

Abstract
Hepatitis C virus (HCV) has infected almost 200 million people worldwide, typically causing chronic liver damage and severe complications such as liver failure. Currently, there are few approved treatments for viral infection. Thus, the HCV RNA-dependent RNA polymerase (gene product NS5B) has emerged as an important target for small molecule therapeutics. Potential therapeutic agents include allosteric inhibitors that bind distal to the enzyme active site. While their mechanism of action is not conclusively known, it has been suggested that certain inhibitors prevent a conformational change in NS5B that is crucial for RNA replication. To gain insight into the molecular origin of long-range allosteric inhibition of NS5B, we employed molecular dynamics simulations of the enzyme with and without an inhibitor bound to the thumb domain. These studies indicate that the presence of an inhibitor in the thumb domain alters both the structure and internal motions of NS5B. Principal components analysis identified motions that are severely attenuated by inhibitor binding. These motions may have functional relevance by facilitating interactions between NS5B and RNA template or nascent RNA duplex, with presence of the ligand leading to enzyme conformations with narrower and thus less accessible RNA binding channels. This study provides the first evidence for a mechanistic basis of allosteric inhibition in NS5B. Moreover, we present evidence that allosteric inhibition of NS5B results from intrinsic features of the enzyme free energy landscape, suggesting a common mechanism for the action of diverse allosteric ligands.
Keywords: allostery, non-nucleoside inhibitor, conformational change, NS5B polymerase, molecular simulation
INTRODUCTION
The hepatitis C virus (HCV) is a single stranded, positive sense RNA virus that has infected ~200 million people worldwide. The currently approved standard treatment, a combination of ribavirin and pegylated interferon, has limited efficacy.1–4 Thus, much effort has been expended to identify new molecules that can serve as therapeutics for HCV infection. Because of its importance for viral propagation, the HCV RNA-dependent RNA polymerase (gene product NS5B) has become a therapeutic drug target.2,5–10 The enzyme has a structure resembling an open right hand with three domains: fingers, palm, and thumb. There are two magnesium ions coordinated in the palm domain by three aspartic acid residues (220, 318, and 319). These ions stabilize key transition states in the nucleotide addition catalyzed by NS5B. To date, five different allosteric binding sites have been identified that are distant from the active site at which chemistry occurs. A number of small, non-nucleoside inhibitors (NNIs) have been shown to bind at these sites.11–33 Several X-ray crystal structures of NS5B have been determined, both in the presence and absence of bound inhibitors and divalent metal ions. Interestingly, the active site of NS5B does not appear significantly perturbed by the presence of allosteric inhibitors. Moreover, neither the structural nor biochemical data obtained thus far explain the mechanism by which these ligands function.
Herein, we employ molecular dynamics (MD) simulations and other computational tools to determine how ligand binding to thumb site NNI2 affects the structure, dynamics, and thermodynamic properties of NS5B (see Fig. 1). Molecular simulations allow us to study these properties with atomic detail, illuminating the molecular origin of allosteric inhibition. We observe structural and dynamic perturbations of NS5B in the presence of inhibitor that are in good agreement with biochemical evidence regarding the impact of inhibitor binding on NS5B function. These results imply that NNI2 inhibitors act to prevent motions required for polymerase activity and induce the enzyme to sample conformations that discourage RNA replication. This is the first study that examines the impact of inhibitor binding on NS5B dynamics using extensive MD simulations, advancing our understanding of the molecular mechanism of allosteric inhibition in this important viral enzyme.
Figure 1.
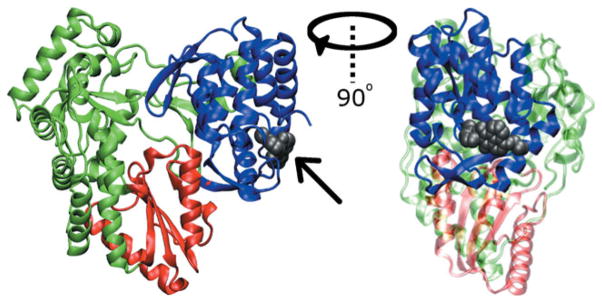
Cartoon representation of NS5B from PDB structure 2WHO. The fingers domain is shown in green, the palm domain in red, and the thumb domain in blue. Allosteric binding site NNI2 is shown occupied by the ligand VGI (gray space filling representation identified by arrow) from the 2WHO structure. The right panel represents a 90° rotation about the vertical axis relative to the left.
MATERIALS AND METHODS
PDB structure selection and preparation
The NS5B enzyme from HCV genotype 1b with PDB identifier 2WHO was employed for our simulations. This structure contains non-nucleoside inhibitor 1H-benzo[-de]isoquinoline-1,3(2H)-dione (PubChem ID 4177750, also known as VGI) bound to site NNI2 of the thumb domain of the enzyme and also contains two manganese (Mn) ions bound to the known divalent ion binding site in NS5B.27 There are two protein chains in the 2WHO unit cell; chain A was selected to perform all simulations. The 2WHO contains 531 amino acids and does not include the 60 C-terminal residues of the enzyme, known as the linker region, that are thought to be membrane associated in vivo. These residues cause protein aggregation in vitro and are not required for in vitro RNA replication.34 Thus, this variant of NS5B has been widely used for biochemical and structural studies. Though 2WHO contains Mn in the divalent ion site, this site is thought to be occupied by magnesium (Mg) under physiological conditions.35–37 Thus, the Mn ions were replaced by Mg and the protein, ions and ligand placed in a truncated octahedral unit cell that was larger than the protein by at least 10 Å in each dimension, generating a cell with edge length 94 Å. To carry out simulations in the absence of inhibitor, the ligand was first deleted from the 2WHO structure. Nineteen chloride ions were added to the unit cell to neutralize the system charge. Finally, TIP3P38 water molecules were added to fill the unit cell and all water molecules overlapping with the protein, ligand or ions were deleted. The resulting simulation systems contained ~60,000 atoms.
Minimization
All molecular dynamics and energy minimization calculations were carried out using the NAMD simulation engine39 and the CHARMM 27 protein force field.40,41 Parameters for the ligand VGI were taken from the CHARMM General Force Field version 2a5.42 Parameters not available in the force field were obtained by following the protocol of MacKerell and coworkers.42,43 One thousand minimization steps were performed for the fully solvated system using the conjugate gradient algorithm available in NAMD.
Molecular dynamics simulations
After minimization, molecular dynamics (MD) simulations were carried out in the NVT ensemble using a 2-fs time step and applying periodic boundary conditions. All covalent bonds to hydrogen atoms were constrained using the SHAKE algorithm. Temperature was maintained at 300 K via velocity reassignment every 100 time steps. The particle-mesh ewald method was employed for electrostatics. The cutoff for nonbonded interactions was 10 Å and a switching function was applied to scale short range interactions to zero starting at 9 Å. The non-bonded pairlist was calculated out to a distance of 11.5 Å. Nearly 50,000 time steps were carried out for a total of 100 ps simulation time. Nonbonded interactions were computed every step and overall momentum was also removed from the system at each step. During this period the positions of Cα atoms were restrained to their initial positions in the minimized structure using force constants of 100 kcal (mol−1 Å−2). Following this initial NVT equilibration, NPT equilibration was performed. All conditions were identical to the NVT simulations except that the pressure was maintained at 1.01 bar using a Berendsen barostat and the restraints on Cα atoms were removed. About 2,500,000 steps were carried out for total simulation time of 5 ns. The final snapshot from the NPT simulations was used to initiate production runs in the NVT ensemble. Conditions applied were identical to those in the initial NVT simulations except that restraints and a barostat were not applied. The production simulations were carried out for a total of 400 ns in 10 ns segments, with coordinates written out every 50,000 steps (100 ps). The final 300 ns of each trajectory were employed for data analysis. During the last 20 ns of the ligand-bound trajectory we observed an unbinding event, with VGI leaving the NNI2 binding pocket after 383 ns and entering the solvent before loosely associating with NS5B again near the top of the palm domain at 388 ns. VGI remains at this location for the remaining 12 ns of the trajectory. However, this event was not observed to alter the enzyme’s structural or dynamic properties including flexibility, average structures, RMSD values, inter-residue distances (e.g., see Figs. 2, 4 and Supporting Information) and covariance maps generated with and without the final 20 ns of trajectory data. Consequently, this event does not alter the conclusions of this work with regard to the impact of inhibitor binding on the intrinsic functional properties of NS5B.
