

Abstract
The regulation of the balance between proliferation and differentiation in the mesenchymal compartment of the lung is largely uncharacterized, unlike its epithelial counterpart. In this study, we determined that miR-142-3p contributes to the proper proliferation of mesenchymal progenitors by controlling the level of WNT signaling. miR-142-3p can physically bind to adenomatous polyposis coli mRNA, functioning to regulate its expression level. In miR-142-3p loss-of-function experiments, proliferation of parabronchial smooth muscle cell progenitors is significantly impaired, leading to premature differentiation. Activation of WNT signaling in the mesenchyme, or Apc loss of function, can both rescue miR-142-3p knockdown. These findings show that in the embryonic lung mesenchyme, the microRNA machinery modulates the level of WNT signaling, adding an extra layer of control in the feedback loop between FGFR2C and β-catenin-mediated WNT signaling.
Keywords: WNT signaling, Mesenchymal cell, miRNA
INTRODUCTION
During lung organogenesis, mesenchymal cells undergo active proliferation to support the continuous growth of the organ. Simultaneously, many cells are actively differentiating into the specific cell types that will populate the adult lung - such as parabronchial and vascular smooth muscle cells, pericytes, nerve cells, lipofibroblasts and interstitial fibroblasts. A specific balance between proliferation and differentiation must exist to guarantee the formation of a functional lung. For example, parabronchial smooth muscle cell (PBSMC) progenitors are kept undifferentiated in the sub-mesothelial mesenchyme while starting to differentiate more proximally (Mailleux et al., 2005). It is crucial to understand this delicate balance in order to better characterize lung diseases involving altered mesenchymal proliferation and differentiation.
WNT signaling plays a crucial role in proper proliferation of mesenchymal cells during lung development (De Langhe et al., 2008; Yin et al., 2008). In particular, through β-catenin (CTNNB1), it promotes G1 phase progression by activation of downstream target genes such as Myc and cyclin D1 (Ccnd1) (Niehrs and Acebron, 2012). High WNT signaling and Myc activation are associated with the hyper-proliferative state of cancer of many organs, including the lung (He et al., 1998; van de Wetering et al., 2002; Van Scoyk et al., 2008).
A main regulator of WNT signaling is adenomatous polyposis coli (APC), which can directly bind to CTNNB1, antagonizing the interaction with T-cell factor (TCF). In combination with AXIN and GSK3B, APC induces ubiquitylation and degradation of CTNNB1 (Clevers and Nusse, 2012). Loss of Apc leads to accumulation of CTNNB1 in the nucleus and hyperactivation of WNT signaling. Apc was first identified as a tumor suppressor gene that, upon mutation, causes intestinal cancer (Groden et al., 1991). APC can also control cytoskeleton structure and cell migration by binding microtubules and actin filaments. For example, loss of Apc in the small intestine reduces the migration of epithelial cells and promotes the formation of polyps (Oshima et al., 1997).
Specific microRNAs (miRNAs) have been implicated in both lung development and disease (Jiang et al., 2010; Ornitz and Yin, 2012). In the epithelium, mice with loss of function of members of the miR-17 family show early lethality and hypoplastic lungs, whereas overexpression results in hyperproliferation and inhibition of differentiation of epithelial progenitors (Lu et al., 2007; Ventura et al., 2008). The miR-302 family regulates epithelial progenitor proliferation and differentiation, as well as apical-basal polarity (Tian et al., 2011). Recently, miR-375 has been shown to target WNT signaling and regulate the differentiation of alveolar epithelial cells by controlling the expression of the frizzled 8 gene (Wang et al., 2013). However, miRNAs regulating WNT signaling in the lung mesenchyme have yet to be reported. miR-142-3p is a miRNA first identified for its function in the development of the lymphoid system (Neilson et al., 2007), and was subsequently implicated in leukemia (Lv et al., 2012). In the lung, miR-142-3p is involved in malignancy and has been reported to be an early marker for aggressive and recurrent lung adenocarcinomas (Kaduthanam et al., 2013).
Herein, we detail miR-142-3p as a specific regulator of Apc expression in the mesenchyme. Using a loss-of-function approach, we analyzed the role of the miR-142-3p-Apc axis in mesenchymal cells. Using both pharmacological and genetic tools, we tested whether WNT signaling upregulation is sufficient to rescue miR-142-3p loss-of-function and whether Apc is a crucial target of this miRNA.
RESULTS
miR-142-3p regulates mesenchymal cell proliferation and differentiation
In order to identify miRNAs with a specific function in the lung mesenchyme, we performed a microarray analysis on embryonic mouse lung tissue (Fig. 1A). We observed that 5 and 14 miRNAs were highly expressed, respectively, in the embryonic lung mesenchyme and the epithelium. Among those in the epithelium, we found several members of the miR-200 family. These miRNAs are involved in epithelial to mesenchymal transition (Brabletz and Brabletz, 2010), and their high expression in the embryonic epithelium suggests they may play a role in the plasticity of the branching tips during lung morphogenesis.
Fig. 1.

miR-142-3p regulates mesenchymal cells proliferation and differentiation. (A) MicroRNA microarray shows miRNAs differentially expressed in E12.5 lung epithelium (blue) versus mesenchyme (red). miR-142-3p (arrowhead) is specifically upregulated in the embryonic lung mesenchyme. (B) Expression of miR-142-3p in the epithelium and mesenchyme of E12.5 lungs analyzed by qPCR. (C-J) miR-142-3p LOF assay on E11.5 lung explants treated with scramble vivo-morpholino (scra; C-F) and miR-142-3p vivo-morpholino (mo142; G-J). (K) Expression of miR-142-3p detected by qPCR 48 hours after treatment with mo142. (L-N) Morphometric analysis 48 hours after miR-142-3p LOF assay on E11.5 lung explants. (O-R) Immunostaining for KI67 showing cell proliferation in scra- (O,P) and mo142 (Q,R)-treated E11.5 lung explants, and quantification of proliferation in the epithelium and mesenchyme (S). (T-W) Immunostaining for ACTA2 showing smooth muscle cells at the tips of scra- (T,U) and mo142 (V,W)-treated E11.5 lung explants. Dashed boxes in O, Q, T and V indicate the magnified areas in P, R, U and W, respectively. White arrowheads indicate ectopic ACTA2 expression. (X) Quantification of ACTA2 expression at the tip of the lung explants from the previous experiment. Scale bars: 250 μm (C,E,G,I); 50 μm (D,F,H,J); 100 μm (O,Q,T,V); 25 μm (P,R,U,W). Data are means ± s.d. See also supplementary material Fig. S1 and Movie 1.
