numbers are relatively low during periods when muscle regeneration is most active.
Fig. 2.

Lineage of myeloid cells that can influence muscle regeneration. Common myeloid progenitors (CMPs) differentiate from multipotent hematopoietic stem cells to give rise to the numerous lines of myeloid cells. CMPs first differentiate into megakaryocyte and erythroid progenitor cells (MEPs) or granulocyte and macrophage progenitor cells (GMPs), with the latter lineage giving rise to the myeloid cell populations that have been most clearly associated with muscle regeneration. GMPs in circulating populations differentiate into either monocyte precursors (MPs) or granulocyte precursors (GPs), which can then undergo terminal differentiation into committed populations that can influence muscle injury, growth or regeneration. MPs give rise to monocytes that release a vast number of soluble factors that modulate the inflammatory response. Monocytes undergo further differentiation to become macrophages, the best characterized population of myeloid cells that influence muscle regeneration. GPs give rise to other populations of myeloid cells that can also influence regeneration. Eosinophils that derive from GPs can either promote muscle damage or muscle regeneration, which varies according to the injury or disease that initiates the inflammatory response. Neutrophils also play a dual role and have the capacity to either amplify muscle damage or promote repair by contributing to the removal of damaged tissue and increasing revascularization. Basophils, which are also derived from GPs, are present in regenerating muscle, although it is not clear whether they have a specific role in muscle regeneration. FAPs, fibro/adipogenic progenitors.
Box 2. Duchenne muscular dystrophy (DMD): a case of chronic muscle injury
DMD is a progressive, lethal, muscle wasting disease caused by null mutation of the dystrophin gene, which encodes a membrane-associated structural protein (Hoffman et al., 1987; Matsumura and Campbell, 1994). In the absence of dystrophin protein, muscle membranes are weaker, causing a lifetime of muscle fiber injury and repair. Although chronic muscle damage in DMD produces an inflammatory response that largely resembles the myeloid cell populations in acutely injured muscle (Villalta et al., 2009; Villalta et al., 2011; Deng et al., 2012), lymphoid cells are also recruited to the inflammatory infiltrate in chronically injured DMD muscle. Furthermore, cytotoxic T-lymphocytes in muscles of multiple DMD patients show T-cell receptor rearrangements that indicate that they recognize a common antigen (Gussoni et al., 1994). Whether this potential autoreactivity of T-cells in dystrophic muscle reflects a breakdown of peripheral tolerance and represents an immune response to chronically injured muscle rather than to DMD muscle per se is unexplored and highlights some of the fundamental questions that remain concerning interactions between injured muscle and the immune system.
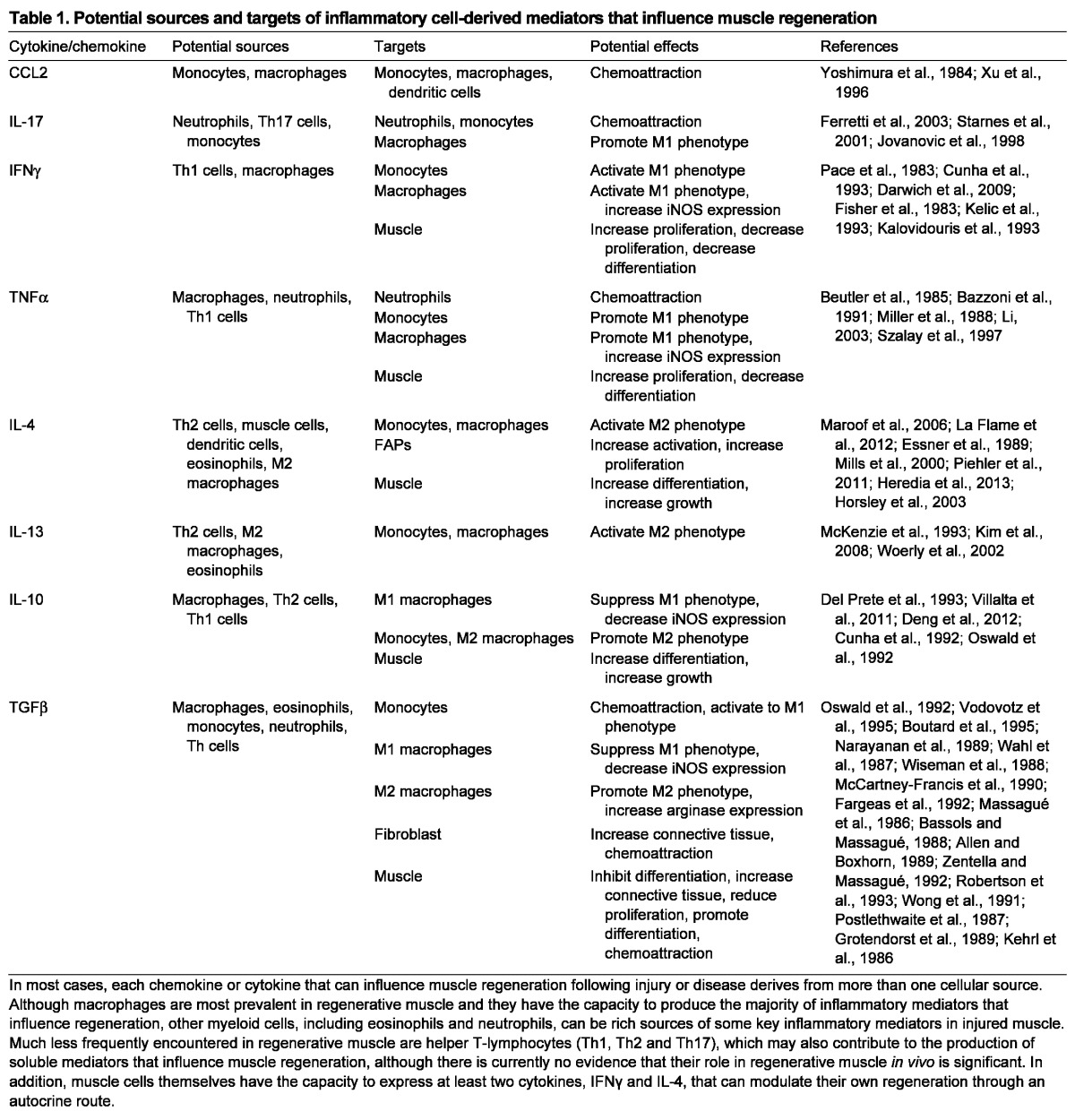
Among the myeloid lineage cell types that enter muscle following injury, macrophages are most clearly demonstrated as positive regulators of regeneration (Table 1). A reduction in the number of circulating macrophages slows the return of normal histology to muscle following acute injury caused by freezing (Summan et al., 2006). In addition, null mutation of the gene encoding chemokine ligand 2 (CCL2), which is a chemoattractant of macrophages to injured tissue, or mutation of its receptor CCR2 reduced macrophage invasion into muscle, delayed recovery of normal muscle architecture and slowed muscle growth following damage caused by toxin injection or ischemia (Contreras-Shannon et al., 2007; Shireman et al., 2007). Importantly, these defects in macrophage invasion and muscle regeneration in Ccr2 null mice were rescued by transplantation of bone marrow from wild-type mice (Sun et al., 2009). Finally, targeted ablation of cells expressing CD11b (also known as Itgam), which include neutrophils, monocytes and macrophages, similarly reduced growth of muscle fibers following acute injury caused by injection of toxin (Arnold et al., 2007).
Table 1.
Potential sources and targets of inflammatory cell-derived mediators that influence muscle regeneration
Macrophage phenotypes: a continuous spectrum
Macrophages display a broad spectrum of phenotypic diversity, determined primarily by the environment in which they are activated. At one end of the spectrum, macrophages can be activated to the M1 (F4/80+/CD68high/CD206-) phenotype by constituents of bacterial cell membranes [e.g. lipopolysaccharides (LPS)] or by proinflammatory cytokines generated by T helper 1 (Th1) cells or other myeloid cells (Mills et al., 2000) (Fig. 1). Interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα) are well-characterized proinflammatory Th1 cytokines that activate macrophages to the M1 phenotype (Pace et al., 1983; Philip and Epstein, 1986). At the opposite end of the spectrum, macrophages can be activated to the M2 (F4/80+/CD68low/CD206+) phenotype by anti-inflammatory Th2 cytokines, including interleukin 4 (IL-4), IL-10 and IL-13 (Stein et al., 1992; de Waal Malefyt et al., 1993; Mills et al., 2000). Although macrophages shift between the M1 and M2 phenotype with changes in cytokine exposure, at least in vitro, the phagocytosis of cellular debris is sufficient to switch macrophages from an M1 to an M2 phenotype without changes in the activation by cytokines (Fadok et al., 1998; Fadok et al., 2001; Arnold et al., 2007; Mounier et al., 2013). Furthermore, M1 and M2 macrophages can originate from distinct monocyte populations, suggesting that some specification of phenotype can occur in relatively undifferentiated, circulating myeloid populations (Nahrendorf et al., 2007).