Figure 2.

RMSD plot illustrating structural fluctuations occurring in molecular dynamics trajectories. RMSD from the initial minimized structure for the protein Cα atoms is shown as a function of time for simulations of the free and ligand-bound enzymes as black and gray lines labeled “Free” and “VGI”, respectively (this nomenclature is maintained in the remaining figures). Regions highlighted by the gray box were considered part of the equilibration period and were not employed for analysis. Note the much larger fluctuations in simulations of the free enzyme and the equilibration time of about 100 ns for each trajectory.
Figure 4.

Solvent accessible surface (SASA) computed for residues along the RNA template channel during equilibrated portion of protein trajectories. Values for ligand-free and ligand bound simulations are shown as solid and dashed lines, respectively. Horizontal lines denote the averages computed for each simulation (also see Table I). Note that the average SASA is significantly greater for the free enzyme simulation. Also note the significant increase in SASA that occurs for this trajectory after about 265 ns of total simulation time (circled region). Increased SASA values coincide with the structural changes described in the text.
Covariance maps
The covariance matrix Cij describes the degree of correlation between motions sampled by atoms i and j during an MD trajectory. The normalized covariance between two atoms is determined by taking the product of the difference between the instantaneous values of the positional coordinates ri or rj and their average values 〈ri〉 or 〈rj〉, where r includes the x, y, and z directions:
| (1) |
The matrix is typically normalized as shown in Eq. (1) so that atoms with motion that is completely positively correlated display Cij values of 1, while those that are completely negatively correlated display Cij values of −1. Values of Cij close to zero indicate the motion of the two atoms is uncorrelated or occurs in orthogonal directions.
Principal components analysis (PCA)
The covariance matrix can be expressed in a simplified form that facilitates interpretation of the underlying motional correlations. The principal components (PCs) of motion represented in the covariance matrix can be obtained by diagonalizing the matrix to generate eigenvalues λ using the eigenvector matrix Q [see Eq. (2)]:
| (2) |
Together the eigenvalues and eigenvectors comprise the PCs of the protein motion. The eigenvectors describe the direction of the motions sampled during the trajectories, while the eigenvalues describe the magnitude of these motions. The PCs are analogous to vibrational modes from a normal mode analysis. Typically the largest eigenvalue accounts for most of the motion, the second accounts for a lesser amount and so on.44–46 These modes can be used to determine the nature of the motions involved in generating specific patterns of correlation.
Filtering PCs to identify specific motions
To focus attention on a given vibrational mode, it is helpful to isolate the correlated motions associated with a given PC. This can be accomplished by reconstructing the covariance matrix using only individual PCs. Given the matrix of PC eigenvectors Q, the covariance matrix C, and eigenvalues λ, one can reconstruct the covariance matrix C as follows:
| (3) |
The last line results because Q is orthogonal. Similarly, one can modify λ to select a subset of eigenvalues λ′ and employ only this subset to reconstruct a filtered covariance matrix C′.
| (4) |
In doing so, the eigenvalues that are not of primary interest are set to zero. This filtering procedure allows one to determine which correlated motions result from a given mode as well as to compute other useful properties.
Projection of a trajectory unto individual PCs
To visualize the conformational space being sampled by the enzyme during the simulations, protein coordinates from the trajectories can be projected onto the eigenvectors Q obtained from PCA. This provides a measure of the overlap between the conformational space present in a given snapshot and that represented in the eigenvector. For a given snapshot Rk, the projection of this snapshot Plk unto eigenvector Ql is given by:
| (5) |
The projections are normalized to fall in the range of −1 to 1, where larger absolute values indicate greater overlap between the conformational space represented by a trajectory snapshot and the conformational space described by the PC eigenvector.
RESULTS AND DISCUSSION
NS5B inherently displays conformational fluctuations on long time scales
We observed that the NS5B requires equilibration times on the order of 100 ns. This can be seen by examining the RMSD from the initial minimized structure for the protein Cα atoms as a function of time (Fig. 2). This observation indicates that the conformational fluctuations of the enzyme are intrinsically slow, occurring on the 100 ns time scale. The RMSD values for the ligand-bound and free enzyme with respect to 2WHO are 2.70 and 1.59 Å respectively, while the two structures display an RMSD of 1.91 Å relative to each other. Thus, the free enzyme remains more similar to the original 2WHO coordinates than the ligand-bound structure, but both simulations become less similar to the crystallographic coordinates. This result is important because it suggests the crystallographic coordinates alone will not provide information about the most likely states of the enzyme in solution.
The need for extended equilibration may arise because the simulations were carried out in aqueous solvent, neutral pH, and at ambient temperature. This is in contrast to the mix of organic and aqueous solvent, low temperature (100 K), and low pH (6.0) at which the crystal structure was generated.27 In addition, crystal packing may limit the number of accessible enzyme conformations in the unit cell. However, despite the long equilibration, the relative flexibility of structural elements in the enzyme is qualitatively consistent between 2WHO and the MD simulations. This can be observed by comparing the relative magnitudes of the crystallographic and computed B-factors along the peptide chain (see Supporting Information). This observation suggests that equilibration does not disrupt the inherent structural elements of NS5B and instead alters their tertiary organization, resulting in new enzyme conformations.
The RMSD plot shown in Figure 2 reveals that large conformational fluctuations persist throughout the trajectory of the free enzyme, with transient excursions to high RMSD values even after 300 ns of simulation time. This observation is further evidence that the characteristic motions in NS5B occur on long time scales. Such long-lived fluctuations may represent motions that are inherent to the enzyme and that have functional significance.
Presence of inhibitor alters the enzyme structure
Given the wealth of structural data already available for NS5B, it is helpful to examine the MD average structures in the context of known NS5B conformational states. NS5B exhibits at least two different conformations: a closed conformation thought to be required for the initiation of RNA replication and an open conformation required for elongation of the nascent RNA strand.47,48 The open and closed conformations are epitomized by the NS5B structures with PDB IDs 1YZ2 and 1YUY respectively.49 The structural differences between the two conformations arise primarily from a rigid body rotation of the thumb domain relative to the fingers and palm domains, in which the thumb domain becomes closer to the fingers domain in the closed conformation. This rotation alters interactions between residues at the tip of the fingers and thumb domains. In contrast to the thumb and fingers, the palm domain remains relatively invariant when different conformational states of NS5B are superimposed. Thus, the open and closed conformations can be discriminated most clearly by aligning the palm domains of various proteins and observing the structural differences in the remaining two domains.