An in vitro loss of function (LOF) assay was optimized to functionally characterize the newly identified miRNAs in the mesenchyme. Embryonic day (E) 11.5 lung explants were grown in the presence of vivo-morpholino against either a scrambled sequence or a specific miRNA. We selected miR-126 as a positive control for our LOF assay because it was previously described to regulate vasculature formation. Using vivo-morpholino against miR-126 (mo126), we were able to impair vasculature formation (supplementary material Fig. S1A-I,J-M,R; n=3, P≤0.05), thus mimicking the phenotype observed in the miR-126 knockdown zebrafish (Fish et al., 2008). As a negative control we selected miR-144, because its inactivation does not lead to a specific lung phenotype in mice (Rasmussen et al., 2010). In our LOF assay, mo144 did not produce any branching defect or alteration of proliferation (supplementary material Fig. S1A-I,N-Q,S; n≥3, P<0.05). These results indicate that our LOF assay is an effective tool for the identification of functionally important miRNAs.
Using this assay we tested whether miR-142-3p plays a functional role in the lung mesenchyme. We first confirmed that miR-142-3p is highly expressed in the embryonic lung mesenchyme by qPCR (Fig. 1B; n=3, P<0.05). Then the LOF assay for miR-142-3p was conducted. qPCR showed reduced miR-142-3p levels (Fig. 1K; n=3, P<0.05) associated with impaired branching (Fig. 1C-J). The number of branching points was decreased (Fig. 1L; n=10, P<0.05) and the lung tips had the tendency to elongate instead of bifurcate, thus resulting in a decreased number of terminal buds (Fig. 1M; n=10, P<0.05; supplementary material Movie 1). Reduction of lung growth, illustrated by a diminished total lung area (Fig. 1N; n=10, P<0.05), was observed. Immunostaining for KI67 at 24 hours post-treatment showed a striking reduction of proliferation that, interestingly, was evident in the mesenchyme but not in the epithelium (Fig. 1O-S; n=4, P<0.05). Furthermore, inhibition of miR-142-3p led to ectopic expression of the SMC marker alpha-SMA (ACTA2) in the distal mesenchyme (Fig. 1T-X; n=3, P<0.05). Together, these results suggest a function for miR-142-3p in the control of proliferation and differentiation of the lung mesenchyme.
miR-142-3p regulates WNT signaling by directly controlling Apc expression
To determine whether specific signaling pathways are involved in miR-142-3p regulation of mesenchymal cell proliferation, we utilized the prediction software miRTar and grouped the candidate genes by pathway. Interestingly, WNT signaling was the first hit (Table 1). To verify that WNT signaling was regulated by miR-142-3p in the mesenchyme, we performed a LOF assay on E11.5 lung explants obtained from a WNT reporter mouse line carrying β-galactosidase (β-Gal) expression under the endogenous control of the Axin2 promoter (Axin2lacZ mice). Staining of β-Gal after 24 hours showed a specific inhibition of WNT signaling in the distal mesenchyme (Fig. 2A-E; n=3, P<0.05). Because miRNAs are negative regulators of gene expression, we aimed to identify WNT genes upregulated upon miR-142-3p knockdown. A qPCR screening for members of the WNT signaling pathway expressed in the lung was carried out at 12 hours and at 24 hours during our LOF assay. At 12 hours, we observed that Apc - a gene encoding a negative regulator of WNT signaling - was highly expressed. At 24 hours, Apc was still upregulated while other members were downregulated (Fig. 2F,G). Analysis of LEF1 expression by immunofluorescence confirmed a strong reduction of mesenchymal WNT signaling 24 hours after LOF assay (Fig. 2H-K). We reasoned that since the effect on proliferation, branching and WNT signaling were visible starting at 24 hours, the upregulation of Apc at 12 hours is an early event that could contribute to the observed phenotype. Immunofluorescence analysis for APC at 12 hours (data not shown) and at 24 hours after our LOF assay confirmed an increase of mesenchymal APC at protein level (Fig. 2L-O). At 12 hours epithelial APC was unchanged (data not shown), but at 24 hours there was a reduction in APC. Considering the timing of this reduction and the fact that miR-142-3p is expressed in the mesenchyme, we assume this effect was secondary to the mesenchymal phenotype. Interestingly, Apc was among the five most highly ranked WNT signaling genes (Apc, Tblx1, Rock2, Rac1, Senp2) that were predicted as targets of miR-142-3p (Table 1). As Tblx1, Rock2 and Rac1 were not included in our first screening, we analyzed their expression 12 hours after the LOF assay. Although the expression of Apc and Senp2 was enhanced, expression of Tblx1, Rock2 and Rac1 showed no significant change (Fig. 2P; n=3, P<0.05). The expression of Senp2 was reduced at 24 hours (Fig. 2G), suggesting that the modulation of its expression by miR-142-3p may not be as specific as for Apc.
Table 1.
miR-142-3p targets pathway enrichment
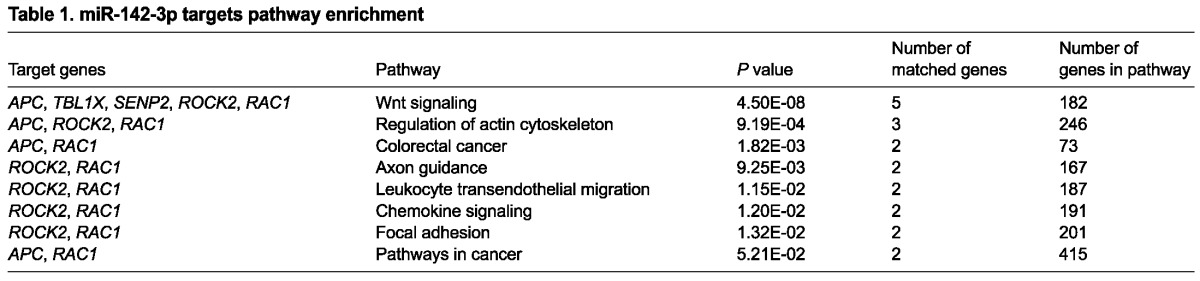
Fig. 2.