Typically, M1 macrophages amplify inflammation by releasing chemoattractants for inflammatory cells. They also express high levels of inducible nitric oxide synthase (iNOS; also known as Nos2), generating nitric oxide (NO) that can ‘tag’ cellular debris for removal by phagocytosis or contribute to cytolysis of neighboring cells (Hibbs et al., 1988). M2 macrophages serve primarily to attenuate inflammation through the release of anti-inflammatory cytokines that deactivate M1 macrophages. They also express enzymes that can promote repair by increasing connective tissue production. Transforming growth factor beta (TGFβ) is a particularly potent pro-fibrotic cytokine, produced by M2 macrophages that can induce the expression of connective tissue proteins (Roberts et al., 1986). M2 macrophage-derived TGFβ can also induce arginase 1 expression (Mills et al., 2000). Arginine metabolism by arginase yields polyamines that can stimulate fibroblast proliferation and increase proline synthesis, leading to increased connective tissue production (Modolell et al., 1995; Hesse et al., 2001; Wehling-Henricks et al., 2010).
Distinct macrophage populations have differential effects on muscle regeneration
Early histological observations (St Pierre and Tidball, 1994) demonstrated that distinct macrophage populations are associated with separate phases of muscle regeneration. Provocatively, the CD68high/CD163- M1 macrophages reached peak numbers at ∼1 to 2 days post-injury, coinciding with the activation and proliferation of satellite cells during early regeneration. Conversely, the CD68low/CD163+ M2 macrophages reached peak numbers by 4 days post-injury, coinciding with early stages of muscle differentiation and were closely associated with the surfaces of regenerative myotubes and nascent muscle fibers (St Pierre and Tidball, 1994). In vitro observations showed similar relationships between macrophage phenotype and stages of myogenesis. Cultures of satellite cells with M1 macrophages increased satellite cell proliferation (Bencze et al., 2012), but decreased the proportion of satellite cells expressing myogenin (Arnold et al., 2007). By contrast, culturing satellite cells with macrophages activated to the M2 phenotype increased myogenin expression and elevated fusion of satellite cells to form myotubes (Arnold et al., 2007).
In vivo observations have now established that perturbations in the normal shift of macrophages from an M1-biased population to an M2-biased population can have major effects on muscle regeneration. F4/80 (also known as Emr1) antibody depletion of macrophages at the time of M1 to M2 transition reduced muscle growth, repair and regeneration and perturbed the expression of muscle-specific transcription factors that drive muscle regeneration (Tidball and Wehling-Henricks, 2007). Similarly, growth of regenerative muscle fibers was slowed when CD11b-expressing inflammatory cells were depleted at the time of peak regeneration following injury by toxin injection (Arnold et al., 2007). Alternative strategies to prevent the M1 to M2 shift in macrophage phenotype during muscle regeneration produced similar effects. Mutation of the Cebpb promoter, which impairs the expression of genes associated with the M2 macrophage phenotype, yielded defects in the growth of nascent, regenerative fibers and slowed the restoration of normal muscle histology following acute injury, without affecting the activity of phagocytic cells (Ruffell et al., 2009). Similarly, administering neutralizing antibodies to macrophage colony stimulating factor (M-CSF; also known as Csf1) to mice following muscle damage by snake cardiotoxin (CTX) injection reduced the number of macrophages in muscle and slowed growth of regenerative muscle fibers (Segawa et al., 2008). M-CSF can bias cells in the myeloid lineage toward an M2 phenotype (Fleetwood et al., 2007), suggesting that the reduction of total macrophages in the muscle of mice with impaired M-CSF signaling reflects a preferential decrease in M2 macrophages. Furthermore, perturbations of the normal timing of the M1 to M2 transition following injury disrupt regeneration. This was clearly demonstrated by Dr Munoz-Canoves and colleagues, who administered IL-10, which deactivates the M1 phenotype and promotes the M2 phenotype, during the period of muscle regeneration that is normally dominated by M1 macrophages. Following injury by CTX injection and subsequent administration of IL-10, the authors observed greatly slowed growth of regenerative fibers in the damaged muscle (Perdiguero et al., 2011). Collectively, these findings show that both the sequence and the timing of the M1 transition to M2 macrophage populations are necessary for normal muscle regeneration.
The complexity of shared signaling systems in muscle regeneration and inflammation
Given the strong evidence that M1 and M2 macrophages are functionally coupled to distinct stages of myogenesis in muscle regeneration, it is tempting to formulate a model in which each macrophage population emerges sequentially and mediates its regenerative effects. However, such a linear scheme is too simplistic because it does not incorporate the simultaneous actions of cytokines on multiple cell types that in turn stimulate the production of additional autocrine and endocrine factors that influence regeneration (Table 1). This complexity in the co-regulation of inflammation and regeneration is particularly well illustrated by focusing on three cytokines, TNFα, IFNγ and IL-10, that play key roles in muscle regeneration (Fig. 3).
Fig. 3.

Coordination of macrophage-mediated signaling and muscle regeneration. Expression of TNFα and IFNγ is elevated during the proliferative stage of myogenesis during muscle regeneration and can contribute to coordinating the inflammatory and myogenic processes by activating macrophages to the M1 phenotype while increasing satellite cell proliferation and inhibiting differentiation. TNFα activation of NFκB in M1 macrophages can promote these effects by further activating TNFα and IFNγ expression in M1 macrophages. Additional positive feedback for M1 activation and satellite cell proliferation can occur through TNFα activation of activin A expression by muscle cells. Increased expression and release of activin A by muscle cells, which has an autocrine effect on muscle cells, inhibits differentiation and maintains muscle cells in the proliferative stage of myogenesis. The released activin A also promotes TNFα expression in macrophages through a SMAD2/3 signaling pathway, maintaining the M1 phenotype. Finally, hypoxia, which is secondary to vascular damage in the injured tissue, activates the expression of genes under the transcriptional control of HIF1α, which promotes the M1 phenotype in macrophages and drives the proliferation of satellite cells. However, TNFα signaling via p38 MAPK can promote the M2 macrophage phenotype, in contrast to the NFκB-mediated activation of the M1 phenotype. As the inflammatory response transitions to a milieu dominated by Th2 cytokines, IL-10 further promotes the M2 phenotype and deactivates M1 macrophages. IL-10 can also act directly on muscle cells through mechanisms that may promote regeneration. IL-4, a Th2 cytokine that is expressed by eosinophils and muscle cells in regenerative muscle, further increases M2 activation through signaling that may be mediated by Kruppel-like factor 4 (KLF4) or PPARγ signaling (Alder et al., 2008; Bouhlel et al., 2007; Bouhlel et al., 2009; Liao et al., 2011). IL-4 can also drive muscle regeneration and growth by direct actions on muscle or by acting through FAPs.
Role of TNFα-mediated signaling
Satellite cells are major targets of TNFα signaling, which can influence their proliferation and differentiation in regenerating muscle. TNFα signaling for this purpose is likely to be mediated primarily by activation of the classical pathway of NFκB signaling, although signaling can also occur via the p38 MAPK (also known as Mapk14) and JNK pathways (see Fig. 3 and Table 1). NFκB is a transcription factor that is normally localized in the cytoplasm in an inactive state as a p50/p65 heterodimer. TNFα binding to its receptor initiates an IKKγ-dependent signaling cascade that causes p50/p65 activation and translocation to the nucleus, where it increases the expression and stability of proteins that influence cell proliferation (Bakkar et al., 2008), increases the degradation of the mRNA for transcription factors that promote muscle differentiation (MyoD) (Guttridge et al., 2000), and decreases the expression of transcriptional repressors that upregulate genes expressed late in muscle regeneration (Wang et al., 2007). Collectively, these TNFα-mediated effects keep satellite cells in the early, proliferative stages of myogenesis, inhibit differentiation and thereby perturb myogenesis. However, once TNFα is removed, muscle may proceed with differentiation and growth founded on an expanded population of satellite cells. Thus, TNFα/NFκB signaling through the classical pathway may yield a net increase in muscle regeneration in some scenarios if the activation by TNFα were transient.
An additional role for TNFα in muscle regeneration might lie in the co-regulation of macrophage phenotype and myogenesis via TNFα-induced activin A release from muscle fibers. Activin A is a mutifunctional growth factor produced by macrophages and muscle. Recent findings showed that stimulation of muscle with TNFα caused activation of TGFβ-activated kinase that induced the release of activin A via two pathways: one requiring NFκB activation and a second requiring activation of p38 MAPK (Trendelenburg et al., 2012). Activin A then mediated autocrine signaling by binding its receptor on the surface of muscle cells, activating SMAD2/3 signaling to inhibit muscle differentiation and fusion (Trendelenburg et al., 2012). Thus, the pathway provides a mechanism through which TNFα can inhibit muscle differentiation and retain muscle cells in the proliferative stage of myogenesis.
Increased release of activin A can also promote the M1 phenotype, at least in vitro. Human monocytes that were biased toward the M1 phenotype by treatment with granulocyte-macrophage colony stimulating factor (GM-CSF; also known as Csf2) produced high levels of activin A and subsequent activin A-mediated signaling via SMAD2/3 resulted in further polarization to the M1 phenotype. In addition, activin A stimulation of macrophages biased toward the M2 phenotype reduced IL-10 production (Sierra-Filardi et al., 2011), which would further reinforce a role for activin A signaling in mediating shifts towards a proinflammatory, M1 phenotype. Together, these results suggest that induction of activin A expression and release via the TNFα/NFκB signaling pathway in muscle provides a mechanism to coordinate the M1 macrophage phenotype with the early proliferative stage of muscle regeneration.