We observe that 2WHO exhibits a structure intermediate to the open and closed conformations (although it is more similar to the closed conformation: see Supporting Information). During the simulations the free enzyme remains similar to the closed conformation while the ligand-bound simulation undergoes a marked conformational change in which the thumb domain reorients into what we refer to as the “hyper-closed” conformation. In the process, a β-hairpin loop in the thumb domain (residues 440–455) becomes much closer to the fingers domain (see Fig. 3). Known as the β-loop, these residues have been implicated in correct positioning of the RNA template.50
Figure 3.

Structural comparison of average structures from the MD simulations with the 2WHO crystallographic coordinates. (Upper Panel) MD average structures from simulations with (orange) and without (blue) ligand superimposed on 2WHO (gray). All three structures were aligned using the protein backbone of residues in the palm and fingers domains. For clarity only the thumb domain of the free enzyme and ligand-bound simulations is shown so structural differences in the thumb domain can be clearly delineated. (Lower Panel) Magnified views of the highlighted region in the top panel comparing the thumb domain structure found in 2WHO with those found in the free enzyme (left) and ligand-bound (right) structures. Note that the β-loop of the free enzyme (left side of the thumb domain) assumes a structure that is similar to that found in the crystallographic coordinates. This loop becomes closer to the fingers domain in the ligand-bound enzyme in what we have designated as a “hyper-closed” conformation. Average structures were computed using all 300 ns of equilibrated trajectory data.
In the “hyper-closed” conformation there is less space between the thumb and finger domains than in the original 2WHO structure. As a result, it is unlikely that RNA can bind to this conformation of NS5B because the RNA template and RNA duplex channels are considerably narrower in this state (see the following section and Supporting Information). Narrowing of the template channel is particularly noteworthy because deletion studies have shown that absence of the C-terminal linker region of NS5B (as is the case in this work) typically results in a wider RNA binding cleft.34 Electron microscopy studies have identified conformational states of NS5B that are more closed than those observed in available crystal structures of the enzyme.47 Our observation of a “hyper-closed” conformation of NS5B when it is bound to VGI is consistent with this finding. It is worth noting that this “hyper-closed” conformation has not previously been characterized at a molecular level of detail, providing a unique opportunity for our studies to provide new insight into its properties.
Narrowing of the RNA template and duplex channels in the “hyper-closed” conformation suggests that the thumb inhibitor VGI disfavors RNA binding. By overstabilizing the closed state, VGI may prevent the transitions between closed and open conformations thought to be necessary for RNA replication. This mechanism of inhibitor action is consistent with biochemical data suggesting that thumb binding inhibitors can prevent a conformational change required for the initial steps of RNA replication.15,22,27,33,51
Conformational fluctuations modulate width of the RNA binding channels
To systematically characterize the environment of the RNA template channel, we examined the solvent accessible surface area (SASA) for amino acid residues lining this channel. These residues were selected by using crystal structure 1NB7 that contains a four-nucleotide fragment of RNA template and identifying all enzyme residues that are <5 Å from any nucleotide atom in the 1NB7 structure. The SASA of these residues was then measured for each snapshot in the two trajectories. The results of this analysis are shown in Figure 4 and Table I. The free simulation displays significantly more SASA overall than the ligand-bound simulation (by ca. 300 Å2) and also displays SASA values that fluctuate more widely (indicated by their larger standard deviation). This result indicates there is more space within the RNA template channel in the ligand-free simulation on average and that the free enzyme undergoes fluctuations that greatly alter channel width.
Table I.
Solvent-Accessible Surface Area (SASA) Values for Residues Lining RNA Template Channel in NS5B
| SASA ± σ (Å2) | |
|---|---|
| Free enzyme | |
| All | 5355 ± 171 |
| 100–220 ns | 5302 ± 109 |
| 221–300 ns | 5512 ± 182 |
| 301–400 ns | 5282 ± 129 |
| Ligand bound | |
| All | 5071 ± 98 |
| 100–240 ns | 5091 ± 93 |
| 241–400 ns | 5054 ± 99 |
σ = one standard deviation.
To get more detailed information about how the environment of the RNA template channel changes during the simulations, the trajectories were divided into different segments that contain snapshots displaying similar SASA values. The free enzyme simulation was partitioned into three time segments: 100–220, 221–300, and 301–400 ns. The simulation of the ligand-bound enzyme was partitioned into two segments: 100–240 and 241–400 ns. The average SASA values computed for each trajectory segment are shown in Table I. It is apparent that the second time slice for the free simulation displays SASA values that are significantly greater than those observed in the other segments of this trajectory. This transient increase suggests that a structural transition occurs in this segment of the simulation that impacts SASA. In contrast, no significant change in SASA is observed during the course of the ligand-bound trajectory.
As shown in Figure 5, we observe that increased SASA in the template channel of the free enzyme is associated with an increase in the separation between fingers and thumb domains. This increased separation is associated with widening of both the RNA template and RNA duplex channels in the enzyme (see Supporting Information). In contrast to the free enzyme, the width of these channels does not vary significantly in ligand-bound NS5B. In addition, template and duplex channels in the ligand-bound enzyme are narrower than those observed in the free enzyme by about 5 and 2 Å, respectively. This is quite remarkable because VGI elicits these effects despite binding to NS5B at least 35 Å from both channels.
Figure 5.

Structural changes that accompany variations of SASA in the free enzyme. The free trajectory can be divided into time segments (labeled) that contain similar SASA values. (Upper panels) Surface representations of the average structures computed for each segment. The fingers domain is drawn in green while the thumb domain is in blue. The viewpoint shown corresponds to a 90° rotation about the horizontal axis relative to that in Figure 1, so that we are looking down on the top of the enzyme. The space between the two domains becomes slightly larger during 221–300 ns. (Lower panel) Conformations of residues at the interface between fingers and thumb domains. Magnified views of region enclosed by red boxes in top panel. Residues P94, H95, R109, V405, P404, and D444 are shown in space filling representation. H95, R109, V405, and D44 are colored cyan, white, light brown and red, respectively, while the two proline residues are displayed in dark brown. The perspective corresponds to that shown on the left side of Figure 1. The large changes in SASA observed for the enzyme are linked to repacking of the side chains of these residues. Note that these residues become much more loosely packed in that segment of the trajectory with larger interdomain separation.
These results are significant because opening of these two RNA channels may represent crucial steps in RNA replication. By hindering the occurrence of these early steps, the presence of inhibitor may make it more difficult for replication to proceed. These observations suggest that the crucial conformational change noted in previous studies to be required for RNA replication and prevented by inhibitor binding15,22,27,33,51 corresponds to fluctuations that widen the cleft between fingers and thumb domains and, simultaneously, the RNA binding channels in the enzyme.
We identified features of the enzyme that differentiate conformations with an increased separation between fingers and thumb domains from those exhibiting reduced separation by examining average structures generated from each of the trajectory segments listed above. In addition to changes in the protein backbone that underlie the motion of secondary structural elements, certain residues undergo side chain repacking associated with the increased SASA values for the 221–300 ns segment of the free protein trajectory. These include: P94, H95, G317, P404, V405, D444, Q446, G449, and C451. In particular, the conformations of residues 94, 95, 404, and 405 at the top of the RNA template channel vary significantly. The former two residues are found at the tip of the fingers domains while the latter two are at the tip of the thumb domains. Magnified views of these residues (shown in Fig. 5) indicate that increased SASA is associated with looser packing between their side chains. These interactions thus may be key to modulating the ability of the enzyme to bind to RNA template. In fact, V405 has been shown to help stabilize the closed conformation required for de novo initiation of NS5B.52 Caillet-Saguy et al. have compiled a list of mutations that occur in response to exposure of HCV to NS5B allosteric inhibitors. Several of the residues described above or those nearby are part of this list (see the section “Relationship to Other Modeling Studies” below).13 Thus, key residues mediating the activity of thumb binding ligands are located in regions of the enzyme engaged in structural rearrangements that affect the separation between fingers and thumb domains and consequently the RNA binding channels. Mutating these residues may prevent the fingers and thumb domains from becoming closely associated. The width of this interdomain cleft may thus be an important factor in regulating the action of NNIs to inhibit NS5B. By reducing separation of the fingers and thumb domains, the presence of an inhibitor at site NNI2 appears to disfavor binding of the RNA template as well as the nascent RNA duplex. These combined effects may act synergistically to enhance the ligand’s inhibition of NS5B.