miR-142-3p regulates WNT signaling by direct control of Apc expression. (A-E) miR-142-3p LOF assay on Axin2lacZ E11.5 lung explants. Lung explants (A,C) and vibratome sections (B,D) are shown 24 hours after miR-142-3p LOF assay. Mesenchymal β-gal is quantified in E. (F,G) Members of the WNT signaling pathway were quantified by qPCR 12 and 24 hours after the miR-142-3p LOF assay. (H-K) LEF1 expression 24 hours after the miR-142-3p LOF assay. (L-O) APC expression after the miR-142-3p LOF assay. Immunofluorescence for APC 24 hours after scra (L,M) and mo142 (N,O) treatment. Dashed boxes indicate the magnified area shown in the panels to the right. Dashed lines in M and O demarcate the epithelium-mesenchyme boundary. (P) Quantification by qPCR of miR-142-3p predicted targets 12 hours after miR-142-3p LOF assay. (Q) Quantification by qPCR of miR-142-3p predicted targets after biotin mRNA-miRNA pull down assay. (R) Luciferase assay for Apc seed sequences on MLg cells. Scale bars: 150 μm (A,C); 100 μm (B,D); 75 μm (H,J,L,N); 25 μm (I,K,M,O). Data are means ± s.d. See also supplementary material Fig. S2.
We then wanted to determine whether miR-142-3p was able to physically bind the mRNA of its predicted target genes. For this purpose, we performed a pull-down experiment using a biotinylated miR-142-3p followed by qPCR to detect specific binding. The biotinylated miR-142-3p was able to cause overexpression of WNT signaling target genes such as Myc and Fgfr2 (supplementary material Fig. S2A,B), suggesting that biotinylation was not altering the property of miR-142-3p. In this assay, performed on a lung mesenchymal cell line (MLg cells), Apc was enriched more than six times compared with the control (Fig. 2M; n=3, P<0.05). The same assay was reproduced in MFLM4 and HEK293 cells, suggesting that indeed miR-142-3p directly interact with Apc to regulate its expression (supplementary material Fig. S2C; n=3, P<0.05). We further wanted to determine if miR-142-3p was binding Apc by specifically interacting with the predicted binding sites located at the 3′-UTR of Apc mRNA. For this purpose, we cloned Apc seed sites into two luciferase (Luc) reporter plasmids containing either a mutated binding site or a wild-type locus. We observed that the ability of miR-142-3p to target Apc was compromised in the presence of the mutated 3′UTR (Fig. 2N; n=3, P<0.05).
miR-142-3p downregulation leads to parabronchial smooth muscle cell progenitor differentiation
APC is a negative regulator of WNT signaling and, as a direct target of miR-142-3p, its increase can explain the drop in WNT signaling following miR-142-3p knockdown. In the embryonic lung mesenchyme, WNT, in combination with FGF signaling, creates a positive feedback loop. In fact, lack of contribution from either FGF (specifically FGF9-FGFR2c) or WNT disrupts this delicate equilibrium (Yin et al., 2011). When this system is operational, WNT signaling maintains Fgfr2c expression in the mesenchyme allowing mesenchymal growth through the regulation of G1 to S phase transition (De Langhe et al., 2008; Yin et al., 2008). Therefore, we decided to test whether the inhibition of WNT signaling observed in our miR-142-3p LOF assay was associated with perturbation of the FGF-WNT sustained regulation. Indeed, qPCR on E11.5 lung explants showed decreased Fgfr2c expression beginning 24 hours into the assay (Fig. 3D-F; n=3, P<0.05). At 48 h, Fgfr2c reduction was associated with a specific decrease in the mesenchyme of phosphorylated ERK (pERK; Fig. 3M-Q; n=4, P<0.05) and phosphorylated MEK (pMEK; Fig. 3R-V; n=3, P<0.05). One of the functions of FGF9-FGFR2c signaling in the lung is to keep mesenchymal PBSMC progenitors undifferentiated and proliferating (Yi et al., 2009). Furthermore, we have previously shown that FGF10 is expressed in, and identifies PBSMC progenitors (Mailleux et al., 2005). Analysis by qPCR after the miR-142-3p LOF assay showed that Fgfr2b expression remained stable, suggesting that epithelial FGF signaling was unaffected (Fig. 3J-L; n=3, P<0.05). However, Fgf10 started decreasing at 24 hours and was significantly reduced at 48 hours (Fig. 3G-I; n=3, P<0.05). This result suggests that Fgf10-positive PBSMC progenitors were specifically affected by the imbalance of FGF-WNT regulation. We therefore decided to analyze the effect of our miR-142-3p LOF assay on SMC differentiation. To directly analyze PBSMC progenitors during miR-142-3p downregulation, conditional red fluorescent protein (RFP) reporter mice (RFPf/f) were crossed with mice carrying Fgf10CreERT2. We will refer to these mice with genotype Fgf10CreERT2/+,RFPf/+ as RFPFgf10.
Fig. 3.

Loss of miR-142-3p affects mesenchymal FGF signaling. (A-L) FGF signaling genes were quantified by qPCR. The expression of Fgf9 (A-C), Fgfr2c (D-F), Fgf10 (G-I) and Fgfr2b (J-L) was analyzed at 12, 24 and 48 hours after LOF assay on E11.5 lung explants. (M-Q) Phosphorylated ERK (pERK) expression was analyzed 24 and 48 hours after LOF assay on E11.5 lung explants. pERK expression in the epithelium and mesenchyme is shown by immunofluorescence at 48 hours (M-P) and was quantified at 24 and 48 hours (Q). pMEK expression in the epithelium and mesenchyme is shown by immunofluorescence at 48 hours (R-U) and quantified at 24 and 48 hours (V). Dashed boxes indicate the magnified areas in the adjacent panels. Dashed lines in N, P, S and U demarcate the epithelium-mesenchyme boundary. Scale bars: 100 μm (M,O,R,T); 50 μm (N,P,S,U). Data are means ± s.d.