TNFα-mediated signaling can also contribute to coordinating the regulation of macrophage phenotype and muscle regeneration through activation of p38 MAPK. Ablation of TNFα expression prevented the activation of p38 MAPK that normally occurs in muscle following injury caused by CTX injection. This disrupted the recovery of normal muscle architecture while causing persistent elevation of CD11b+ cell numbers, suggesting a prolonged Th1 inflammatory response (Chen et al., 2005). In addition, CTX-injured muscle showed coinciding peaks of p38 MAPK activation and TNFα expression at early stages of muscle inflammation and regeneration followed by coinciding peaks of Th2 cytokines (IL-10 and TGFβ) and activation of MAPK1 phosphatase (MKP1; also known as Dusp1) as regeneration proceeded (Perdiguero et al., 2011). Because MKP1 is a negative regulator of p38 MAPK, the findings suggest that suppression of p38 MAPK signaling in regenerative muscle could cause a switch in macrophage phenotype, as well as regulating myogenesis. Null mutation of Mkp1 also disrupted the timing and magnitude of expression of both Th1 and Th2 cytokines in injured muscle, while slowing the growth of regenerative fibers, further supporting a role for p38 MAPK in coordinating stages of inflammation and regeneration. However, in vitro observations show that MKP1-mediated functions in muscle are more complex than simply promoting muscle differentiation. For example, overexpression of MKP1 in myogenic cells in vitro inhibited differentiation (Bennett and Tonks, 1997), whereas Mkp1 null mutation in cultured myoblasts slowed proliferation and accelerated differentiation of the cells (Shi et al., 2010).
Although many of the features of the complex and apparently conflicting roles of p38 MAPK/MKP1 signaling still await discovery, the work of Dr Puri and colleagues has provided important insights (Palacios et al., 2010). They observed that administration of neutralizing antibodies to TNFα to mdx mice reduced inflammation-activated p38 MAPK signaling and increased the numbers of Pax7-expressing cells while slowing the growth of the regenerating fibers. They also found that the increase in Pax7 expression was due to inhibition of p38 MAPK-mediated phosphorylation of EZH2, which is the enzymatic subunit of polycomb repressive complex 2 (PRC2), and prevention of the formation of a repressive complex on the Pax7 promoter (Palacios et al., 2010). This evidence has provided an epigenetic link between inflammation-activated p38 MAPK signaling and the control of Pax7 expression in satellite cells during muscle regeneration.
Role of IFNγ-mediated signaling
During the early stages of muscle regeneration, elevated expression of IFNγ in myeloid cells, lymphoid cells or perhaps in muscle cells themselves (Cheng et al., 2008) can promote the M1 phenotype and directly influence muscle regeneration. When regenerative muscle transitions from the proliferative stage to early differentiation, IFNγ levels decline as regeneration and growth proceed. However, the mechanisms through which modulating IFNγ levels influence muscle regeneration by direct actions on muscle are only beginning to be understood. Although proliferative muscle cells in vitro experience reduced cycling time when IFNγ-mediated signaling is blocked with neutralizing antibodies to the IFNγ receptor (Cheng et al., 2008), addition of exogenous IFNγ to muscle cell cultures also reduces cell proliferation (Villalta et al., 2011). These apparently conflicting findings might reflect dose-dependent differences in how IFNγ affects myoblast cycling. Relatively low levels of IFNγ increase myoblast proliferation (Kelić et al., 1993), whereas high concentrations inhibit proliferation (Fisher et al., 1983). However, IFNγ also influences post-mitotic myocytes by inhibiting their differentiation, as shown in vitro by reductions in satellite cell fusion, lower expression levels of genes associated with terminal differentiation and lower levels of transcription factors that drive terminal differentiation (Kalovidouris et al., 1993; Villalta et al., 2011; Londhe and Davie, 2011). In particular, myogenin expression by muscle cells is greatly decreased by IFNγ treatments, which contributes to the inhibition of differentiation (Villalta et al., 2011; Londhe and Davie, 2011). However, IFNγ also slows differentiation at later stages of myogenesis, after the myocytes have fused to form myotubes and express myogenin. At this later stage of development, IFNγ induces expression of the major histocompatibility complex class II transactivator (CIITA), which in turn interacts with myogenin to repress its activation of muscle-specific genes required for differentiation (Londhe and Davie, 2011). Together, these observations show that the decline in IFNγ in regenerative muscle could play a role in coordinating the shift in macrophage phenotype with the elimination of a block to muscle differentiation.
Role of IL-10-mediated signaling
Although a function for IL-10 in suppressing M1 activation and promoting activation to the M2 phenotype has been long established, we have learned more recently that IL-10 can act directly on muscle cells to affect their response to stimulation with proinflammatory cytokines and thereby influence their differentiation. For example, treating myoblasts with IL-10 prevented the induction of JNK phosphorylation by TNFα-activated pathways (Strle et al., 2007). Because TNFα can promote satellite cell proliferation and inhibit differentiation through a JNK-mediated pathway in addition to the NFκB and p38 MAPK pathways (Alter et al., 2008), IL-10 may contribute to the transition from the proliferative stage of myogenesis to early differentiation by inhibiting TNFα induction of JNK in myogenic cells.
In vivo observations show that stages of inflammation and stages of muscle regeneration can be synchronized by IL-10 that is likely to be derived from macrophages. For example, ablation of IL-10 expression disrupts the normal transition of macrophages from an M1 to M2 phenotype and diminishes muscle fiber growth during increased muscle use after periods of atrophy (Deng et al., 2012). Although the apparently beneficial effects of IL-10 on muscle regeneration might reflect direct actions of IL-10 on macrophages or myogenic cells (Villalta et al., 2011), recent findings demonstrated that macrophage-derived IL-10 can affect muscle regeneration by promoting mesoangioblast differentiation into the myogenic lineage (Bosurgi et al., 2012). Mesoangioblasts are a population of stem cells associated with the walls of the microvasculature in skeletal muscle that can contribute to regeneration (Sampaolesi et al., 2003). When mesoangioblasts were transplanted into muscle that had been injured by CTX injection, their differentiation was significantly reduced when IL-10 signaling was blocked by an IL-10 receptor (IL-10R)-neutralizing antibody (Bosurgi et al., 2012). Similarly, culturing mesoangioblasts in the presence of M2 macrophages increased the differentiation of mesoangioblasts into myotubes, an effect that was blocked by antibodies to IL-10R. Although these findings support a positive role for macrophage-derived IL-10 in muscle regeneration, the timing of increased IL-10-mediated signaling in muscle might be important. Injecting IL-10 into regenerative muscle before the stage at which endogenous IL-10 was upregulated disrupted the inflammatory response and caused impaired regeneration, as assessed by histology (Perdiguero et al., 2011).
The role of other myeloid cells in regeneration
Most attention to the role of myeloid cells in muscle regeneration has focused on macrophages and their monocyte precursors because they constitute the vast majority of myeloid cells in regenerating muscle. However, eosinophils are also significantly elevated in injured muscle and can reach concentrations as high as 8000 cells/mm3 of skeletal muscle in dystrophic mice (Wehling-Henricks and Tidball, 2011). In the context of chronic muscular dystrophy, eosinophils exacerbate muscle damage through direct cytotoxic effects on the muscle cells, as well as promoting cellular immune responses involving cytotoxic T-cells that can further increase damage (Wehling-Henricks et al., 2008). But in the acute injury setting, eosinophils might play a role in the early stages of muscle regeneration, possibly by secreting IL-4. This is supported by a recent study by Heredia and colleagues, who reported a population of CD11b+/Siglec-F+ (also known as Siglec5) eosinophils that invaded injured muscle and expressed high levels of IL-4 following CTX injections into muscle (Heredia et al., 2013). Ablation of IL-4 expression slowed the recovery of normal muscle architecture (Heredia et al., 2013), at least during the first 8 days following toxin injection. Furthermore, the investigators found that mutation of the IL-4 receptor in macrophages did not affect muscle histology at 8 days following CTX injection, suggesting that the role of M2 macrophages in driving muscle regeneration following CTX-induced damage does not require their activation by IL-4 or IL-13. However, phagocytosis of muscle debris by macrophages is sufficient to induce M1 to M2 phenotype switching in the absence of IL-4 or IL-13 (Arnold et al., 2007; Mounier et al., 2013). In addition, IL-10 signaling is sufficient for transition to the M2 macrophage phenotype that drives muscle regeneration in other injury models (Deng et al., 2012), and there might be a similar IL-10-mediated role in muscle regeneration following injury by toxins.
Much of the beneficial effect of eosinophil-derived IL-4 in regeneration appears to be mediated through a population of stromal cells called fibro/adipogenic progenitors (FAPs), which normally reside in mature muscle in a quiescent state. FAPs derive from a non-myogenic lineage and do not form muscle fibers, but they can nonetheless have a tremendous influence on myogenesis following injury by modulating the microenvironment (Joe et al., 2010). Muscle injury activates FAPs, causing their rapid proliferation within 3 days of the injury, followed by a rapid return to pre-damage levels. Activated FAPs produce high levels of IL-6 and insulin-like growth factor 1, either of which can promote myogenesis (Joe et al., 2010). The study by Heredia et al. (Heredia et al., 2013) suggests that IL-4 activates FAPs, which in turn promote muscle regeneration. However, IL-4-mediated effects on muscle regeneration might also occur through direct actions on muscle cells via autocrine signaling. Muscle cells express IL-4 and IL-4 receptor α (IL-4Rα) throughout myogenesis (Horsley et al., 2003) and IL-4 treatment of muscle cells in vitro increased myocyte fusion and muscle growth. Furthermore, systemic ablation of IL-4Rα expression produced defects in the growth of regenerative muscle fibers that were significant at 25 days post-injury, although undetectable at 10 days post-injury. These observations contrast with the more recent report (Heredia et al., 2013) in which an inducible mutation of IL-4Rα in satellite cells and their progeny produced no detectable defects in histology at 8 days following CTX injection. Whether differences between the findings in the two investigations are attributable to differences in the injury model, sampling times or other variables remains to be determined.