Correlated enzyme motions are eliminated by ligand binding
In addition to distinct average structures being generated in each simulation (see Fig. 3), the fluctuations about these averages differ. This is most apparent if one examines covariance matrices for the protein fluctuations as shown in Figure 6. While there are similar correlated motions that occur in both cases, the free enzyme displays higher levels of correlation (i.e., more intense colors in the plot) and these correlations involve the concerted motions of a larger number of residues. Thus, ligand binding eliminates intrinsic correlated fluctuations that occur in NS5B. Among correlations that differ between the two simulations are those that involve relative movement of the thumb and fingers domains. The free enzyme displays pronounced anticorrelated movement between the fingers and thumb domains, indicated by the dark blue colors in the free enzyme plot at the intersection of residues 400–475 and 75–275, which correspond to the thumb and fingers domains, respectively. In contrast, the ligand-bound enzyme does not exhibit these extensive anticorrelated movements. If these motions are functionally important, reduction of these motions when VGI is bound would be consistent with inhibitory action of the molecule. Thus, it is important to understand the fundamental nature of these motions and how they are altered by ligand binding. To facilitate this process, we performed principal components analysis (PCA) by diagonalizing the covariance matrix.
Figure 6.
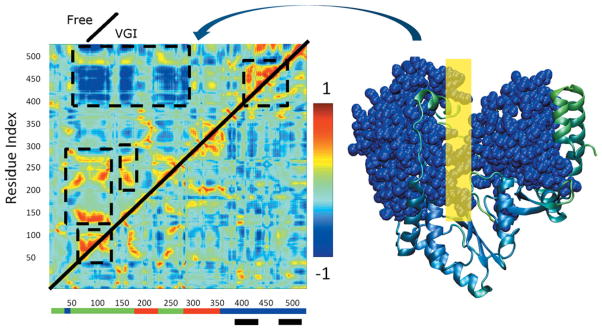
(Left) Normalized covariance matrices showing correlated motion between Cα atoms in NS5B. Indices along the axes denote position along the amino acid sequence. Color scale shown on right ranges from −1 for maximally anticorrelated motion through 1 for maximally correlated motion. Upper diagonal: free enzyme simulation. Lower diagonal: ligand-bound simulation. Regions where correlation in the free enzyme differs from that in the ligand bound simulations are indicated by dashed black lines. Colored bars at base of plot indicate locations of the fingers (green), thumb (blue), and palm (red) domains. Black bars indicate the location of the inhibitor binding site. A space filling representation of the regions engaged in large scale anticorrelated movement involving thumb and fingers domains are shown in blue in the representation of the protein on the far right. The corresponding region of the covariance plot is indicated by the curved arrow. The yellow rectangle on the right indicates the approximate location where the RNA template strand binds to the enzyme.
In comparing the principal components (PCs) obtained from the two sets of simulations, we are particularly interested in the 1% that correspond to the largest amplitude, lowest frequency motions,44–46,53 as there is much evidence to suggest that such motions are more likely to be functionally relevant.45,46,54 Each PC represents a vector that encodes the unique fluctuations occurring in a given trajectory. The projection of a protein snapshot along this vector (see Methods) defines a similarity metric, with larger values of this metric indicating greater similarity between the conformational space represented in the snapshot and that described by the PC. Thus, to systematically compare the conformations in each trajectory we projected every snapshot from the two simulations unto each of the 1% of largest amplitude PCs. PCs that display unique projection distributions for each of the two simulations represent motions that differ between the two.
As a result of this procedure, we identified the largest amplitude PC from each simulation that displays a unique projection distribution for both trajectories. These correspond to mode 1 for the free enzyme and mode 2 for the ligand-bound enzyme. In this nomenclature mode 1 refers to the largest amplitude PC and mode 2 to the second largest. Mode 1 of the free enzyme represents a collective opening of the cleft between fingers and thumb domains while mode 2 of the ligand-bound enzyme represents a breathing motion in which the finger loop interacts with the thumb domain and prevents the relative motion of the two domains. These PCs identify the key low frequency motions that differentiate the two simulations.
One would expect that these two PCs are associated with the unique patterns of correlated motion that occur in the free and ligand-bound enzyme. By filtering the PCs of the free enzyme to isolate mode 1 and reconstructing the covariance matrix (see Methods), we essentially reproduced the anticorrelation between the thumb and finger domains shown in Figure 6, confirming that this mode plays an important role in generating this pattern. The reconstructed covariance matrix is provided as Supporting Information, in addition to movies illustrating the motions occurring for these and other PCs that differentiate the two simulations.
Conformational distributions of the free and ligand-bound enzyme differ
The protein coordinates can be simultaneously projected against multiple PCs: each point in such a plot represents a distinct location in the protein conformation space. Thus, one can quantify structural differences between protein conformations by projecting the coordinates unto a defined set of PCs. This approach provides an objective way to assess how the ensemble of conformations sampled by the free enzyme is altered by the presence of ligand. Figure 7 shows both trajectories projected unto mode 1 from the free (x axis) and mode 2 from the ligand-bound (y axis) simulations. From the figure it is apparent that structural ensembles from the two simulations occur in distinct regions of conformation space, with little overlap between the two.
Figure 7.
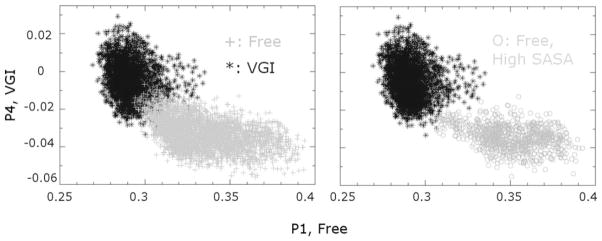
Using principal components to categorize the conformational space explored in MD simulations of the free and ligand-bound enzyme. Coordinates for each snapshot of the two trajectories are projected unto the lowest frequency principal component identified in simulations of the free (x axis) and ligand-bound (y axis) enzyme that discriminates between the two trajectories. (Left) Each point in the plot indicates a distinct position in the protein conformational space for the free and ligand-bound trajectories. Note that there is little overlap between the conformations sampled in the two sets of simulations. (Right) Conformations of the free enzyme with increased SASA values for the RNA template channel (circles) are found in a distinct region of conformational space, indicating that specific conformations are associated with larger SASA values and thus greater separation between the fingers and thumb domains. Also note that, in general, these conformations are less similar to conformations of the ligand-bound protein than the conformations of the free enzyme.
The conformational changes that accompany widening of the cleft between fingers and thumb domains are apparent if we locate coordinates that display higher SASA in the vector space shown in Figure 7. Doing so reveals that NS5B conformations with higher SASA values in the free enzyme are less similar to conformations from the ligand-bound simulations than other conformations from the free enzyme trajectory.