Live imaging of RFPFgf10 E11.5 lung explants treated with miR-142-3p vivo-morpholino (mo142; supplementary material Movie 2) showed that labeled PBSMC progenitors formed ring-like structures, reminiscent of smooth muscle cell phenotype, at the tip of the epithelial buds (Fig. 4A-D). These data are in agreement with the ectopic alpha-SMA (ACTA2) expression at the tip of the epithelial buds during mo142 treatment (Fig. 1T-W). Additionally, in mo142-treated explants, labeled cells displayed reduced motility and proliferation, but in scrambled-treated explants, they were highly motile and proliferative (supplementary material Movie 2; Fig. 4E-G; n=3, P<0.05). Our results suggest that PBSMC progenitors prematurely differentiate upon mo142 treatment. To further demonstrate that this was due to loss of FGF9-FGFR2c signaling, primary embryonic lung mesenchymal cells were cultured in vitro in different experimental conditions. Scrambled-treated primary mesenchymal cells grown in the presence of FGF9 were prevented from differentiating into SMCs (Fig. 4H,I). After miR-142-3p knockdown, FGF9 was no longer effective and thus differentiation into SMCs occurred (Fig. 4J-L). The role of miR-142-3p in SMC differentiation was further evaluated using a gain of function approach. We used mesenchymal cells from a mouse lung mesenchymal cell line (MLg cells). Under normal in vitro culture conditions, MLg cells differentiate into ACTA2-positive cells. Overexpression of miR-142-3p by a synthetic form of miR-142-3p that mimics its function (mi142) led to reduced ACTA2 expression and diminished elongation (Fig. 4M-O). Because Apc is a specific target for miR-142-3p (Fig. 2H-N), we wanted to determine if Apc overexpression is sufficient to mimic miR-142-3p knockdown. Primary mesenchymal cells isolated from RFPFgf10 lungs were electroporated with either control plasmids (pGFP) or experimental plasmids overexpressing APC (pAPC). Apc overexpression was sufficient to mimic loss of miR-142-3p, producing reduced proliferation and motility of PBSMC progenitors (Fig. 4P-V; n=3, P<0.05; supplementary material Movie 3).
Fig. 4.

Loss of miR-142-3p leads to lung mesenchyme differentiation. (A-D) Analysis of RFPFgf10 lung explants after scra (A,B) and mo142 (C,D) administration, and quantification of cell motility (E,F) and proliferation (G). (H-K) Immunostaining for ACTA2 showing lung primary mesenchymal cells after administration of BSA or FGF9 in the presence of scra (H,I) or mo142 (J,K). (L) Quantification of ACTA2-positive cells. (M,N) MLg cells were analyzed for ACTA2 expression by immunostaining after scra (M) or mi142 (N) administration. (O) Quantification of cellular morphology as a ratio of length to width. (P-S) RFPFgf10 primary mesenchymal cells were electroporated with a plasmid expressing GFP (pGFP) or APC (pAPC) and analyzed between 0 and 45 hours. (T-V) Quantification of the cell motility (T,U) and proliferation (V) for pGFP- and pAPC-treated cells. Dashed boxes indicate the magnified areas shown in the insets. Dashed lines demarcate the epithelium-mesenchyme boundary. Scale bars: 250 μm (A,C); 50 μm (B,D); 50 μm (H-K insets); 20 μm (H-K main image); 50 μm (M,N); 75 μm (P-S). Data are means ± s.d. See also supplementary material Movies 2 and 3.
Constitutive WNT signaling rescues miR-142-3p knockdown
The inhibition of WNT signaling in the miR-142-3p LOF assay prompted us to attempt a rescue experiment in which WNT signaling was stimulated at different levels of the pathway. We began with WNT3a, a commercially available WNT ligand. Lungs treated with WNT3a display expansion of the epithelium and mesenchyme compared with BSA-treated lungs (data not shown and supplementary material Fig. S3C,D). Upon miR-142-3p attenuation, WNT3a was unable to affect mesenchymal growth (supplementary material Fig. S3A,B,D; n=3, P<0.05). Similar results were obtained by using FGF9 (Fig. 5A-D; n=3, P<0.05) and FGF9 in combination with WNT3a (supplementary material Fig. S3E-H; n=3, P<0.05). Under these conditions FGF9 still caused expansion of the epithelium. This result further confirms that when miR-142-3p expression is inhibited the positive feedback loop between FGF and WNT signaling in the mesenchyme is impaired. To intervene downstream of WNT signaling, we used mice harboring a floxed allele in the Ctnnb1 gene (Ctnnb1(ex3)f) to generate a stable form of Ctnnb1. Crossing this line with Tbx4LMECreERT2 mice, we were able to obtain inducible constitutive activation of Ctnnb1, specifically in the mesenchyme. We will refer to the genotype Tbx4LMECreERT2/+,Ctnnb1(ex3)f/+ as Ctnnb1f/+;Tbx4. Ctnnb1f/+;Tbx4 and Ctnnb1f/f lungs from the same mating represented WNT-activated and control lungs, respectively, and were used in the LOF assay (Fig. 5E). Lungs from Tbx4LMECreERT2/+ mice injected with tamoxifen intraperitoneally were also used as controls in the LOF assay, showing similar results to the Ctnnb1f/f lungs (data not shown). Forty-eight hours after the miR-142-3p LOF assay, the activation of CTNNB1 signaling in Ctnnb1f/+;Tbx4 lungs, compared to Ctnnb1f/f lungs (supplementary material Fig. S3M-P), led to the disappearance of mo142-induced MYH11 (supplementary material Fig. S3I-L) and ACTA2 ectopic expression (Fig. 5F-I,N; n=3, P<0.05) at the tip of the epithelial buds. We also observed increased pMEK expression in mo142-treated Ctnnb1f/+;Tbx4 compared with Ctnnb1f/f lungs, suggesting recovery of FGF signaling in the mesenchyme (Fig. 5J-M,O; n=3, P<0.05).
Fig. 5.