Neutrophils are also prominent constituents of the inflammatory infiltrate of injured muscle, invading within hours of injury (Fielding et al., 1993). Neutrophils produce high levels of free radicals that can lyse muscle membranes and exacerbate damage caused by M1 macrophages (Nguyen and Tidball, 2003). Neutrophils may also contribute to muscle regeneration by phagocytosis of tissue debris and by releasing cytokines that regulate the inflammatory response. Depletion of neutrophils from mice by injections of antibodies to Gr1 (also known as Ly6g) before muscle injury by toxin injection slowed phagocytosis of the damaged muscle and disrupted muscle regeneration, as assessed by the recovery of normal histology following injury (Teixeira et al., 2003). However, the apparent disruption of histology might be attributable in part to the reduced recruitment of macrophages to the injured tissue, secondary to the neutrophil depletion (Teixeira et al., 2003). Neutrophils are rich sources of chemokines and cytokines, such as IL-17 (Ferretti et al., 2003), that can attract and activate macrophages that modulate the regenerative process. Neutrophils also have the capacity to regulate tissue repair more directly; for example, by the release of cytokines that act on muscle cells to influence growth and differentiation. Although this possibility has not been tested explicitly, neutrophils do express and release TNFα (Bazzoni et al., 1991), showing that they might potentially promote the early, proliferative stage of myogenesis. In fact, they reach their highest concentrations in injured muscle during that early stage of myogenesis.
Myeloid cells can modulate muscle regeneration by interactions with the muscle vasculature
Injuries can cause severe damage to the blood supply of muscle, which further exacerbates muscle damage, attributable in part to the reduction in tissue oxygenation that is required to maintain homeostasis. Several experimental findings have demonstrated a relationship between the presence of myeloid cells in injured muscle and revascularization of the tissue, which can thereby influence regeneration through indirect, permissive processes. For example, null mutation of Ccl2 or its receptor Ccr2 reduced macrophage invasion into skeletal muscle following ischemic injury, which delayed restoration of normal vascular perfusion and slowed regeneration (Shireman et al., 2007; Ochoa et al., 2007). Feasibly, the defect in angiogenesis might be due to a reduction in the number of myeloid cells that contribute to revascularization via transdifferentiation. This possibility is supported by the finding that Gr1low/CD11b+ macrophages were observed in the vascular wall following their direct injection into muscle experiencing ischemic injury, and that this was accompanied by improved vascular perfusion (Kim et al., 2010). By contrast, other investigators observed no detectable donor-derived bone marrow cells in the vascular wall of new blood vessels formed in host muscle injured by ischemia (Zentilin et al., 2006) or by CTX injection (Ieronimakis et al., 2012). The discrepancies might be partly attributable to differences in the method of delivery of the cells to the injured muscle or perhaps to differences in the stage of differentiation of the cells at the time of delivery. Regardless of the extent to which donor-derived cells became located in the vascular wall, the studies reported a beneficial effect of the transplanted cells on angiogenesis in vivo (Kim et al., 2010) and in vitro (Ieronimakis et al., 2012).
Neutrophils might also have a role in influencing angiogenesis during muscle repair. Pharmacological inhibition of plasminogen activator inhibitor 1 (PAI1; also known as Serpine1) was recently found to promote angiogenesis in muscle injured by ischemia and reperfusion (I/R) and this was accompanied by elevation of Gr1+ neutrophils in circulation and in muscle (Tashiro et al., 2012). Surprisingly, transplantation of neutrophils from PAI1-inhibited mice to control mice with I/R injury improved revascularization and muscle histology in the recipients (Tashiro et al., 2012), suggesting that the transplanted neutrophils mediated the improved regeneration. In part, the beneficial effect mediated by neutrophils was attributed to increased production of MMP9, which increased angiogenesis. However, the transplanted neutrophils also showed increased levels of expression of vascular endothelial growth factor (VEGF), which is of pre-eminent importance in driving angiogenesis in injured muscle (Tang et al., 2004; Kamba et al., 2006).
The hypoxic environment of injured muscle can also change patterns of gene expression in muscle and myeloid cells that influence the course of muscle repair and regeneration. A key component in this pathway is hypoxia inducible transcription factor 1 (HIF1), which is a heterodimer of α and β subunits (Semenza, 2001). Whereas HIF1β is constitutively expressed, HIF1α is inducible and oxygen responsive, with low oxygen tension increasing its stability. Although HIF1 can activate the expression of hundreds of genes that influence metabolism, its strong induction of VEGF is expected to be particularly important in improving angiogenesis and regeneration (Forsythe et al., 1996).
Evidence from muscle and other tissues indicates that HIF1α-mediated signaling can provide an additional mechanism to coordinate the inflammatory process with aspects of regeneration. Recent findings show that ablation of HIF1α specifically in myeloid cells, but not muscle cells, delayed the invasion of macrophages into regenerating muscle and slowed muscle growth and repair following contusion injury (Scheerer et al., 2013). HIF1α-mediated signaling may also regulate macrophage phenotype. Null mutation of Hif1a increased the expression of M2 macrophage markers and reduced expression of M1 markers, indicating that HIF1α supports the M1 phenotype (Werno et al., 2010). Interestingly, M1 macrophages might also promote vascularization through effects on stem cell differentiation, as suggested by the finding that the differentiation of embryonic stem cells into CD31+ (also known as Pecam1) endothelial cells was decreased in co-cultures with Hif1a-/- macrophages, compared with co-cultures with wild-type macrophages (Werno et al., 2010). However, this effect of M1 macrophages on adult stem cell populations has not yet been explored. Collectively, these findings indicate that reduced oxygen tension in injured muscle results in activation of HIF1α signaling, which then has multiple downstream effects, such as increased inflammation, promotion of the M1 macrophage phenotype and possibly increased vascularization, all of which can affect regeneration.
Myeloid cells contribute to fibrosis during muscle regeneration
Muscle injury followed by regeneration is invariably accompanied by turnover of the extracellular matrix (ECM) as tissue debris is removed and replaced. The accumulation of fibrinogen or its proteolytic fragment fibrin (both forms are collectively referred to herein as fibrin) in injured muscle may play a key role in determining whether subsequent regeneration is facilitated or impaired by connective tissue production. Excessive accumulation of the fibrin matrix or defects in its removal lead to increased fibrosis in both the acute and chronic injury settings, causing defects in muscle regeneration (Suelves et al., 2002; Suelves et al., 2007). Furthermore, perturbation of fibrin turnover also affects the inflammatory response to muscle injury, which may in turn influence regeneration. Initial reports showed that the loss of plasmin activity that caused fibrin accumulation in acutely injured muscle also decreased Mac1+ (CD11b) myeloid cell invasion into the muscle (Suelves et al., 2002). By contrast, reversal of fibrin accumulations in mdx muscle by treatments with a fibrinolytic agent also reduced myeloid cell numbers in muscles (Vidal et al., 2008). The apparent incongruity of the findings might result from differences in the relative importance of different fibrin-mediated functions in the response of muscle to acute injury versus chronic injury. Increased accumulation of fibrin in acutely injured muscles of plasmin null mice might reflect impaired myeloid cell migration into injured muscle. Alternatively, reduced myeloid cell activation in fibrin-depleted mdx mice might reflect the loss of proinflammatory effects of fibrin on myeloid cells. Observations supporting this latter possibility include the finding that stimulation of mdx muscle macrophages with fibrinogen in vitro biases them toward the M1 phenotype, increasing the expression of TNFα and IL-1β (Vidal et al., 2008). However, prevention or reversal of fibrin accumulation in mdx muscle reduces M2 macrophage numbers in muscle (Vidal et al., 2008), suggesting that the net effect of fibrin-mediated signaling on macrophage phenotypes in vivo might be under more complex regulation.
The finding that accumulations of fibrin in mdx muscle can cause elevations in M2 macrophages suggests the underpinnings of a feed-forward mechanism through which pathological fibrosis impedes muscle regeneration. Activated M2 macrophages express TGFβ and arginase 1, either of which can play important roles in driving fibrosis and thereby impede muscle regeneration. Thus, driving macrophages to the M2 phenotype in regenerating muscle might improve tissue growth and repair, as noted above, or impede the regenerative process by increasing ECM production. This overinduction of M2 macrophages in injured muscle can have significant pathological consequences. For example, during the late, progressive stages of muscle or cardiac pathology in mdx mice, fibrosis is a major pathological feature that contributes substantially to lethality. In these mice, the muscles and heart show continuously elevated populations of M2 macrophages expressing arginase 1 at progressive stages of the disease (Wehling-Henricks et al., 2010). However, ablation of arginase 1 expression greatly attenuates mdx muscle fibrosis, showing that the beneficial effects of M2 macrophage functions on muscle regeneration can also have significant negative consequences in chronic disease states (Wehling-Henricks et al., 2010).