We can relate our observations to two representative models of protein allostery: the conformational selection and induced fit models. In the conformational selection model, ligand binding stabilizes and promotes pre-existing enzyme conformations that occur at low levels even in the absence of ligand.55,56 Allostery thus results from a shift in the relative populations of protein conformations when ligand is bound. In contrast, the induced fit model invokes concerted structural changes that result from ligand binding. Thus, in the presence of ligand, entirely new enzyme conformations (and dynamics) are observed compared to the free enzyme.57,58 Our results support the induced fit model as the more applicable description of allosteric inhibition in NS5B. Figure 7 illustrates that there is negligible overlap between the ligand-bound and free enzyme conformational distributions, suggesting that ligand binding induces the formation of distinct conformations not observed in the free enzyme. For conformational selection to be at play, one would expect to see greater overlap between the two conformational distributions in the enzyme’s free energy landscape. While this observation does not preclude the possibility that pre-existing ligand-bound conformations occur at low levels in the free enzyme,59,60 it does suggest that conformational selection is not the dominant mechanism by which allostery occurs in NS5B.
Ligand binding reduces overall flexibility of NS5B
The PC eigenvalues were used to compute the vibrational entropy of Cα atoms in NS5B to assess the overall flexibility of the enzyme. This was done using two different methods: the approach described by Brooks et al.44 and another due to Andricioaei and Karplus.61 Both give similar results and indicate that there is more low frequency motion when the ligand is bound. Consequently, even though the free enzyme displays an increased number of correlated regions, the ligand-bound enzyme displays lower frequency motions overall. This occurs because the ligand-bound enzyme moves to a lesser degree than the free enzyme, leading to lower frequency vibrational modes on average. In this regard, the bound ligand acts like a molecular staple by preventing NS5B from carrying out its innate motions. As is apparent from Figures 6 and 7, not only is the enzyme moving to a reduced degree when ligand is bound, it is also moving in a different manner compared to the free enzyme simulation. Thus, studying only the ligand-bound enzyme is not likely to inform one about properties of the functional enzyme and vice versa.
Relationship to experimental studies
Steady state kinetics and order of addition measurements using several different classes of inhibitors demonstrate that benzimidazole inhibitors (which bind to the same NNI2 site in the thumb domain as VGI) prevent a step that is crucial for the initiation of RNA replication.24,32 Liu et al. showed that this class of molecules inhibit conformational changes that occur during RNA synthesis.62 This data suggests that thumb binding inhibitors at site NNI2 prevent functional motions or specific conformations that occur early in the replication cycle and thus decrease the competency of the enzyme to replicate RNA. Such a model is consistent with the observations made in our work, as the altered conformations we observe to be mediated by inhibitor binding may correspond to the altered conformational states induced by the binding of benzimidazole inhibitors to NS5B.
Relationship to other molecular dynamics studies of viral polymerases
Recent simulation studies by Moustafa et al. on several viral RNA polymerases bear relation to our work.63 These authors used MD to determine the intrinsic motions of polymerases from poliovirus, coxsackievirus B3, human rhinovirus type 16 and foot-and-mouth disease virus. Each of these viruses possesses a genome containing a single strand of positive sense RNA and thus employs an RNA-dependent RNA polymerase (RdRp) in a similar manner to that in which HCV employs NS5B for RNA replication. These RdRps share similar structural motifs and overall structural organization with NS5B. However, these enzymes are distinct from NS5B in terms of amino acid sequence. They are part of the positive-stranded RNA virus supergroup I, while flaviviruses such as HCV are found in supergroup II.64 Thus, they contain only ~460–480 residues compared to the 531 residues found in 2WHO, with sequence identity to 2WHO ranging from 8 to 11%. In addition, group I viruses lack the β-loop composed of residues 440–455 that we observe to modulate interaction between the thumb and finger domains in NS5B. Comparisons of the sequence and secondary structure of NS5B with respect to the RdRps studied by Moustafa et al. is provided as Supporting Information.
These authors63 observed well-defined patterns of motion occurring in the group I RdRp enzymes. These include anticorrelated movement of the fingers and thumb domain as well as fluctuations that modulate the widths of the nucleotide binding region, the RNA template groove and the RNA duplex channel on the nanosecond time scale. Thus, our studies of NS5B demonstrate patterns of motional correlation that appear analogous to those observed previously, supporting the general observations made for group I RdRps in the context of HCV. The anticorrelation we observe from residues 400–475 to residues 75–275 appears similar to the anticorrelation observed from residues 400–450 to residues 100–150 and 325–375 in group I RdRps.63 In both cases the anti-correlated motions correspond to movement of the thumb and fingers domains in a pincer like fashion. Note that residue numbers cannot be directly compared between the two studies because of the structural differences between NS5B and group I RdRps.
The shared observations of the two studies with respect to the patterns of motion described above suggest that these motions may be innate to positive-stranded RNA polymerases because they play important functional roles. These observations also suggest that conformational fluctuations modulating the width of the RNA template and nascent RNA duplex binding channels are intrinsic features of RdRp enzymes. The fact that these intrinsic fluctuations are disrupted by inhibitor binding in NS5B would suggest that these motions are, in fact, functional and that inhibitors reduce the activity of the enzyme by altering these motions. This is an important finding, as it implies that allosteric ligands bound at thumb site NNI2 inhibit NS5B by inducing conformational states with less accessible RNA binding channels.
Relationship to other modeling studies
A recent modeling study by Caillet-Saguy et al.13 analyzed the conformational diversity in several NS5B crystal structures with various amino acid sequences and from different viral genotypes. They identified likely hinge sites for NS5B conformational changes by predicting which residues display low flexibility based on high reproducibility of their locations within aligned NS5B structures.13 Several of the locations they identified as hinges are also sites where mutations are known to occur when HCV is challenged by non-nucleoside inhibitors in vitro.15,32,49,65–68 Such residues may lie along allosteric communication pathways involving the ligand binding site. Thus, these mutations may induce resistance by altering hinge regions to increase overall enzyme flexibility and decrease the rigidification caused by inhibitor binding. Of particular interest are residues 95, 138, 162, and 445 that are part of the RNA template binding channel. Of these, H95 was identified in our molecular dynamics simulations as being involved in side chain repacking that modulates the separation between the fingers and thumb domains (see Fig. 5).
We observe many of the same elements of structural flexibility identified previously, including several similar to the identified hinge points (see Supporting Information). The fact that two independent studies with markedly different methods (analysis of an ensemble of static crystal structures13 versus molecular dynamics simulations herein) lead to similar observations lends support to the notion that the patterns of flexibility observed in NS5B are robust properties of the enzyme and gives credence to the assertion that these flexibility patterns have functional significance.63
In a similar manner to Liu et al.,62 Caillet-Saguy et al.13 suggest that allosteric NS5B inhibitors prevent the thumb domain from achieving a conformation appropriate for the initiation of RNA replication. These authors note that inhibitor binding to any of the three sites they examined perturbs the conformation of the NS5B thumb. This observation supports the notion that different inhibitors may share a common mechanism of action. The results obtained in our present study thus may be relevant to understanding the effects of diverse types of NS5B inhibitors.
CONCLUSIONS
Our studies support previous findings13 with regard to predicting the flexibility of different regions of NS5B and identifying amino acids likely to serve as hinges for intrinsic motions. However, the long equilibration times observed in our studies suggest that it would be difficult or impossible to understand the effects of inhibitor binding on the structural properties of NS5B solely by examining static crystal structures of the enzyme. Our studies are also consistent with recent studies of supergroup I RNA polymerases,63 in that they indicate NS5B experiences similar patterns of correlated motion not until now demonstrated for supergroup II enzymes such as NS5B. This observation suggests that dynamics, structure, and function have coevolved in NS5B and similar RNA polymerases.