Stabilized Ctnnb1 expression or decreased Apc expression in the mesenchyme counteracts the effect of miR-142-3p inhibition on proliferation and differentiation. (A-D) FGF9 (200 ng/μl) was administrated to E11.5 lung explants cultured in the presence of scra (A) or mo142 (B), and mesenchyme thickness (C) and epithelial and mesenchymal surfaces (D) were measured. (E) Schematic showing the time line of the experimental treatment. The green and red lines depict tamoxifen and mo142 effects, respectively. (F-O) E11.5 lung explants harboring a mesenchymal stabilized form of Ctnnb1 obtained by Tbx4-driven Cre expression, and control lungs were treated with mo142 and immunostained for ACTA2 (F-I) or pMEK (J-M). Quantification of ACTA2 and pMEK are shown in N and O, respectively. (P-II) E11.5 lung explants with mesenchymal deletion of Apc by Tbx4-driven Cre expression (P-S) and control lungs (T-W) were treated with mo142 and their morphometry was analyzed (X,Y). Immunostaining for ACTA2 and APC (Z-CC) or KI67 and APC (DD-GG) from the previous experiment and its relative quantification (HH,II). Dashed boxes indicate the magnified areas in the adjacent panels. Dashed lines demarcate the epithelium-mesenchyme boundary. Scale bars: 250 μm (A,B); 75 μm (F,H,J,L); 25 μm (G,I,K,M); 250 μm (P,R,T,V); 50 μm (Q,S,U,W); 75 μm (Z,BB,DD,FF); 25 μm (AA,CC,EE,GG). Data are means ± s.d. See also supplementary material Fig. S3.
Reduction of Apc expression rescues miR-142-3p knockdown
Having identified Apc as a direct target of miR-142-3p, we decided to test whether reduction of Apc by ablation of one of the two copies of the gene was able to rescue the effect of miR-142-3p inhibition. For this purpose we used Apcf/f mice crossed with Tbx4LMECreERT2 mice. We will refer to the genotype Tbx4LMECreERT2/+; Apcf/+ as Apcf/+;Tbx4. Apcf/f lungs were used as controls. Tbx4LMECreERT2/+ were also used as controls with similar results to Apcf/f lungs in the LOF assay (data not shown). Forty-eight hours after the miR-142-3p LOF assay, the reduction of APC expression in Apcf/+;Tbx4 compared with Apcf/f lung explants was confirmed by qPCR (data not shown) and immunohistochemistry (Fig. 5DD-GG). This reduction led to the disappearance of mo142-induced ectopic expression of ACTA2 at the tip of the epithelial buds (Fig. 5Z-CC,HH; n=3, P<0.05), as well as increased KI67 expression in the mesenchyme (Fig. 5DD-GG,II; n=3, P<0.05). In addition, Apcf/+;Tbx4 lungs were able to recover growth and branching (Fig. 5P-Y; n=3, P<0.05). These results suggest that the inhibition of Apc alone is sufficient to counteract miR-142-3p knockdown.
DISCUSSION
In this study we describe a new function of the microRNA machinery in the lung embryonic mesenchyme. We determined that miR-142-3p is a crucial regulator of WNT signaling in this compartment. By directly controlling Apc expression, miR-142-3p can fine-tune its inhibition of WNT signaling. Furthermore, miR-142-3p controls proliferation and differentiation of mesenchymal progenitor in the embryonic lung (Fig. 6A,B).
Fig. 6.
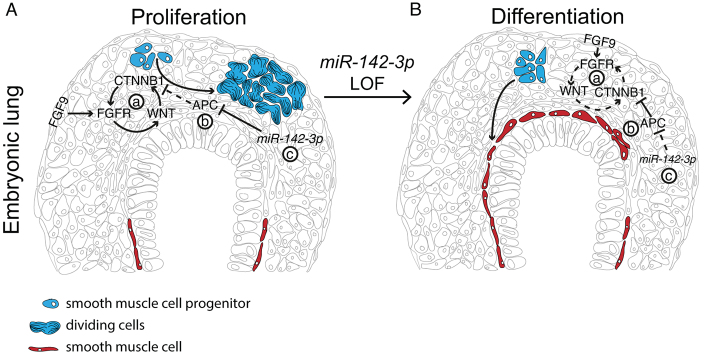
Schematic of miR-142-3p regulation of WNT signaling by direct control of Apc expression. (A) A fine balance of mesenchymal FGF and WNT signaling (a) is necessary to maintain proper mesenchymal proliferation. In this context, APC (b) contributes to control the level of β-catenin (CTNNB1), and miR-142-3p (c) controls the inhibitory effect of APC. Changes in miR-142-3p expression can therefore modulate the balance between proliferation (A) and differentiation (B) of mesenchymal progenitors. Blue, mesenchymal progenitors; red, differentiated smooth muscle cells.
A fine balance between FGF and WNT signaling helps maintain the pool of mesenchymal progenitor cells in the lung embryonic mesenchyme. For example, evidence suggests that FGF9 from the mesothelium can signal to Fgf10-positive cells located in the submesothelial mesenchyme to prevent differentiation (Colvin et al., 2001). In this system, WNT signaling helps maintain proper levels of FGF signaling, because reduced WNT signaling creates disequilibrium in the feedback loop that maintains mesenchymal FGF-WNT signaling (De Langhe et al., 2008; Yin et al., 2011). Here, we found that miR-142-3p is implicated in the modulation of the WNT and FGF balance, adding an extra layer of control that increases the robustness of this system.
We show that miR-142-3p fulfills this function by directly controlling Apc expression, therefore acting as a negative regulator of an inhibitor of WNT signaling. Deregulation of miR-142-3p expression uncouples WNT and FGF signaling in the mesenchyme, affecting mesenchymal progenitor cells differentiation. Specifically, the Fgf10-positive lineage which contributes to the formation of parabronchial smooth muscle cells is affected by the deregulation of miR-142-3p expression, resulting in premature differentiation and proximalization of the mesenchyme.
Recently, miR-142-3p was found to control hemangioblast specification and vasculogenesis in Xenopus (Nimmo et al., 2013). In our LOF assay we observed impaired angiogenesis of the lung explants, which was associated with enrichment of markers for undifferentiated hemangioblasts such as CD31 (data not shown). This suggests that a similar regulation could also contribute to the vasculogenesis in the lung. It will be interesting to determine if miR-142-3p controls similar pathways - for instance WNT signaling - or if the two systems are completely independent.
Although our screening identified Apc as a main target of miR-142-3p, other genes are likely to be affected. In particular, other members of WNT signaling were found to bind to biotinylated miR-142-3p to a lower degree than Apc. Nonetheless, rescue experiments with ApcTbx4 mice showed that the inhibition of Apc in the mesenchyme is sufficient to rescue the lung from miR-142-3p downregulation, suggesting that Apc is a crucial molecule regulated by miR-142-3p in the lung mesenchyme. We also found that in the parenchyma of the adult lungs, miR-142-3p and APC are mutually exclusive (data not shown). This suggests that the regulation here described may not be limited to the embryonic lung. In addition, our finding is consistent with the observation that inactivation of Apc in the lung mesenchyme using a Tbx4-rtTA driver mouse line leads to reduced and disorganized smooth muscle cell differentiation in the lung (personal communication from Wei Shi). This suggests that APC per se, has a critical role in the regulation of proliferation, survival, and differentiation of smooth muscle progenitor cells.