Perspectives and conclusions
Although it has been recognized for decades that myeloid cells are present in regenerative muscle, the crucial role they play in the regenerative process is a relatively recent observation. Despite this, progress in the field has been dramatic and the role of specific myeloid populations and the factors that regulate their activity is becoming increasingly understood. For example, a key observation is that the roles played by various myeloid cells can vary greatly over the course of muscle regeneration, with cells that promote damage at one stage of the regenerative process contributing to repair at other stages. Evidence is also accumulating to show that the roles played by myeloid cells in muscle regeneration can vary with the injury or with the disease context in which injury occurs.
The studies to date have relied extensively on observing the effects of manipulating single cells or molecules with a binary ‘readout’ as either good or bad for regeneration. Although this approach might be simplistic, it has nevertheless been useful in identifying a role for various myeloid cells in muscle regeneration. Such approaches might still be valuable for providing insights into the poorly understood role of non-macrophage/monocyte myeloid populations, such as eosinophils and neutrophils, in muscle growth and regeneration. More information is also needed regarding the role played by other, non-myeloid effector populations. In this regard, the discovery that FAPs, a cell type identified only recently, could play a key role in regeneration suggests that other as yet unknown players might emerge as centrally important in regeneration.
This Review provides an overview of some of the signaling pathways that are activated by myeloid cells as they mediate their degenerative or regenerative functions. However, we have only scratched the surface in terms of signaling complexity, and much more must be learned before targeted approaches to activate or inactivate specific pathways in selected cell types can be contemplated. Ultimately, the challenge, which is not unique to the field of muscle regeneration, is to develop systems and/or analytical tools that will allow us to delineate the cascade of events and intricate intercellular interactions involved in a complex biological process. With regard to muscle regeneration, this means that systems to probe complex interactions between different myeloid effectors, soluble mediators and regenerating muscle cells are needed. It might not be possible to achieve this using a single model. Instead, results from in vitro and in vivo approaches will probably need to be employed and the results integrated.
The goal of the research reviewed here is to translate the reported findings into treatments for muscle pathologies and injuries. The discoveries that have been outlined have illuminated new potential strategies through which the hematopoietic system may be manipulated to improve muscle regeneration. Of these, several approaches are worth particular consideration, as discussed below.
Administration of soluble factors
Although there are beneficial effects of inflammation in the short term, a chronic inflammatory response can be deleterious. Thus, the systemic administration of soluble factors that affect the duration of inflammation could have a salutary effect on muscle regeneration. This strategy has been tested in preclinical models for the treatment of DMD by attempting to reduce the activation of cytolytic inflammatory cells, such as M1 macrophages, via ablation of TNFα signaling (Grounds and Torrisi, 2004; Hodgetts et al., 2006). Such treatments have mitigated inflammation-associated tissue damage in other conditions such as rheumatoid arthritis. However, a particular caution for blocking TNFα signaling in chronic muscle diseases is that TNFα also plays important positive roles in myogenesis. Although short-term administration to reduce inflammatory cell-mediated damage may be beneficial, these effects might be outweighed by long-term negative effects on muscle regeneration, as well as off-target effects that result from systemic delivery.
Direct injection of macrophages
The direct injection of select populations of myeloid cells into injured or diseased muscle at the site of damage could modulate the inflammatory response and minimize off-target effects. This strategy has shown some promise in recent pre-clinical work, again using the mdx mouse model. Here, incorporation of GFP-labeled Pax7+ cells into mdx muscles was significantly greater if the Pax7+ cells were co-injected with F4/80+ mature macrophages (Lesault et al., 2012). The co-injection of macrophages also improved dispersal of the transplanted muscle cells in the recipient muscles. These observations suggest that the presence of elevated numbers of macrophages improves the survival or proliferation of the transplanted muscle cells, which could help overcome a major obstacle to some cell-based strategies for the treatment of muscle disease. Interestingly, at 5 days post-injection, the great majority of the transplanted macrophages were CD206+ (also known as Mrc1) M2 macrophages, suggesting that bias toward the M2 phenotype might be beneficial. However, an additional investigation showed that co-injection of muscle cells with human M1 macrophages into immunodeficient [Rag2-/- γC-/- (also known as Il2rg)] mdx mice improved the proliferation and dispersal of transplanted muscle cells, although co-injection with M2 macrophages did not affect these outcomes (Bencze et al., 2012). In that study, macrophages were activated to the M1 phenotype with LPS and IFNγ or to the M2 phenotype with dexamethasone and IL-10, prior to injection. Together, the two sets of findings support the possibility that amplification of myeloid populations can improve the delivery of cells for therapeutic use, although the contexts in which M1 or M2 macrophages are most beneficial need to be determined.
Manipulation of cellular phenotype
Recent findings indicate that the manipulation of myeloid cell phenotypes might also be exploited to improve the viability of transplanted materials following the large-scale loss of muscle tissue due to trauma or presence of a tumor. This type of muscle loss, called volumetric muscle loss, can be partially overcome using implants of biological origin consisting largely of ECM scaffolding materials that support the growth of restorative tissues. However, implantation of these materials frequently induces a foreign body reaction that causes destruction or encapsulation of the transplanted material. Because macrophages are the major effector cell type in foreign body reactions, recent investigations have assessed whether differences in macrophage phenotype during engraftment of scaffolding materials can reduce the foreign body reaction and thus improve muscle regeneration. Indeed, the success of implantation of a biological scaffold to replace skeletal muscle was strongly influenced by whether the inflammatory response to the implant was biased toward M1 or M2 macrophages (Brown et al., 2009). The investigators had previously shown that apparently minor features of the preparation of the scaffolding material could determine the macrophage phenotype that predominated in the inflammatory response and the success of the implant. For example, when using porcine small intestinal submucosa (SIS) as a transplanted material, they found that crosslinked SIS-ECM elicited an inflammatory response dominated by M1 macrophages, whereas non-crosslinked SIS-ECM generated an infiltrate that was primarily M2 macrophages and produced a superior regenerative response (Badylak et al., 2008). However, the extent to which the improved histology was attributable to the predominance of M2 macrophages and the underlying mechanisms through which different scaffolds yield distinct inflammatory responses await discovery.
Injection of non-macrophages into muscle
Although their in vivo numbers are limited, additional myeloid cells, such as eosinophils, might have therapeutic potential. For example, signaling initiated by eosinophil-derived IL-4 is associated with improved muscle regeneration following toxin injection (Heredia et al., 2013), but whether interventions to increase eosinophil numbers in diseased muscle would have a net beneficial effect is unknown. Notably, depletion of eosinophils from dystrophic muscle reduced pathology rather than impairing regeneration, suggesting that the role of eosinophils in muscle injury and repair might vary with the severity or type of injury or disease (Wehling-Henricks et al., 2008). Similarly, neutrophils have the capacity to contribute to regeneration, suggesting that amplification of their functions in muscle injury or disease could have a beneficial effect, although they can also promote injury in at least some injury models.
Future challenges
The above interventions are well within the realm of possibility. It is now possible to produce unlimited volumes of various cytokines, and the engineering of hematopoietic cells that express specific receptors to mediate a desired function is an active area of investigation. Thus, continued progress in how myeloid cells affect muscle regeneration and how to translate this information can be expected. Of course, progress might not be linear: we have limited knowledge of how the activities of regenerating muscle cells themselves can modulate myeloid cell functions. We also know little about whether feedback from other systems, such as the nervous or endocrine systems, plays a role in regulating muscle interactions with inflammatory cells during muscle regeneration. Therefore, while optimism that progress will be made is warranted, there is also little doubt that surprises lie ahead.
Acknowledgments
We are grateful to the anonymous reviewers for their helpful critiques of this Review.
Footnotes
Competing interests
The authors declare no competing financial interests.
Funding
During the preparation of this work, support was received from the National Institutes of Health and the Muscular Dystrophy Association, USA, to J.G.T. Deposited in PMC for release after 12 months.