Our studies clearly describe how the structural and dynamic properties of NS5B are perturbed by the presence of an allosteric ligand bound to the thumb domain of the enzyme. This is a distinctly new contribution to the field, as the impact of allosteric inhibitors on the structural and dynamic properties of NS5B has not yet been fully determined. We find that the presence of the ligand not only changes the average structure of the enzyme, but also reduces the magnitude of fluctuations occurring in the protein and alters the directions of the remaining motions. In particular, large-scale anticorrelated motions involving the thumb and fingers domains are abrogated when the inhibitor is bound.
The correlated motions removed due to inhibitor binding may have functional relevance by facilitating interactions between NS5B and RNA template or nascent RNA duplex. The presence of ligand leads to enzyme conformations with narrower and thus less accessible RNA binding channels. We identified specific residues involved in structural changes modulating the width of these channels, providing a concrete mechanism for how inhibitor binding inhibits the activity of NS5B. When put together these results suggest that ligand binding modulates the activity of the polymerase by disfavoring its ability to bind to RNA.
The ligand VGI bound to site NNI2 of NS5B acts as a molecular staple, significantly reducing overall enzyme motion. Because of its general nature, one would anticipate that such an effect could be readily replicated by any ligand able to rigidify the protein. Thus, the impact of allosteric ligands on NS5B may result from general features of the protein–ligand free energy landscape that are intrinsic to this enzyme. This may be one reason why multiple allosteric inhibitors with disparate structural and chemical features have been identified for NS5B and would suggest that it should be relatively easy to identify novel allosteric inhibitors of the enzyme.
In addition to providing fundamental insight into how allosteric inhibitors reduce the activity of NS5B, this knowledge may allow us to understand how NS5B is able to escape the effects of inhibitors via mutation. The simulations allow us to predict specific residues involved in mediating important interactions in the enzyme. In this regard, amino acids in regions that display significant structural stability in the ligand bound simulations may play a role in allowing inhibitors to immobilize the enzyme, freezing it in an inactive conformational state. We expect that mutating such residues will reduce the inhibitory capacity of allosteric ligands. Conversely, mutating amino acids in regions engaged in large scale motions in the ligand-free enzyme may reduce the activity of the enzyme even when it is not bound to any inhibitor. These ideas can be experimentally tested and we plan to carry out such studies in the near future.
Supplementary Material
Acknowledgments
Grant sponsor: US National Science Foundation; Grant number: CNS-0821258; Grant sponsor: SCREMS Program; Grant number: DMS-0821311; Grant sponsor: National Science Foundation; Grant number: OCI-1053575; Grant sponsor: University of Maryland, Baltimore County (UMBC)
Much of the hardware used in this study is part of the UMBC High Performance Computing Facility (HPCF). In addition, this work used the Extreme Science and Engineering Discovery Environment (XSEDE).
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Maass P, Schulz-Gasch T, Stahl M, Rarey M. Recore: a fast and versatile method for scaffold hopping based on small molecule crystal structure conformations. J Chem Inf Model. 2007;47:390–399. doi: 10.1021/ci060094h. [DOI] [PubMed] [Google Scholar]
- 2.Lindenbach B, Rice C. Unraveling hepatitis c virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 3.Mchutchison J, Bartenschlager R, Patel K, Pawlotsky JM. The face of future hepatitis c antiviral drug development: recent biological and virologic advances and their translation to drug development and clinical practice. J Hepatol. 2006;44:411–421. doi: 10.1016/j.jhep.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS. Hepatitis c viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 5.Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis c virus. Structure. 1999;7:1417–1426. doi: 10.1016/s0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 6.Behrens S, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis c virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 7.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey F. Crystal structure of the RNA-dependent RNA polymerase of hepatitis c virus. Proc Natl Acad Sci USA. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol. 2006;16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Lescar J, Canard B. RNA-dependent RNA polymerases from flaviviruses and picornaviridae. Curr Opin Struct Biol. 2009;19:759–767. doi: 10.1016/j.sbi.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Ortin J, Parra F. Structure and function of RNA replication. Annu Rev Microbiol. 2006;60:305–326. doi: 10.1146/annurev.micro.60.080805.142248. [DOI] [PubMed] [Google Scholar]
- 11.Beaulieu P, Jolicoeur E, Gillard J, Brochu C, Coulombe R, Dansereau N, Duan J, Garneau M, Jakalian A, Kuhn P, Lagace L, Laplante S, Mckercher G, Perrault S, Poirier M, Poupart MA, Stammers T, Thauvette L, Thavonekham B, Kukolj G. N-acetamideindolecarboxylic acid allosteri ‘finger-loop’ inhibitors of the hepatitis c virus ns5b polymerase: discovery and initial optimization studies. Bioorg Med Chem Lett. 2010;20:857–861. doi: 10.1016/j.bmcl.2009.12.101. [DOI] [PubMed] [Google Scholar]
- 12.Biswal BK, Wang M, Cherney MM, Chan L, Yannopoulos CG, Bilimoria D, Bedard J, James MNG. Non-nucleoside inhibitors binding to hepatitis c virus ns5b polymerase reveal a novel mechanism of inhibition. J Mol Biol. 2006;361:33–45. doi: 10.1016/j.jmb.2006.05.074. [DOI] [PubMed] [Google Scholar]
- 13.Caillet-Saguy C, Simister PC, Bressanelli S. An objective assessment of conformational variability in complexes of hepatitis c virus poly-merase with non-nucleoside inhibitors. J Mol Biol. 2011;414:370–384. doi: 10.1016/j.jmb.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 14.De Vicente J, Hendricks RT, Smith DB, Fell JB, Fischer J, Spencer SR, Stengel PJ, Mohr P, Robinson JE, Blake JF, Hilgenkamp RK, Yee C, Adjabeng G, Elworthy TR, Tracy J, Chin E, Li J, Wang B, Bamberg JT, Stephenson R, Oshiro C, Harris SF, Ghate M, Leveque V, Najera I, Le Pogam S, Rajyaguru S, Ao-Ieong G, Alexandrova L, Larrabee S, Brandl M, Briggs A, Sukhtankar S, Farrell R, Xu B. Non-nucleoside inhibitors of hcv polymerase ns5b. Part 2: synthesis and structure-activity relationships of benzothiazine-substituted quinolinediones. Bioorg Med Chem Lett. 2009;19:3642–3646. doi: 10.1016/j.bmcl.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Di Marco S, Volpari C, Tomei L, Altamura S, Harper S, Narjes F, Koch U, Rowley M, De Francesco R, Migliaccio G, Carfi A. Interdomain communication in hepatitis c virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J Biol Chem. 2005;280:29765–29770. doi: 10.1074/jbc.M505423200. [DOI] [PubMed] [Google Scholar]