In summary, our work shows that miR-142-3p is intimately linked to mesenchymal activated WNT signaling by specific regulation of Apc. It will be of interest to determine if the miR-142-3p-Apc-β-catenin axis plays a role in lung diseases such as idiopathic pulmonary fibrosis, in which cellular proliferation and differentiation of mesenchymal cells is unbalanced.
MATERIALS AND METHODS
Mouse strains
C57BL/6 wild-type, Apcflox/+, Tomatoflox/flox and Axin2lacZ mice were obtained from Jackson Laboratories. Fgf10CreERT2/+ mice were generated in our laboratory as previously described (El Agha et al., 2012). Ctnnb1(ex3)f mice were generated in the Taketo laboratory (Harada et al., 1999). Tbx4LMECreERT2/+ mice were generated in the Krasnow laboratory (M.E.K., Patrick E. Bogard, F. Hernán Espinoza, Douglas B. Menke, David M. Kingsley and Mark A. Krasnow, unpublished data). For rescue experiments, tamoxifen was administrated by intraperitoneal injection (0.1 mg/g of body weight) 24 hours before the LOF assay. Animals were housed at room temperature with a 12 hour/12 hour light/dark cycle and free access to food and water. The Federal Authorities for Animal Research of the Regierungspraesidium Giessen (Hessen, Germany) approved the study protocol (miR-142-3p protocol 52/2012).
MicroRNA array and qPCR
Epithelium from E12.5 lung buds was isolated as previously described (Carraro et al., 2009); mesenchyme from E12.5 lung buds was physically isolated using 0.12 mm tungsten needles (Fine Science Tools). Total RNA was extracted using a miRNeasy Mini Kit (Qiagen). The quality of the total RNA was verified using an Agilent 2100 Bioanalyzer profile. Total RNA (1000 ng) was labeled with fluorescent Hy3 and Hy5 using the miRCURY LNA Array power labeling kit (Exiqon) following the procedure described by the manufacturer. The labeled RNAs were mixed pair-wise and hybridized to the miRCURY LNA Array version 5th Generation (Exiqon). The hybridization was performed according to the miRCURY LNA array manual using an HS4800 hybridization station (Tecan). The slides were scanned using the Agilent G2565BA Microarray Scanner System (Agilent Technologies) and the image analysis was carried out using ImaGene 8.0 software (BioDiscovery). The quantified signals were background corrected (Normexp with offset value 10) (Ritchie et al., 2007), and normalized using the global Lowess (LOcally WEighted Scatterplot Smoothing) regression algorithm. miRNAs that were more than fourfold differentially expressed were considered for validation by qPCR. Statistical analyses were performed using Student’s t-tests. RT-PCR for mRNA was carried out using SuperScript II reverse transcriptase (Invitrogen) with random primers, and in RT-PCR for miRNA the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) was used. In both cases, reactions were assembled following the manufacturer’s recommendations. qPCR was performed on a LightCycler 480 system (Roche). The universal probe library (Roche) was used for the analysis of mRNA expression. The Taqman microRNA assay (Applied Biosystems) was used for screening the differential expression of miRNAs. qPCR reactions and data analysis were performed as previously described (Carraro et al., 2009). Results are presented as relative expression compared with control. Results were collected from at least three lungs or pools of lungs from independent experiments.
Culture of embryonic lung explants
Timed-pregnant wild-type or transgenic mice were sacrificed on post-coitum (embryonic) day 11.5 (E11.5), and the embryos were harvested. Lung primordia were placed on 8 μm Nucleopore Track-Etch membranes (Whatman) and cultured in DMEM:F12 medium (Gibco) and 0.5% fetal bovine serum (FBS). Vivo-morpholinos (Gene Tools) were added at 1-4 μM to the lung explants. Recombinant FGF9 and WNT3a (R&D) were added at 200 ng/ml and 250 ng/ml, respectively. The data are representative of at least three lungs from different timed pregnant mice.
Computational targets prediction
The miR-142-3p targets were selected on the basis of the on-line available prediction database miRTar (mirtar.mbc.nctu.edu.tw). The 5′-UTR and the 3′-UTR of target genes were included in the screening. A minimum free energy (MFE) ≤11 Kcal/mol and Score ≥140 were used. A Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis for pathway enrichment was performed.
β-galactosidase staining
Axin2lacZ lung explants were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) at 4°C for 5 minutes with rocking, washed twice for 5 minutes in PBS at 4°C, then transferred into freshly prepared X-gal solution and stained at 37°C until a clear precipitate formed; the method was modified from that of Hogan et al. (Hogan et al., 1994). After rinsing with PBS, epithelial explants were post-fixed in 4% PFA in PBS. For vibratome sections, samples were embedded in a mixture of 300 mg/ml albumin, 5 mg/ml gelatin and 0.6% glutaraldehyde, and sectioned. The data are representative of three lungs from independent experiments.
Immunofluorescence
Tissues were fixed in 4% PFA, gradually dehydrated in ethanol, impregnated with toluene, embedded in paraffin, and sectioned into 5 μm slices on poly-L-lysine-coated slides. Antigen retrieval was performed by boiling the sample for 10 minutes in sodium citrate buffer (10 mM, pH 6.0; Vector). The following antibodies (Abs) were used: APC Ab (Abcam), KI67 Ab (Novabios), phospho-ERK (pERK) Ab (Cell Signaling), phospho-MEK Ab (Cell Signaling), LEF1 Ab (Cell Signaling), phospho-Ser552-CTNNB1 Ab (Cell Signaling), ACTA2 Ab (Sigma), MYH11 Ab (Sigma). All antibodies were used at a 1:200 dilution. The data are representative of at least three lungs from independent experiments.