References
- Alder J. K., Georgantas R. W., III, Hildreth R. L., Kaplan I. M., Morisot S., Yu X., McDevitt M., Civin C. I. (2008). Kruppel-like factor 4 is essential for inflammatory monocyte differentiation in vivo. J. Immunol. 180, 5645–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R. E., Boxhorn L. K. (1989). Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J. Cell. Physiol. 138, 311–315 [DOI] [PubMed] [Google Scholar]
- Alter J., Rozentzweig D., Bengal E. (2008). Inhibition of myoblast differentiation by tumor necrosis factor alpha is mediated by c-Jun N-terminal kinase 1 and leukemia inhibitory factor. J. Biol. Chem. 283, 23224–23234 [DOI] [PubMed] [Google Scholar]
- Arnold L., Henry A., Poron F., Baba-Amer Y., van Rooijen N., Plonquet A., Gherardi R. K., Chazaud B. (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badylak S. F., Valentin J. E., Ravindra A. K., McCabe G. P., Stewart-Akers A. M. (2008). Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Eng. Part A 14, 1835–1842 [DOI] [PubMed] [Google Scholar]
- Bakkar N., Wang J., Ladner K. J., Wang H., Dahlman J. M., Carathers M., Acharyya S., Rudnicki M. A., Hollenbach A. D., Guttridge D. C. (2008). IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J. Cell Biol. 180, 787–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassols A., Massagué J. (1988). Transforming growth factor beta regulates the expression and structure of extracellular matrix chondroitin/dermatan sulfate proteoglycans. J. Biol. Chem. 263, 3039–3045 [PubMed] [Google Scholar]
- Bazzoni F., Cassatella M. A., Laudanna C., Rossi F. (1991). Phagocytosis of opsonized yeast induces tumor necrosis factor-alpha mRNA accumulation and protein release by human polymorphonuclear leukocytes. J. Leukoc. Biol. 50, 223–228 [DOI] [PubMed] [Google Scholar]
- Bencze M., Negroni E., Vallese D., Yacoub-Youssef H., Chaouch S., Wolff A., Aamiri A., Di Santo J. P., Chazaud B., Butler-Browne G., et al. (2012). Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol. Ther. 20, 2168–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett A. M., Tonks N. K. (1997). Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278, 1288–1291 [DOI] [PubMed] [Google Scholar]
- Beutler B., Greenwald D., Hulmes J. D., Chang M., Pan Y. C., Mathison J., Ulevitch R., Cerami A. (1985). Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 316, 552–554 [DOI] [PubMed] [Google Scholar]
- Bosurgi L., Corna G., Vezzoli M., Touvier T., Cossu G., Manfredi A. A., Brunelli S., Rovere-Querini P. (2012). Transplanted mesoangioblasts require macrophage IL-10 for survival in a mouse model of muscle injury. J. Immunol. 188, 6267–6277 [DOI] [PubMed] [Google Scholar]
- Bouhlel M. A., Derudas B., Rigamonti E., Dièvart R., Brozek J., Haulon S., Zawadzki C., Jude B., Torpier G., Marx N., et al. (2007). PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143 [DOI] [PubMed] [Google Scholar]
- Bouhlel M. A., Brozek J., Derudas B., Zawadzki C., Jude B., Staels B., Chinetti-Gbaguidi G. (2009). Unlike PPARgamma, PPARalpha or PPARbeta/delta activation does not promote human monocyte differentiation toward alternative macrophages. Biochem. Biophys. Res. Commun. 386, 459–462 [DOI] [PubMed] [Google Scholar]
- Boutard V., Havouis R., Fouqueray B., Philippe C., Moulinoux J. P., Baud L. (1995). Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J. Immunol. 155, 2077–2084 [PubMed] [Google Scholar]
- Brown B. N., Valentin J. E., Stewart-Akers A. M., McCabe G. P., Badylak S. F. (2009). Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 30, 1482–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. E., Gerken E., Zhang Y., Zhan M., Mohan R. K., Li A. S., Reid M. B., Li Y. P. (2005). Role of TNF-alpha signaling in regeneration of cardiotoxin-injured muscle. Am. J. Physiol. 289, C1179–C1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Nguyen M. H., Fantuzzi G., Koh T. J. (2008). Endogenous interferon-gamma is required for efficient skeletal muscle regeneration. Am. J. Physiol. 294, C1183–C1191 [DOI] [PubMed] [Google Scholar]
- Contreras-Shannon V., Ochoa O., Reyes-Reyna S. M., Sun D., Michalek J. E., Kuziel W. A., McManus L. M., Shireman P. K. (2007). Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2-/- mice following ischemic injury. Am. J. Physiol. 292, C953–C967 [DOI] [PubMed] [Google Scholar]
- Cornelison D. D., Wold B. J. (1997). Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol. 191, 270–283 [DOI] [PubMed] [Google Scholar]
- Cunha F. Q., Moncada S., Liew F. Y. (1992). Interleukin-10 (IL-10) inhibits the induction of nitric oxide synthase by interferon-gamma in murine macrophages. Biochem. Biophys. Res. Commun. 182, 1155–1159 [DOI] [PubMed] [Google Scholar]
- Cunha F. Q., Assreuy J., Xu D., Charles I., Liew F. Y., Moncada S. (1993). Repeated induction of nitric oxide synthase and leishmanicidal activity in murine macrophages. Eur. J. Immunol. 23, 1385–1388 [DOI] [PubMed] [Google Scholar]
- Darwich L., Coma G., Peña R., Bellido R., Blanco E. J., Este J. A., Borras F. E., Clotet B., Ruiz L., Rosell A., et al. (2009). Secretion of interferon-γ by human macrophages demonstrated at the single-cell level after costimulation with interleukin (IL)-12 plus IL-18. Immunology 126, 386–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waal Malefyt R., Figdor C. G., Huijbens R., Mohan-Peterson S., Bennett B., Culpepper J., Dang W., Zurawski G., de Vries J. E. (1993). Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J. Immunol. 151, 6370–6381 [PubMed] [Google Scholar]
- Del Prete G., De Carli M., Almerigogna F., Giudizi M. G., Biagiotti R., Romagnani S. (1993). Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. J. Immunol. 150, 353–360 [PubMed] [Google Scholar]
- Deng B., Wehling-Henricks M., Villalta S. A., Wang Y., Tidball J. G. (2012). IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 189, 3669–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essner R., Rhoades K., McBride W. H., Morton D. L., Economou J. S. (1989). IL-4 down-regulates IL-1 and TNF gene expression in human monocytes. J. Immunol. 142, 3857–3861 [PubMed] [Google Scholar]
- Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V. A., Bratton D. L., Guthrie L., Henson P. M. (2001). Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J. Immunol. 166, 6847–6854 [DOI] [PubMed] [Google Scholar]
- Fargeas C., Wu C. Y., Nakajima T., Cox D., Nutman T., Delespesse G. (1992). Differential effect of transforming growth factor beta on the synthesis of Th1- and Th2-like lymphokines by human T lymphocytes. Eur. J. Immunol. 22, 2173–2176 [DOI] [PubMed] [Google Scholar]
- Ferretti S., Bonneau O., Dubois G. R., Jones C. E., Trifilieff A. (2003). IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170, 2106–2112 [DOI] [PubMed] [Google Scholar]
- Fielding R. A., Manfredi T. J., Ding W., Fiatarone M. A., Evans W. J., Cannon J. G. (1993). Acute phase response in exercise. III. Neutrophil and IL-1 beta accumulation in skeletal muscle. Am. J. Physiol. 265, R166–R172 [DOI] [PubMed] [Google Scholar]
- Fisher P. B., Miranda A. F., Babiss L. E., Pestka S., Weinstein I. B. (1983). Opposing effects of interferon produced in bacteria and of tumor promoters on myogenesis in human myoblast cultures. Proc. Natl. Acad. Sci. USA 80, 2961–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleetwood A. J., Lawrence T., Hamilton J. A., Cook A. D. (2007). Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J. Immunol. 178, 5245–5252 [DOI] [PubMed] [Google Scholar]
- Forsythe J. A., Jiang B. H., Iyer N. V., Agani F., Leung S. W., Koos R. D., Semenza G. L. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füchtbauer E. M., Westphal H. (1992). MyoD and myogenin are coexpressed in regenerating skeletal muscle of the mouse. Dev. Dyn. 193, 34–39 [DOI] [PubMed] [Google Scholar]
- Grotendorst G. R., Smale G., Pencev D. (1989). Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J. Cell. Physiol. 140, 396–402 [DOI] [PubMed] [Google Scholar]
- Grounds M. D., Torrisi J. (2004). Anti-TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 18, 676–682 [DOI] [PubMed] [Google Scholar]
- Grounds M. D., Garrett K. L., Lai M. C., Wright W. E., Beilharz M. W. (1992). Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res. 267, 99–104 [DOI] [PubMed] [Google Scholar]
- Gussoni E., Pavlath G. K., Miller R. G., Panzara M. A., Powell M., Blau H. M., Steinman L. (1994). Specific T cell receptor gene rearrangements at the site of muscle degeneration in Duchenne muscular dystrophy. J. Immunol. 153, 4798–4805 [PubMed] [Google Scholar]
- Guttridge D. C., Mayo M. W., Madrid L. V., Wang C. Y., Baldwin A. S., Jr (2000). NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366 [DOI] [PubMed] [Google Scholar]
- Heredia J. E., Mukundan L., Chen F. M., Mueller A. A., Deo R. C., Locksley R. M., Rando T. A., Chawla A. (2013). Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse M., Modolell M., La Flamme A. C., Schito M., Fuentes J. M., Cheever A. W., Pearce E. J., Wynn T. A. (2001). Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J. Immunol. 167, 6533–6544 [DOI] [PubMed] [Google Scholar]
- Hibbs J. B., Jr, Taintor R. R., Vavrin Z., Rachlin E. M. (1988). Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 157, 87–94 [DOI] [PubMed] [Google Scholar]
- Hodgetts S., Radley H., Davies M., Grounds M. D. (2006). Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul. Disord. 16, 591–602 [DOI] [PubMed] [Google Scholar]
- Hoffman E. P., Brown R. H., Jr, Kunkel L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 [DOI] [PubMed] [Google Scholar]