- 16.Hendricks RT, Fell JB, Blake JF, Fischer JP, Robinson JE, Spencer SR, Stengel PJ, Bernacki AL, Leveque VJP, Le Pogam S, Rajyaguru S, Najera I, Josey JA, Harris JR, Swallow S. Non-nucleoside inhibitors of hcv ns5b polymerase. Part 1: synthetic and computational exploration of the binding modes of benzothiadiazine and 1,4-benzothiazine hcv ns5b polymerase inhibitors. Bioorg Med Chem Lett. 2009;19:3637–3641. doi: 10.1016/j.bmcl.2009.04.119. [DOI] [PubMed] [Google Scholar]
- 17.Ikegashira K, Oka T, Hirashima S, Noji S, Yamanaka H, Hara Y, Adachi T, Tsuruha JI, Doi S, Hase Y, Noguchi T, Ando I, Ogura N, Ikeda S, Hashimoto H. Discovery of conformationally constrained tetracyclic compounds and potent hepatitis c virus ns5b RNA polymerase inhibitors. J Med Chem. 2006;49:6950–6953. doi: 10.1021/jm0610245. [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Kim K, Kim D, Chong Y. Identification of novel hcv RNA-dependent RNA polymerase inhibitors using pharmacophore-guided virtual screening. Chem Biol Drug Des. 2008;72:585–591. doi: 10.1111/j.1747-0285.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- 19.Laplante S, Jakalian A, Aubry N, Bousquet Y, Ferland JM, Gillard J, Lefebvre S, Poirier M, Tsantrizos YS, Kukolj G, Beaulieu P. Binding modes determination of benzimidazole inhibitors of the hepatitis c virus RNA polymerase by a structure and dynamics strategy. Drug Des. 2004;43:4306–4311. doi: 10.1002/anie.200460326. [DOI] [PubMed] [Google Scholar]
- 20.Lee G, Piper D, Wang Z, Anzola J, Walker J, Li Y. Novel inhibitors of hepatitis c virus RNA-dependent RNA polymerases. J Mol Biol. 2006;357:1051–1057. doi: 10.1016/j.jmb.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Linton A, Tatlock J, Gonzalez J, Borchardt A, Abreo M, Jewell T, Patel L, Drowns M, Ludlum S, Goble M, Yang M, Blazel J, Rahavendran R, Skor H, Shi S, Lewis C, Fuhrman S. Allosteric inhibitors of hepatitis c polymerase: discovery of potent and orally bioavailable carbon-linked dihydropyrones. J Med Chem Lett. 2007;50:3969–3972. doi: 10.1021/jm0704447. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Tatlock J, Linton A, Gonzalez J, Borchardt A, Dragovich P, Jewell T, Prins T, Zhou R, Blazel J, Parge H, Love R, Hickey M, Doan C, Shi S, Duggal R, Lewis C, Fuhrman S. Identification and structure-based optimization of novel dihydropyrones as potent hcv RNA polymerase inhibitors. Bioorg Med Chem Lett. 2006;16:4834–4838. doi: 10.1016/j.bmcl.2006.06.065. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Tatlock J, Linton A, Gonzalez J, Jewell T, Patel L, Ludlum S, Drowns M, Raghavendran S, Skor H, Hunter R, Shi S, Herlihy K, Parge H, Hickey M, Yu X, Chau F, Nonomiya J, Lewis C. Discovery of (r)-6-cyclopentryl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-di-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (pf-00868554) as a potent and orally available hepatitis c virus polymerase inhibitor. J Med Chem. 2009;52:1255–1258. doi: 10.1021/jm8014537. [DOI] [PubMed] [Google Scholar]
- 24.Mckercher G, Beaulieu PL, Lamarre D, Laplante S, Lefebvre S, Pellerin C, Thauvette L, Kukolj G. Specific inhibitors of hcv polymerase identified using an ns5b with lower affinity for template/primer substrate. Nucleic Acids Res. 2004;32:422–431. doi: 10.1093/nar/gkh160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meladraki G, Afantitis A, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O. Identification of a series of novel derivatives as potent hcv inhibitors by a ligand-based virtual screening optimized procedure. Bioorgan Med Chem. 2007;15:7237–7247. doi: 10.1016/j.bmc.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 26.Nyanguile O, Pauwels F, Van Den Broeck W, Boutton CW, Quirynen L, Ivens T, Van Der Helm L, Vandercruyssen G, Mostmans W, Delouvroy F, Dehertogh P, Cummings MD, Bonfanti JF, Simmen KA, Raboisson P. 1,5-benzodiazepines, a novel class of hepatitis c virus polymerase non-nucleoside inhibitors. Antimicrob Agents Ch. 2008;52:4420–4431. doi: 10.1128/AAC.00669-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ontoria J, Rydberg E, Di Marco S, Tomei L, Atenni B, Malancona S, Hernando J, Gennari N, Koch U, Narjes F, Rowley M, Summa V, Carroll S, Olsen D, De Francesco R, Altamura S, Migliaccio G, Carfi A. Identification and biological evaluation of a series of 1h-benzo[-de]isoquinoline-1,3(2h)-diones as hepatitis c virus ns5b polymerase inhibitors. J Med Chem. 2009;52:5217–5227. doi: 10.1021/jm900517t. [DOI] [PubMed] [Google Scholar]
- 28.Pfefferkorn JA, Greene ML, Nugent RA, Gross RJ, Mitchell MA, Finzel BC, Harris MS, Wells PA, Shelly JA, Anstadt RA, Kilkuskie RE, Kopta LA, Schwende FJ. Inhibitors of hcv ns5b polymerase. Part 1: evaluation of the southern region of (2z)-2-(benzoylamino)-3-(5-phenyl-2-furyl)acrylic acid. Bioorg Med Chem Lett. 2005;15:2481–2486. doi: 10.1016/j.bmcl.2005.03.066. [DOI] [PubMed] [Google Scholar]
- 29.Rydberg H, Cellucci A, Bartholomew L, Mattu M, Barbato G, Ludmerer S, Graham DJ, Altamura S, Paonessa G, De Francesco R, Migliaccio G, Carfi A. Structural basis for resistance of the genotype 2b hepatitis c virus ns5b polymerase to site a non-nucleoside inhibitors. J Mol Biol. 2009;390:1048–1059. doi: 10.1016/j.jmb.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Ryu K, Kim N, Choi SI, Han CK, Yoon JH, No KT, Kim K, Seong BL. Identification of novel inhibitors of hcv RNA-dependent RNA polymerase by pharmacophore-based virtual screening and in vitro evaluation. Bioorgan Med Chem. 2009;17:2975–2982. doi: 10.1016/j.bmc.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Shaw A, Tedesco R, Bambal R, Chai D, Concha N, Darcy M, Dhanak D, Duffy K, Fitch D, Gates A, Johnston V, Keenan R, Lin-Georke J, Liu N, Sarisky R, Wiggall K, Zimmerman M. Substituted benzothiadiazine inhibitors of hepatitis c virus polymerase. Bioorg Med Chem Lett. 2009;19:4350–4353. doi: 10.1016/j.bmcl.2009.05.091. [DOI] [PubMed] [Google Scholar]
- 32.Tomei L, Altamura S, Bartholomew L, Biroccio A, Ceccacci A, Pacini L, Narjes F, Gennari N, Bisbocci M, Incitti I, Orsatti L, Harper S, Stansfield I, Rowley M, De Francesco R, Migliaccio G. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis c virus RNA-dependent RNA polymerase. J Virol. 2003;77:13225–13231. doi: 10.1128/JVI.77.24.13225-13231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M, Ng KK, Cherney MM, Chan L, Yannopoulos CG, Bedard J, Morin N, Nguyen-Ba N, Alaoui-Ismaili MH, Bethell RC, James MN. Non-nucleoside analogue inhibitors bind to an allosteric site on hcv ns5b polymerase. Crystal structures and mechanism of inhibition. J Biol Chem. 2003;278:9489–9495. doi: 10.1074/jbc.M209397200. [DOI] [PubMed] [Google Scholar]
- 34.Adachi T, Ago H, Habuka N, Okuda K, Komatsu M, Ikeda S, Yatsunami K. The essential role of c-terminal residues in regulating the activity of hepatitis c virus RNA-dependent RNA polymerase. BBA-Proteins Proteom. 2002;1601:38–48. doi: 10.1016/s1570-9639(02)00433-8. [DOI] [PubMed] [Google Scholar]
- 35.Benzaghou I, Bougie I, Bisaillon M. Effect of metal ion binding on the structural stability of the hepatitis c virus RNA polymerase. J Biol Chem. 2004;279:49755–49761. doi: 10.1074/jbc.M409657200. [DOI] [PubMed] [Google Scholar]