Image analysis
Photomicrographs of immunofluorescence staining were taken using a Leica DMRA fluorescence microscope with a Leica digital camera. Live-cell and lung explant time-lapse experiments were performed with a Leica AF6000 fluorescence imaging system. Digital analysis of staining intensity and distribution, and of live-imaging experiments was performed with MetaMorph (Molecular Devices) or ImageJ software. At least three different samples for each condition were analyzed. Phase-contrast images of the samples were recorded using a digital camera (Diagnostic Instruments) connected to a reversed phase-contrast microscope (Leica). The public domain software ImageJ was broadened by routines specifically developed by us and used to process and analyze the images (Guidolin et al., 2004). The number of branches, percentage of tissue area involved in branching, and branching complexity (i.e. the number of branching points of the binary skeleton and the distribution of branching orders) were the morphometric parameters estimated from each sample. Data were processed by using statistical analysis software (GraphPad Prism 3.03, GraphPad Software). The data are representative of at least three lungs from independent experiments.
miRNA pull-down assay
MLg, MFLM4 or HEK293 cells were cultured in six-well plates and transfected in triplicate with 3′-biotinylated miR-142-3p (Bio-miR-142) or 3′-biotinylated scramble (Bio-scramble; Dharmacon), at a final concentration of 30 nM using Lipofectamine RNAimax (Invitrogen) following the manufacturer’s protocol. After 48 hours, the cells were pelleted at 1000 rpm for 5 minutes. After washing twice, cell pellets were resuspended in 0.5 ml lysis buffer [50 mM Tris-HCl, 2 mM EDTA, 0.1% NP40, 10% glycerol, 2 mM EGTA, diethylpyrocarbonate (DEPC)-treated water, 50 U RNasin (Promega) and complete mini-protease inhibitor cocktail (Roche Applied Science)], and incubated at 4°C for 10 minutes. The cytoplasmic extract was isolated by centrifugation at 10,000 rpm for 10 minutes. Streptavidin-coated magnetic beads (Invitrogen) were blocked for 1 hour at 4°C in blocking buffer (10 mM Tris-HCl pH 6.5, 1 mM EDTA, 1 mg/ml yeast tRNA and 1 mg/ml BSA) and washed twice with 1 ml washing buffer (10 mM Tris-HCl pH 6.5, 1 mM EDTA 0.5 M NaCl). Beads were resuspended in 0.5 ml washing buffer. Cytoplasmic extract was then added to the beads and incubated for 1 hour at 4°C with slow rotation. The beads were then washed five times with 1 ml washing buffer. RNA bound to the beads (pull-down RNA) or from 10% of the extract (input RNA), was isolated using Trizol reagent LS (Invitrogen). The level of mRNA in the Bio-miR-142-3p or Bio-scramble control pull-down was quantified by qPCR. mRNA levels were normalized to a housekeeping gene (Gapdh, H4). The enrichment ratio of the control-normalized pull-down RNA to the control-normalized input levels was then calculated. The data for each cell line are representative of three independent experiments.
Luciferase reporter assay
The predicted binding sequence on the 3′ UTR of Apc was synthetically produced by PCR and transferred into a luciferase reporter vector. To determine the specificity of the interaction, a reporter vector containing a ‘seed’ mutated sequence was generated using specific primers. The vector backbone was a pmiRGLO luciferase reporter (Promega) carrying a synthetic firefly luciferase (luc2) and a Renilla luciferase. Our constructs were inserted downstream of the luciferase gene into a unique XbaI restriction site. The Renilla luciferase was used as a control to normalize luminescence levels. Experiments were carried out using a dual-luciferase reporter assay system (Promega) and analyzed with a Victor3 Luminometer (PerkinElmer). The data are representative of three independent experiments.
Cell culture
For the preparation of primary mesenchymal cells, whole lungs were dissected at E14.5 and subjected to trypsin digestion to give rise to single cells. Mesenchymal cells were separated from epithelial cells by differential adhesion as previously described (Lebeche et al., 1999). Fifty minutes after plating, 200 ng/ml FGF9 was added to the mesenchymal cells. The cells were cultured in DMEM/F12 medium containing penicillin, streptomycin and 0.5% FBS in the presence or absence of FGF9. MFLM4 (Seven Hills), MLg (ATCC) and HEK293 (ATCC) cells were grown in DMEM with 10% FBS. Plasmids expressing GFP (pGFP) and APC (pAPC) were introduced into the cells by electroporation (Lonza). MLg cells were electroporated with 30 nM scramble (scra) or miR-142-3p mimic (mi142) molecules (Dharmacon). The data are representative of three independent experiments.
Statistical analysis
All data are reported as means ± s.d. Statistical analyses were performed using Student’s t-tests, with P≤0.05 considered significant.
Supplementary Material
Acknowledgments
We thank Matthew Jones for critical reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
G.C. developed the approach, performed experiments and prepared the manuscript. A.S., J.R., A.C., C.C., E.A. and B.M. performed experiments. S.D. and D.G. performed data analysis. M.M.T. and M.E.K. provided mouse lines. W.S. and A.G. edited the manuscript. G.B. and S.D.L. developed the approach and edited the manuscript. S.B. developed the approach and prepared the manuscript prior to submission.