- Horsley V., Jansen K. M., Mills S. T., Pavlath G. K. (2003). IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 113, 483–494 [DOI] [PubMed] [Google Scholar]
- Ieronimakis N., Hays A., Reyes M. (2012). Bone marrow-derived cells do not engraft into skeletal muscle microvasculature but promote angiogenesis after acute injury. Exp. Hematol. 40, 238-249, e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe A. W., Yi L., Natarajan A., Le Grand F., So L., Wang J., Rudnicki M. A., Rossi F. M. (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic D. V., Di Battista J. A., Martel-Pelletier J., Jolicoeur F. C., He Y., Zhang M., Mineau F., Pelletier J. P. (1998). IL-17 stimulates the production and expression of proinflammatory cytokines, IL-β and TNF-α, by human macrophages. J. Immunol. 160, 3513–3521 [PubMed] [Google Scholar]
- Kalovidouris A. E., Plotkin Z., Graesser D. (1993). Interferon-gamma inhibits proliferation, differentiation, and creatine kinase activity of cultured human muscle cells. II. A possible role in myositis. J. Rheumatol. 20, 1718–1723 [PubMed] [Google Scholar]
- Kamba T., Tam B. Y., Hashizume H., Haskell A., Sennino B., Mancuso M. R., Norberg S. M., O’Brien S. M., Davis R. B., Gowen L. C., et al. (2006). VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am. J. Physiol. 290, H560–H576 [DOI] [PubMed] [Google Scholar]
- Kehrl J. H., Wakefield L. M., Roberts A. B., Jakowlew S., Alvarez-Mon M., Derynck R., Sporn M. B., Fauci A. S. (1986). Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 163, 1037–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelić S., Olsson T., Kristensson K. (1993). Interferon-gamma promotes proliferation of rat skeletal muscle cells in vitro and alters their AChR distribution. J. Neurol. Sci. 114, 62–67 [DOI] [PubMed] [Google Scholar]
- Kellner A., Robertson T. (1954). Selective necrosis of cardiac and skeletal muscle induced experimentally by means of proteolytic enzyme solutions given intravenously. J. Exp. Med. 99, 387–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. Y., Battaile J. T., Patel A. C., You Y., Agapov E., Grayson M. H., Benoit L. A., Byers D. E., Alevy Y., Tucker J., et al. (2008). Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat. Med. 14, 633–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. A., March K., Chae H. D., Johnstone B., Park S. J., Cook T., Merfeld-Clauss S., Broxmeyer H. E. (2010). Muscle-derived Gr1(dim)CD11b(+) cells enhance neovascularization in an ischemic hind limb mouse model. Blood 116, 1623–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Flamme A. C., Kharkrang M., Stone S., Mirmoeini S., Chuluundorj D., Kyle R. (2012). Type II-activated murine macrophages produce IL-4. PLoS ONE 7, e46989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesault P. F., Theret M., Magnan M., Cuvellier S., Niu Y., Gherardi R. K., Tremblay J. P., Hittinger L., Chazaud B. (2012). Macrophages improve survival, proliferation and migration of engrafted myogenic precursor cells into MDX skeletal muscle. PLoS ONE 7, e46698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. P. (2003). TNF-alpha is a mitogen in skeletal muscle. Am. J. Physiol. 285, C370–C376 [DOI] [PubMed] [Google Scholar]
- Liao X., Sharma N., Kapadia F., Zhou G., Lu Y., Hong H., Paruchuri K., Mahabeleshwar G. H., Dalmas E., Venteclef N., et al. (2011). Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Invest. 121, 2736–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londhe P., Davie J. K. (2011). Gamma interferon modulates myogenesis through the major histocompatibility complex class II transactivator, CIITA. Mol. Cell. Biol. 31, 2854–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof A., Penny M., Kingston R., Murray C., Islam S., Bedford P. A., Knight S. C. (2006). Interleukin-4 can induce interleukin-4 production in dendritic cells. Immunology 117, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J., Cheifetz S., Endo T., Nadal-Ginard B. (1986). Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc. Natl. Acad. Sci. USA 83, 8206–8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K., Campbell K. P. (1994). Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve 17, 2–15 [DOI] [PubMed] [Google Scholar]
- Mauro A. (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney-Francis N., Mizel D., Wong H., Wahl L., Wahl S. (1990). TGF-β regulates production of growth factors and TGF-β by human peripheral blood monocytes. Growth Factors 4, 27–35 [DOI] [PubMed] [Google Scholar]
- McKenzie A. N., Culpepper J. A., de Waal Malefyt R., Brière F., Punnonen J., Aversa G., Sato A., Dang W., Cocks B. G., Menon S., et al. (1993). Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc. Natl. Acad. Sci. USA 90, 3735–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. C., Ito H., Blau H. M., Torti F. M. (1988). Tumor necrosis factor inhibits human myogenesis in vitro. Mol. Cell. Biol. 8, 2295–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills C. D., Kincaid K., Alt J. M., Heilman M. J., Hill A. M. (2000). M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 164, 6166–6173 10843666 [Google Scholar]
- Modolell M., Corraliza I. M., Link F., Soler G., Eichmann K. (1995). Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur. J. Immunol. 25, 1101–1104 [DOI] [PubMed] [Google Scholar]
- Moss F. P., Leblond C. P. (1971). Satellite cells as the source of nuclei in muscles of growing rats. Anat. Rec. 170, 421–435 [DOI] [PubMed] [Google Scholar]
- Mounier R., Théret M., Arnold L., Cuvellier S., Bultot L., Göransson O., Sanz N., Ferry A., Sakamoto K., Foretz M., et al. (2013). AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 18, 251–264 [DOI] [PubMed] [Google Scholar]
- Nahrendorf M., Swirski F. K., Aikawa E., Stangenberg L., Wurdinger T., Figueiredo J. L., Libby P., Weissleder R., Pittet M. J. (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A. S., Page R. C., Swanson J. (1989). Collagen synthesis by human fibroblasts. Regulation by transforming growth factor-beta in the presence of other inflammatory mediators. Biochem. J. 260, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. X., Tidball J. G. (2003). Interactions between neutrophils and macrophages promote macrophage killing of rat muscle cells in vitro. J. Physiol. 547, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa O., Sun D., Reyes-Reyna S. M., Waite L. L., Michalek J. E., McManus L. M., Shireman P. K. (2007). Delayed angiogenesis and VEGF production in CCR2-/- mice during impaired skeletal muscle regeneration. Am. J. Physiol. 293, R651–R661 [DOI] [PubMed] [Google Scholar]
- Oswald I. P., Gazzinelli R. T., Sher A., James S. L. (1992). IL-10 synergizes with IL-4 and transforming growth factor-beta to inhibit macrophage cytotoxic activity. J. Immunol. 148, 3578–3582 [PubMed] [Google Scholar]
- Pace J. L., Russell S. W., Schreiber R. D., Altman A., Katz D. H. (1983). Macrophage activation: priming activity from a T-cell hybridoma is attributable to interferon-gamma. Proc. Natl. Acad. Sci. USA 80, 3782–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios D., Mozzetta C., Consalvi S., Caretti G., Saccone V., Proserpio V., Marquez V. E., Valente S., Mai A., Forcales S. V., et al. (2010). TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7, 455–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiguero E., Sousa-Victor P., Ruiz-Bonilla V., Jardí M., Caelles C., Serrano A. L., Muñoz-Cánoves P. (2011). p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol. 195, 307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip R., Epstein L. B. (1986). Tumour necrosis factor as immunomodulator and mediator of monocyte cytotoxicity induced by itself, gamma-interferon and interleukin-1. Nature 323, 86–89 [DOI] [PubMed] [Google Scholar]
- Piehler D., Stenzel W., Grahnert A., Held J., Richter L., Köhler G., Richter T., Eschke M., Alber G., Müller U. (2011). Eosinophils contribute to IL-4 production and shape the T-helper cytokine profile and inflammatory response in pulmonary cryptococcosis. Am. J. Pathol. 179, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwaite A. E., Keski-Oja J., Moses H. L., Kang A. H. (1987). Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J. Exp. Med. 165, 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. B., Sporn M. B., Assoian R. K., Smith J. M., Roche N. S., Wakefield L. M., Heine U. I., Liotta L. A., Falanga V., Kehrl J. H., et al. (1986). Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 83, 4167–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson T. A., Maley M. A., Grounds M. D., Papadimitriou J. M. (1993). The role of macrophages in skeletal muscle regeneration with particular reference to chemotaxis. Exp. Cell Res. 207, 321–331 [DOI] [PubMed] [Google Scholar]
- Ruffell D., Mourkioti F., Gambardella A., Kirstetter P., Lopez R. G., Rosenthal N., Nerlov C. (2009). A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 106, 17475–17480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaolesi M., Torrente Y., Innocenzi A., Tonlorenzi R., D’Antona G., Pellegrino M. A., Barresi R., Bresolin N., De Angelis M. G., Campbell K. P., et al. (2003). Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 301, 487–492 [DOI] [PubMed] [Google Scholar]
- Scheerer N., Dehne N., Stockmann C., Swoboda S., Baba H. A., Neugebauer A., Johnson R. S., Fandrey J. (2013). Myeloid hypoxia-inducible factor-1α is essential for skeletal muscle regeneration in mice. J. Immunol. 191, 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P., Sabourin L. A., Girgis-Gabardo A., Mansouri A., Gruss P., Rudnicki M. A. (2000). Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786 [DOI] [PubMed] [Google Scholar]