- 36.Bougie I, Bisaillon M. Metal ion-binding studies highlight important differences between flaviviral RNA polymerases. Biochim Biophys Acta. 2009;1794:50–60. doi: 10.1016/j.bbapap.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Bougie I, Charpentier S, Bisaillon M. Characterization of the metal ion binding properties of the hepatitis c virus RNA polymerase. J Biol Chem. 2003;278:3868–3875. doi: 10.1074/jbc.M209785200. [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential function for simulating liquid water. J Phys Chem. 1983;79:926–935. [Google Scholar]
- 39.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Charmm: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 41.Brooks BR, Brooks CL, III, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui W, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. Charmm: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1615. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD. Charmm general force field: a force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackerell AD. Atomistic models and force fields. In: Becker OM, Mackerell AD, Roux B, Watanabe M, editors. Computational biochemistry and biophysics. New York: Marcel Dekker; 2001. pp. 7–38. [Google Scholar]
- 44.Brooks BR. Harmonic analysis of large systems. J Comput Chem. 1995;16:1522–1542. [Google Scholar]
- 45.Lange OF, Grubmuller H. Can principle components yield a dimension reduced description of protein dynamics on long time scales? J Phys Chem B. 2006;110:22842–22852. doi: 10.1021/jp062548j. [DOI] [PubMed] [Google Scholar]
- 46.Ivosev G, Burton L, Bonner R. Dimensionality reduction and visualization in principle component analysis. Anal Chem. 2008;80:4933–4944. doi: 10.1021/ac800110w. [DOI] [PubMed] [Google Scholar]
- 47.Chinnaswamy S, Murali A, Cai H, Yi G, Palaninathan S, Kao CC. Conformations of the monomeric hepatitis c virus RNA-dependent RNA polymerase. Virus Adapt Treat. 2010;2:21–39. [Google Scholar]
- 48.Caillet-Saguy C, Simister PC, Bressanelli S. An objective assessment of conformational variability in complexes of hepatitis c virus polymerase with non-nucleoside inhibitors. J Mol Biol. 2011;414:370–384. doi: 10.1016/j.jmb.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 49.Biswal BK, Cherney MM, Wang M, Chan L, Yannopoulos CG, Bilimoria D, Nicolas O, Bedard J, James MNG. Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis c virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J Biol Chem. 2005;280:18202–18210. doi: 10.1074/jbc.M413410200. [DOI] [PubMed] [Google Scholar]
- 50.Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol. 2006;16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 51.Love R, Parge H, Yu X, Hickey M, Diehl W, Gao J, Wriggers H, Ekker A, Wang L, Thomson JA, Dragovich P, Fuhrman S. Crystallographic identification of a noncompetitive inhibitor binding site on the hepatitis c virus ns5b RNA polymerase enzyme. J Virol. 2003;77:7575–7581. doi: 10.1128/JVI.77.13.7575-7581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitt M, Scrima N, Radujkovic D, Caillet-Saguy CL, Simister PC, Friebe P, Al E. A comprehensive structure function comparison of hepatitis c virus strain jfh1 and j6 polymerases reveals a key residue stimulating replication in cell culture across genotypes. J Virol. 2011;85:2565–2581. doi: 10.1128/JVI.02177-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurylowicz M, Yu C, Pomes R. Systematic study of anharmonic features in a principle component analysis of gramicidin a. Biophys J. 2010;98:386–395. doi: 10.1016/j.bpj.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramanathan A, Agarwal PK. Computational identification of slow conformational fluctuations in proteins. J Phys Chem B. 2009;113:16669–16680. doi: 10.1021/jp9077213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kar G, Keskin O, Gursoy A, Nussinov R. Allostery and population shift in drug discovery. Curr Opin Pharmacol. 2010;10:715–722. doi: 10.1016/j.coph.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang S, Yang S, An BY, Wang SC, Yin YJ, Lu Y, Xu Y, Hao DY. Molecular dynamics analysis reveals structural insights into mechanism of nicotine n-demethylation catalyzed by tobacco cytochrome p450 mono-oxygenase. PLoS ONE. 2011;6:e23342. doi: 10.1371/journal.pone.0023342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pieniazek SN, Hingorani MM, Beveridge DL. Dynamical allosterism in the mechanism of action of DNA mismatch repair protein muts. Biophys J. 2011;101:1730–1739. doi: 10.1016/j.bpj.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kadirvelraj R, Sennett NC, Polizzi SJ, Weitzel S, Wood ZA. Role of packing defects in the evolution of allostery and induced fit in human udp-glucose dehydrogenase. Biochemistry. 2011;50:5780–5789. doi: 10.1021/bi2005637. [DOI] [PubMed] [Google Scholar]
- 59.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weikl TR, Von Deuster C. Selected-fit versus induced-fit protein binding: kinetic differences and mutational analysis. Proteins. 2009;75:104–110. doi: 10.1002/prot.22223. [DOI] [PubMed] [Google Scholar]
- 61.Andricioaei I, Karplus M. On the calculation of entropy from covariance matrices of the atomic fluctuations. J Chem Phys. 2001;115:6289–6292. [Google Scholar]
- 62.Liu Y, Jiange WW, Pratt J, Rockway T, Harris K, Vasavanonda S. Mechanistic study of hcv polymerase inhibitors at individual steps of the polymerization reaction. Biochemistry US. 2006;45:11312–11323. doi: 10.1021/bi060511j. [DOI] [PubMed] [Google Scholar]
- 63.Moustafa IM, Shen H, Morton B, Colina CM, Cameron CE. Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity. J Mol Biol. 2011;410:159–181. doi: 10.1016/j.jmb.2011.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buck KW. Comparison of the replication of positive-stranded RNA viruses of plants and animals. Adv Virus Res. 1996;47:159–251. doi: 10.1016/S0065-3527(08)60736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howe AYM, Cheng H, Johann S, Mullen S, Chunduru SK, Young DC, Bard J, Chopra R, Krishnamurthy G, Mansour T, O’connell J. Molecular mechanism of hepatitis c virus replicon variants with reduced susceptibility to a benzofuran inhibitor, hcv-796. Antimicrob Agents Ch. 2008;52:3327–3338. doi: 10.1128/AAC.00238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nyanguile O, Devogelaere B, Vijgen L, Van Den Broeck W, Pauwels F, Cummings MD, De Bondt HL, Vos AM, Berke JM, Lenz O, Vandercruyssen G, Vermeiren K, Mostmans W, Dehertogh P, Delouvroy F, Vendeville S, Vandyck K, Dockx K, Cleiren E, Raboisson P, Simmen KA, Fanning GC. 1a/1b subtype profiling of nonnucleoside polymerase inhibitors of hepatitis c virus. J Virol. 2010;84:2923–2934. doi: 10.1128/JVI.01980-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Labonte P, Axelrod V, Agarwal A, Aulabaugh A, Amin A, Mak P. Modulation of hepatitis c virus RNA-dependent RNA polymerase activity by structure-based site-directed mutagenesis. J Biol Chem. 2002;277:38838–38846. doi: 10.1074/jbc.M204657200. [DOI] [PubMed] [Google Scholar]
- 68.Beaulieu P. Recent advances in the development of ns5b polymerase inhibitors for the treatment of hepatitis c virus infection. Expert Opin Ther Pat. 2009;49:145–164. doi: 10.1517/13543770802672598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.