Funding
This work was supported by the National Institutes of Health (NIH) [RO1 grants HL086322 and HL107307 to S.B., and HL092967 to S.D.L.]; the Excellence Cluster in Cardio-Pulmonary system, Deutsche Forschungsgemeinschaft, LOEWE (S.B.); LOEWE [III L 4 - 518/15.004 2009 to G.B.]; and the Deutsche Forschungsgemeinschaft [BA 4036/1-1 to G.B.]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.105908/-/DC1
References
- Brabletz S., Brabletz T. (2010). The ZEB/miR-200 feedback loop - a motor of cellular plasticity in development and cancer? EMBO Rep. 11, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro G., El-Hashash A., Guidolin D., Tiozzo C., Turcatel G., Young B. M., De Langhe S. P., Bellusci S., Shi W., Parnigotto P. P., et al. (2009). miR-17 family of microRNAs controls FGF10-mediated embryonic lung epithelial branching morphogenesis through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev. Biol. 333, 238–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H., Nusse R. (2012). Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 [DOI] [PubMed] [Google Scholar]
- Colvin J. S., White A. C., Pratt S. J., Ornitz D. M. (2001). Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development 128, 2095–2106 [DOI] [PubMed] [Google Scholar]
- De Langhe S. P., Carraro G., Tefft D., Li C., Xu X., Chai Y., Minoo P., Hajihosseini M. K., Drouin J., Kaartinen V., et al. (2008) Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signalling. PLoS ONE 3, e1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Agha E., Al Alam D., Carraro G., MacKenzie B., Goth K., De Langhe S. P., Voswinckel R., Hajihosseini M. K., Rehan V. K., Bellusci S. (2012). Characterization of a novel fibroblast growth factor 10 (Fgf10) knock-in mouse line to target mesenchymal progenitors during embryonic development. PLoS ONE 7, e38452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish J. E., Santoro M. M., Morton S. U., Yu S., Yeh R. F., Wythe J. D., Ivey K. N., Bruneau B. G., Stainier D. Y., Srivastava D. (2008). miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 15, 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groden J., Thliveris A., Samowitz W., Carlson M., Gelbert L., Albertsen H., Joslyn G., Stevens J., Spirio L., Robertson M., et al. (1991). Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66, 589–600 [DOI] [PubMed] [Google Scholar]
- Guidolin D., Vacca A., Nussdorfer G. G., Ribatti D. (2004). A new image analysis method based on topological and fractal parameters to evaluate the angiostatic activity of docetaxel by using the Matrigel assay in vitro. Microvasc. Res. 67, 117–124 [DOI] [PubMed] [Google Scholar]
- Harada N., Tamai Y., Ishikawa T., Sauer B., Takaku K., Oshima M., Taketo M. M. (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T. C., Sparks A. B., Rago C., Hermeking H., Zawel L., da Costa L. T., Morin P. J., Vogelstein B., Kinzler K. W. (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 [DOI] [PubMed] [Google Scholar]
- Hogan B. L., Blessing M., Winnier G. E., Suzuki N., Jones C. M. (1994). Growth factors in development: the role of TGF-beta related polypeptide signalling molecules in embryogenesis. Dev. Suppl. 1994, 53–60 [PubMed] [Google Scholar]
- Jiang Q., Hao Y., Wang G., Juan L., Zhang T., Teng M., Liu Y., Wang Y. (2010). Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 4 Suppl. 1, S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaduthanam S., Gade S., Meister M., Brase J. C., Johannes M., Dienemann H., Warth A., Schnabel P. A., Herth F. J., Sültmann H., et al. (2013). Serum miR-142-3p is associated with early relapse in operable lung adenocarcinoma patients. Lung Cancer 80, 223–227 [DOI] [PubMed] [Google Scholar]
- Lebeche D., Malpel S., Cardoso W. V. (1999). Fibroblast growth factor interactions in the developing lung. Mech. Dev. 86, 125–136 [DOI] [PubMed] [Google Scholar]
- Lu Y., Thomson J. M., Wong H. Y., Hammond S. M., Hogan B. L. (2007). Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev. Biol. 310, 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv M., Zhang X., Jia H., Li D., Zhang B., Zhang H., Hong M., Jiang T., Jiang Q., Lu J., et al. (2012). An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-α and cAMP/PKA pathways. Leukemia 26, 769–777 [DOI] [PubMed] [Google Scholar]
- Mailleux A. A., Kelly R., Veltmaat J. M., De Langhe S. P., Zaffran S., Thiery J. P., Bellusci S. (2005). Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development 132, 2157–2166 [DOI] [PubMed] [Google Scholar]
- Neilson J. R., Zheng G. X., Burge C. B., Sharp P. A. (2007). Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Dev. 21, 578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C., Acebron S. P. (2012). Mitotic and mitogenic Wnt signalling. EMBO J. 31, 2705–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmo R., Ciau-Uitz A., Ruiz-Herguido C., Soneji S., Bigas A., Patient R., Enver T. (2013). MiR-142-3p controls the specification of definitive hemangioblasts during ontogeny. Dev. Cell 26, 237–249 [DOI] [PubMed] [Google Scholar]
- Ornitz D. M., Yin Y. (2012). Signaling networks regulating development of the lower respiratory tract. Cold Spring Harb. Perspect. Biol. 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima H., Oshima M., Kobayashi M., Tsutsumi M., Taketo M. M. (1997). Morphological and molecular processes of polyp formation in Apc(delta716) knockout mice. Cancer Res. 57, 1644–1649 [PubMed] [Google Scholar]
- Rasmussen K. D., Simmini S., Abreu-Goodger C., Bartonicek N., Di Giacomo M., Bilbao-Cortes D., Horos R., Von Lindern M., Enright A. J., O’Carroll D. (2010). The miR-144/451 locus is required for erythroid homeostasis. J. Exp. Med. 207, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E., Silver J., Oshlack A., Holmes M., Diyagama D., Holloway A., Smyth G. K. (2007). A comparison of background correction methods for two-colour microarrays. Bioinformatics 23, 2700–2707 [DOI] [PubMed] [Google Scholar]
- Tian Y., Zhang Y., Hurd L., Hannenhalli S., Liu F., Lu M. M., Morrisey E. E. (2011). Regulation of lung endoderm progenitor cell behavior by miR302/367. Development 138, 1235–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M., Sancho E., Verweij C., de Lau W., Oving I., Hurlstone A., van der Horn K., Batlle E., Coudreuse D., Haramis A. P., et al. (2002). The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111, 241–250 [DOI] [PubMed] [Google Scholar]
- Van Scoyk M., Randall J., Sergew A., Williams L. M., Tennis M., Winn R. A. (2008). Wnt signaling pathway and lung disease. Transl. Res. 151, 175–180 [DOI] [PubMed] [Google Scholar]
- Ventura A., Young A. G., Winslow M. M., Lintault L., Meissner A., Erkeland S. J., Newman J., Bronson R. T., Crowley D., Stone J. R., et al. (2008). Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132, 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Huang C., Reddy Chintagari N., Bhaskaran M., Weng T., Guo Y., Xiao X., Liu L. (2013). miR-375 regulates rat alveolar epithelial cell trans-differentiation by inhibiting Wnt/β-catenin pathway. Nucleic Acids Res. 41, 3833–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L., Domyan E. T., Lewandoski M., Sun X. (2009). Fibroblast growth factor 9 signaling inhibits airway smooth muscle differentiation in mouse lung. Dev. Dyn. 238, 123–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., White A. C., Huh S. H., Hilton M. J., Kanazawa H., Long F., Ornitz D. M. (2008). An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev. Biol. 319, 426–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., Wang F., Ornitz D. M. (2011). Mesothelial- and epithelial-derived FGF9 have distinct functions in the regulation of lung development. Development 138, 3169–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.