- Segawa M., Fukada S., Yamamoto Y., Yahagi H., Kanematsu M., Sato M., Ito T., Uezumi A., Hayashi S., Miyagoe-Suzuki Y., et al. (2008). Suppression of macrophage functions impairs skeletal muscle regeneration with severe fibrosis. Exp. Cell Res. 314, 3232–3244 [DOI] [PubMed] [Google Scholar]
- Semenza G. L. (2001). HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 13, 167–171 [DOI] [PubMed] [Google Scholar]
- Shafiq S. A., Gorycki M. A., Mauro A. (1968). Mitosis during postnatal growth in skeletal and cardiac muscle of the rat. J. Anat. 103, 135–141 [PMC free article] [PubMed] [Google Scholar]
- Shi H., Boadu E., Mercan F., Le A. M., Flach R. J., Zhang L., Tyner K. J., Olwin B. B., Bennett A. M. (2010). MAP kinase phosphatase-1 deficiency impairs skeletal muscle regeneration and exacerbates muscular dystrophy. FASEB J. 24, 2985–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shireman P. K., Contreras-Shannon V., Ochoa O., Karia B. P., Michalek J. E., McManus L. M. (2007). MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J. Leukoc. Biol. 81, 775–785 [DOI] [PubMed] [Google Scholar]
- Sierra-Filardi E., Puig-Kröger A., Blanco F. J., Nieto C., Bragado R., Palomero M. I., Bernabéu C., Vega M. A., Corbí A. L. (2011). Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood 117, 5092–5101 [DOI] [PubMed] [Google Scholar]
- Snow M. H. (1977). Myogenic cell formation in regenerating rat skeletal muscle injured by mincing. I. A fine structural study. Anat. Rec. 188, 181–199 [DOI] [PubMed] [Google Scholar]
- St Pierre B. A., Tidball J. G. (1994). Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J. Appl. Physiol. 77, 290–297 [DOI] [PubMed] [Google Scholar]
- Starnes T., Robertson M. J., Sledge G., Kelich S., Nakshatri H., Broxmeyer H. E., Hromas R. (2001). Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J. Immunol. 167, 4137–4140 [DOI] [PubMed] [Google Scholar]
- Stein M., Keshav S., Harris N., Gordon S. (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strle K., McCusker R. H., Tran L., King A., Johnson R. W., Freund G. G., Dantzer R., Kelley K. W. (2007). Novel activity of an anti-inflammatory cytokine: IL-10 prevents TNFalpha-induced resistance to IGF-I in myoblasts. J. Neuroimmunol. 188, 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suelves M., López-Alemany R., Lluís F., Aniorte G., Serrano E., Parra M., Carmeliet P., Muñoz-Cánoves P. (2002). Plasmin activity is required for myogenesis in vitro and skeletal muscle regeneration in vivo. Blood 99, 2835–2844 [DOI] [PubMed] [Google Scholar]
- Suelves M., Vidal B., Serrano A. L., Tjwa M., Roma J., López-Alemany R., Luttun A., de Lagrán M. M., Díaz-Ramos A., Jardí M., et al. (2007). uPA deficiency exacerbates muscular dystrophy in MDX mice. J. Cell Biol. 178, 1039–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summan M., Warren G. L., Mercer R. R., Chapman R., Hulderman T., Van Rooijen N., Simeonova P. P. (2006). Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am. J. Physiol. 290, R1488–R1495 [DOI] [PubMed] [Google Scholar]
- Sun D., Martinez C. O., Ochoa O., Ruiz-Willhite L., Bonilla J. R., Centonze V. E., Waite L. L., Michalek J. E., McManus L. M., Shireman P. K. (2009). Bone marrow-derived cell regulation of skeletal muscle regeneration. FASEB J. 23, 382–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay K., Rázga Z., Duda E. (1997). TNF inhibits myogenesis and downregulates the expression of myogenic regulatory factors myoD and myogenin. Eur. J. Cell Biol. 74, 391–398 [PubMed] [Google Scholar]
- Tang K., Breen E. C., Gerber H. P., Ferrara N. M., Wagner P. D. (2004). Capillary regression in vascular endothelial growth factor-deficient skeletal muscle. Physiol. Genomics 18, 63–69 [DOI] [PubMed] [Google Scholar]
- Tashiro Y., Nishida C., Sato-Kusubata K., Ohki-Koizumi M., Ishihara M., Sato A., Gritli I., Komiyama H., Sato Y., Dan T., et al. (2012). Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice. Blood 119, 6382–6393 [DOI] [PubMed] [Google Scholar]
- Teixeira C. F., Zamunér S. R., Zuliani J. P., Fernandes C. M., Cruz-Hofling M. A., Fernandes I., Chaves F., Gutiérrez J. M. (2003). Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected with Bothrops asper snake venom. Muscle Nerve 28, 449–459 [DOI] [PubMed] [Google Scholar]
- Tidball J. G., Wehling-Henricks M. (2007). Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol. 578, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg A. U., Meyer A., Jacobi C., Feige J. N., Glass D. J. (2012). TAK-1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skelet. Muscle 2, 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal B., Serrano A. L., Tjwa M., Suelves M., Ardite E., De Mori R., Baeza-Raja B., Martínez de Lagrán M., Lafuste P., Ruiz-Bonilla V., et al. (2008). Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 22, 1747–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta S. A., Nguyen H. X., Deng B., Gotoh T., Tidball J. G. (2009). Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 18, 482–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta S. A., Rinaldi C., Deng B., Liu G., Fedor B., Tidball J. G. (2011). Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet. 20, 790–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovotz Y., Bogdan C., Paik J., Xie Q. W., Nathan C. (1993). Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor beta. J. Exp. Med. 178, 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann R. (1893). Uber die regeneration des quergestreiften muskelgewebes beim menschen und saugetier. Beitr. Pathol. Anat. 12, 233–332 [Google Scholar]
- Wahl S. M., Hunt D. A., Wakefield L. M., McCartney-Francis N., Wahl L. M., Roberts A. B., Sporn M. B. (1987). Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc. Natl. Acad. Sci. USA 84, 5788–5792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeyer W. (1865). Uber die veranderungen der quesgestreiften muskeln bie der entzundung und dem Typhus prozess, sowie uber ide regeneration dorselben nach Sustanz defecten. Virchows Arch. 34, 473–514 [Google Scholar]
- Wang H., Hertlein E., Bakkar N., Sun H., Acharyya S., Wang J., Carathers M., Davuluri R., Guttridge D. C. (2007). NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol. Cell. Biol. 27, 4374–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M., Spencer M. J., Tidball J. G. (2001). A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 155, 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling-Henricks M., Tidball J. G. (2011). Neuronal nitric oxide synthase-rescue of dystrophin/utrophin double knockout mice does not require nNOS localization to the cell membrane. PLoS ONE 6, e25071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling-Henricks M., Sokolow S., Lee J. J., Myung K. H., Villalta S. A., Tidball J. G. (2008). Major basic protein-1 promotes fibrosis of dystrophic muscle and attenuates the cellular immune response in muscular dystrophy. Hum. Mol. Genet. 17, 2280–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling-Henricks M., Jordan M. C., Gotoh T., Grody W. W., Roos K. P., Tidball J. G. (2010). Arginine metabolism by macrophages promotes cardiac and muscle fibrosis in mdx muscular dystrophy. PLoS ONE 5, e10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werno C., Menrad H., Weigert A., Dehne N., Goerdt S., Schledzewski K., Kzhyshkowska J., Brüne B. (2010). Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 31, 1863–1872 [DOI] [PubMed] [Google Scholar]
- Wiseman D. M., Polverini P. J., Kamp D. W., Leibovich S. J. (1988). Transforming growth factor-beta (TGF beta) is chemotactic for human monocytes and induces their expression of angiogenic activity. Biochem. Biophys. Res. Commun. 157, 793–800 [DOI] [PubMed] [Google Scholar]
- Woerly G., Lacy P., Younes A. B., Roger N., Loiseau S., Moqbel R., Capron M. (2002). Human eosinophils express and release IL-13 following CD28-dependent activation. J. Leukoc. Biol. 72, 769–779 [PubMed] [Google Scholar]
- Wong D. T., Elovic A., Matossian K., Nagura N., McBride J., Chou M. Y., Gordon J. R., Rand T. H., Galli S. J., Weller P. F. (1991). Eosinophils from patients with blood eosinophilia express transforming growth factor beta 1. Blood 78, 2702–2707 [PubMed] [Google Scholar]
- Xu L. L., Warren M. K., Rose W. L., Gong W., Wang J. M. (1996). Human recombinant monocyte chemotactic protein and other C-C chemokines bind and induce directional migration of dendritic cells in vitro. J. Leukoc. Biol. 60, 365–371 [DOI] [PubMed] [Google Scholar]
- Yablonka-Reuveni Z., Rivera A. J. (1994). Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev. Biol. 164, 588–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T., Honda M., Hayashi H. (1984). Selective chemotaxis of Ia antigen-positive blood monocytes in response to a macrophage chemotactic lymphokine extractable from PPD-induced delayed hypersensitivity reaction site in guinea-pigs. Immunology 52, 269–280 [PMC free article] [PubMed] [Google Scholar]
- Zentella A., Massagué J. (1992). Transforming growth factor beta induces myoblast differentiation in the presence of mitogens. Proc. Natl. Acad. Sci. USA 89, 5176–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentilin L., Tafuro S., Zacchigna S., Arsic N., Pattarini L., Sinigaglia M., Giacca M. (2006). Bone marrow mononuclear cells are recruited to the sites of VEGF-induced neovascularization but are not incorporated into the newly formed vessels. Blood 107, 3546–3554 [DOI] [PubMed] [Google Scholar]